
In situ polymerization induced supramolecular hydrogels of chitosan and poly(acrylic acid-acrylamide) with high toughness†
Jin
Li
,
Zhilong
Su
,
Xiaodong
Ma
,
Hongjie
Xu
,
Zixing
Shi
,
Jie
Yin
and
Xuesong
Jiang
*
School of Chemistry & Chemical Engineering, State Key Laboratory for Metal Matrix Composite Materials, Shanghai Jiao Tong University, Shanghai 200240, People's Republic of China. E-mail: ponygle@sjtu.edu.cn; Fax: +86-21-54747445; Tel: +86-21-54743268
First published on 10th August 2016
Abstract
A novel type of supramolecular hydrogel was developed by in situ polymerization of acrylic acid (AA) and acrylamide (AM) monomers in the aqueous solution of chitosan (CS), in which nanofiber-structured CS and polyelectrolyte chains of poly(acrylic acid-acrylamide) P(AA-r-AM) form the dynamic cross-linked network through the electrostatic interaction of ions. 1H NMR, WAXD, SEM and TEM were used to trace the formation of this supramolecular hydrogel, and revealed that CS chains aggregate into a nanofiber when the copolymerization of AA and AM proceeded. The obtained hydrogels exhibited good comprehensive mechanical properties. With the excellent fatigue resistance and a high toughness of ∼500 J m−3, the tensile strength and elongation of hydrogel-5 are 120 kPa and 1600%, respectively. The obtained supramolecular hydrogel can self-heal and exhibited no residual strain after continuous deformation-resting processes and loading–unloading tests. Furthermore, the obtained hydrogels can be used to adsorb metal ions in water, interestingly their tensile modulus enhanced several times after adsorption of metal ions.
Introduction
As an attractive class of soft and wet materials with a 3D network structure, synthetic hydrogels are of great interest due to their tunable physical and chemical properties and inherent similarities to biological materials. The hydrogels possess a great potential range from biomedical fields to various practical applications. With consideration of their intrinsic inhomogeneity structure or poor energy dissipation capacity, there still exist tremendous challenges in many applications which require high strength and toughness of hydrogels. As one of the most meaningful solutions, Gong et al. have reported a method to obtain strong hydrogels with a special double-network structure consisting of two interpenetrating networks, which provided a new sight into the generation of soft and wet materials.1 Encouragingly, many strategies have been explored to enhance the mechanical properties of hydrogels through the incorporation of some specific structures like nanocomposites2–7 and nanostructured hydrogels,8 topological slide-ring hydrogels,9,10 microgel-reinforced hydrogels,11 polyelectrolyte hydrogels,12,13 tetra-arm poly(ethylene glycol) hydrogels14 and others.15–18 Chu et al. developed a type of nano-structured hydrogel with activated nanogels as nano-crosslinkers which exhibited both high elasticity with 12 times elongation and high strength with a modulus of 130 kPa.8 Zhang et al. have developed high strength nanofibrous architecture hydrogels and tough, tunable mechanical property hydrogels based on chitosan.19,20Chitosan, which is produced by deacetylation of chitin, is soluble in aqueous acidic media. Being the only pseudonatural cationic polymer, chitosan finds many applications as solutions, gels, or films and fibers due to its unique character of excellent biocompatibility, antimicrobial activity, biodegradability, and good complexing ability.21 Various efforts have been made to prepare chitosan-based hydrogels over the past few decades. However, the weak mechanical toughness restricted their application in certain fields. Many methods have been used to enhance their mechanical strength such as chemical cross-linking,22,23 nanofiller reinforcement,24,25 and blending with other polymers26–28 to resolve this problem.
One important way to improve the comprehensive mechanical properties of supramolecular hydrogels is developing dynamic cross-linked networks or reversible sacrificial bonds. Due to the dynamic character of supramolecular interaction such as hydrogen-bonding,29–33 hydrophobic associations,34,35 ionic interaction,9,36 host–guest interactions37,38 and metal–ligand interactions,36,39–41 the supramolecular hydrogels exhibit some advantages over conventional covalent hydrogels. These dynamic cross-linked networks provide some examples of supramolecular hydrogels with excellent strength and good self-healing ability.38,41 On the other hand, it is also necessary and important for supramolecular hydrogels to improve the durability in stress-bearing applications,33,42,43 which might mimic complex biological materials such as cartilage and tissues. Various dynamic covalent bonds44–49 and non-covalent bonds30,32,34–38 have been formed in synthetic hydrogels and significant achievements were made, however, it is still challenging to develop supramolecular hydrogels with good comprehensive performances, especially the quick recovery with no residual strain.
In this paper, we present a novel class of supramolecular nanofiber-structured hybrid hydrogels through the ionic coordination between CS and P(AA-r-AM) by in situ copolymerization of AA and AM monomers in the aqueous solution of CS (Scheme 1). CS, which tends to aggregate into a nanofiber through a parallel arrangement at a high concentration or temperature, is an ideal component to form the rigid part of the hybrid hydrogel. While the soft P(AA-r-AM) chain may provide both the elasticity and hydrophilicity to the hybrid hydrogel. As a natural polymer and cationic polyelectrolyte, CS has a large number of amino groups on its polymer chains, and can form a cross-linked network with the anionic polyelectrolyte P(AA-r-AM) via ionic bonds. In the process of in situ polymerization of AA and AM, the CS chains aggregated into a nanofiber due to the temperature increase. The obtained supramolecular hydrogels exhibited high tensile strength, large elongation, good self-healing property, excellent fatigue resistance with no residual strain and fast recovery performances. Due to the abundant carboxyl and amino groups, the hydrogel can be used to adsorb metal ions in waste water through telechelation. The telechelation can also provide the further cross-linking of hydrogels, resulting in the enhancement of the mechanical properties of the obtained supramolecular hydrogel.
 | ||
| Scheme 1 Whole strategy for the fabrication of supramolecular hydrogels via in situ polymerization of AA and AM in the aqueous solution of CS. | ||
Experimental section
Materials
Chitosan (CS, deacetylation degree ≥90.0%), acrylic acid (AA), acrylamide (AM), potassium persulfate (KPS), chromic nitrate (Cr(NO3)3·9H2O), cadmium nitrate (Cd(NO3)2·4H2O), lead nitrate (Pb(NO)2), copper nitrate (Cu(NO3)2·3H2O), nickel nitrate (Ni(NO3)2·6H2O), perchloride iron (FeCl3·6H2O), stannic chloride (SnCl4·5H2O), hydrochloric acid (HCl), and sodium hydroxide (NaOH) were supplied by Sinopharm Chemical. All the reagents were used without further purification.Methods
1H nuclear magnetic resonance spectroscopy (1H-NMR) was carried out with the help of a Varian Mercury Plus 400 MHz spectrometer in D2O.Scanning electron microscopy (SEM) was carried out on a NOVA NanoSEM 430 (FEI Company) field emission scanning electron microscope operating at an acceleration voltage of 5 kV, and all of the hydrogels were fractured with liquid nitrogen and coated with gold in a vacuum before observation.
Transmission electron microscopy (TEM) was conducted on a JEM-2100 (JEOL, Ltd, Japan) microscope, operating at 200 kV. The sample was prepared by dropping the hydrogel-5 solution (before the hydrogel developed) onto copper grids coated with a thin carbon film, and dried at 25 °C for 48 h. And the ultra-thin section of the hydrogel-5 was analysed after hydrogel formation. In this case no staining treatment was performed during the measurements.
Wide Angle X-ray Diffraction (WAXD) patterns were obtained on a D/max-220 0/PC (Japan Rigaku Corp.) using CuKα radiation (λ = 1.5418 Å). The formation of hydrogel-5 at different times of in situ polymerization was traced.
Tensile and compressive stress–strain measurements of CS/P(AA-r-AM) were conducted at room temperature using an Instron 4465 instrument at a cross-head speed of 10 mm min−1. The speed of unloading was kept at 5 mm min−1, and at least three samples were evaluated for each test. The sample was prepared with a diameter of 0.8 cm and a length of 2.0 cm in the tensile test, and a diameter of 1.8 cm in and a height of 2.0 cm in the compressive test.
Preparation of ion-crosslinked hydrogels
A series of different concentrations of acrylic acid (AA) solution were prepared according to Table 1 at first. Then a certain amount of chitosan (CS) was dispersed into an AA aqueous solution and stirred overnight to get a homogeneous solution. After that, variable contents of acrylamide (AM) (Table 1) and the thermal initiator KPS were added subsequently to the CS solution. The homogeneous solution was heated to 60 °C to initiate the polymerization after purging with nitrogen for 30 min. After heating for 7 h, an ion crosslinking was introduced by the association between CS and polyelectrolyte P(AA-r-AM) through the subsequent thermal polymerization in situ. With the hydrogel-5 as an example, 0.2 g of chitosan (CS) was directly dissolved in an aqueous solution of acrylic acid (AA, 10 wt%, 10 mL), and then acrylamide (AM, 0.5 g) monomers with a trace amount of K2S2O8 (1% of the total moles AA and AM) initiator were added to the solution and stirred to get a homogeneous solution. The as-prepared solution was heated at 60 °C for 7 hours to obtain the hydrogels after degassing with nitrogen.| Samples | Chitosan (CS) (wt%) | Acrylic acid (AA) (wt%) | Acrylamide (AM) (wt%) | Tensilea | Compressiona | |||
|---|---|---|---|---|---|---|---|---|
| Tensile strength (kPa) | Elongation at break (%) | Young's modulus (kPa) | Compressive strength at 80% strain (kPa) | Compressive modulus (kPa) | ||||
| a Each dataset is obtained here after at least three parallel samples. | ||||||||
| Hydrogel-1 | 2.0 | 1.0 | 5.0 | 4.2 ± 0.1 | 180 ± 5 | 4.8 ± 0.1 | 24.5 ± 0.2 | 67.9 ± 0.4 |
| Hydrogel-2 | 2.0 | 1.0 | 10.0 | 9.3 ± 0.2 | 496 ± 13 | 4.8 ± 0.6 | 72.7 ± 0.2 | 16.7 ± 0.5 |
| Hydrogel-3 | 2.0 | 5.0 | 5.0 | 7.2 ± 0.2 | 632 ± 9 | 1.7 ± 0.3 | 27.4 ± 0.6 | 69.2 ± 0.4 |
| Hydrogel-4 | 2.0 | 5.0 | 10.0 | 22.6 ± 0.9 | 648 ± 10 | 17.6 ± 0.7 | 94.3 ± 0.3 | 155.0 ± 3.3 |
| Hydrogel-5 | 2.0 | 10.0 | 5.0 | 122.0 ± 4.5 | 1617 ± 25 | 41.7 ± 0.6 | 42.2 ± 0.4 | 125.0 ± 9.1 |
| Hydrogel-6 | 2.0 | 10.0 | 10.0 | 73.4 ± 0.7 | 1130 ± 12 | 35.6 ± 0.1 | 120.0 ± 0.2 | 240.0 ± 2.7 |
Adsorption of metal ions
We chose seven metal ions to study the adsorption behavior on the hydrogels. The equilibrium adsorption capacity (Qeq) of metal ions onto hydrogels was calculated using the following equation:where c0 and ce (mol L−1) are the initial and equilibrium concentration of metal ions, respectively. V (mL) is the volume of the solution and M (mg) is the mass of dried hydrogel. All tests were carried out in triplicate, and the average was used in the analysis.

The adsorption experiments were conducted with an initial ion concentration of 0.005 mol L−1 and 30 mg of wet hydrogels were added to each 15 mL metal ion solution for 3 days.
A quantitative determination of ions was made by inductively coupled plasma (ICP) atomic emission spectrometry using iCAP6300 equipment (Thermo, America).
Relative swelling ratio (RSR): the RSR of hydrogels in different pH values was measured at 25 °C and defined as:

Results and discussion
Preparation of hydrogels by polymerization
The whole strategy for the preparation of nanofiber-structured supramolecular hydrogels is illustrated in Scheme 1. A certain amount of chitosan (CS) was directly dissolved in acrylic acid aqueous solution (AA, 10 wt%), and then acrylamide (AM) monomers with a trace amount of K2S2O8 initiator (1% of the total moles AA and AM) were added to the solution and stirred to get a homogeneous solution. After degassing with nitrogen, the transparent solution was heated at 60 °C for 7 hours to obtain the supramolecular hydrogel. It is worth noting that, as will be discussed later, the clear solution became turbid in several minutes and maintained this appearance for hours before the formation of hydrogels. Upon further heating, the semitransparent hydrogel was formed. The abundant –COOH groups of P(AA-r-AM) polyelectrolyte chains coupled with –NH2 of CS to form a dynamic ionic cross-linked supramolecular network. Because of –COOH and double bonds, the AA monomer played two main roles: making CS soluble in aqueous solution and forming anionic polyelectrolyte chains through in situ polymerization. Additionally, the poly(acrylic acid)/polyacrylamide copolymer is an excellent water superabsorbent which prevents the hydrogel from losing water. Also the acrylamide component shows high flexibility under wet conditions and has been widely used in the preparation of highly elastic hydrogels.50Through this strategy, a series of hydrogels with different formulations were prepared through ionic interactions between CS and in situ generated P(AA-r-AM) chains. The feed ratios are summarized in Table 1.
The structure of the hydrogels was carefully studied. First, the internal morphology of the obtained supramolecular hydrogel was examined by SEM. The typical SEM images of the cross-section morphological structure of freeze-dried hydrogels are presented in Fig. 1 and Fig. S1 (ESI†), revealing the highly porous microstructures. The pore size of the hydrogel varied with the content of monomers and various crosslinking densities, and was around 1–5 µm in diameter. The pores formed in these supramolecular hydrogels have some obvious advantages in transporting guest molecules such as water and metal ions across the hydrogels. It is interesting that the nano-fibrous structure across the micro-pores can be found in SEM images of various hydrogels, especially hydrogel-5. These nano-fibrous structures may be ascribed to the parallel aggregates of CS.
 | ||
| Fig. 1 (a) The representative cross-sectional SEM images of the obtained hydrogel-5 and (b) shows the magnified marked area of (a). | ||
To check the formation of these nano-fibrous structures, TEM was used to reveal the CS aggregation during the process of in situ polymerization (Fig. 2). At first, the protonation of –NH2 on the C-2 position of the D-glucosamine repeat unit converted the polysaccharide into a soluble polyelectrolyte. As a result, AA and CS complexes formed a sheath-like structure on the surface of CS chains in solution. With the temperature increasing, the interactions between AA and CS weakened which decreased the hydrophobicity of CS, resulting in a rapid aggregation into nanospheres (Fig. 2a). Upon further heating, the nanosphere shaped CS tends to disassemble and formed the nanofibers through parallel arrangement (Fig. 2b–d). The nanofiber aggregate of chitin/chitosan in NaOH/urea aqueous solution at high temperature has been reported by Zhang et al., in which any chemical reactions were absent.51 On the other hand, AA and AM were co-polymerized in situ, resulting in the negatively charged P(AA-r-AM). The system finally developed into a “crosslinked” nano-fibrous structured hydrogel (Fig. 2e), while CS formed the nanofiber and P(AA-r-AM) acted as the crosslinking point. It should be specifically noted that no chemical crosslinkers were added to this system. The strong interaction of CS and P(AA-r-AM) should be ascribed to the various –COOH groups in P(AA-r-AM). The multiple ionic bonding between P(AA-r-AM) and CS made the interaction very strong.
 | ||
| Fig. 2 TEM and WAXD tracing of the formation of hydrogel-5. Representative TEM images of the solution of hydrogel-5 upon heating at 60 °C: (a) 1 h, (b) 3 h, (c) 5 h, and (d) 6 h; and the ultra-thin section of the hydrogel-5 (7 h) (e). (f) WAXD tracing of the formation of hydrogel-5 at different times of polymerization. | ||
The aggregation of CS chains along with in situ polymerization of AA and AM was further confirmed and characterized by WAXD (Fig. 2f). Before polymerization, CS was dissolved in the aqueous solution of AA to form a homogeneous solution. When polymerization proceeded, a new peak at 2θ = 12° assigned to the crystallization of CS chains appeared and its intensity increased, while the broad peak ranging from 25° to 45° became narrowed in the WAXD diffraction pattern. This strongly indicated that CS chains aggregate during the process of polymerization, which is in agreement with the results of TEM.
Also, 1H NMR was used to trace the whole process. As shown in Fig. 3, proton signals attributed to CS (∼2.70–3.90 ppm) disappeared after heating for 20 min, which indicated that CS chains are phase-separated from water. On the other hand, proton signals (∼5.5–6.2 ppm) assigned to the double bond of AA and AM monomers disappeared gradually with the increasing time of polymerization, while new signals at ∼1.5 and 2.1 ppm which belonged to the –CH2 of polyelectrolyte chains P(AA-r-AM) appeared. After heating for 100 min, the polymerization of AA and AM completed. This process agrees well with the TEM and XRD. It should be noted that the total concentration of the solution for NMR (30 mg in 0.6 mL) is much lower than that for hydrogel-5 (17 wt%), which may result in different polymerization times.
 | ||
| Fig. 3 1H NMR spectra of hydrogel-5 through in situ polymerization in D2O. | ||
To further identify the effect of AA, AM and CS chains on the formation of supramolecular hydrogels, we conducted a series of control experiments to prepare a hydrogel with the similar formulation to hydrogel-5 (Fig. S2b, ESI†). When acetic acid (HAc) was used instead of AA, no gel can be formed after heating for 7 h (Fig. S2b(1), ESI†). In the presence of AA as the only monomer, an imperfect gel was formed without the second monomer (AM) (Fig. S2b(2), ESI†). This may be explained by the fact that the strong interaction between the cationic polyelectrolyte (CS) and the anion polyelectrolyte (PAA) results in a highly dense structure during the thermal polymerization. In other words, to a certain extent, the existing AM increased the distance between the polyelectrolyte chains, resulting in the dilution of the crosslinking density between CS and AA, and thus formed a preferable hydrogel. Based on these results, we concluded that the generation of an anionic polyelectrolyte P(AA-r-AM) is critical to the formation of a hydrogel. By using N,N′-methylenebisacrylamide (MBA) and polyallyamine (PAAM) to replace CS, we prepared a chemical P(AA-r-AM) hydrogel and a PAAM/P(AA-r-AM) supramolecular hydrogel, respectively (Fig. S2b(3) and (4), ESI†). The resulting chemical P(AA-r-AM) hydrogel using MBA as a cross-linker is too fragile, and cannot withstand compressive or tensile stresses (Fig. S2c, ESI†). Unlike the hydrogel-5, the hydrogel generated with cationic polyelectrolyte PAAM was too weak that it could not bear the 200 g weight (Fig. S2d and e, ESI†). These observations suggested that the crystallized fibrous aggregation of CS chains was of great significance in enhancing the mechanical properties of our hydrogels.
Based on the above experimental results, we proposed the possible formation mechanism of our supramolecular hydrogel of CS/P(AA-r-AM), which is presented in Scheme 1. CS is first protonated by AA and is dissolved in water to form a homogeneous solution. With the temperature increasing, the interactions between AA and CS weakened which decreased the hydrophilicity of CS. Consequently, CS chains become less soluble and aggregate into nanospheres in water. Upon further heating, the nanosphere shaped CS tends to disassemble and formed the nanofibers through parallel arrangement. Simultaneously, AA and AM were co-polymerized in situ, resulting in the negatively charged P(AA-r-AM). Following the polymerization, the nano-fibrous CS turns into a network intertwined with polyelectrolyte P(AA-r-AM) through the ionic bond. Like other nanofiber based hydrogels,52 the hybrid of the CS nano-fiber and the soft P(AA-r-AM) chain through ionic interaction might lead to the excellent mechanical performance of the CS/P(AA-r-AM) supramolecular hydrogel.
Mechanical performance of the hydrogel
Fig. 4 presents the digital photographs of the obtained supramolecular hydrogels of CS/P(AA-r-AM) (hydrogel-5), which are highly stretchable and flexible. These hydrogels are so tough that they can even resist slicing with a blade. This may be because of the intricate structure of the hybrid supramolecular hydrogel. The rigid nanofibers of CS resisted the slicing of the knife edge, avoiding the formation of a notch; while the stress can be dissipated effectively to a large number of flexible P(AA-r-AM)bridge chains connecting nanofibers in the supramolecular hydrogels.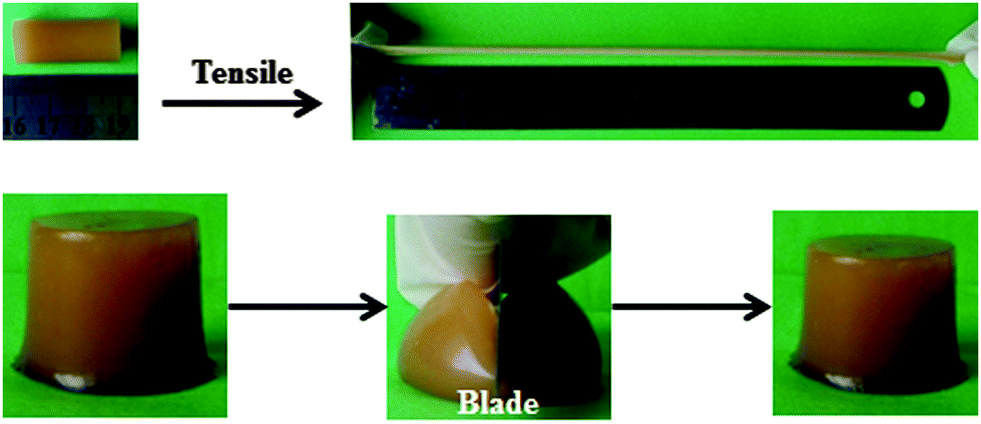 | ||
| Fig. 4 Digital photographs showing that the resulting hydrogel-5 is very elastic to resist a high stretching and slicing with a blade. | ||
Given the consideration of the nano-fibrous structure and reversible ionic cross-linking and excellent elasticity, we investigated the mechanical properties of the obtained supramolecular hydrogels in detail (Fig. 5–7) and the specific mechanical parameters are summarized in Table 1.
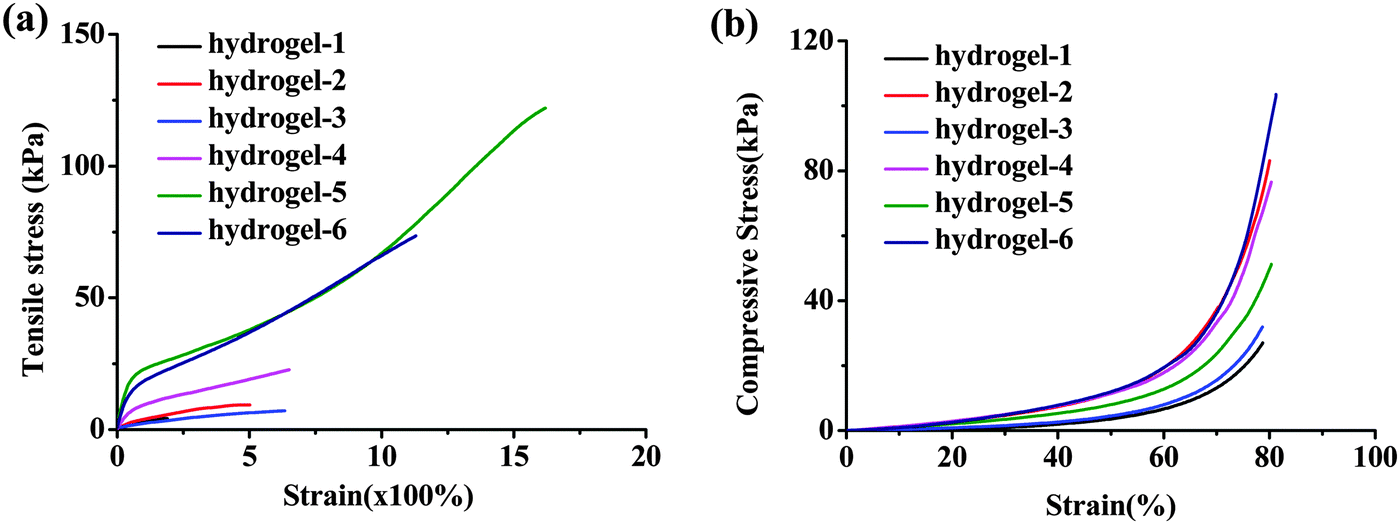 | ||
| Fig. 5 Mechanical properties of the hydrogels. Tensile stress–strain (a) and compressive stress–strain (b) curves of hydrogels in different formulations. | ||
 | ||
| Fig. 6 Recovery properties of the hydrogel-5. (a) Loading–unloading curves for different waiting times performed by cyclic tensile tests. (b) The time-dependent recovery of dissipated energy and the elastic modulus estimated from the hysteresis area change in (a). | ||
 | ||
| Fig. 7 Fatigue resistance of the obtained hydrogel-5. (a) Cyclic compression tests of hydrogel-5 undergoing 10 cycles of compression to 80% strain and (b) the calculated total toughness and the dissipated toughness of hydrogel-5 during loading–unloading tests based on (a). | ||
Fig. 5 shows the typical tensile stress–strain and compressive stress–strain curves of the obtained hydrogels with different formulations. What the results showed was that all of the hydrogels possessed high Young's modulus and fracture strength. When the content of AA remained unchanged but AM increased, the tensile and compressive strength enhanced, proving the assumption that AM can enlarge the distance of CS chains and PAA. And it can be elongated under stress until straight and recover to the lowest energy state when the stress was taken away similar to the functional soft segment. The same results have been reported by Chu et al., the flexible PNIPAM bridge chains connecting nanogels in the nano-structured hydrogels can dissipate the stress effectively.8 The largest elongation was over 1600% with tensile strength higher than 120 kPa as for hydrogel-5 (Fig. 5a and Movie S1, ESI†). Both the long macromolecular backbones and the density of the ionic crosslink contributed to their extraordinary mechanical performances in this supramolecular nanofiber-structured hydrogel. Fig. 5b presents the representative compressive stress–strain curves of as-prepared hydrogels. We controlled the stress at 80% strain, and the hydrogels did not fracture during the whole compression tests. Because of the enhanced stiffness by the mentioned ionic cross-linked supramolecular interaction and the unique nanofiber-structure, the hydrogel-6 exhibited high toughness with compressive stress over 120 kPa. It was notable that the hydrogel was able to regain its original shape immediately after unloading (Movie S2, ESI†), which implied the excellent recoverability of the dynamic ionic interactions. The excellent performance of the obtained in situ hydrogel was even comparable to high performance ionic chemical dual cross-linked hydrogels.36
The measurement of stress cycles was conducted to further understand the effect of sacrificial bond recovery on the dynamic mechanical properties of our system. Taking hydrogel-4 for example, we performed six successive loading–unloading cycles on it. The hydrogel was less weak if the second loading was applied immediately after the first unloading (Fig. S3, ESI†). Apparent hysteresis loops were observed in this whole test, indicating the obtained hydrogel dissipated energy effectively. After six loading–unloading cycles, the dissipated energy and the elastic modulus were maintained at ∼45% and ∼95% relative to those of the first loading. If we left the hydrogel at room temperature for a while, the internal damage was much better recovered for some time before reloading (Fig. 6a and b). Upon increasing for 20 min, the dissipated energy recovered to ∼80% of the first loading, and the elastic modulus remained almost unchanged in this process. Compared with the recent results of references, our supramolecular hydrogel exhibited very fast recovery. The chemically cross-linked P(AAm-co-AAc) hydrogel with ionic coordination with Fe(III) developed by Zhou et al. possessed fast self-healing, and its elastic modulus reached 87.6% of its original value after 4 hours at room temperature.39 A mechanically strong supramolecular polymer hydrogel reported by Liu et al. could be completely healed with the help of a 90 °C water bath for 3 hours.42 Noting that there was no residual strain after continuous deformation-resting processes and different lapse times, loading–unloading tests for our hydrogels were carried out, and compared with the ionic cross-linking copolymer hydrogels,36,40 hydrogen bonding elastic hydrogels,29 or nanocomposite hydrogels.6 Those observations suggested that no bond breakage or permanent damage appeared in our hydrogel after each repeated load. The recovery of the strength in the material might be attributed to the unique supramolecular ionic cross-linked structure of our hydrogels, in which the CS nano-fibrous network prevented the permanent deformation and the ionic cross-linking network dissipated a huge amount of energy when stretched and assisted in re-formation to recover the original shape after the stress moved.
We further conducted continuous loading–unloading tests on hydrogel-5 (Fig. 7 and Movie S2, ESI†) and hydrogel-4 (Fig. S4, ESI†) 10 times. The recovery was fast and complete under repeated stress. The same loading–unloading hysteresis loop, toughness and energy dissipated properties revealed the excellent fatigue resistance and fast recovery of our hydrogels. The CS backbone and the introduced ionic bonds played highly important roles in the enhancement of fatigue resistance bearing reduplicative stress.
The dynamic ionic interactions might provide the self-healing ability to our supramolecular hydrogels. We performed self-healing experiments on these series of hydrogels via compressive stress tests and good self-healing was observed in these hydrogels as expected (Fig. S5 and S6, ESI†). The hydrogel-4 and hydrogel-5 were cut into two parts firstly and they were brought into contact in a constant temperature and humidity chamber for 24 h. Fig. S5a and S6a (ESI†) display the compressive stress–strain curves of the original and repaired hydrogel, respectively. The samples were reloaded three times with almost no change in the loading–unloading behavior after self-healing. It exhibited a similar loading–unloading hysteresis loop, toughness and energy dissipated properties (Fig. S5b and S6b, ESI†), which revealed that dynamic reaction occurred in this system rather than the simple adhesion at the interfaces of broken hydrogels. Namely, when the hydrogel bears the loading, the ionic bonds were broken and the energy dissipated effectively. The rebuilding of ionic bonds took place after removing the loading. The destruction and reconstruction of the dynamic ionic bonds occurred repeatedly in the whole process of loading–unloading cycles. So the existence of dynamic ionic bonds in the hydrogel was thus crucial for the self-healing properties. Note that the strength of the hydrogels after self-healing was a little higher than that of original hydrogels. It might be resulted from some loss of water during the self-healing process.
Besides the excellent mechanical properties, the as-prepared supramolecular hydrogels are very stable. For example, hydrogel-5 did not disassemble in aqueous solutions with pH from 0 to 14 (Fig. S7, ESI†). This may be ascribed to the fact that under very acidic conditions (pH ≤ 2), a screening effect of the counterions, i.e. Cl−, shields the charge of the ammonium cations and prevents the hydrogel from disassembling. The relative swell ratio (RSR) is 23.6% when the hydrogel-5 was immersed into 1 M HCl after 7 days. At higher pH values, the carboxylic acid groups become ionized and the electrostatic repulsive force between the charged sites (COO−) causes increased swelling. However, when pH > 12, a screening effect of the counterions (Na+) may also limit the swelling. As a result, the gel remained well under extreme conditions with 278.9% of RSR in 1 M NaOH solution after 7 days. A similar observation has been reported by Mahdavinia.53
Absorption of metal ions
Chitosan is known to have good complexing ability with –NH2 groups on the chain involved in specific interactions with metals. Many papers are concerned with complexation for the recovery of heavy metals from various waste waters.21,54,55 Because of the excellent mechanical performance, high stability, easy preparation and abundant amino and carboxyl groups, CS/P(AA-r-AM) supramolecular hydrogels are expected to have potential to remove pollutants in water treatment. Recently, water pollution increased seriously the environmental damage and carcinogenic effect for biological diversities with the development of industry, especially pollutants from the difficultly handled heavy metal ions.56,57 Adsorption by hydrogels is one of the most useful and simple methods to remove heavy metal ions.41,58 With the consideration of the porous structure (Fig. 1 and Fig. S1, ESI†) and the abundant –OH, –COOH and –NH2 in our system, we investigated the adsorption behavior of the obtained hydrogel-5 for various metal ions and measured the saturated adsorption capacity (Qeq) (Fig. S8a, ESI†). As we know, in highly acidic media, no adsorption occurred owing to the strong protonation of the amine group, which resulted in positively charged pendant groups causing electrostatic repulsion. In the adsorption experiments, we adjusted the solution pH to the desired value (pH = 6.0–6.5) by adding hydrochloric acid according to the previous reports.59–61 The hydrogel-5 had an effective adsorption on the metal ions as expected. The Qeq value of Cd2+, Ni2+, and Pb2+ reached 859, 637, and 461 mg per gram of dry hydrogel, respectively, which is better than many literature results.62–64It is interesting that the mechanical performance of hydrogel-5 was enhanced to some extent after adsorption of metal ions. As shown in Fig. 8, after being soaked in Fe3+ solution with different concentrations, the compressive stress and Young's modulus were strengthened over 5 and 3 times, respectively. This might be due to the complex interactions between Fe3+ and the residual –COOH and –NH2 groups in the hydrogel network, which provides the further cross-linking (Fig. 8b). It was worth noting that the compressive stress decreased when the concentration was higher than 0.01 mol L−1. This might be explained by the fact that much more Fe3+ was adsorbed and deposited on the surface of the hydrogel to form a stiff cover, which affected the adsorption efficiency and the mechanical properties of the hydrogel seriously. Moreover, a similar behavior has been observed when the hydrogels were soaked with the other metal ion solutions. Taking the 0.001 mol L−1 concentration as an example, all the compressive stresses of hydrogels were enhanced obviously (Fig. S8b, ESI†). This indicated that further cross-linking with metal ions can affect and enhance the mechanical properties of the material after immersing the hydrogels into the metal ion solutions.
 | ||
| Fig. 8 Mechanical properties of the hydrogel-5 after adsorption of metal ions. The compressive stress–strain curves of hydrogel-5 after being soaked in different concentrations of Fe3+ solution (a); the proposed mechanism for the cross-linked structure of the hydrogel after absorption of metal ions through the complexation between metal ions and –COOH and –NH2, in which Mn+ are representative metal ions (b). | ||
Conclusion
In summary, we demonstrated a new type of supramolecular hydrogel (CS/P(AA-r-AM)), which was synthesized by in situ copolymerization of acrylic acid (AA) and acrylamide (AM) monomers in the aqueous solution of chitosan (CS). Nano-fibrous aggregates of CS were formed in the hydrogel along with the polymerization, which is critical for the excellent mechanical performance. With the high toughness, the tensile strength and elongation of hydrogel-5 are 120 kPa and 1600%, respectively. The crystalline aggregations of CS prevented the permanent damage while bearing the load; the ionic bonds between nano-fibrous CS and polyelectrolyte P(AA-r-AM) acted as revisable sacrificial bonds and dissipated energy upon repeated loading–unloading cycles. These hydrogels displayed high compressive and tensile stresses without fracture or residual strain and exhibited excellent fatigue resistance and quick recovery upon circulatory load. In addition, the obtained hydrogels can be used to adsorb metal ions in water, and their tensile modulus was enhanced several times after adsorption of metal ions. It is believed that the CS/P(AA-r-AM) supramolecular hydrogel with good comprehensive mechanical performance, simple preparation and fast recovery offered a new insight into the design of the functional hydrogels and provided some opportunities in practical applications.Acknowledgements
The authors thank the National Basic Research Program (2013CB834506) and National Nature Science Foundation of China (21274088, 51373098, and 21522403) for their financial support.References
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- J. Ning, G. Li and K. Haraguchi, Macromolecules, 2013, 46, 5317–5328 CrossRef CAS.
- W.-C. Lin, W. Fan, A. Marcellan, D. Hourdet and C. Creton, Macromolecules, 2010, 43, 2554–2563 CrossRef CAS.
- C. Yao, Z. Liu, C. Yang, W. Wang, X.-J. Ju, R. Xie and L.-Y. Chu, Adv. Funct. Mater., 2015, 25, 2980–2991 CrossRef CAS.
- J. Wang, L. Lin, Q. Cheng and L. Jiang, Angew. Chem., Int. Ed., 2012, 51, 4676–4680 CrossRef CAS PubMed.
- J. Yang, C.-R. Han, X.-M. Zhang, F. Xu and R.-C. Sun, Macromolecules, 2014, 47, 4077–4086 CrossRef CAS.
- K. Haraguchi, K. Murata and T. Takehisa, Macromolecules, 2012, 45, 385–391 CrossRef CAS.
- L.-W. Xia, R. Xie, X.-J. Ju, W. Wang, Q. Chen and L.-Y. Chu, Nat. Commun., 2013, 4, 2226–2236 Search PubMed.
- A. Bin Imran, K. Esaki, H. Gotoh, T. Seki, K. Ito, Y. Sakai and Y. Takeoka, Nat. Commun., 2014, 5, 5124–5131 CrossRef PubMed.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- J. Hu, K. Hiwatashi, T. Kurokawa, S. M. Liang, Z. L. Wu and J. P. Gong, Macromolecules, 2011, 44, 7775–7781 CrossRef CAS.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- F. Luo, T. L. Sun, T. Nakajima, T. Kurokawa, Y. Zhao, K. Sato, A. B. Ihsan, X. Li, H. Guo and J. P. Gong, Adv. Mater., 2015, 27, 2722–2727 CrossRef CAS PubMed.
- H. Kamata, Y. Akagi, Y. Kayasuga-Kariya, U.-I. Chung and T. Sakai, Science, 2014, 343, 873–875 CrossRef CAS PubMed.
- T. Bai, P. Zhang, Y. Han, Y. Liu, W. Liu, X. Zhao and W. Lu, Soft Matter, 2011, 7, 2825–2831 RSC.
- S. Lin, C. Cao, Q. Wang, M. Gonzalez, J. E. Dolbow and X. Zhao, Soft Matter, 2014, 10, 7519–7527 RSC.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- V. X. Truong, M. P. Ablett, S. M. Richardson, J. A. Hoyland and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 1618–1622 CrossRef CAS PubMed.
- J. Duan, X. Liang, Y. Cao, S. Wang and L. Zhang, Macromolecules, 2015, 48, 2706–2714 CrossRef CAS.
- J. You, S. Xie, J. Cao, H. Ge, M. Xu, L. Zhang and J. Zhou, Macromolecules, 2016, 49, 1049–1059 CrossRef CAS.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- Y. Zhang, L. Tao, S. Li and Y. Wei, Biomacromolecules, 2011, 12, 2894–2901 CrossRef CAS PubMed.
- C. M. Valmikinathan, V. J. Mukhatyar, A. Jain, L. Karumbaiah, M. Dasari and R. V. Bellamkonda, Soft Matter, 2012, 8, 1964–1976 RSC.
- Y. Xu, X. Ren and M. A. Hanna, J. Appl. Polym. Sci., 2006, 99, 1684–1691 CrossRef CAS.
- J. Roosen, J. Spooren and K. Binnemans, J. Mater. Chem. A, 2014, 2, 19415–19426 CAS.
- M. He, Z. Wang, Y. Cao, Y. Zhao, B. Duan, Y. Chen, M. Xu and L. Zhang, Biomacromolecules, 2014, 15, 3358–3365 CrossRef CAS PubMed.
- J. Fukuda, A. Khademhosseini, Y. Yeo, X. Yang, J. Yeh, G. Eng, J. Blumling, C.-F. Wang, D. S. Kohane and R. Langer, Biomaterials, 2006, 27, 5259–5267 CrossRef CAS PubMed.
- J. Li, Z. Su, H. Xu, X. Ma, J. Yin and X. Jiang, Macromolecules, 2015, 48, 2022–2029 CrossRef CAS.
- B. Zhu, N. Jasinski, A. Benitez, M. Noack, D. Park, A. S. Goldmann, C. Barner-Kowollik and A. Walther, Angew. Chem., Int. Ed., 2015, 54, 8653–8657 CrossRef CAS PubMed.
- X. Hu, M. Vatankhah-Varnoosfaderani, J. Zhou, Q. Li and S. S. Sheiko, Adv. Mater., 2015, 27, 6899–6905 CrossRef CAS PubMed.
- Y. Zhang, Y. Li and W. Liu, Adv. Funct. Mater., 2015, 25, 471–480 CrossRef CAS.
- Z. S. Kean, J. L. Hawk, S. Lin, X. Zhao, R. P. Sijbesma and S. L. Craig, Adv. Mater., 2014, 26, 6013–6018 CrossRef CAS PubMed.
- J. A. Neal, D. Mozhdehi and Z. Guan, J. Am. Chem. Soc., 2015, 137, 4846–4850 CrossRef CAS PubMed.
- D. C. Tuncaboylu, M. Sari, W. Oppermann and O. Okay, Macromolecules, 2011, 44, 4997–5005 CrossRef CAS.
- M. A. Haque, T. Kurokawa, G. Kamita and J. P. Gong, Macromolecules, 2011, 44, 8916–8924 CrossRef CAS.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- K. Miyamae, M. Nakahata, Y. Takashima and A. Harada, Angew. Chem., Int. Ed., 2015, 54, 8984–8987 CrossRef CAS PubMed.
- P. Lin, S. Ma, X. Wang and F. Zhou, Adv. Mater., 2015, 27, 2054–2059 CrossRef CAS PubMed.
- K. J. Henderson, T. C. Zhou, K. J. Otim and K. R. Shull, Macromolecules, 2010, 43, 6193–6201 CrossRef CAS.
- Z. Sun, F. Lv, L. Cao, L. Liu, Y. Zhang and Z. Lu, Angew. Chem., Int. Ed., 2015, 54, 7944–7948 CrossRef CAS PubMed.
- X. Dai, Y. Zhang, L. Gao, T. Bai, W. Wang, Y. Cui and W. Liu, Adv. Mater., 2015, 27, 3566–3571 CrossRef CAS PubMed.
- T.-C. Tseng, L. Tao, F.-Y. Hsieh, Y. Wei, I.-M. Chiu and S.-H. Hsu, Adv. Mater., 2015, 27, 3518–3524 CrossRef CAS PubMed.
- G. Deng, F. Li, H. Yu, F. Liu, C. Liu, W. Sun, H. Jiang and Y. Chen, ACS Macro Lett., 2012, 1, 275–279 CrossRef CAS.
- J. A. Yoon, J. Kamada, K. Koynov, J. Mohin, R. Nicolaÿ, Y. Zhang, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2012, 45, 142–149 CrossRef CAS.
- M. Pepels, I. Filot, B. Klumperman and H. Goossens, Polym. Chem., 2013, 4, 4955–4965 RSC.
- L. He, D. E. Fullenkamp, J. G. Rivera and P. B. Messersmith, Chem. Commun., 2011, 47, 7497–7499 RSC.
- Z. Wei, J. H. Yang, X. J. Du, F. Xu, M. Zrinyi, Y. Osada, F. Li and Y. M. Chen, Macromol. Rapid Commun., 2013, 34, 1464–1470 CrossRef CAS PubMed.
- K. L. Tan and E. N. Jacobsen, Angew. Chem., Int. Ed., 2007, 46, 1315–1317 CrossRef CAS PubMed.
- L. Xiong, X. Hu, X. Liu and Z. Tong, Polymer, 2008, 49, 5064–5071 CrossRef CAS.
- B. Duan, X. Zheng, Z. Xia, X. Fan, L. Guo, J. Liu, Y. Wang, Q. Ye and L. Zhang, Angew. Chem., Int. Ed., 2015, 54, 5152–5156 CrossRef CAS PubMed.
- Z. Yang, L. Wang, J. Wang, P. Gao and B. Xu, J. Mater. Chem., 2010, 20, 2128–2132 RSC.
- G. R. Mahdavinia, A. Pourjavadi, H. Hosseinzadeh and M. J. Zohuriaan, Eur. Polym. J., 2004, 40, 1399–1407 CrossRef CAS.
- T. Puspitasari, Oktaviani, D. S. Pangerteni, E. Nurfilah and D. Darwis, Macromol. Symp., 2015, 353, 168–177 CrossRef CAS.
- E. Guibal, Sep. Purif. Technol., 2004, 38, 43–74 CrossRef CAS.
- Z. Geng, H. Zhang, Q. Xiong, Y. Zhang, H. Zhao and G. Wang, J. Mater. Chem. A, 2015, 3, 19455–19460 CAS.
- R. Sharma, N. Singh, A. Gupta, S. Tiwari, S. K. Tiwari and S. R. Dhakate, J. Mater. Chem. A, 2014, 2, 16669–16677 CAS.
- F. Peng, G. Li, X. Liu, S. Wu and Z. Tong, J. Am. Chem. Soc., 2008, 130, 16166–16167 CrossRef CAS PubMed.
- D. R. Jansen, J. Rijn Zeevaart, A. Denkova, Z. I. Kolar and G. C. Krijger, Langmuir, 2009, 25, 2790–2796 CrossRef CAS PubMed.
- G. Zhang, R. Qu, C. Sun, C. Ji, H. Chen, C. Wang and Y. Niu, J. Appl. Polym. Sci., 2008, 110, 2321–2327 CrossRef CAS.
- R. Youda, H. Nishihara and K. Aramaki, Electrochim. Acta, 1990, 35, 1011–1017 CrossRef CAS.
- D. H. K. Reddy and S.-M. Lee, Adv. Colloid Interface Sci., 2013, 201–202, 68–93 CrossRef CAS PubMed.
- H. Liu and C. Wang, RSC Adv., 2014, 4, 3864–3872 RSC.
- Y. Chen, L. Chen, H. Bai and L. Li, J. Mater. Chem. A, 2013, 1, 1992–2001 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00002a |
| This journal is © the Partner Organisations 2017 |