
Synthesis and biological evaluation of simplified pironetin analogues with modifications in the side chain and the lactone ring†
Steven
Roldán‡
a,
Adrià
Cardona§
a,
Laura
Conesa
a,
Juan
Murga
*a,
Eva
Falomir
a,
Miguel
Carda
*a and
J. Alberto
Marco
b
aDepart. de Q. Inorgánica y Orgánica, Univ. Jaume I, Castellón, E-12071 Castellón, Spain. E-mail: jmurga@uji.es
bDepart. de Q. Orgánica, Univ. de Valencia, E-46100 Burjassot, Valencia, Spain. E-mail: alberto.marco@uv.es
First published on 22nd November 2016
Abstract
The preparation of several new analogues of the natural dihydropyrone pironetin is described. They differ from the natural product mainly in the nature of the side chain and the lactone ring. Their cytotoxic activity has been measured. In addition, their interaction with tubulin, their ability to inhibit the secretion of the vascular endothelial growth factor (VEGF) and the expression of angiogenesis and telomerase-related genes have been determined. Unexpectedly, and unlike pironetin, the lactones studied in this work do not interact with tubulin. Two of the compounds have been found to downregulate the expression of the hTERT and VEGF genes. Furthermore, one of them causes an appreciably decrease in the secretion of the VEGF protein.
Introduction
It is widely known that cancer, one leading cause of death in developed countries, may be induced by a plethora of both external and internal factors, including genetic mutations. Accordingly, a number of types of therapeutic attack have been investigated.1 One of these involves the use of cytotoxic drugs, which exert their effect in many cases by means of inducing various mechanisms of cell death.2 As a matter of fact, many such drugs owe this property to interaction with the microtubule network. Microtubules are dynamic polymers that play a central role in a number of cellular processes, most particularly cell division, as they are key constituents of the mitotic spindle.3 Microtubules are constituted of a protein named tubulin, the functional form of which, and the most abundant component, is a heterodimer formed through non-covalent binding of two monomeric constituents, called α- and β-tubulin. For cell division to occur in a normal way, microtubules must be in a constant state of formation and disruption, a process named microtubule dynamic instability.4 Molecules which influence microtubule instability will also influence the cell division process, not only of normal cells but also of tumoral ones. Therefore, it is not surprising that tubulin-binding molecules (TBMs) constitute a very important class of anticancer agents.5TBMs are able to interfere with microtubule assembly and functions, either by causing disruption of the microtubules or else through their stabilization. Most of the hitherto described active drugs are natural products or derivatives thereof.6 Many drugs can already be found on the market and many other promising compounds are in clinical trials.
TBMs may be divided in two broad categories, those that bind to α-tubulin and those that bind to β-tubulin. The latter group is presently by far the most numerous and contains products which cause either disruption7 or stabilization8 of microtubules. The number of products that bind to α-tubulin is, however, very small,9 the naturally occurring 5,6-dihydro-α-pyrone pironetin (Fig. 1) being the first-reported example. Pironetin is a potent inhibitor of tubulin assembly and has been found to arrest cell cycle progression in the G2/M phase.10 This feature has motivated a number of groups to undertake total syntheses of this natural compound.11 It is worth mentioning here that 5,6-dihydro-α-pyrones constitute an ample group of natural products endowed with a broad variety of pharmacologically useful properties, most likely related to the presence of the Michael acceptor moiety in the pyrone ring.12
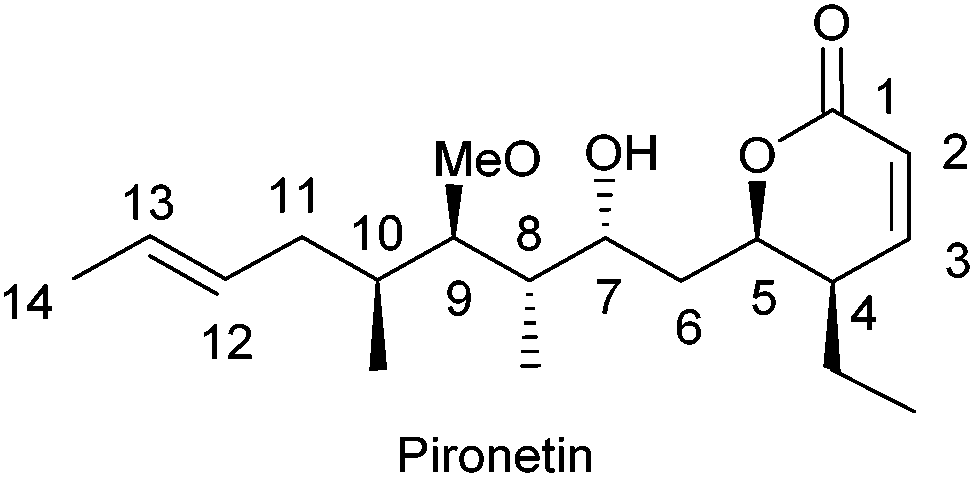 | ||
| Fig. 1 Structure of pironetin, a highly cytotoxic natural pyrone. | ||
Some structure–activity (SAR) studies on pironetin have been reported.10 These studies have shown that the presence of the conjugated double bond in the lactone ring and of the hydroxyl group at C-9, either free or methylated, are essential for the biological activity.9 The epoxidation of the C12![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C13 double bond has been shown to cause a decrease in the activity.
C13 double bond has been shown to cause a decrease in the activity.
As a member of the up to now small group of products that bind to α-tubulin, pironetin constitutes a pharmacologically interesting target. Thus, a key purpose of our research is the preparation of pironetin analogues that retain a substantial proportion of the biological activity of the natural metabolite while displaying a more simplified structure. In order to develop SAR studies based upon the pironetin framework, we designed several years ago13 a simplified model structure where all elements that had not yet proven to be essential for the biological activity were removed. The target structures I/II are schematically shown in Fig. 2. The elements that were maintained are the conjugated dihydropyrone ring and the side chain with the methoxy group at C-9. The hydroxyl group at C-7 was removed in some substrates (I) and retained in others (II), in order to see its influence on the activity. All alkyl pendants (methyl groups at C-8 and C-10, ethyl at C-4) and the isolated C12–C13 double bond were removed. The configurations of the two/three remaining stereocentres were then varied in a systematic way. Thus, all four possible stereoisomers with general constitution I, with no hydroxyl group at C-7, were prepared. Likewise, all eight stereoisomers exhibiting general structure II, with a hydroxyl group at C-7, were synthesized. Subsequently, the cytotoxic activity of these analogues and their interactions with tubulin were investigated.13
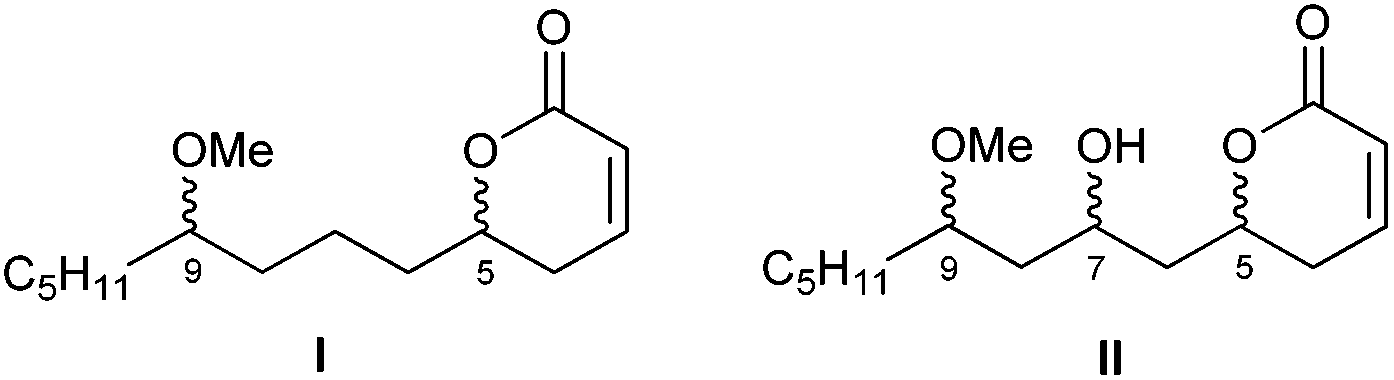 | ||
| Fig. 2 General structures of the first generation of simplified pironetin analogues (ref. 13). | ||
In continuation of this line of research, we concentrated our attention on the importance of the alkyl pendants in the pironetin molecule for the biological properties of the natural compound. In line with this reasoning, we prepared the six pironetin analogues III–VIII (Fig. 3). In all these compounds, the configurations at the oxygenated carbons C-5, C-7 and C-9 are as in natural pironetin. With respect to general structure II (Fig. 2), compounds III and IV contain an additional methyl residue at C-10 with either configuration, whereas in compounds V and VI, the extra methyl pendant is allocated at C-8. Finally, compounds VII and VIII display an extra alkyl residue (methyl or ethyl) at C-4, in both cases with the same configuration as in natural pironetin.14
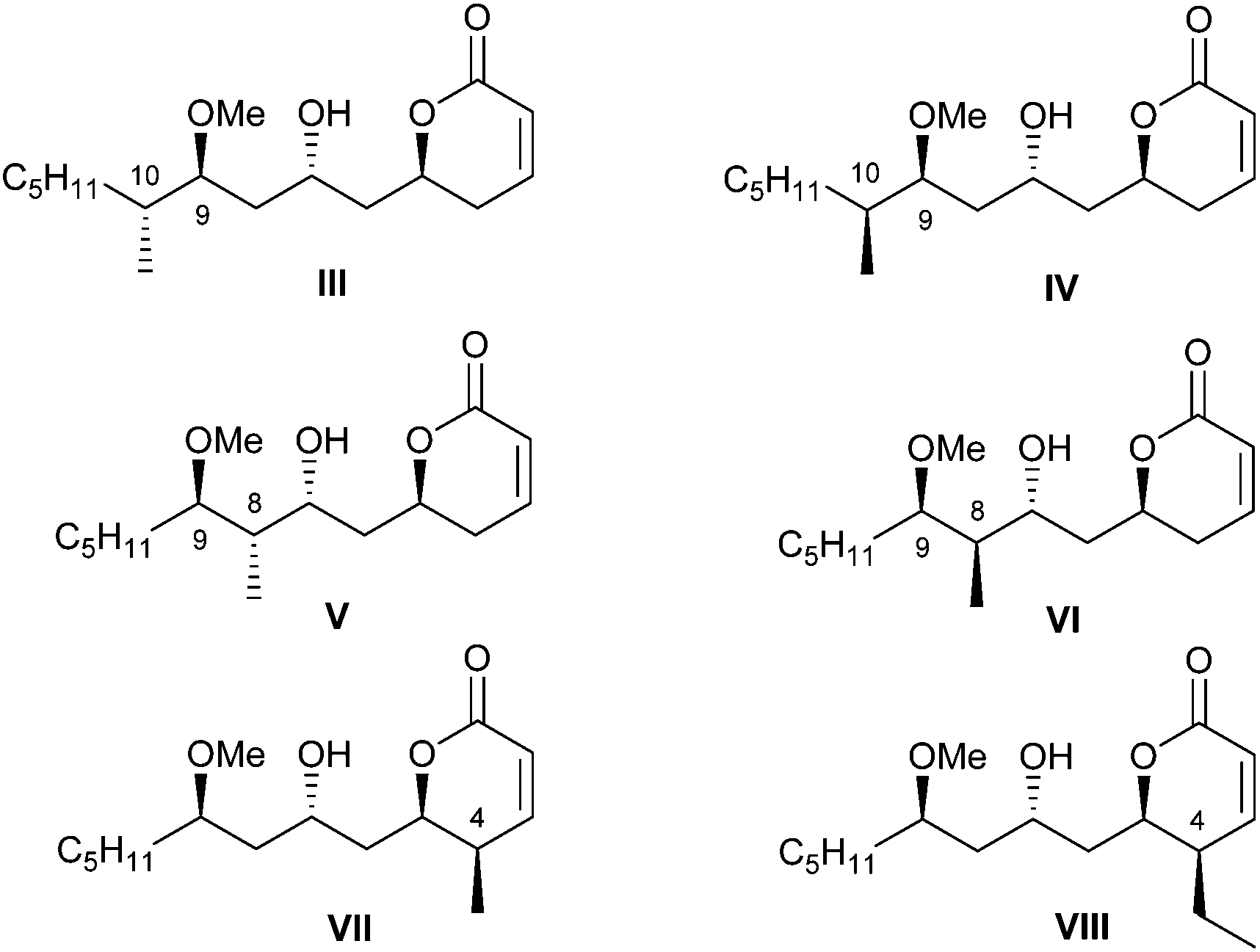 | ||
| Fig. 3 Structures III–VIII of the second series of pironetin analogues (ref. 13). | ||
The cytotoxic activities of pironetin analogues III–VIII were then investigated. Most compounds proved cytotoxic in the low micromolar range, therefore about two–three orders of magnitude less active than pironetin itself.14 These results suggest that all alkyl pendants are important for the full biological activity, this being most likely due to the fact that the alkyl groups restrict the conformational mobility of the molecule and reduce the number of available conformations.15,16 This in turn makes more probable that the molecule adopts a shape that fits better into the active site of α-tubulin.
In view of these results, we decided to prepare a new group of pironetin analogues with a higher degree of alkylation in the side chain but still retaining a simplified structure. Fig. 4 shows the eight compounds we have prepared and evaluated for their biological properties.
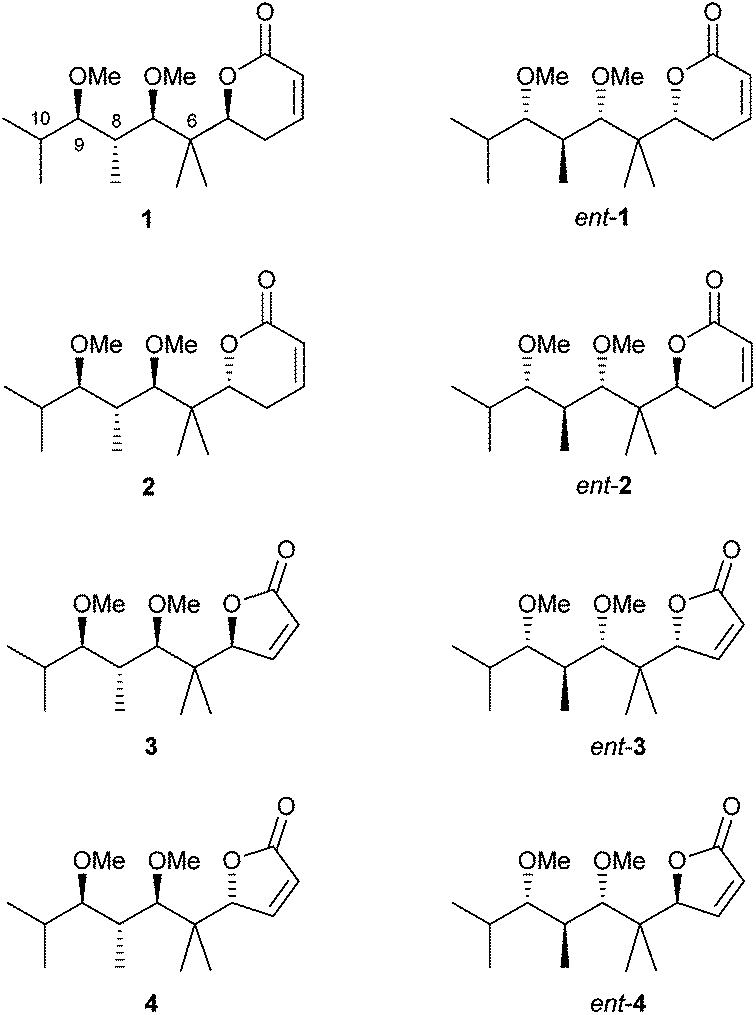 | ||
| Fig. 4 Structures of compounds of the third series of pironetin analogues (this work). | ||
In comparison to pironetin, pyrones 1, ent-1, 2 and ent-2 in Fig. 4 display a shorter carbon chain, two stereocentres less (C-4 and C-10 in pironetin numbering) and an additional gem-dimethyl moiety (at C-6 in pironetin numbering). Furthermore, and in order to investigate the importance of the lactone ring size, analogues 3, ent-3, 4 and ent-4 having a furanone system were also prepared.
In recent times, we have not limited our biological investigations on bioactive molecules to solely measurements of their cytotoxic activity, expressed as IC50 values. Indeed, while mechanisms of anticancer activity are often related to interference with microtubule assembly and functions, other mechanisms may also be operative. In most solid tumors, for example, angiogenesis is an important process for tumor growth and metastasis. Many different mediators are involved in this process, including the vascular endothelial growth factor (VEGF), which has been shown to play a critical role in pathological angiogenesis.17
Another relevant mechanism in cancer genesis is related to the role of the chromosomal telomers. Most cancer cells exhibit telomerase activity. The latter maintains the length of the telomeres, thus preserving genomic stability.18 Telomerase is a ribonucleoprotein composed of two main subunits which, in the case of human beings, are called human telomerase RNA (hTR) and human telomerase protein (hTERT). Many studies have demonstrated that interference in the expression of the hTERT gene can efficiently inhibit the growth and tumorigenicity of cancer cells, as the hTERT gene is a rate-limiting factor in telomerase synthesis and activity. Equally important is the c-Myc gene, which has been found to be amplified in various types of human cancers. The result of the expression of this gene, the c-Myc protein, is a transcriptional factor with an important role in cell proliferation, differentiation, invasion and adhesion of tumor cells.19 It is also involved in the activation of hTERT gene transcription.
Since on one hand tumoral cell secretion of VEGF is an important factor in metastasis and, on the other hand, telomerase is responsible for the inmortality of tumoral cells, the potential multiple ability20 of some compounds to perturb microtubule dynamics and, at the same time, to inhibit VEGF secretion by tumoral cells and the expression of the VEGF, hTERT and c-Myc genes was considered a goal worth pursuing. For that reason, we have also included the last types of biological activities in our investigation of the general pharmacological profile of our compounds.
Results and discussion
Synthesis of compounds 1–4 and their enantiomers
For our purposes, we aimed at performing a simple synthetic sequence in which stereochemical complexity is rapidly achieved through a convergent methodology. Thus, the synthesis of compounds 1–4 was carried out as depicted in Scheme 1. Creation of chirality was achieved by means of an adaptation of a published organocatalytic procedure.21 Thus, propionaldehyde and isobutyraldehyde were allowed to react in DMF in the presence of D-proline. This gave a crossed aldol product which was subjected in situ to Barbier-type, indium-mediated prenylation to yield diol 5 in a fair yield and high enantio- and diastereoselectivity.21 Methylation to 6 was followed by ozonolysis to yield an unstable intermediate aldehyde which, without isolation, was allowed to react with allylmagnesium bromide in THF. This sequence furnished a mixture of diastereoisomeric alcohols 7 and 8 (dr 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), which proved amenable to chromatographic separation. Both compounds were then esterified with acryloyl chloride, and the resulting acrylates, 9 and 10, were subjected to ruthenium-catalyzed ring-closing metathesis22 to afford the target dihydropyranones 1 and 2, respectively. Their enantiomers ent-1 and ent-2 were obtained by means of an identical synthetic sequence with the only difference of using L-proline as the organocatalyst (see Experimental).
:1), which proved amenable to chromatographic separation. Both compounds were then esterified with acryloyl chloride, and the resulting acrylates, 9 and 10, were subjected to ruthenium-catalyzed ring-closing metathesis22 to afford the target dihydropyranones 1 and 2, respectively. Their enantiomers ent-1 and ent-2 were obtained by means of an identical synthetic sequence with the only difference of using L-proline as the organocatalyst (see Experimental).
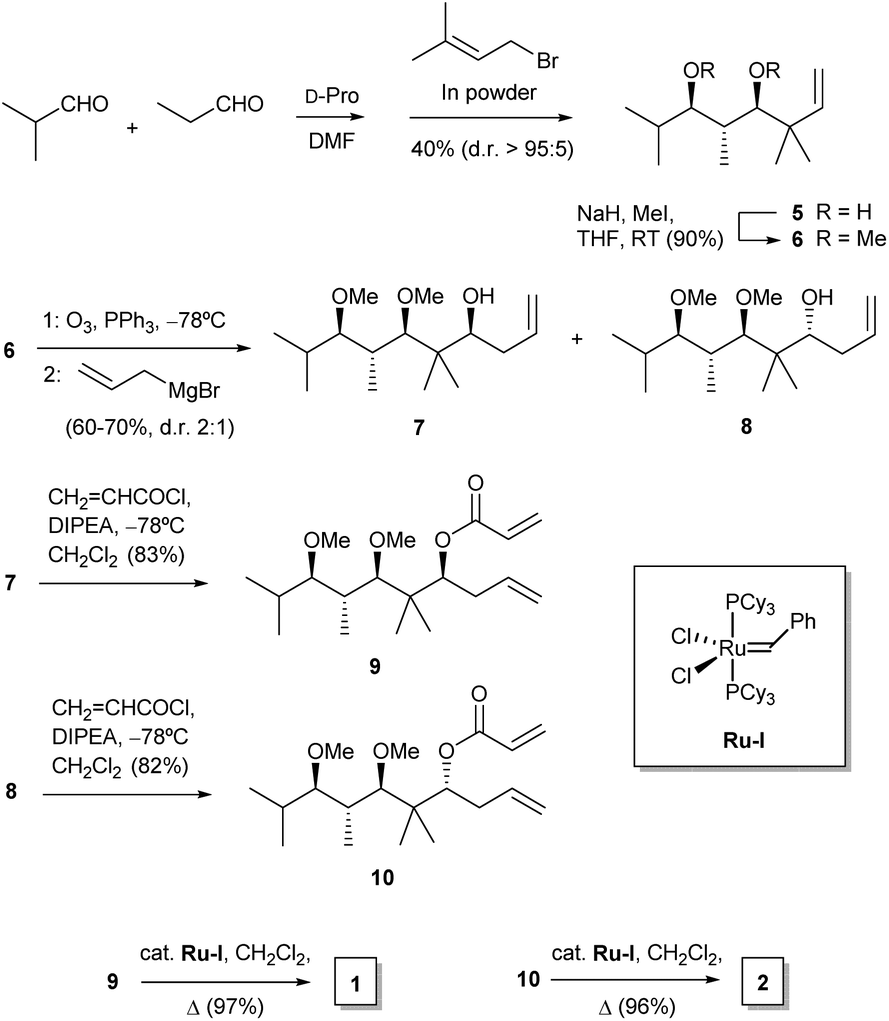 | ||
| Scheme 1 Synthesis of dihydropyranones 1 and 2. Abbreviations: D-pro, D-proline; DIPEA, ethyl N,N-diisopropylamine. | ||
Furanones 3 and 4 were prepared by means of a similar reaction sequence starting from olefin 6 (Scheme 2). Thus, the latter compound was subjected to ozonolysis followed by treatment of the crude unstable aldehyde with vinylmagnesium chloride to yield alcohols 11 and 12. These were then separated and subjected to esterification to acrylates 13 and 14, respectively. Ring-closing metathesis of the latter compounds required the use of a second generation Grubbs ruthenium catalyst22 in hot toluene as the solvent, and provided the target furanones 3 and 4. Their enantiomers ent-3 and ent-4 were obtained from ent-6 alongside the same reaction sequence (see Experimental).
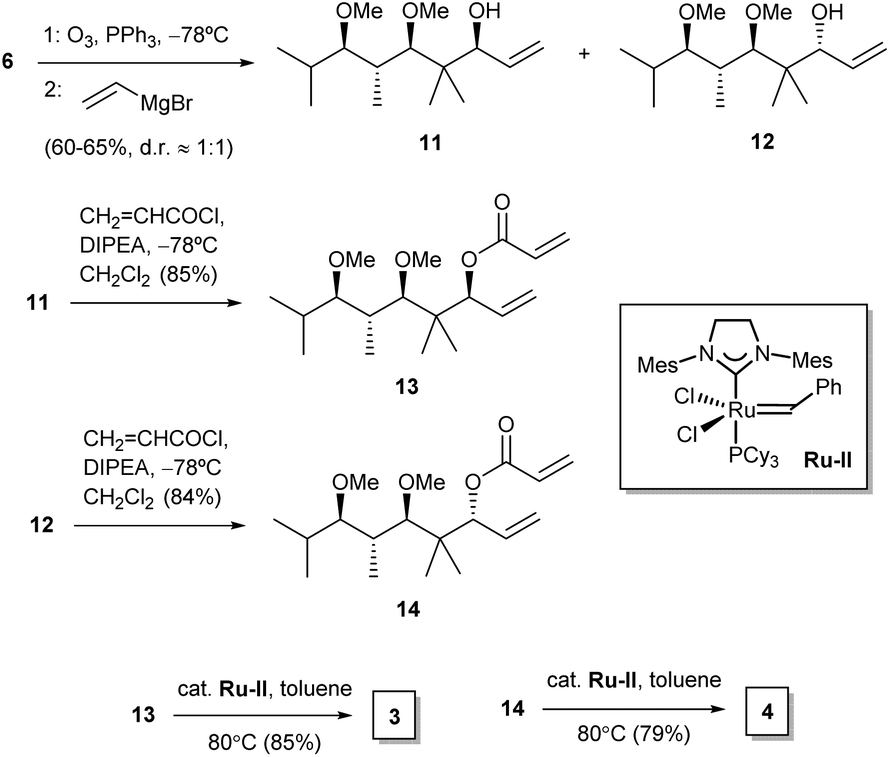 | ||
| Scheme 2 Synthesis of furanones 3 and 4. | ||
In order to check the influence of having hydroxy instead of methoxy groups in the side chain (pironetin and compounds in Fig. 3 have one methoxy group and one hydroxyl group), we also tried to prepare analogues of compounds 1–4 with two hydroxy groups. To that purpose, diol 5 was doubly silylated to 15, and the latter subjected to the same ozonolysis/allylation or alternatively ozonolysis/vinylation sequence to yield the diastereoisomeric pairs 16/18 and 24/26, respectively (Scheme 3). After esterification with acryloyl chloride to 17/19 and 25/27, ring-closing metathesis using in this case a Hoveyda–Grubbs-type ruthenium catalyst22 afforded 20/21 and 28/29, respectively. Unfortunately, all attempts at desilylation of the latter compounds under many different conditions to the desired lactones 22, 23, 30 and 31 only led to either no reaction, decomposition or formation of complex inseparable mixtures.23
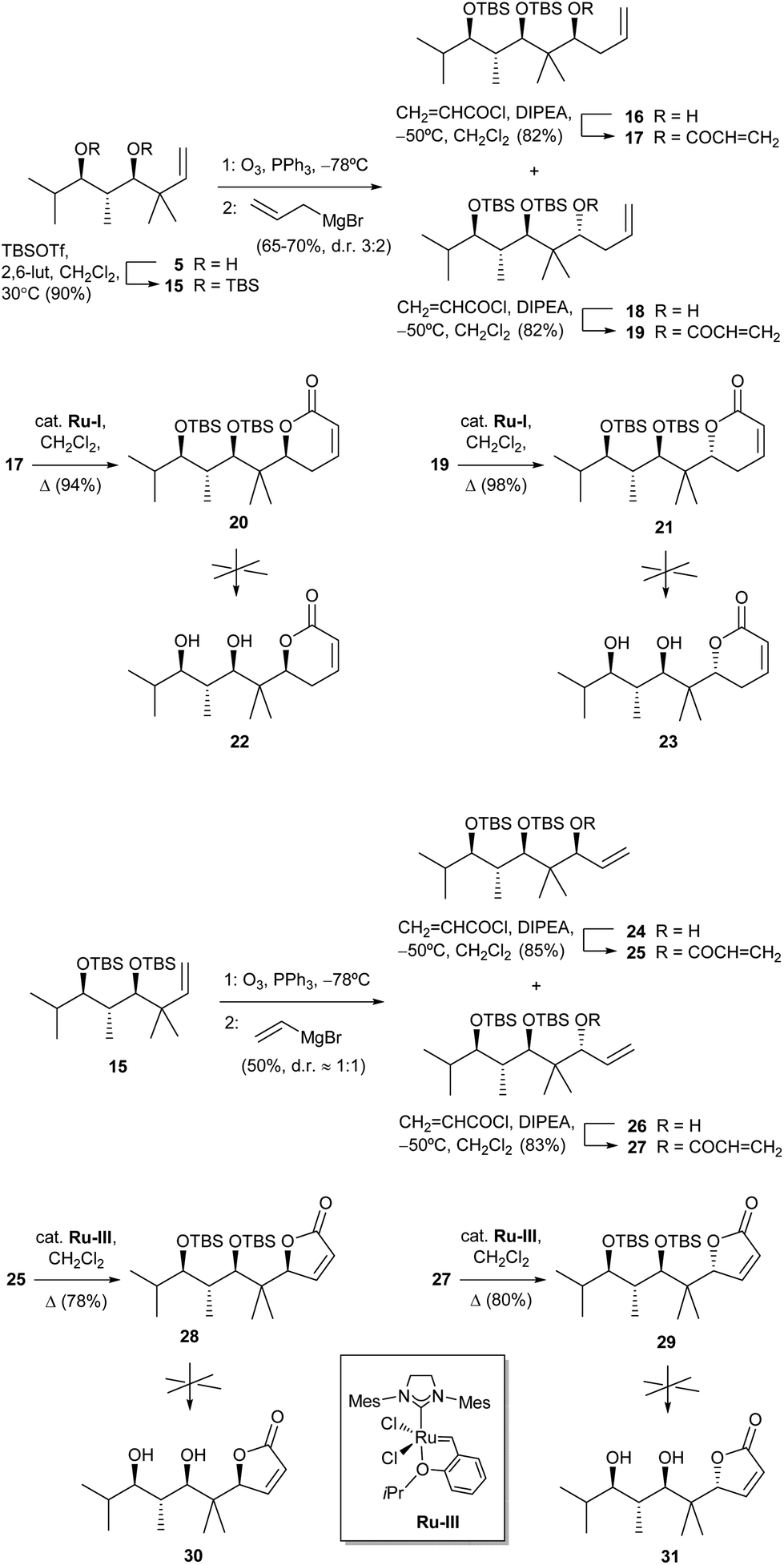 | ||
| Scheme 3 Attempts at the synthesis of dihydropyranones 22 and 23 and furanones 30 and 31. Abbreviation: 2,6-lut, 2,6-lutidine. | ||
Biological properties of pironetin analogues 1–4 and their enantiomers
| Comp. | HT-29 | HTC-116 | MCF-7 | HeLa | HL-60 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a IC50 values (μM) are the mean ± standard error of three independent experiments. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Pironetin | 0.0071 ± 0.0004 | 0.0083 ± 0.0005 | 0.0068 ± 0.0006 | 0.0092 ± 0.0008 | 0.0126 ± 0.0009 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 4.2 ± 0.4 | 30 ± 1 | 22.25 ± 0.18 | 38 ± 3 | 3.4 ± 0.7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 28.5 ± 0.5 | 62 ± 2 | 21 ± 2 | 60.5 ± 0.3 | 4.0 ± 0.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | >100 | >100 | >100 | 95 ± 5 | >100 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | >100 | >100 | >100 | >100 | >100 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ent-1 | 47 ± 2 | 62.0 ± 0.6 | 50 ± 2 | 54.2 ± 0.4 | 33 ± 7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ent-2 | 5.9 ± 0.8 | 36 ± 1 | 12.9 ± 0.9 | 53.8 ± 0.1 | 1.38 ± 0.15 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ent-3 | >100 | >100 | >100 | >100 | >100 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ent-4 | >100 | >100 | >100 | >100 | >100 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The first conclusion that can be drawn from the IC50 values is that furanones 3 and 4 and their respective enantiomers ent-3 and ent-4 show almost no cytotoxicity. Pyranones did prove cytotoxic in the low micromolar range, thus about two–three orders of magnitude less active than pironetin itself. Among pyranones the most active compounds are 1 and ent-2. These two compounds share a common structural feature, the configuration of the lactone stereocenter, which is the same as in pironetin.
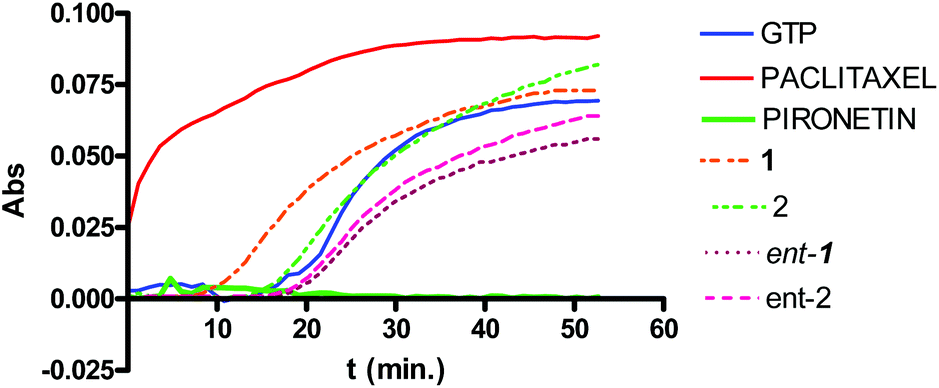 | ||
| Fig. 5 Effects of colchicine, pironetine and compounds 1, 2, ent-1 and ent-2, as well as paclitaxel and pironetin, on the in vitro microtubule assembly. The lines in the figure show the turbidimetric time course of polymerization of tubulin in the presence of GTP, and in the presence of 27.5 μM of each of the indicated compound. | ||
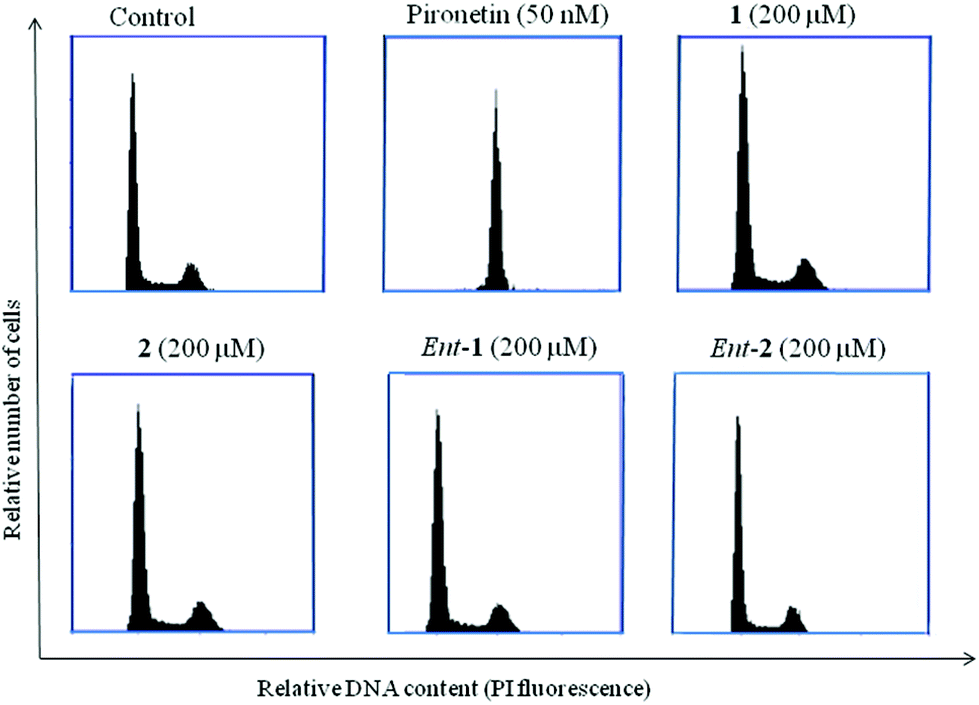 | ||
| Fig. 6 Cell cycle histograms of A549 lung carcinoma cells untreated and treated with pironetin and pironetin analogues 1, 2, ent-1 and ent-2. | ||
| Comp. | Concent. | hTERT (%) | c-Myc (%) | VEGF (%) | VEGF proteina (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a At least three measurements were performed in each case. Experiments were performed on HT-29 cells. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 5 μM | 49 ± 4 | 18 ± 1 | 45 ± 5 | 29 ± 4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 25 μM | 42 ± 7 | 25 ± 2 | 26 ± 1 | 36 ± 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ent-1 | 25 μM | 59 ± 8 | 36.5 ± 1.5 | 36 ± 2 | 63 ± 7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ent-2 | 5 μM | 39 ± 5 | 25 ± 4 | 19 ± 0.2 | 76 ± 4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In regards to the inhibition of the hTERT and c-Myc genes, compounds 1 and ent-2 are the most active ones, especially if one considers that the concentration of these two compounds is five times lower than that of compounds 2 and ent-1. Particularly appealing is the activity of compound 1 on the inhibition in the expression of the c-Myc gene, which is decreased to 18% of the control value. Regarding the VEGF gene expression, compound ent-2 shows the greatest inhibition (19% of the control value). However, this high decrease in gene expression is not accompanied by a similar decrease in VEGF protein secretion as compound ent-2 downregulates protein secretion to 76% of the control value. In this sense, the most active compound is pyranone 1, which downregulates VEGF protein secretion to 29% of the control value.
Summary
Pironetin analogues 1–4 and their enantiomers were synthesized with the aim of exploring the influence of the alkyl pendants as well as their stereochemistry and lactone ring size in their biological activity. Pyranones were shown to be cytotoxic at the micromolar level while furanones showed no cytotoxicity. Among the pyranones the most cytotoxic were 1 and ent-2, which have the same configuration at the lactone stereocenter as pironetin. The influence of pyranones in tubulin polymerization was also measured but, in contrast to pironetin, they seem to have little influence in the tubulin polymerization process. It thus seems that removal of the methyl pendants at the side chain and introduction of a gem-dimethylated pattern causes a strong decrease in the interaction of the compounds with tubulin. Contrary to what is expected for antimitotic compounds, pironetin analogues 1, 2, ent-1 and ent-2 have practically no effect on the number of G2/M cells. This suggests that, in spite of their structural similarity with pironetin, the analogues exert their cytotoxicity through a mechanism different to that of the natural compound.When we initiated our pironetin project several years ago,13 the exact nature of the interaction between the natural compound and tubulin was not yet known. Indeed, a 2004 publication10 postulated the occurrence of a Michael addition of the nitrogen atom of the Lys352 residue at the α-tubulin subunit to the C3 carbon of the α-pyrone moiety in pironetin, with the formation of a C–N covalent bond. This view was still present in our minds when we initiated the design and synthesis of the compounds described here. After our research was finished, however, two recent publications have appeared which show that what actually happens is a Michael addition of the sulfur atom of the Cys316 residue at the α-tubulin subunit.24 The detailed X-ray studies described in these publications have permitted a deeper insight into the bonding interactions that develop between tubulin and the pironetin molecule. For instance, these investigations have highlighted the importance of the ethyl group at C4 and the two methyl groups at C8 and C10 of, respectively the pyrome ring and the side chain of the pironetin molecule. These alkyl residues are inserted into specific hydrophobic pockets of the α-tubulin subunit and their removal or modification is accompanied by marked decreases of the cytotoxic activity. It is worth noting here that this is in a good concordance with the results described by us here and in a previous publication.14
As regards the inhibition of the c-Myc and VEGF genes, pyranones 1 and ent-2 proved to be the most active compounds with 1 showing the strongest inhibition of VEGF protein secretion.
Experimental
Chemical procedures
NMR spectra were recorded at 500 MHz (1H NMR) and 125 MHz (13C NMR) in CDCl3 solution at 25 °C, with the solvent signals as internal reference. 13C NMR signal multiplicities were determined with the APT pulse sequence. Mass spectra were run in the electrospray (ESMS) mode. IR data, which were measured as films on NaCl plates (oils) or as KBr pellets (solids), are given only when relevant functions (CO, OH) are present. Optical rotations were measured at 25 °C. Reactions which required an inert atmosphere were carried out under dry N2 with flame-dried glassware. Commercial reagents were used as received. THF and Et2O were freshly distilled from sodium–benzophenone ketyl. Dichloromethane was freshly distilled from CaH2. Toluene was freshly distilled from sodium wire. Tertiary amines were freshly distilled from KOH.
:5 to 80:20). This yielded diol 5 (801 mg, 40% based on propionaldehyde) as off-white crystals (from Et2O–CHCl3): mp 76–77 °C (from Et2O–CH2Cl2), [α]D +17.6 (c 1; CHCl3). Spectral data were consistent with those published17 (see ESI†).
The procedure described above represents the maximum scale at which we were able to obtain reasonable yields. Attempts at increasing the scale only led to a decrease in the yield.
The procedure was repeated under the same conditions with L-proline to yield ent-5: [α]D −18.1 (c 1; CHCl3). Physical and spectral data identical to those of 5.
The stereostructures of 5 and ent-5 have been secured by means of an X-ray diffraction analysis.25
:5). This yielded 6 (822 mg, 90%): oil, [α]D +1.8 (c 1; CHCl3); 1H NMR δ 5.99 (1H, dd, J = 17.5, 11 Hz), 4.98 (1H, dd, J = 17.5, 1.5 Hz), 4.94 (1H, dd, J = 11, 1.5 Hz), 3.44 (3H, s), 3.38 (3H, s), 3.06 (1H, dd, J = 6, 3.5 Hz), 2.88 (1H, d, J = 5 Hz), 1.98 (1H, m), 1.88 (1H, br m), 1.08 (3H, s), 1.07 (3H, s), 0.99 (3H, d, J = 7.5 Hz), 0.93 (3H, d, J = 7 Hz), 0.90 (3H, d, J = 7 Hz); 13C NMR δ 43.2 (C), 146.6, 91.6, 86.9, 38.8, 30.3 (CH), 110.8 (CH2), 61.4, 59.5, 25.7, 23.3, 22.0, 17.1, 16.4 (CH3); HR ESMS m/z 251.1991 (M + Na+), calcd for C14H28NaO2, 251.1987.
(ent-6): oil, [α]D −1.8 (c 1; CHCl3). Physical and spectral data identical to those of 6.
:1. After removal of the volatiles under reduced pressure, the crude oily aldehyde was then directly used as such in the next allylation step (for weight calculations, the yield of the ozonolysis step was assumed to be quantitative).
The oily material from above was dissolved under N2 in dry THF (15 mL) and cooled in an ice bath. After this, a 1 M solution of allylmagnesium bromide in THF (4 mL, 4 mmol) was added dropwise, and the mixture was allowed to reach room temperature, followed by stirring for 3 h (TLC monitoring). The reaction mixture was then poured onto saturated ammonium chloride and extracted several times with Et2O. The organic layers were then dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure. The resulting oil was the subjected to a slow and careful chromatography on silica gel (hexane–Et2O, 9:1) to yield 7 (367 mg, 45%) and 8 (182 g, 22%).
(7): oil, [α]D −23.3 (c 1; CHCl3); IR νmax (cm−1): 3400 (br, OH); 1H NMR δ 5.90 (1H, ddt, J = 17, 10.5, 7 Hz), 5.01 (1H, dm, J ∼ 17 Hz), 4.94 (1H, dm, J ∼ 10.5 Hz), 4.90 (1H, br d, J ∼ 3 Hz, OH), 3.68 (1H, dt, J = 10, 3 Hz), 3.45 (6H, s), 3.02 (1H, dd, J = 10, 1 Hz), 2.74 (1H, d, J = 2.5 Hz), 2.38 (1H, m), 2.14 (1H, m), 2.04 (1H, m), 1.81 (1H, m), 0.99 (3H, d, J = 7 Hz), 0.90 (3H, d, J = 7.5 Hz), 0.89 (3H, s), 0.87 (3H, s), 0.80 (3H, d, J = 7 Hz); 13C NMR δ 43.4 (C), 137.9, 97.1, 87.3, 73.0, 36.2, 30.3 (CH), 115.1, 35.9 (CH2), 62.0, 60.3, 25.5, 22.2, 21.1, 17.2, 14.5 (CH3); HR ESMS m/z 273.2429 (M + H+), calcd for C16H33O3, 273.2430.
(ent-7): oil, [α]D +21.5 (c 1; CHCl3). Physical and spectral data identical to those of 7.
(8): oil, [α]D +9.5 (c 1; CHCl3); IR νmax (cm−1): 3480 (br, OH); 1H NMR δ 5.94 (1H, ddt, J = 17, 10.5, 7 Hz), 5.08 (1H, dm, J ∼ 17 Hz), 5.04 (1H, dm, J ∼ 10.5 Hz), 3.70 (2H, m), 3.47 (3H, s), 3.41 (3H, s), 3.02 (2H, m), 2.22 (1H, m), 2.10 (2H, m), 1.90 (1H, m), 1.03 (3H, d, J = 7.5 Hz), 0.99 (3H, s), 0.98 (3H, d, J = 7 Hz), 0.90 (3H, d, J = 7 Hz), 0.88 (3H, s); 13C NMR δ 42.9 (C), 137.4, 95.1, 86.8, 75.7, 37.0, 30.5 (CH), 115.9, 36.7 (CH2), 61.9, 59.9, 22.5, 21.7, 21.0, 19.7, 16.0 (CH3); HR ESMS m/z 273.2430 (M + H+), calcd for C16H33O3, 273.2430.
(ent-8): oil, [α]D −11.8 (c 1; CHCl3). Physical and spectral data identical to those of 8.
:2) to afford 9 (81 mg, 83%) and 10 (80 mg, 82%), respectively.
(9): oil, [α]D +6.8 (c 1; CHCl3); IR νmax (cm−1): 1724 (CO); 1H NMR δ 6.38 (1H, dd, J = 17.5, 1.5 Hz), 6.10 (1H, dd, J = 17.5, 10.5 Hz), 5.79 (1H, dd, J = 10.5, 1.5 Hz), 5.75 (1H, m), 5.20 (1H, dd, J = 10, 2.5 Hz), 5.02 (1H, br dd, J = 17, 1.5 Hz), 4.97 (1H, br dd, J ∼ 10, 1.5 Hz), 3.43 (3H, s), 3.39 (3H, s), 3.05–3.00 (2H, m), 2.59 (1H, m), 2.24 (1H, m), 2.04 (1H, d quint, J = 7, 4 Hz), 1.90 (1H, d quint, J = 7, 2.5 Hz), 1.02 (3H, d, J = 7.5 Hz), 0.98 (3H, d, J = 7.5 Hz), 0.96 (3H, s), 0.95 (3H, s), 0.90 (3H, d, J = 7 Hz); 13C NMR δ 165.8, 43.6 (C), 135.6, 128.9, 90.2, 86.4, 77.7, 37.5, 30.5 (CH), 130.1, 116.7, 35.5 (CH2), 60.9, 59.7, 21.7, 20.8, 20.3, 18.8, 16.1 (CH3); HR ESMS m/z 349.2359 (M + Na+), calcd for C19H34NaO4, 349.2355.
(ent-9): oil, [α]D −9.1 (c 1; CHCl3). Physical and spectral data identical to those of 9.
(10): oil, [α]D −22.8 (c 1; CHCl3); IR νmax (cm−1): 1726 (CO); 1H NMR δ 6.38 (1H, dd, J = 17.5, 1.5 Hz), 6.11 (1H, dd, J = 17.5, 10.5 Hz), 5.80 (1H, dd, J = 10.5, 1.5 Hz), 5.76 (1H, m), 5.23 (1H, dd, J = 10, 3 Hz), 5.01 (1H, br dd, J = 17, 1.5 Hz), 4.97 (1H, br dd, J ∼ 10, 1.5 Hz), 3.39 (3H, s), 3.33 (3H, s), 3.02 (1H, dd, J = 8.5, 2 Hz), 2.94 (1H, d, J = 3.5 Hz), 2.45 (1H, m), 2.24 (1H, m), 1.98 (1H, d quint, J = 6.5, 3.5 Hz), 1.89 (1H, d quint, J = 7, 2.5 Hz), 1.02 (3H, d, J = 7 Hz), 0.97 (3H, d, J = 7 Hz), 0.96 (3H, s), 0.94 (3H, s), 0.88 (3H, d, J = 6.5 Hz); 13C NMR δ 165.8, 43.4 (C), 135.5, 129.1, 88.3, 86.2, 76.8, 37.6, 30.5 (CH), 130.1, 116.8, 35.1 (CH2), 60.7, 59.8, 21.7, 19.1, 19.0, 18.5, 15.6 (CH3); HR ESMS m/z 349.2360 (M + Na+), calcd for C19H34NaO4, 349.2355.
(ent-10): oil, [α]D +17.8 (c 1; CHCl3).
:1) furnished the desired metathesis products 1 (57 mg, 97%) and 2 (56 mg, 96%), respectively.
(1): oil, [α]D −78.2 (c 1.05; CHCl3); IR νmax (cm−1): 1725 (CO); 1H NMR δ 6.92 (1H, ddd, J = 9.5, 6.5, 2.5 Hz), 6.00 (1H, dd, J = 9.5, 2 Hz), 4.38 (1H, dd, J = 12.5, 3.5 Hz), 3.42 (3H, s), 3.39 (3H, s), 3.20 (1H, d, J = 3 Hz), 3.00 (1H, dd, J = 8, 3 Hz), 2.49 (1H, ddt, J = 18, 12.5, 2.5 Hz), 2.36 (1H, ddd, J = 18, 6.5, 3.5 Hz), 2.00–1.85 (2H, m), 1.02 (3H, d, J = 7 Hz), 0.97 (3H, d, J = 7 Hz), 0.91 (3H, s), 0.86 (3H, d, J = 7 Hz), 0.85 (3H, s); 13C NMR δ 164.9, 42.7 (C), 146.3, 121.1, 89.6, 86.3, 82.7, 37.3, 30.4 (CH), 25.4 (CH2), 61.1, 59.8, 21.7, 20.3, 19.8, 19.1, 15.7 (CH3); HR ESMS m/z 321.2040 (M + Na+), calcd for C17H30NaO4, 321.2042.
(ent-1): oil, [α]D +71.4 (c 1; CHCl3). Physical and spectral data identical to those of 1.
(2): off-white solid, mp 67–69 °C (from Et2O–CH2Cl2), [α]D −10.6 (c 1; CHCl3); IR νmax (cm−1): 1727 (CO); 1H NMR δ 6.93 (1H, ddd, J = 9.5, 6.5, 2 Hz), 6.00 (1H, dd, J = 9.5, 2 Hz), 4.58 (1H, dd, J = 13, 3.5 Hz), 3.43 (3H, s), 3.37 (4H overall, an OMe singlet overlapping an one-proton signal), 2.99 (1H, dd, J = 8, 2.5 Hz), 2.37 (1H, ddt, J = 18, 13, 2.5 Hz), 2.25 (1H, ddd, J = 18, 6.5, 3.5 Hz), 1.90–1.80 (2H, m), 1.02 (3H, d, J = 7 Hz), 0.97 (3H, d, J = 7 Hz), 0.91 (3H, s), 0.86 (3H, d, J = 7 Hz), 0.85 (3H, s); 13C NMR δ 164.8, 42.5 (C), 146.1, 121.1, 87.4, 86.2, 81.1, 37.2, 30.4 (CH), 24.4 (CH2), 61.1, 59.8, 21.7, 19.8, 18.2, 17.4, 15.5 (CH3); HR ESMS m/z 321.2040 (M + Na+), calcd for C17H30NaO4, 321.2042.
(ent-2): off-white solid, [α]D +6.1 (c 1; CHCl3). Physical and spectral data identical to those of 2.
The stereostructures of 2 and ent-2 have been secured by means of an X-ray diffraction analysis.25
:1. After removal of the volatiles under reduced pressure, the crude oily aldehyde was then directly used as such in the next allylation step (for weight calculations, the yield of the ozonolysis step was assumed to be quantitative).
The oily material from above was dissolved under N2 in dry THF (15 mL) and cooled in an ice bath. After this, a 1.6 M solution of vinylmagnesium chloride in THF (2.5 mL, 4 mmol) was added dropwise, and the mixture was allowed to reach room temperature, followed by stirring for 2 h (TLC monitoring). The reaction mixture was then poured onto saturated ammonium chloride and extracted several times with Et2O. The organic layers were then dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure. The resulting oil was the subjected to slow and careful chromatography on silica gel (hexane–Et2O, from 98:2 to 95:5) to yield 11 (255 mg, 33%) and 12 (240 mg, 31%).
(11): oil, [α]D −26.4 (c 1; CHCl3); IR νmax (cm−1): 3380 (br, OH); 1H NMR δ 5.85 (1H, ddd, J = 17, 10.5, 6.5 Hz), 5.22 (1H, br d, J ∼ 17 Hz), 5.20 (1H, br s, OH), 5.09 (1H, br d, J ∼ 10.5 Hz), 4.17 (1H, m), 3.47 (6H, s), 3.05 (1H, dd, J = 10, 1.5 Hz), 2.80 (1H, br d, J ∼ 3 Hz), 2.38 (1H, m), 1.83 (1H, m), 1.00 (3H, s), 0.91 (3H, d, J = 7.5 Hz), 0.88 (3H, s), 0.85 (3H, s), 0.81 (3H, d, J = 7 Hz); 13C NMR δ 43.1 (C), 138.0, 96.5, 87.2, 75.1, 36.2, 30.3 (CH), 115.3 (CH2), 61.9, 60.3, 25.6, 22.0, 21.1, 17.1, 14.5 (CH3); HR ESMS m/z 281.2094 (M + Na+), calcd for C15H30NaO3, 281.2093.
(ent-11): oil, [α]D +22.4 (c 1; CHCl3). Physical and spectral data identical to those of 11.
(12): oil, [α]D +17.9 (c 1; CHCl3); IR νmax (cm−1): 3450 (br, OH); 1H NMR δ 5.90 (1H, ddd, J = 17, 10.5, 6 Hz), 5.27 (1H, br d, J ∼ 17 Hz), 5.15 (1H, br d, J ∼ 10.5 Hz), 4.11 (1H, m), 3.95 (1H, br d, J ∼ 4 Hz, OH), 3.45 (3H, s), 3.41 (3H, s), 3.08 (1H, d, J = 4 Hz), 3.01 (1H, d, J = 8.5, 2 Hz), 2.06 (1H, m), 1.89 (1H, m), 1.03 (3H, d, J = 7 Hz), 0.98 (3H, d, J = 7 Hz), 0.96 (3H, s), 0.89 (3H, s), 0.88 (3H, d, J = 7 Hz); 13C NMR δ 42.6 (C), 138.0, 94.4, 86.7, 78.4, 37.1, 30.4 (CH), 115.9 (CH2), 61.4, 60.0, 22.3, 21.6, 21.2, 19.6, 15.6 (CH3); HR ESMS m/z 281.2095 (M + Na+), calcd for C15H30NaO3, 281.2093.
(ent-12): oil, [α]D −16.9 (c 1; CHCl3). Physical and spectral data identical to those of 12.
(13): oil, [α]D −20.7 (c 1; CHCl3); IR νmax (cm−1): 1728 (CO); 1H NMR δ 6.42 (1H, dd, J = 17.5, 1.5 Hz), 6.15 (1H, dd, J = 17.5, 10.5 Hz), 5.90 (1H, ddd, J = 17.5, 10.5, 7 Hz), 5.83 (1H, dd, J = 10.5, 1.5 Hz), 5.37 (1H, br d, J ∼ 7 Hz), 5.30–5.20 (2H, m), 3.41 (3H, s), 3.40 (3H, s), 3.07 (1H, d, J = 3.5 Hz), 3.03 (1H, dd, J = 8, 2 Hz), 2.00 (1H, d quint, J = 7, 3.5 Hz), 1.88 (1H, d quint, J = 7, 2.5 Hz), 1.03 (3H, d, J = 8 Hz), 1.02 (3H, s), 0.95 (3H, d, J = 8 Hz), 0.94 (3H, s), 0.88 (3H, d, J = 6.5 Hz); 13C NMR δ 165.3, 43.5 (C), 133.8, 128.9, 88.9, 86.2, 79.7, 37.5, 30.5 (CH), 130.4, 118.2 (CH2), 60.4, 59.8, 21.7, 19.8 (×2), 18.6, 15.7 (CH3); HR ESMS m/z 335.2201 (M + Na+), calcd for C18H32NaO4, 335.2198.
(ent-13): oil, [α]D +20.8 (c 1; CHCl3). Physical and spectral data identical to those of 13.
(14): oil, [α]D +16.7 (c 1; CHCl3); IR νmax (cm−1): 1729 (CO); 1H NMR δ 6.42 (1H, dd, J = 17.5, 1.5 Hz), 6.16 (1H, dd, J = 17.5, 10.5 Hz), 5.90–5.80 (2H, m), 5.42 (1H, br d, J ∼ 7 Hz), 5.30–5.20 (2H, m), 3.41 (3H, s), 3.31 (3H, s), 3.06 (1H, d, J = 3 Hz), 3.03 (1H, dd, J = 8, 2 Hz), 1.93 (1H, d quint, J = 7, 3.5 Hz), 1.85 (1H, d quint, J = 7, 2.5 Hz), 1.00 (3H, d, J = 7 Hz), 0.94 (3H, d, J = 7.5 Hz), 0.91 (3H, s), 0.89 (3H, s), 0.84 (3H, d, J = 7 Hz); 13C NMR δ 165.2, 42.8 (C), 133.4, 128.9, 87.7, 86.2, 78.6, 37.5, 30.4 (CH), 130.3, 118.3 (CH2), 60.5, 59.8, 21.6, 18.7, 18.6 (×2), 15.5 (CH3); HR ESMS m/z 335.2198 (M + Na+), calcd for C18H32NaO4, 335.2198.
(ent-14): oil, [α]D −17.2 (c 1; CHCl3). Physical and spectral data identical to those of 14.
:1) furnished the desired metathesis products 3 (48 mg, 85%) and 4 (45 mg, 79%), respectively.
(3): oil, [α]D −98.3 (c 1; CHCl3); IR νmax (cm−1): 1758 (CO); 1H NMR δ 7.61 (1H, dd, J = 6, 1.5 Hz), 6.01 (1H, dd, J = 6, 2 Hz), 5.00 (1H, dd, J = 2, 1.5 Hz), 3.39 (3H, s), 3.31 (3H, s), 2.99 (1H, dd, J = 9.5, 2.5 Hz), 2.94 (1H, d, J = 3.5 Hz), 2.00 (1H, m), 1.89 (1H, m), 1.07 (3H, s), 1.04 (3H, d, J = 7.5 Hz), 0.99 (3H, d, J = 7.5 Hz), 0.94 (3H, s), 0.86 (3H, d, J = 7 Hz); 13C NMR δ 173.6, 44.3 (C), 157.3, 119.4, 89.6, 88.8, 86.1, 37.3, 30.4 (CH), 60.3, 60.0, 21.4, 21.1, 20.8, 19.0, 15.3 (CH3); HR ESMS m/z 307.1882 (M + Na+), calcd for C16H28NaO4, 307.1885.
(ent-3): off-white solid, [α]D +91 (c 1; CHCl3). Physical and spectral data identical to those of 3.
(4): off-white solid, mp 57–59 °C (from Et2O–CH2Cl2), [α]D +47 (c 1; CHCl3); IR νmax (cm−1): 1759 (CO); 1H NMR δ 7.50 (1H, dd, J = 6, 1.5 Hz), 6.10 (1H, dd, J = 6, 2 Hz), 5.17 (1H, dd, J = 2, 1.5 Hz), 3.47 (3H, s), 3.37 (3H, s), 3.22 (1H, d, J = 4 Hz), 2.97 (1H, dd, J = 8, 2.5 Hz), 1.90–1.80 (2H, m), 1.00 (3H, d, J = 7 Hz), 0.97 (3H, s), 0.93 (3H, d, J = 7 Hz), 0.84 (3H, d, J = 7 Hz), 0.69 (3H, s); 13C NMR δ 173.2, 43.5 (C), 155.8, 122.2, 88.9, 87.8, 86.1, 37.2, 30.4 (CH), 60.8, 59.8, 21.5, 19.4, 17.9, 17.6, 15.3 (CH3); HR ESMS m/z 307.1888 (M + Na+), calcd for C16H28NaO4, 307.1885.
(ent-4): off-white solid, [α]D −47.4 (c 1; CHCl3). Physical and spectral data identical to those of 4.
The stereostructures of 4 and ent-4 have been secured by means of an X-ray diffraction analysis.25
(ent-15): oil, [α]D −6.6 (c 1; CHCl3). Physical and spectral data identical to those of 15.
:1. After removal of volatiles under reduced pressure, the crude oily aldehyde was then directly used as such in the next allylation step (for weight calculations, the yield of the ozonolysis step was assumed to be quantitative).
The oily material from above was dissolved under N2 in dry THF (15 mL) and cooled in an ice bath. After this, a 1 M solution of allylmagnesium bromide in THF (4 mL, 4 mmol) was added dropwise, and the mixture was allowed to reach room temperature, followed by stirring for 3 h (TLC monitoring). The reaction mixture was then poured onto saturated ammonium chloride and extracted several times with Et2O. The organic layers were then dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure. The resulting oil was the subjected to slow and careful chromatography on silica gel (hexane–Et2O, 99:1) to yield 16 (540 mg, 38%) and 18 (369 mg, 26%).
(16): oil, [α]D +5.5 (c 1; CHCl3); IR νmax (cm−1): 3480 (br, OH); 1H NMR δ 5.85 (1H, ddt, J = 17, 10.5, 7 Hz), 5.20–5.15 (2H, m), 3.92 (1H, d, J = 6 Hz), 3.87 (1H, d, J = 2 Hz), 3.65 (1H, d, J = 10 Hz), 2.44 (1H, m), 2.20 (1H, m), 2.15–2.05 (2H, m), 1.95 (1H, br s, OH), 1.07 (3H, d, J = 7 Hz), 0.99 (3H, s), 0.95 (9H, s), 0.94 (3H, s), 0.93 (9H, s), 0.91 (3H, d, J = 7 Hz), 0.89 (3H, d, J = 7 Hz), 0.16 (3H, s), 0.11 (3H, s), 0.10 (3H, s), 0.08 (3H, s); 13C NMR δ 43.5, 18.8, 18.6 (C), 136.2, 80.7, 77.1, 75.1, 43.4, 30.8 (CH), 118.2, 37.1 (CH2), 26.5 (×3), 26.3 (×3), 22.0, 21.1, 20.3, 16.8, 14.3, −2.2, −3.3, −4.3, −4.4 (CH3); HR ESMS m/z 495.3667 (M + Na+), calcd for C26H56NaO3Si2, 495.3666.
(ent-16): oil, [α]D −6.1 (c 1; CHCl3). Physical and spectral data identical to those of 16.
(18): oil, [α]D +23.6 (c 1; CHCl3); IR νmax (cm−1): 3470 (br, OH); 1H NMR δ 5.93 (1H, ddt, J = 17, 10.5, 7 Hz), 5.09 (1H, dm, J ∼ 17 Hz), 5.05 (1H, dm, J ∼ 10.5 Hz), 4.35 (1H, br s, OH), 4.00 (1H, dd, J = 10, 1.5 Hz), 3.92 (1H, d, J = 2 Hz), 3.86 (1H, dd, J = 7.5, 1 Hz), 2.18 (1H, m), 2.10–2.00 (3H, m), 1.14 (3H, d, J = 7 Hz), 1.04 (3H, s), 0.92 (24H, br s), 0.88 (3H, d, J = 7 Hz), 0.15 (3H, s), 0.13 (3H, s), 0.12 (3H, s), 0.09 (3H, s); 13C NMR δ 42.6, 18.8, 18.3 (C), 137.0, 83.7, 78.1, 76.1, 45.0, 31.5 (CH), 116.3, 36.9 (CH2), 26.4 (×3), 26.2 (×3), 23.1, 21.8, 20.5, 16.4, 13.7, −3.0, −3.4, −4.3 (×2) (CH3); HR ESMS m/z 495.3666 (M + Na+), calcd for C26H56NaO3Si2, 495.3666.
(ent-18): oil, [α]D −16.6 (c 1; CHCl3). Physical and spectral data identical to those of 18.
:2) to afford, respectively, 17 (130 mg, 82%) and 19 (130 mg, 82%), respectively.
(17): oil, [α]D +10.9 (c 1; CHCl3); IR νmax (cm−1): 1727 (CO); 1H NMR δ 6.37 (1H, dd, J = 17.3, 1.5 Hz), 6.10 (1H, dd, J = 17.3, 10.5 Hz), 5.80 (1H, dd, J = 10.5, 1.5 Hz), 5.74 (1H, dddd, J = 17, 10, 8, 6 Hz), 5.30 (1H, dd, J = 10.5, 2.5 Hz), 5.03 (1H, dd, J = 17, 1.5 Hz), 4.98 (1H, dd, J = 10, 1.5 Hz), 3.90 (1H, dd, J = 6.3, 1.5 Hz), 3.77 (1H, d, J = 2.2 Hz), 2.68 (1H, m), 2.30–2.15 (2H, m), 2.00 (1H, d quint, J = 7.5, 2.2 Hz), 1.07 (3H, d, J = 7.5 Hz), 1.04 (3H, s), 0.99 (3H, s), 0.96 (9H, s), 0.93 (9H, s), 0.92 (3H, d, J = 7 Hz), 0.90 (3H, d, J = 7 Hz), 0.15 (3H, s), 0.13 (6H, s), 0.10 (3H, s); 13C NMR δ 165.7, 43.9, 18.7, 18.6 (C), 135.2, 128.8, 79.7, 77.4, 77.1, 43.9, 31.1 (CH), 130.1, 117.0, 35.8 (CH2), 26.5 (×3), 26.2 (×3), 22.2, 21.4, 20.4, 17.2, 14.6, −2.5, −3.4, −4.2, −4.3 (CH3); HR ESMS m/z 549.3774 (M + Na+), calcd for C29H58NaO3Si2, 549.3771.
(ent-17): oil, [α]D −6.3 (c 1; CHCl3). Physical and spectral data identical to those of 17.
(19): oil, [α]D +9.1 (c 1; CHCl3); IR νmax (cm−1): 1728 (CO); 1H NMR δ 6.40 (1H, dd, J = 17.3, 1.5 Hz), 6.13 (1H, dd, J = 17.3, 10.5 Hz), 5.81 (1H, dd, J = 10.5, 1.5 Hz), 5.72 (1H, ddt, J = 17, 10, 7 Hz), 5.21 (1H, dd, J = 10, 2.5 Hz), 5.03 (1H, dd, J = 17, 1.5 Hz), 4.99 (1H, dd, J = 10, 1.5 Hz), 4.05 (1H, d, J = 5 Hz), 3.63 (1H, d, J = 1.5 Hz), 2.34 (1H, hept, J = 7 Hz), 2.30–2.20 (2H, m), 1.92 (1H, m), 1.13 (3H, d, J = 7.5 Hz), 1.03 (3H, s), 0.97 (3H, s), 0.96 (9H, s), 0.94 (9H, s), 0.89 (3H, d, J = 7 Hz), 0.88 (3H, d, J = 7 Hz), 0.17 (3H, s), 0.16 (3H, s), 0.15 (3H, s), 0.11 (3H, s); 13C NMR δ 165.7, 43.7, 19.0, 18.4 (C), 134.3, 128.8, 81.5, 76.9, 75.7, 42.9, 30.4 (CH), 130.3, 117.5, 34.8 (CH2), 26.6 (×3), 26.1 (×3), 22.9, 22.6, 20.1, 17.2, 14.8, −1.9, −3.7, −4.3, −4.7 (CH3); HR ESMS m/z 549.3774 (M + Na+), calcd for C29H58NaO3Si2, 549.3771.
(ent-19): oil, [α]D −8.1 (c 1; CHCl3). Physical and spectral data identical to those of 19.
:1) furnished the desired metathesis products 20 (94 mg, 94%) and 21 (98 mg, 98%), respectively.
(20): off-white solid, mp 134–135 °C (from Et2O–CH2Cl2), [α]D −30.5 (c 1; CHCl3); IR νmax (cm−1): 1731 (CO); 1H NMR δ 6.92 (1H, ddd, J = 9.5, 6.3, 2.2 Hz), 6.01 (1H, dd, J = 9.5, 2 Hz), 4.55 (1H, dd, J = 12.2, 4 Hz), 3.90 (1H, d, J = 2.5 Hz), 3.80 (1H, dd, J = 5.5, 1 Hz), 2.50–2.35 (2H, m), 2.20 (1H, br quint, J ∼ 7 Hz), 2.05 (1H, m), 1.12 (3H, s), 1.08 (3H, d, J = 7 Hz), 1.00 (3H, s), 0.93 (9H, s), 0.90 (9H, s), 0.89 (3H, d, J = 7 Hz), 0.88 (3H, d, J = 7 Hz), 0.14 (3H, s), 0.09 (3H, s), 0.04 (3H, s), 0.02 (3H, s); 13C NMR δ 164.4, 42.8, 18.8, 18.5 (C), 145.6, 121.3, 82.3, 79.3, 77.1, 43.4, 30.9 (CH), 26.4 (×3), 26.1 (×3), 25.5, 22.1, 21.8, 19.8, 17.2, 14.4, −2.4, −3.5, −4.3, −4.4 (CH3); HR ESMS m/z 521.3451 (M + Na+), calcd for C27H54NaO4Si2, 521.3458.
(ent-20): oil, [α]D +31.1 (c 1; CHCl3). Physical and spectral data identical to those of 20.
The stereostructures of 20 and ent-20 have been secured by means of an X-ray diffraction analysis.25
(21): oil, [α]D +16.8 (c 1; CHCl3); IR νmax (cm−1): 1737 (CO); 1H NMR δ 6.89 (1H, ddd, J = 9.5, 6.5, 2 Hz), 5.98 (1H, dd, J = 9.5, 2 Hz), 4.45 (1H, dd, J = 13, 3.5 Hz), 4.09 (1H, d, J = 2.5 Hz), 3.88 (1H, dd, J = 5.5, 1 Hz), 2.33 (1H, ddt, J = 18, 13, 2.5 Hz), 2.25–2.15 (2H, m), 1.93 (1H, m), 1.08 (3H, d, J = 7.5 Hz), 1.02 (3H, s), 0.92 (3H, s), 0.89 (9H, s), 0.86 (9H, s, overlapping two methyl doublets), 0.12 (3H, s), 0.05 (3H, s), 0.03 (3H, s), 0.005t (3H, s); 13C NMR δ 164.4, 42.7, 18.8, 18.4 (C), 145.6, 121.2, 80.6, 76.8, 76.3, 43.5, 30.6 (CH), 26.4 (×3), 26.1 (×3), 24.0, 22.2, 19.7, 18.6, 17.0, 14.4, −2.3, −3.6, −4.5, −4.8 (CH3); HR ESMS m/z 521.3456 (M + Na+), calcd for C27H54NaO4Si2, 521.3458.
(ent-21): oil, [α]D −18.2 (c 1; CHCl3). Physical and spectral data identical to those of 21.
:1) afforded 24 (26%) and 26 (24%).
(24): oil, [α]D −15 (c 1; CHCl3); IR νmax (cm−1): 3460 (br, OH); 1H NMR δ 5.95 (1H, ddd, J = 17, 10, 6.5 Hz), 5.26 (1H, br dt, J ∼ 17, 1.5 Hz), 5.20 (1H, br dt, J ∼ 10, 1.5 Hz), 4.14 (1H, dt, J = 6.5, 1.5 Hz), 4.01 (1H, d, J = 2.5 Hz), 3.89 (1H, dd, J = 10, 1.5 Hz), 2.50 (1H, br s, OH), 2.10 (2H, m), 1.05 (3H, d, J = 7 Hz), 0.99 (3H, s), 0.94 (9H, s), 0.93 (9H, s, overlapping a methyl doublet), 0.90 (3H, s), 0.89 (3H, d, J = 7 Hz), 0.17 (3H, s), 0.14 (6H, s), 0.09 (3H, s); 13C NMR δ 43.4, 18.7, 18.6 (C), 138.0, 80.3, 79.6, 77.8, 44.7, 31.3 (CH), 116.7 (CH2), 26.3 (×6), 21.1, 21.0, 19.9, 16.6, 13.8, −2.5, −3.0, −4.3 (×2) (CH3); HR ESMS m/z 481.3513 (M + Na+), calcd for C25H54NaO3Si2, 481.3509.
(ent-24): oil, [α]D +19.3 (c 1; CHCl3). Physical and spectral data identical to those of 24.
(26): oil, [α]D +8.9 (c 1; CHCl3); IR νmax (cm−1): 3450 (br, OH); 1H NMR δ 5.84 (1H, ddd, J = 17, 10.5, 6.5 Hz), 5.26 (1H, br ddd, J ∼ 17, 2, 1.5 Hz), 5.15 (1H, br ddd, J ∼ 10, 2, 1.5 Hz), 4.70 (1H, br s, OH), 4.46 (1H, d, J = 6.5 Hz), 4.01 (1H, d, J = 2.2 Hz), 3.87 (1H, dd, J = 8.2, 1.5 Hz), 2.08 (1H, d quint, J = 8, 2 Hz), 2.02 (1H, d quint, J = 7, 1.5 Hz), 1.16 (3H, d, J = 7.5 Hz), 1.04 (3H, s), 0.95 (3H, d, J = 7 Hz), 0.94 (9H, s), 0.93 (9H, s), 0.91 (3H, s), 0.90 (3H, d, J = 7 Hz), 0.16 (3H, s), 0.15 (3H, s), 0.14 (3H, s), 0.10 (3H, s); 13C NMR δ 42.3, 18.8, 18.3 (C), 137.9, 83.1, 78.5, 78.2, 45.3, 31.6 (CH), 116.6 (CH2), 26.4 (×3), 26.1 (×3), 23.2, 22.2, 20.2, 16.3, 13.3, −2.8, −3.4, −4.3, −4.4 (CH3); HR ESMS m/z 481.3506 (M + Na+), calcd for C25H54NaO3Si2, 481.3509.
(ent-26): oil, [α]D −8.1 (c 1; CHCl3). Physical and spectral data identical to those of 26.
:2) furnished respectively, 25 (85%) and 27 (83%).
(25): oil, [α]D −14.2 (c 1; CHCl3); IR νmax (cm−1): 1732 (CO); 1H NMR δ 6.40 (1H, dd, J = 17.3, 1.5 Hz), 6.14 (1H, dd, J = 17.3, 10.5 Hz), 5.92 (1H, ddd, J = 17.3, 10.5, 6.2 Hz), 5.82 (1H, dd, J = 10.5, 1.5 Hz), 5.43 (1H, d, J = 6.2 Hz), 5.30–5.20 (2H, m), 3.93 (1H, d, J = 5.5 Hz), 3.78 (1H, d, J = 2 Hz), 2.25 (1H, hept, J = 7 Hz), 1.93 (1H, m), 1.06 (3H, d, J = 7 Hz), 1.05 (3H, s), 1.00 (3H, s), 0.96 (9H, s), 0.92 (9H, s), 0.91 (3H, d, J = 7 Hz), 0.89 (3H, d, J = 7 Hz), 0.15 (3H, s), 0.13 (3H, s), 0.12 (3H, s), 0.10 (3H, s); 13C NMR δ 164.9, 43.3, 18.9, 18.5 (C), 133.7, 128.7, 79.7, 78.9, 76.7, 43.4, 30.9 (CH), 130.3, 118.2 (CH2), 26.5 (×3), 26.2 (×3), 22.0 (×2), 20.1, 17.3, 14.7, −2.2, −3.6, −4.2, −4.4 (CH3); HR ESMS m/z 535.3618 (M + Na+), calcd for C28H56NaO4Si2, 535.3615.
(ent-25): oil, [α]D +13.3 (c 1; CHCl3). Physical and spectral data identical to those of 25.
(27): oil, [α]D +26.5 (c 1; CHCl3); IR νmax (cm−1): 1731 (CO); 1H NMR δ 6.38 (1H, dd, J = 17.3, 1.5 Hz), 6.15 (1H, dd, J = 17.3, 10.5 Hz), 5.83 (1H, dd, J = 10.5, 1.5 Hz), 5.77 (1H, ddd, J = 17.3, 10.5, 6.2 Hz), 5.37 (1H, d, J = 6.2 Hz), 5.30–5.20 (2H, m), 3.97 (1H, d, J = 6 Hz), 3.70 (1H, d, J = 2 Hz), 2.28 (1H, hept, J = 7 Hz), 2.00 (1H, m), 1.09 (3H, d, J = 7 Hz), 1.04 (3H, s), 0.99 (3H, s), 0.96 (9H, s), 0.93 (9H, s), 0.90 (3H, d, J = 7 Hz), 0.89 (3H, d, J = 7 Hz), 0.13 (6H, s), 0.11 (3H, s), 0.06 (3H, s); 13C NMR δ 165.3, 43.3, 19.0, 18.5 (C), 132.9, 128.9, 79.4, 78.7, 76.2, 43.5, 30.5 (CH), 130.4, 119.1 (CH2), 26.6 (×3), 26.1 (×3), 22.6, 21.6, 19.4, 17.2, 14.5, −2.2, −3.5, −4.5, −4.8 (CH3); HR ESMS m/z 535.3613 (M + Na+), calcd for C28H56NaO4Si2, 535.3615.
(ent-27): oil, [α]D −28.4 (c 1; CHCl3). Physical and spectral data identical to those of 27.
The stereostructure of ent-27 has been secured by means of an X-ray diffraction analysis.25
:1) furnished the desired metathesis products 28 (75 mg, 78%) and 29 (77 mg, 80%), respectively.
(28): oil, [α]D −42 (c 1; CHCl3); IR νmax (cm−1): 1763 (CO); 1H NMR δ 7.62 (1H, dd, J = 6, 1.5 Hz), 6.12 (1H, dd, J = 6, 2 Hz), 5.25 (1H, dd, J = 2, 1.5 Hz), 3.95 (1H, d, J = 2.5 Hz), 3.80 (1H, dd, J = 7.5, 2 Hz), 2.08 (2H, m), 1.08 (3H, d, J = 7.5 Hz), 1.05 (3H, s), 0.95 (3H, d, J = 7 Hz), 0.93 (9H, s), 0.92 (9H, s), 0.94 (3H, s), 0.90 (3H, d, J = 7 Hz), 0.15 (3H, s), 0.11 (3H, s), 0.10 (3H, s), 0.07 (3H, s); 13C NMR δ 173.1, 44.7, 18.7, 18.5 (C), 155.8, 121.8, 87.7, 78.3, 77.8, 45.3, 31.5 (CH), 26.3 (×3), 26.2 (×3), 22.4, 20.3 (×2), 16.8, 13.8, −2.8, −3.1, −4.3, −4.4 (CH3); HR ESMS m/z 507.3296 (M + Na+), calcd for C26H52NaO4Si2, 507.3302.
(ent-28): oil, [α]D +44.5 (c 1; CHCl3). Physical and spectral data identical to those of 28.
(29): oil, [α]D +34.5 (c 1; CHCl3); IR νmax (cm−1): 1763 (CO); 1H NMR δ 7.46 (1H, dd, J = 6, 1.5 Hz), 6.15 (1H, dd, J = 6, 2.2 Hz), 5.10 (1H, dd, J = 2.2, 1.5 Hz), 4.14 (1H, d, J = 2.5 Hz), 3.80 (1H, dd, J = 7, 1.5 Hz), 2.13 (1H, d quint, J = 7, 1.5 Hz), 1.98 (1H, d quint, J = 7.5, 2.5 Hz), 1.09 (3H, s), 1.07 (3H, d, J = 7.5 Hz), 0.92 (3H, d, J = 7 Hz), 0.91 (9H, s), 0.90 (9H, s), 0.89 (3H, d, J = 7 Hz), 0.82 (3H, s), 0.17 (3H, s), 0.13 (3H, s), 0.12 (3H, s), 0.05 (3H, s); 13C NMR δ 173.0, 44.4, 18.8, 18.7 (C), 155.0, 123.1, 87.8, 77.5, 77.0, 45.0, 31.5 (CH), 26.5 (×3), 26.4 (×3), 21.2, 20.2, 18.1, 17.2, 14.1, −2.6, −3.1, −4.1, −4.3 (CH3); HR ESMS m/z 507.3304 (M + Na+), calcd for C26H52NaO4Si2, 507.3302.
(ent-29): oil, [α]D −36.6 (c 1; CHCl3). Physical and spectral data identical to those of 29.
Biological procedures
Cell culture
Cell culture media were purchased from Gibco (Grand Island, NY, USA). Fetal bovine serum (FBS) was a product of Harlan-Seralab (Belton, UK). Supplements and other chemicals not listed in this section were obtained from Sigma Chemicals Co. (St Louis, MO, USA). Plastics for cell culture were supplied by Thermo Scientific™ BioLite. All tested compounds were dissolved in DMSO at a concentration of 10 μg mL−1 and stored at −20 °C until use.Cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) containing glucose (1 g L−1), glutamine (2 mM), penicillin (50 U mL−1), streptomycin (50 μg mL−1) and amphotericin B (1.25 μg mL−1), supplemented with 10% FBS.
Cytotoxicity assays
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma Chemical Co., St Louis, MO) dye reduction assay in 96-well microplates was used, as previously described.26 Some 5 × 103 cells of HT-29, HTC-116, MCF-7 and HL-60 and 2.5 × 103 cells of HEK-293 and Hela cells in a total volume of 100 μL of their respective growth media were incubated with serial dilutions of the tested compounds. After 2 days of incubation (37 °C, 5% CO2 in a humid atmosphere), 10 μl of MTT (5 mg ml−1 in PBS) were added to each well and the plate was incubated for a further 4 h (37 °C). The resulting formazan was dissolved in 150 μL of 0.04 N HCl/2-propanol and read at 550 nm. All determinations were carried out in triplicate.Tubulin polymerization
Purified tubulin was used for these measurements. Tubulin polymerization was carried out in a 96 well plate. In each well 50 μL of a solution of 25 μM of tubulin in GAB buffer was added to 50 μL of 27.5 μM solution of the corresponding compounds in GAB buffer (20 mM sodium phosphate, 10 mM MgCl2, 1 mM EGTA, 30% glycerol) and 0.1 mM GTP at pH = 6.5. Then, the plate was incubated at 37 °C in Multiskan® and absorbance at 340 nM was registered every 30 seconds during 2 hours.Cell cycle
Progression of the cell cycle was analysed by DNA determination by means of flow cytometry with propidium iodide. A549 cells were fixed, treated with RNase and stained with propidium iodide following instructions of BD Cycletest™ DNA Kit. Analysis was performed with a BD Accuri™ C6 flow cytometer.ELISA analysis
HT-29 cells at 70–80% confluence were collected and 1.5 × 105 cells were placed in a six well plate in 1.5 mL of medium. After 24 h, cells were incubated with the corresponding compounds for 72 h. Culture supernatants were collected and VEGF secreted by HT-29 cells was determined using Invitrogen Human Vascular Endothelial Growth Factor ELISA Kit according to the manufacturer's instructions.RT-qPCR analysis
HT-29 cells at 70–80% confluence were collected and 1.5 × 105 cells were placed in a six well plate in 1.5 mL of medium. After 24 h, cells were incubated with the corresponding compounds for 72 h. Cells were collected and the total cellular RNA from HT-29 cells was isolated using an Ambion RNA extraction kit according to the manufacturer's instructions. The cDNA was synthesized by MMLV-RT with 1–21 μg of extracted RNA and oligo(dT)15 according to the manufacturer's instructions.Genes were amplified by use of a thermal cycler and StepOnePlus™ Taqman® probes. TaqMan® Gene Expression Master Mix Fast containing the appropriate buffer for the amplification conditions, dNTPs, thermostable DNA polymerase enzyme and a passive reference probe were used. To amplify each of the genes the predesigned primers were used and sold by Life Technologies TaqMan® Gene Expression Assays, Hs99999903-m1 (β-actin), Hs00900055-m1 (VEGF), Hs00972646-m1 (hTERT) and Hs00153408-m1 (c-Myc).
Acknowledgements
Financial support has been granted to M. C. by the Spanish Government (Ministerio de Economía y Competitividad of Spain, project CTQ2014-52949-P), by the Consellería d'Empresa, Universitat i Ciencia de la Generalitat Valenciana (project PROMETEO/2013/027) and by the University Jaume I (project P1·1B2015-75). The authors further thank Prof. P. M. Pihko for fruitful discussions.References
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- F. Torres-Andón and B. Fadeel, Acc. Chem. Res., 2013, 46, 733–742 CrossRef PubMed.
- The Role of Microtubules in Cell Biology, Neurobiology and Oncology, ed. T. Fojo, Humana Press, Totowa, NJ, 2008 Search PubMed.
- G. M. Alushin, G. C. Lander, E. H. Kellogg, R. Zhang, D. Baker and E. Nogales, Cell, 2014, 157, 1117–1129 CrossRef CAS PubMed.
- (a) T. Beckers and S. Mahboobi, Drugs Future, 2003, 28, 767–785 CrossRef CAS; (b) J. A. Hadfield, S. Ducki, N. Hirst and A. T. McGown, Prog. Cell Cycle Res., 2003, 5, 309–325 Search PubMed; (c) M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS PubMed; (d) S.-H. Chen and J. Hong, Drugs Future, 2006, 31, 123–150 CrossRef CAS; (e) E. Pasquier and M. Kavallaris, IUBMB Life, 2008, 60, 165–170 CrossRef CAS PubMed; (f) P. Singh, K. Rathinasamy, R. Mohan and D. Panda, IUBMB Life, 2008, 60, 368–375 CrossRef CAS PubMed; (g) P. G. Morris and M. N. Fornier, Clin. Cancer Res., 2008, 14, 7167–7172 CrossRef CAS PubMed; (h) E. A. Perez, Mol. Cancer Ther., 2009, 8, 2086–2095 CrossRef CAS PubMed; (i) S. M. Chen, L.-H. Meng and J. Ding, Expert Opin. Invest. Drugs, 2010, 19, 329–343 CrossRef CAS PubMed; (j) D. Calligaris, P. Verdier-Pinard, F. Devred, C. Villard, D. Braguer and D. Lafitte, Cell. Mol. Life Sci., 2010, 67, 1089–1104 CrossRef CAS PubMed.
- (a) K.-H. Altmann and J. Gertsch, Nat. Prod. Rep., 2007, 24, 327–357 RSC; (b) D. G. I. Kingston, J. Nat. Prod., 2009, 72, 507–515 CrossRef CAS PubMed.
- J. Chen, T. Liu, X. Dong and Y. Hu, Mini-Rev. Med. Chem., 2009, 9, 1174–1190 CrossRef CAS PubMed.
- Y. Fu, S. Li, Y. Zu, G. Yang, Z. Yang, M. Luo, S. Jiang, M. Wink and T. Efferth, Curr. Med. Chem., 2009, 16, 3966–3985 CrossRef CAS PubMed.
- F. Sarabia, M. García-Castro and A. Sánchez-Ruiz, Curr. Bioact. Compd., 2006, 2, 269–299 CrossRef CAS.
- T. Usui, H. Watanabe, H. Nakayama, Y. Tada, N. Kanoh, M. Kondoh, T. Asao, K. Takio, H. Watanabe, K. Nishikawa, T. Kitahara and H. Osada, Chem. Biol., 2004, 11, 799–806 CrossRef CAS PubMed.
- (a) K. Yasui, Y. Tamura, K. Nakatani, K. Kawada and M. Ohtani, J. Org. Chem., 1995, 60, 7567–7574 CrossRef CAS; (b) M. K. Gurjar, J. T. Henri Jr., D. S. Bose and A. V. R. Rao, Tetrahedron Lett., 1996, 37, 6615–6618 CrossRef CAS; (c) N. Chida, M. Yoshinaga, T. Tobe and S. Ogawa, Chem. Commun., 1997, 1043–1044 RSC; (d) H. Watanabe, H. Watanabe, M. Bando, M. Kido and T. Kitahara, Tetrahedron, 1999, 55, 9755–9776 CrossRef CAS; (e) G. E. Keck, C. E. Knutson and S. A. Wiles, Org. Lett., 2001, 3, 707–710 CrossRef CAS PubMed; (f) L. C. Dias, L. G. de Oliveira and M. A. de Sousa, Org. Lett., 2003, 5, 265–268 CrossRef CAS PubMed; (g) X. Shen, A. S. Wasmuth, J. Zhao, C. Zhu and S. G. Nelson, J. Am. Chem. Soc., 2006, 128, 7438–7439 CrossRef CAS PubMed; (h) D. Enders, S. Dhulut, D. Steinbusch and A. Herrbach, Chem. – Eur. J., 2007, 13, 3942–3949 CrossRef CAS PubMed; (i) C. Bressy, J.-P. Vors, S. Hillebrand, S. Arseniyadis and J. Cossy, Angew. Chem., Int. Ed., 2008, 47, 10137–10140 CrossRef CAS PubMed; (j) M. T. Crimmins and A.-M. R. Dechert, Org. Lett., 2009, 11, 1635–1638 CrossRef CAS PubMed.
- For two comprehensive, recent reviews, see: (a) J. A. Marco, M. Carda, J. Murga and E. Falomir, Tetrahedron, 2007, 63, 2929–2958 CrossRef CAS; (b) J. A. Marco and M. Carda, Recent advances in the field of naturally occurring 5,6-dihydropyran-2-ones, in Natural Lactones and Lactams. Synthesis, Occurrence and Biological Activity, ed. T. Janecki, Wiley-VCH, 2014, pp. 51–100 Search PubMed.
- J. A. Marco, J. García-Pla, M. Carda, J. Murga, E. Falomir, C. Trigili, S. Notararigo, J. F. Díaz and I. Barasoain, Eur. J. Med. Chem., 2011, 46, 1630–1637 CrossRef CAS PubMed.
- J. Paños, S. Díaz-Oltra, M. Sánchez-Peris, J. García-Pla, J. Murga, E. Falomir, M. Carda, M. Redondo-Horcajo, J. F. Díaz, I. Barasoain and J. A. Marco, Org. Biomol. Chem., 2013, 11, 5809–5826 Search PubMed.
- R. W. Hoffmann, Chem. Rev., 1989, 89, 1841–1860 CrossRef CAS.
- For two interesting reviews on the conformational aspects of open-chain carbon backbones, see: (a) R. W. Hoffmann, M. Stahl, U. Schopfer and G. Frenking, Chem. – Eur. J., 1998, 4, 559–566 CrossRef CAS; (b) R. W. Hoffmann, Angew. Chem., Int. Ed., 2000, 39, 2054–2070 CrossRef.
- J. Welti, S. Loges, S. Dimmeler and P. Carmeliet, J. Clin. Invest., 2013, 123, 3190–3200 CAS.
- C. Yinnan and Z. Yanmin, Pharmacol. Ther., 2016, 163, 24–47 CrossRef PubMed.
- (a) H. Huang, H. Weng, H. Zhou and L. Qu, Curr. Pharm. Des., 2014, 20, 6543–6554 CrossRef CAS PubMed; (b) B.-J. Chen, Y.-L. Wu, Y. Tanaka and W. Zhang, Int. J. Biol. Sci., 2014, 10, 1084–1096 CrossRef PubMed.
- This corresponds to the concept of polypharmacology: (a) J.-U. Peters, J. Med. Chem., 2013, 56, 8955–8971 CrossRef CAS PubMed; (b) A. Anighoro, J. Bajorath and G. Rastelli, J. Med. Chem., 2014, 57, 7874–7887 CrossRef CAS PubMed.
- S. Källström, A. Erkkilä, P. M. Pihko, R. Sjöholm, R. Sillanpää and R. Leino, Synlett, 2005, 751–756 Search PubMed . In order to work efficiently at a much higher scale than in the original paper, the experimental procedure had to be carried out as described in the Experimental.
- For recent reviews on metathesis, see: (a) Metathesis in Natural Product Synthesis, ed. J. Cossy, S. Arseniyadis and C. Meyer, Wiley-VCH, Weinheim, 2010 Search PubMed; (b) A. H. Hoveyda, J. Org. Chem., 2014, 79, 4763–4792 CrossRef CAS PubMed.
- In some cases, products from intramolecular Michael addition of one hydroxyl group in the side chain to the conjugated CC bond were detected.
- (a) A. E. Prota, J. Setter, A. B. Waight, K. Bargsten, J. Murga, J. F. Díaz and M. O. Steinmetz, J. Mol. Biol., 2016, 428, 2981–2988 CrossRef CAS PubMed; (b) J. Yang, Y. Wang, T. Wang, J. Jiang, C. H. Botting, H. Liu, Q. Chen, J. Yang, J. H. Naismith, X. Zhu and L. Chen, Nat. Commun., 2016, 7, 12103 CrossRef CAS PubMed.
- The crystal structures have been deposited at the Cambridge Crystallographic Data Centre and allocated the following deposition numbers: 2 (CCDC 1431101), ent-2 (CCDC 1431108), 4 (CCDC 1431102), ent-4 (CCDC 1431106), 5 (CCDC 1431104), ent-5 (CCDC 1431105), 20 (CCDC 1431103), ent-20 (CCDC 1431107) and ent27 (CCDC 1431109).
- S. Rodríguez-Nieto, M. A. Medina and A. R. Quesada, Anticancer Res., 2001, 21, 3457–3460 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Graphical NMR spectra of all final and intermediate compounds (one PDF file). CCDC 1431101–1431109. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob01585a |
| ‡ Current address: Depart. de Química. Univ.de Girona, E-17003 Girona, Spain. E-mail: steven.roldan@udg.edu |
| § Current address: Depart. de Q. Analítica y Q. Orgánica. Univ. Rovira i Virgili, E-43007 Tarragona, Spain. E-mail adrian.cardona@urv.cat |
| This journal is © The Royal Society of Chemistry 2017 |