DOI:
10.1039/C6QM00025H
(Research Article)
Mater. Chem. Front., 2017,
1, 780-788
Fabrication of functionalized graphene filled carboxylated nitrile rubber nanocomposites as flexible dielectric materials†
Received
21st April 2016
, Accepted 17th October 2016
First published on 14th November 2016
Abstract
Hexadecylamine (HDA) was used to covalently functionalize graphite oxide followed by its reduction to form functionalized graphene nanosheets (GNS–HDA). Subsequently, GNS–HDA has been used as filler in fabricating carboxylated nitrile rubber (XNBR) nanocomposites via solution mixing. Mechanical measurements of XNBR/GNS–HDA-2 nanocomposites compared to neat XNBR exhibited 60% improvement in tensile strength (TS), 62% improvement in elongation at break (EB) and 13% reduction in Young's modulus (YM). Thermogravimetric studies demonstrate that incorporation of GNS–HDA into a XNBR polymer matrix increases the thermal stability of composites with GNS–HDA loading. Further investigations show significant enhancement in the dielectric constant of XNBR/GNS–HDA-2 (127.6) compared to neat XNBR (8.9) at 100 Hz. The corresponding value of the dielectric loss of the composite was enhanced only ∼2 times. Such enhanced TS, EB, reduced YM and dielectric constants while retaining low dielectric loss enables the possible application of XNBR/GNS–HDA as flexible dielectric materials in electronic devices.
1. Introduction
Graphene is a two-dimensional material which is composed of several atomic sheets of sp2 carbon atoms arranged in a hexagonal lattice. It remains one of the thinnest material compared to other allotropes of carbon (graphite, CNTs, CF, fullerenes etc.). The high specific surface area, high aspect ratio, superior high Young's modulus, fracture strength, and thermal conductivity of graphene make it an attractive material for many important applications.1–3 Graphene has successfully been used as a reinforcing filler in polymers, exhibiting its outstanding structural, electrical, and mechanical properties. The low price and availability of pristine graphite, followed by a simple methodology applied in the formation of graphene make it one of the potential conductive fillers in polymers. However, poor dispersion of graphene in a polymer matrix remains a major barrier and can be overcome by covalent and noncovalent functionalization.4–6 Furthermore, the low dielectric constants (less than 10) of most of the polymers restricts their direct application in many electrical fields. Therefore, it is desirable to find out ways to enhance the dielectric constant of polymers, while retaining a low dielectric loss and good flexibility.
Dielectric elastomers (DEs) are electroactive-polymers (EAPs), which have the ability to convert electrical energy to mechanical energy over a broad frequency range.7,8 The lightweight, low modulus, large strain, fast response, high energy density and high electromechanical coupling efficiency9,10 of DEs account for their application in artificial muscles, sensors, micro-air vehicles, flat-panel speakers, micro-robotics and responsive prosthetics.11–14 In order to obtain DEs in the application of high actuated strain and at a low electric field, a high value of electromechanical sensitivity, β (β = dielectric constant, k)/(elastic modulus, Y) is required. Accordingly, a high dielectric constant and low elastic modulus are the solution for this problem. When a conducting filler is incorporated into polymer matrices, k as well as Y values usually increase simultaneously.
The dielectric constant of the polymer can be enhanced by incorporating high dielectric constant ceramic materials such as BaTiO3 and TiO2. But a high amount of ceramic material (50 vol%) is required to enhance the desired dielectric constant, resulting in an enhancement of the elastic modulus, lowering of flexibility and poor processability, which narrow down the applications of DEs. Alternatively, DEs can also be fabricated through percolative composites by incorporating conducting fillers such as graphene sheets and carbon nanotubes.15–18 The choice of graphene in such work is also guided by its easy synthesis from natural graphite compared to carbon nanotubes. The dispersion of covalent19–23 and non-covalent modified24–29 graphene and filler–polymer interfacial interactions account for its reinforcing ability, especially at low filler loading. Interestingly, the dielectric constant of such polymer composites increases abruptly near the percolation threshold.18 When the percolation threshold is reached, an undesirable enhancement in the dielectric loss of composites is also inevitable.4 The existing literature also highlights the inferior extensibility of these polymer nanocomposites.30,31 In view of this, several rubbers filled with graphene oxide or graphene have been investigated as DE materials. Poly(isobutylene-co-isoprene)/reduced graphene oxide nanocomposites were prepared by a solution blending method at 5 wt% loading. It introduces the dielectric properties over a broad frequency range and at different temperatures. The result shows that the dielectric constant at room temperature for reduced graphene oxide nanocomposites increases 10 times (29 at 10−2 Hz) when compared with that of expanded graphite composites (2.9).32 Al-Hartomy et al.33 studied the dielectric and microwave properties of graphene nanoplatelets (1 to 5 phr)/carbon black (50 phr) filled natural rubber composites in the 1–12 GHz frequency range. Graphene oxide-based dielectric elastomer composite materials exhibited high dielectric constants and electric breakdown strength and low dielectric loss.4,34–39 Potts et al.34 fabricated NR/2 wt% reduced graphene oxide (RGO) nanocomposites by co-coagulating latex composites followed by mixing in two roll mill. Subsequently, they observed reduction in the elongation at break by 50% in the corresponding XNBR nanocomposite. It is reported that increased dispersion level and interactions between the rubber chain and filler materials render the dissipation of external stress throughout the polymer matrix. Liu et al.35 reported that dissipation of external stress can occur through alignment and slippage of rubber chains adsorbed on the surface of nanofiller. Poly(dimethyl) siloxane embedded with 2 wt% thermally expanded graphene sheets showed an increment in k from 2.7 to 23 at frequency of 10 Hz.4 The low increment in the dielectric constant than expected could be ascribed to the poor dispersion and restacking of graphene sheets. In another interesting work, Jiang et al.14 prepared 5 wt% loaded hyper branched aromatic polyamide functionalized graphene sheets (GS–HBA) into a TPU matrix and achieved an enhancement in the k value to 850 at 1000 Hz. However, the low dielectric loss is retained (<1.5) due to the suppression of leakage current by hyperbranched aromatic polyamide functional groups. Noncovalent-modified graphene (3%) polyurethane nanocomposite showed a dielectric constant of 350, a dielectric loss of 0.20 and a loss modulus of 200 MPa at room temperature in 1 kHz.36 According to Ning et al.,37 poly(dopamine) (PDA) coated GO (GO-PDA) assembled around a carboxylated nitrile rubber (XNBR) latex showed that dielectric loss and elastic modulus decrease, and breakdown strength increases upon increasing the thickness of the PDA shells. Tian et al.38 reported carboxylated nitrile rubber (XNBR)/graphene oxide nanosheet encapsulated carbon sphere (GON–CNS) nanocomposites by latex. The compounding dielectric composites exhibited a low dielectric loss and improved breakdown strength and actuated strain at a low electric field facilitating the wide application of dielectric elastomers. Another work involved the formation of dielectric composites prepared by using the combined strategy of encapsulating graphene oxide nanosheets (GONS) on XNBR latex particles and the in situ thermal reduction in GONS at a moderate temperature.39 The corresponding nanocomposites exhibited high dielectric constant, low dielectric loss and low percolation threshold. The available literature also suggests that a high aspect ratio and low content of graphene37–39 are enough to enhance the k value compared to ceramic dielectric fillers,7,8 and retains a low elastic modulus and better processability. It is to be noted that carboxylation of the butadiene-acrylonitrile latex confers colloidal stability.40 In addition, XNBR latex is also stabilised with at least one external surface-active agent.40 However, the drawbacks of latex stabilization during storage, mixing, and processing are determined by time, temperature, phase separation, pH drift, shear in mixing etc.41 Therefore, it would be interesting to use raw rubbers as alternatives in order to overcome all these undesirable hurdles in the fabrication of flexible dielectric elastomeric nanocomposites. However, no significant advances have been made in fabricating mechanically improved and thermally enhanced XNBR/graphene dielectric materials directly from XNBR rubber and overcoming undesirable hurdles in fabrication imposed by the corresponding latex.
Motivated by this, we focused our work on the functionalization of graphene nanosheet (GNS) surfaces using hexadecylamine (HDA). It is anticipated that such a modification of graphene could also reduce the dielectric loss by preventing the direct current conductance and enhance the compatibility of functionalized reduced graphene oxide with the polymer matrix. Furthermore, the low elastic modulus of the composite could overall lead to its application as a flexible dielectric material. The choice of carboxylated nitrile rubber (XNBR) in our work is mainly guided by the presence of strong polar carboxyl and cyano groups, which could form a strong adhesion between GNS–HDA and XNBR. Finally, these XNBR/GNS–HDA nanocomposites have been studied for their thermal stability, mechanical and dielectric properties.
2. Experimental
2.1 Materials
XNBR (Krynac® X 740, LANXESS) and graphite Micro-850 were procured from Germany and Asbury Graphite Mills, Inc., Asbury, NJ, respectively. HDA (98%), ethanol, dicumyl peroxide (DCP), and sodium nitrate (NaNO3) were supplied by SRL (Mumbai), Hercules (USA) and S. D. Fine Chemicals (India) respectively. Potassium permanganate (KMnO4), concentrated sulfuric acid (H2SO4), hydrogen peroxide (H2O2), and hydrazine hydrate (N2H4) were purchased from Merck, India. Tetrahydrofuran (THF) was obtained from RFCL Limited, New Delhi.
2.2 Preparation of graphene oxide
GO was prepared by the modified Hummers’ method reported earlier.42 According to this method, graphite (500 mg) along with sodium nitrate (250 mg) were taken in a beaker, containing 30 ml concentrated H2SO4, followed by addition of 1.5 g KMnO4 under stirring for 30 min. The resulting mixture was diluted with deionized water and then 10 ml of H2O2 was added to this. As a result, the color of the solution turned to brilliant yellow, indicating the formation of fully exfoliated graphene oxide. Finally, the product free from metal ions was obtained by washing it several times with dilute HCl followed by deionized water to remove the free acid.
2.3 Preparation of hexadecylamine functionalized graphene
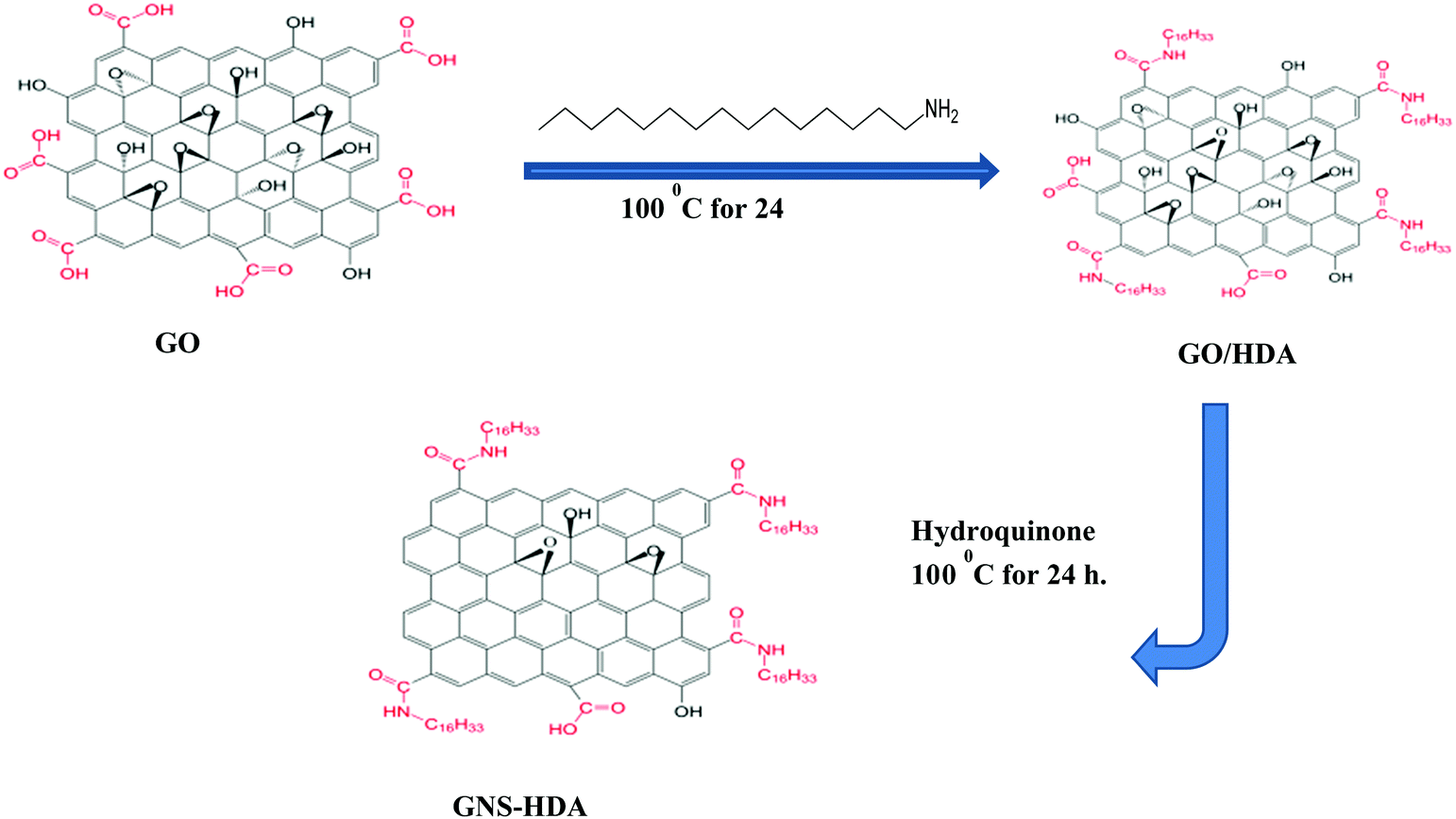
Graphene oxide (200 mg) nanosheets prepared earlier by Hummers’ method were dispersed in 30 ml of water by sonication for 30 min. Subsequently, HDA modified graphene was prepared based on an idea from earlier work.43,44 Accordingly, HDA (300 mg) was dissolved in ethanol (60 ml) followed by heating and cooling. This cooled solution of HDA was added to the dispersion of graphite oxide in water prepared earlier with sonication continuing for 30 min. The resultant mixture was subsequently refluxed at 100 °C in the presence of hydroquinone (400 mg) for 24 hours. The formed solid product (GNS–HDA) was centrifuged followed by washing with ethanol and drying at 65 °C in a vacuum for 24 h. The prepared GNS–HDA is indicated by its dispersion in organic nonpolar solvents (tetrahydrofuran, cyclohexane, toluene, and ethanol etc.).
2.4 Preparation of GNS–HDA/XNBR composites
XNBR/GNS–HDA nanocomposites were prepared by a solution mixing method. Initially, GNS–HDA nanofillers (0, 0.5, 1, 1.5, 2 phr) were dispersed in THF with the assistance of an ultrasonicator for 5 min. Accordingly, 5 g of XNBR was dissolved in 50 ml THF in a 100 cc beaker. In another beaker, the required amount of GNS–HDA nanofiller was dispersed in THF under ultrasonic treatment for 5 min. Following this, it was added to XNBR solution at room temperature and magnetically stirred for half an hour, followed by the addition of 2 phr DCP. The stirring was continued for another 6 h. Finally, the solution was cast in a glass petri dish to evaporate the solvent, THF, at room temperature. Finally, the cast films were cured at 160 °C for 10 min, according to the cure time obtained from oscillating disc rheometer analysis (ODR). In a similar manner, XNBR composites filled with 0.5, 1.0, 1.5 and 2 phr of GNS–HDA were also fabricated. The obtained sheets were taken for further characterization and application.
2.5 Characterization
The structure of GNS–HDA was analyzed by Fourier transform infrared spectroscopy (FTIR) on a PerkinElmer RXI FTIR spectrometer, USA, in the frequency range of 400–4000 cm−1. The crystalline/amorphous nature of GNS–HDA was obtained by carrying out X-ray diffraction analysis on a Bruker D8, Advance X-ray diffractometer with Cu Kα (λ = 0.1542 nm) radiation in the 2θ range of 5° to 80° and at a scan rate of 10° min−1. The morphology of the samples was analyzed by TEM using a JEOL2100 microscope at a voltage of 200 keV. The sample preparation for TEM observation was carried out by cutting the composite film with an ultramicrotome into thin sections of 100 nm thickness with a diamond knife and the sample was collected on 200-mesh copper grids. The morphology of the tensile fractured surface of the samples was studied using a Novanano SEM 430 scanning electron microscope (Germany). Atomic force microscopy (AFM) measurement was carried out on a Veeco Multimode V scanning probe microscope in the tapping mode. Samples for AFM images of GNS–HDA and GNS were prepared by spin coating on a mica surface using 0.1 mg ml−1 dispersion in THF. The thermal stability of the samples was determined on a TGA Q5000 instrument in the temperature range of 60–600 °C at a heating rate of 10 °C min−1 in nitrogen gas purging. Differential scanning calorimetry (DSC) measurement of the samples was recorded on a TA instruments Q-20 from −50 °C to room temperature. Dielectric properties were obtained using a QuadTech 7600 precision LCR meter in the frequency range of 10 Hz to 1 MHz.
3. Results and discussion
3.1 Structure and morphology of GNS–HDA
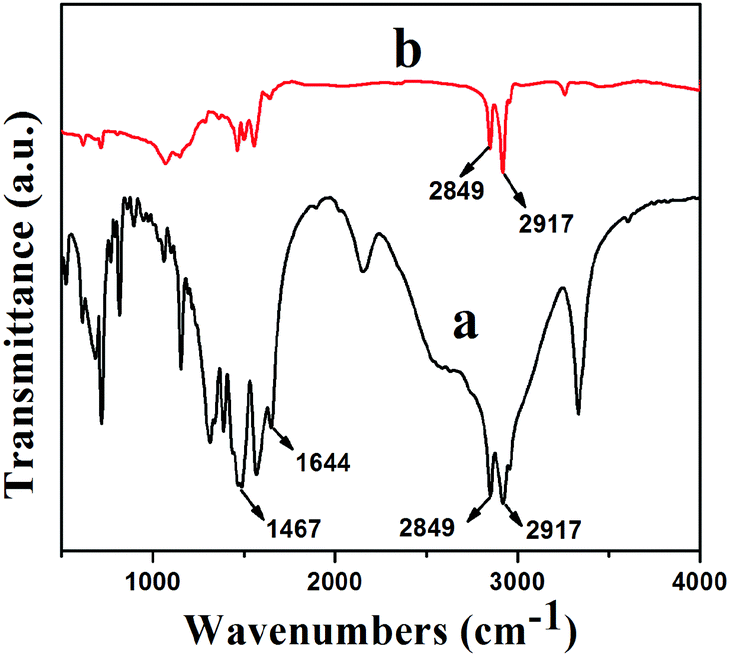
Fourier transform infrared (FTIR) spectra of HDA and GNS–HDA are displayed in Fig. 1. It is clearly evident that two asymmetric bands appeared at about 2849 cm−1 and 2917 cm−1 in HDA as well as the presence of an amine group (–NH2) in GNS–HDA, clearly signifying the functionalization of graphene nanosheets by HDA. Furthermore, the appearance of the carbonyl stretching vibration at 1644 cm−1 established the formation of a linkage between the reduced graphite nanosheets and HDA. It is known that graphene oxide (GO) consists of hydroxyl (–OH) and epoxy groups lying above and below the basal planes and the carboxyl groups (–COOH) bound to the edges.37 Therefore, it is anticipated that functionalization of GO takes place via covalent bonding between the –NH2 (HDA) and –COOH (GO) groups as proposed in Scheme 1.
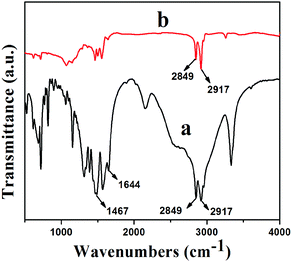 |
| | Fig. 1 FTIR spectra of (a) GNS–HDA and (b) HDA. | |
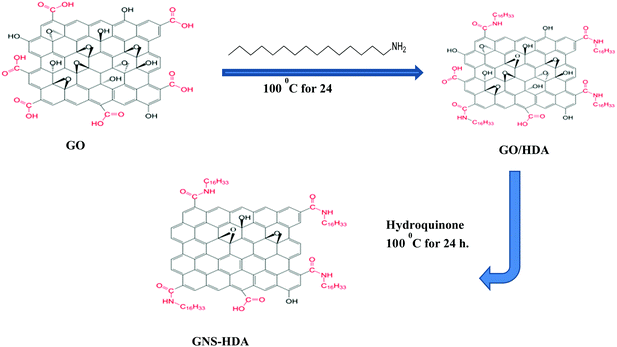 |
| | Scheme 1 Chemical functionalization of HDA on graphene sheets. | |
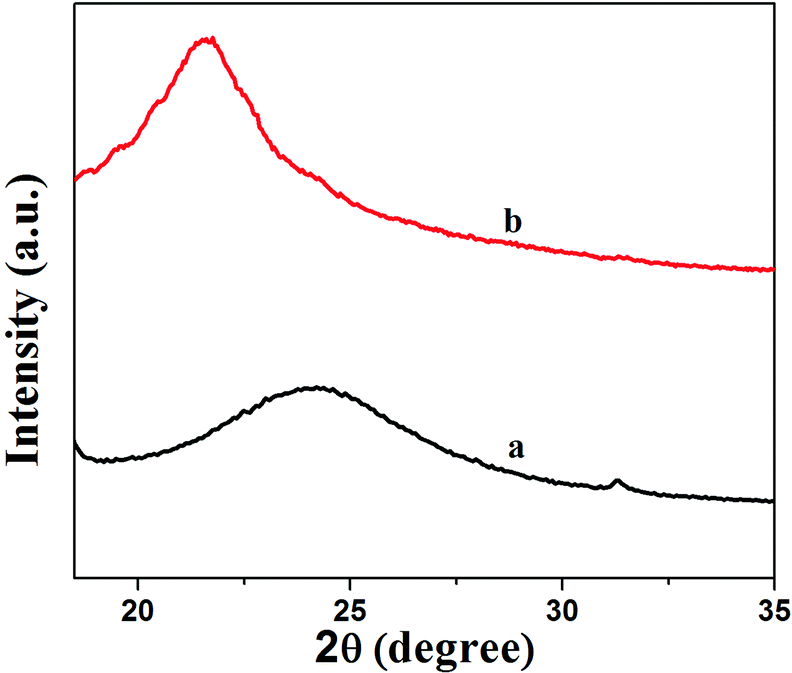
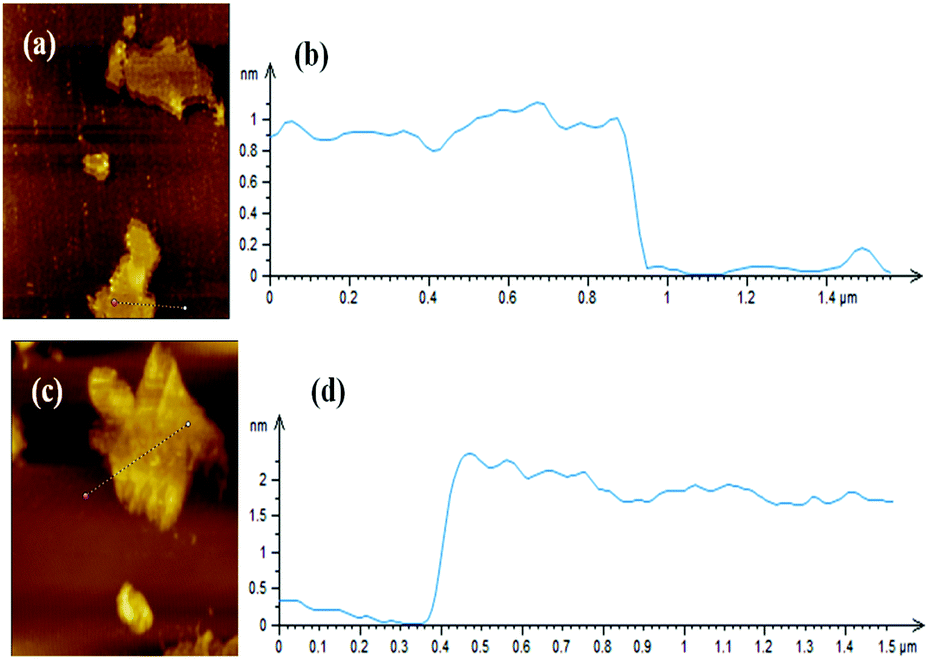
Fig. 2 shows XRD patterns of GNS and GNS–HDA. It is noted that a (002) diffraction peak appeared for GNS at ∼24°, corresponding to a d-spacing of 0.34 nm. However, a (002) diffraction peak of GNS–HDA appeared at 21.5° (d002 = 0.41 nm). These findings further strengthened our contention based on FTIR about the attachment of an amine group (HDA) on the graphene nanosheets. In order to achieve further validation, AFM analysis of GNS and GNS–HDA was performed in the tapping mode and is displayed in Fig. 3. The multilayers with broad distribution in thickness and size are clearly evident from this. The height of the GNS layers was found to be ∼1 nm and less than that of graphite oxide due to the expulsion of different oxygen functionalities (>C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –COOH, –OH, –O–).
O, –COOH, –OH, –O–).
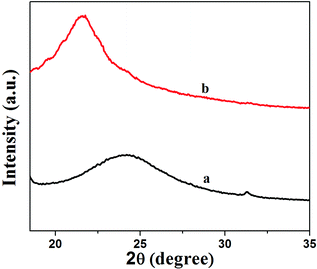 |
| | Fig. 2 XRD patterns of (a) GNS and (b) GNS–HDA. | |
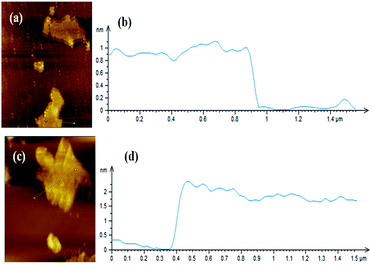 |
| | Fig. 3 AFM image of (a) GNS, (b) height profile of GNS, (c) AFM image of GNS–HDA, and (d) height profile of GNS–HDA. | |
However, HDA functionalized graphene sheets exhibited relatively higher thickness (∼2 nm).5 Our findings are also in agreement with AFM studies reported on individually dispersed graphene and rhodamine B noncovalently modified graphene exhibiting a thickness of about 1 and 1.6 nm respectively.45
The morphology of GNS and GNS–HDA has been studied by FESEM and TEM analysis and the corresponding findings are displayed in Fig. 4. It is clearly evident that GNS–HDA exists as aggregated, thin crumpled nanosheets which are largely associated with each other. The TEM image of GNS–HDA showed the appearance of a flat and rippled morphology. The presence of light gray color in this image corresponds to the formation of dispersed GNS–HDA thin layers, while darker regions indicated the stacking of the GNS–HDA layers.
 |
| | Fig. 4 (a) FESEM and (b) TEM images of GNS–HDA. | |
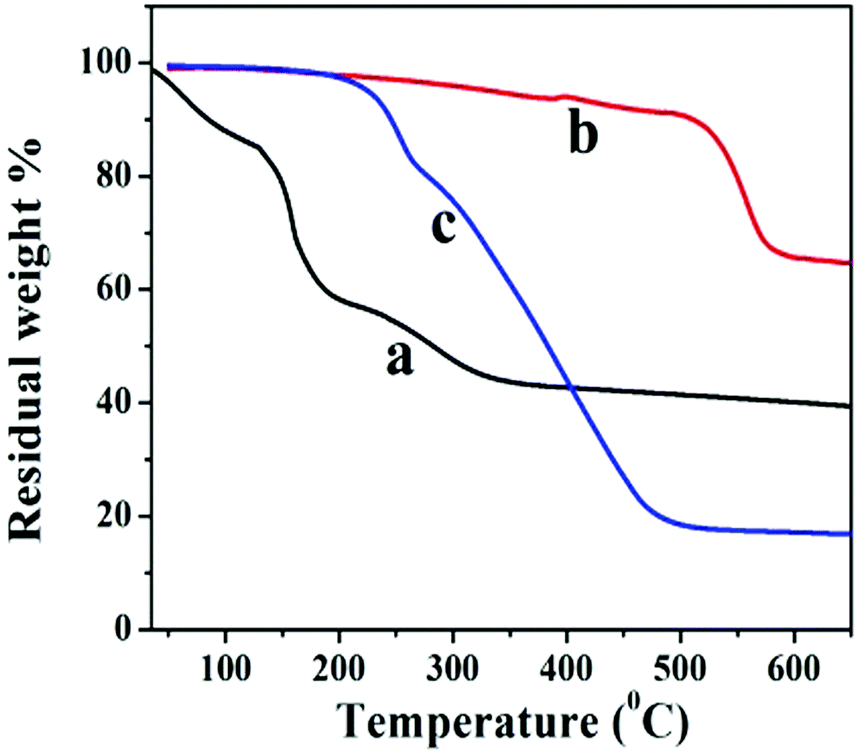
The thermal stability behavior of GO, GNS, and GNS–HDA has been investigated in the temperature range of 60–600 °C under a nitrogen atmosphere and the corresponding findings are displayed in Fig. 5. The initial weight loss (∼12%) in GO between 60 °C and 130 °C can be ascribed to the adsorbed water. At further higher temperatures, TG shows a weight loss of 56% in the range of 130–325 °C due to pyrolysis of hydroxyl, carbonyl, epoxy, and carboxylic acid groups in GO. After 325 °C no significant weight loss is noticed due to the expulsion of oxygen functionalities in GO. It is also noted that GNS and GNS–HDA are both thermally stable up to 195 °C. This suggested that neither GNS nor GNS–HDA is sensitive towards moisture adsorption. This could be ascribed to the enhanced hydrophobic nature of GNS–HDA achieved by effective functionalization of HDA on the graphene surface. At higher temperatures, GNS undergoes a significantly low weight loss of ∼5% due to incomplete reduction of the remaining oxygen functionalities in GO. In contrast, degradation of GNS–HDA takes place much faster and achieves a weight loss of ∼80% (495 °C). This is attributed to the complete degradation of hexadecylamine due to the functionalization of GNS. However, the possibility of a fraction of HDA adsorbed on the surface of graphene also cannot be ruled out.35 The room temperature dispersion stability of GNS–HDA and GNS has been investigated for THF (10 mg ml−1) for about 1-month duration by recording its digitized image. Fig. 6 shows that chemically functionalized GNS–HDA has significantly higher dispersion stability in THF compared to GNS.
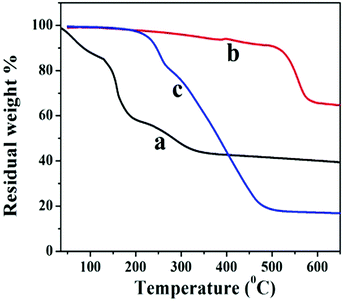 |
| | Fig. 5 TGA of (a) GO, (b) GNS, and (c) GNS–HDA. | |
 |
| | Fig. 6 Digital picture of the dispersion stability of (a) GNS–HDA and (b) GNS. | |
3.2 Structure and morphology of GNS–HDA/XNBR composites
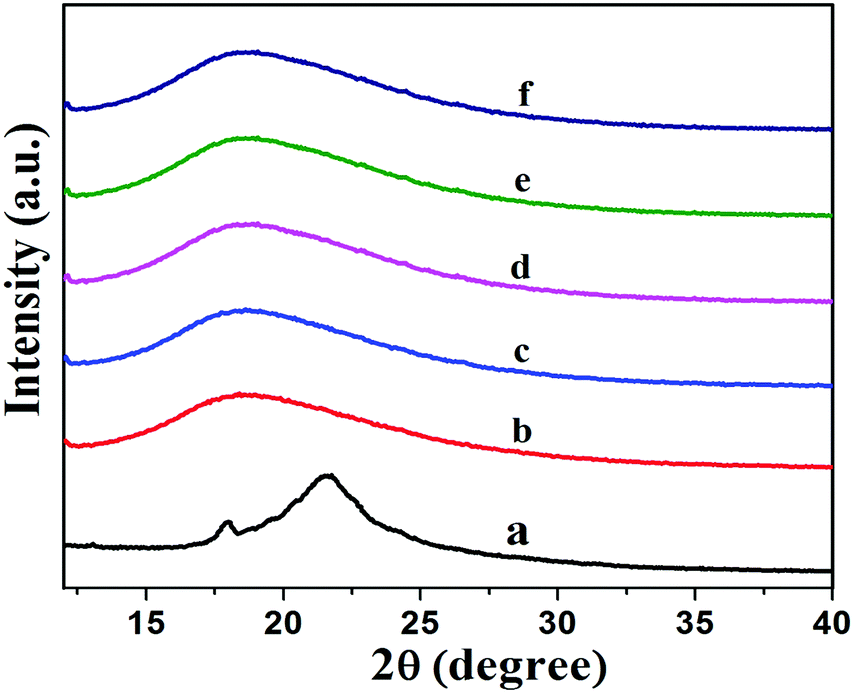
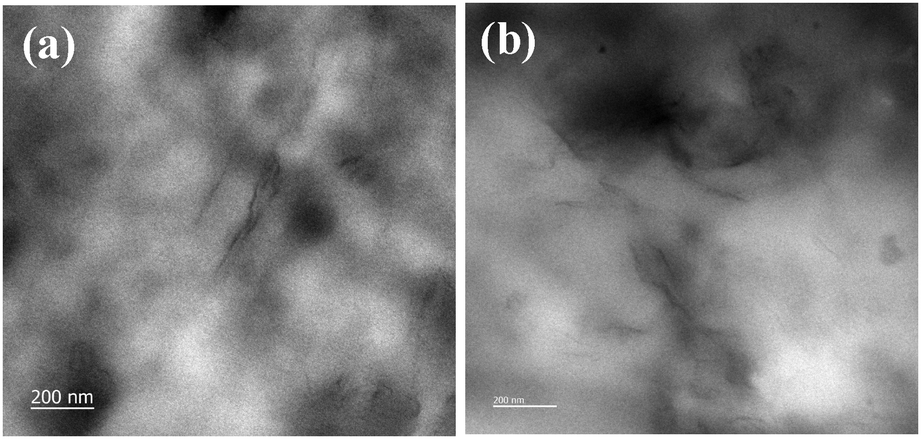
Fig. 7 shows XRD patterns of XNBR and its 0.5, 1, 1.5 and 2 phr GNS–HDA loaded nanocomposites. It is noted that the crystalline and broad diffraction peak of GNS–HDA (2θ = 21.50) completely disappeared for all nanocomposites. Such observations demonstrated the possible exfoliation of GNS–HDA in the XNBR matrix.39,46 In order to further validate this, HRTEM analysis of XNBR/GNS–HDA composites has also been carried out and the findings are displayed in Fig. 8. It clearly shows that the wrinkled and folded sheets of GNS–HDA exhibit good dispersion in the XNBR matrix. This is in all probability due to the compatibility between GNS–HDA and the XNBR matrix and accounts for the significantly enhanced properties of XNBR/GNS–HDA nanocomposites.
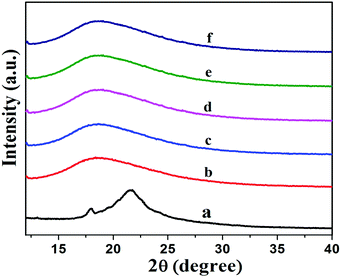 |
| | Fig. 7 XRD spectra of (a) GNS–HDA, (b) neat XNBR, (c) 0.5 phr, (d) 1 phr, (e) 1.5 phr, (f) 2 phr GNS–HDA filled XNBR composites. | |
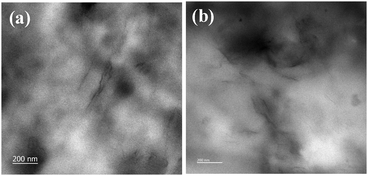 |
| | Fig. 8 HRTEM of (a) 1 phr and (b) 1.5 phr GNS–HDA filled XNBR composites. | |
3.3 TGA of XNBR composites
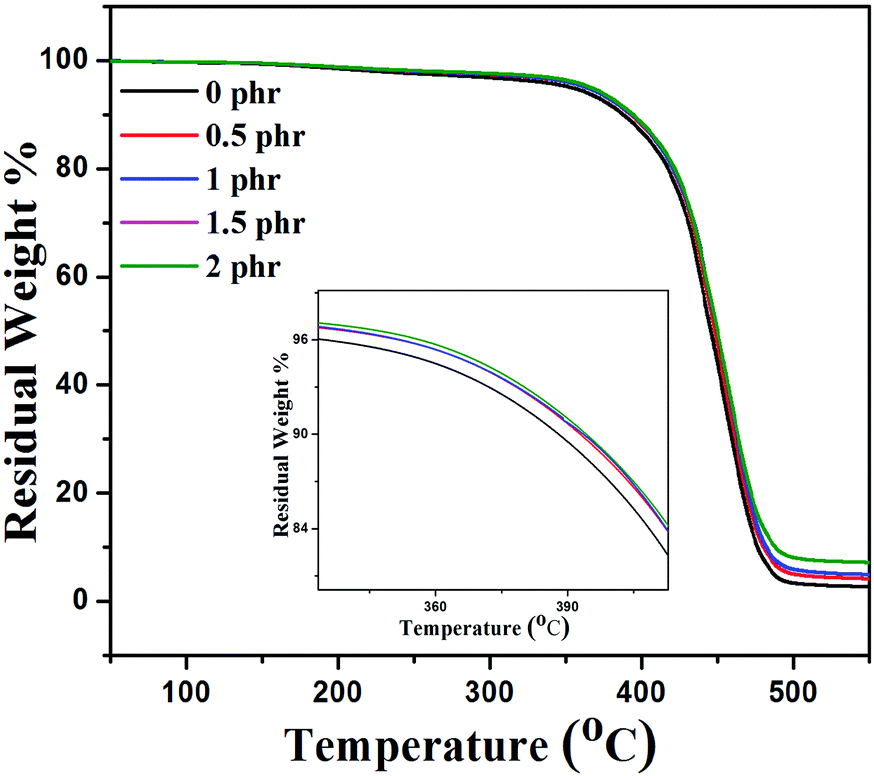
There are various studies which demonstrate that the incorporation of graphene sheets into the polymer matrix enhances the thermal stability of the composites under nitrogen atmospheric conditions.47Fig. 9 shows the thermal stability behavior of neat XNBR, 0.5, 1, 1.5, and 2 phr GNS–HDA filled XNBR under a nitrogen atmosphere and the corresponding thermal data referring to thermal degradation temperature at 10 wt% loss (T10) are 388, 392, 393, 393, and 394 °C for 0, 0.5, 1, 1.5, and 2 phr GNS–HDA loadings respectively. These findings clearly demonstrate that incorporation of GNS–HDA into the XNBR polymer matrix increases the thermal stability of composites with GNS–HDA loading. However, XNBR/GNS–HDA (0.5 wt%) shows significant improvement in thermal stability compared to neat XNBR. Such an enhancement in the thermal stability of XNBR/GNS–HDA nanocomposites is ascribed to the high aspect ratio of graphene and complete exfoliation of GNS–HDA in the XNBR polymer matrix. As a result, emission of volatile decomposed gaseous products and supply of oxygen from the surface to the bulk are hindered due to an effective barrier effect of GNS–HDA.47
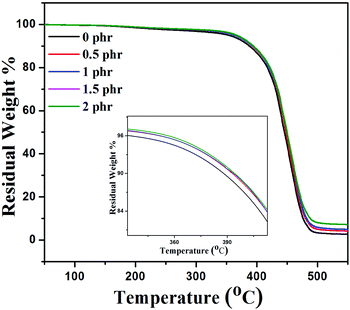 |
| | Fig. 9 TGA curve of XNBR and various GNS–HDA/XNBR composites. | |
3.4 DSC
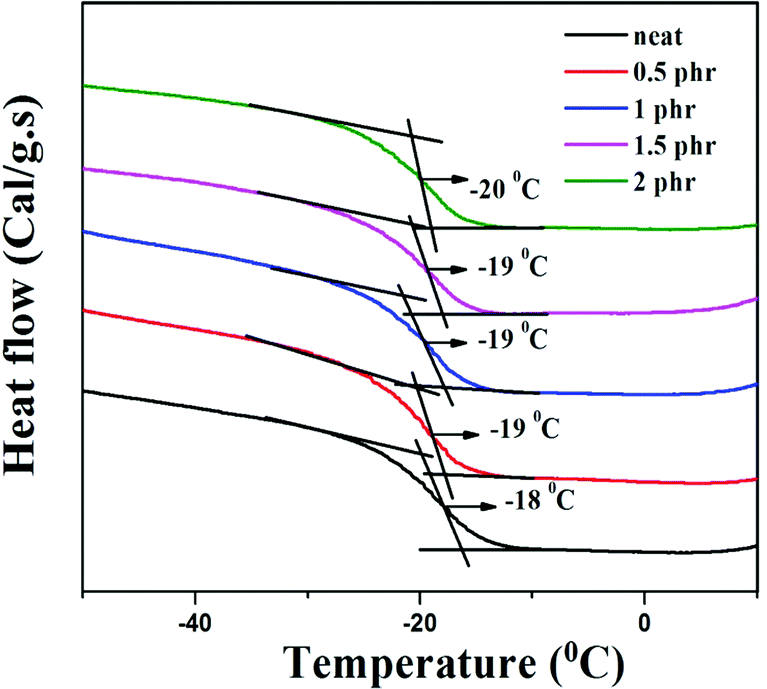
Chemically modified graphene loaded polymers usually show an enhancement in glass transition temperature (Tg).48,49 This is attributed to the close proximity of embedded nanoparticles in the polymer macromolecules, which restricts the chain mobility of polymer chains at the interface.48–50 In contrast, DSC analysis of neat XNBR and its GNS–HDA filled nanocomposites in Fig. 10 showed that Tg of XNBR decreased from −18 °C in all XNBR/GNS–HDA nanocomposites. The maximum decrease in Tg of −20 °C is attained in 2 phr GNS–HDA loaded XNBR. This could be ascribed to the enhanced chain mobility due to the presence of free surfaces and repulsive interfaces in XNBR/GNS–HDA nanocomposites.51,52 Alternatively, lowering of Tg in XNBR/GNS–HDA nanocomposites could be attributed to the plasticization effect53 or free volume existing at the polymer surface interface.54
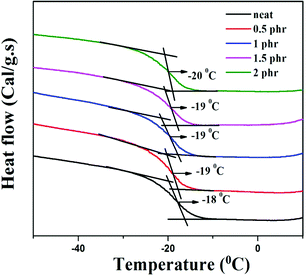 |
| | Fig. 10 DSC curve of XNBR and various GNS–HDA/XNBR composites. | |
3.5 Mechanical properties
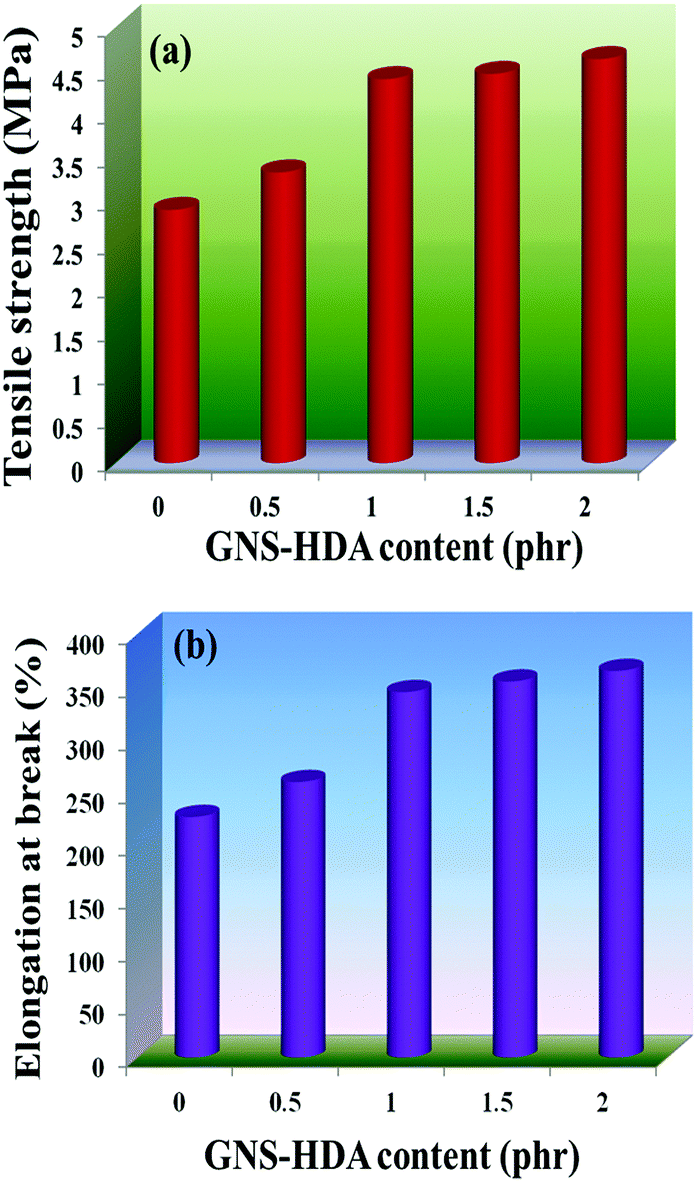
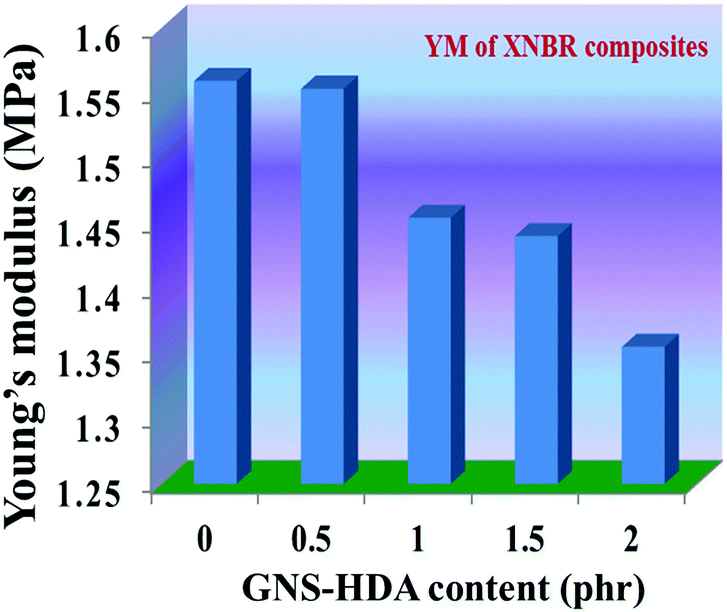
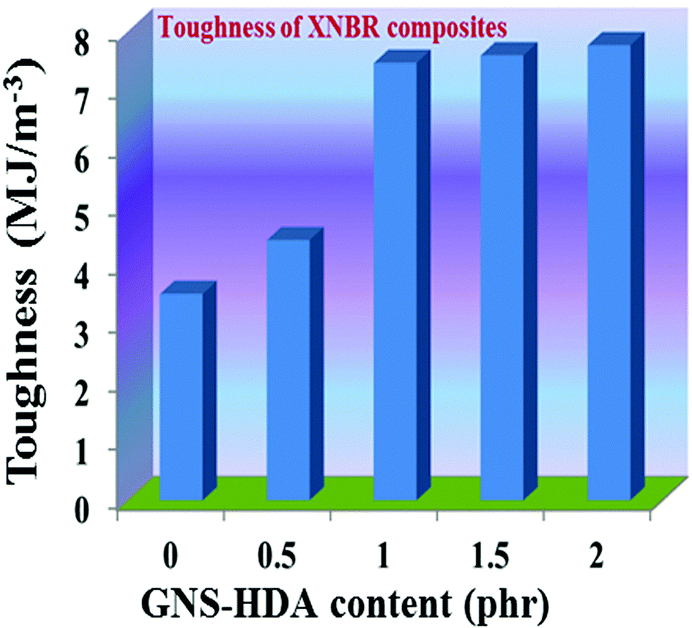
The reinforcing effect of GNS–HDA on the mechanical properties of neat XNBR has also been investigated. Tensile strength (TS), elongation at break (EB), toughness (W), and Young's modulus (YM) data have been generated from a stress–strain plot (ESI†) and are displayed in Fig. 11–13, respectively. It is noted that TS of neat XNBR (2.9 MPa) increases with the incorporation of filler loading and attained maximum improvements (4.63 MPa) in 2 phr GNS–HDA loaded XNBR. The variation in EB also shows improvement in 1, 1.5 and 2 phr filled GNS–HDA in XNBR compared to neat XNBR. It is also noted that EB of neat XNBR (227%) increased substantially to 365% in 2 phr loaded GNS–HDA in XNBR. The toughness of the GNS–HDA/XNBR composites also increases significantly with filler loading. It is well anticipated that higher dispersion of nanomaterials in the polymer matrix renders better performance of its nanocomposites. Such an enhancement in the mechanical properties of XNBR/GNS–HDA nanocomposites could be attributed to the interfacial interaction between GNS–HDA and the XNBR matrix and effective load transfer from the filler to the polymer matrix.46,50 Alternatively, the role of molecular level dispersion of GNS–HDA and its wrinkled shape leading to mechanical interlocking and transmitting the applied stress to XNBR also cannot be ruled out.51 In addition, crumpled GNS–HDA adds some extra reinforcement at high elongation when it is stretched, which is attributed to the enhancement in toughness and elongation at break.51Fig. 14 shows FESEM images of the tensile fracture surface of XNBR and its GNS–HDA filled nanocomposites. It demonstrates that XNBR exhibits a smooth surface in contrast to the corresponding GNS–HDA composites, exhibiting divergence on the surface. The extent of divergence increases with an increase in GNS–HDA loading in the XNBR matrix. The YM defines the slope of the curve in the elastic region. It is inferred from Fig. 13 that the YM of the composites is reduced remarkably in GNS–HDA incorporated XNBR. This clearly indicates the reduced stiffness and enhanced flexibility of fabricated XNBR/GNS–HDA nanocomposites. It is also noted that 2 phr of GNS–HDA loaded XNBR showed a maximum decrease of about 13% in YM compared to neat XNBR. Such findings are found to be considerably superior compared to those reported for TRG/XNBR nanocomposites earlier.39 In most of the graphene polymer composites, the modulus enhances with the incorporation of filler loading,38,39 which is contrary to our study (ESI†). The increase in toughness and decrease in YM suggest that GNS–HDA existing as almost a single sheet adopting a wrinkled morphology imparts a measure of entropic and rubber like elasticity itself.51 Furthermore, the low modulus of wrinkled GNS–HDA single sheets and the high compatibility between XNBR and GNS–HDA can also account for the lowering of the modulus in its XNBR nanocomposites.
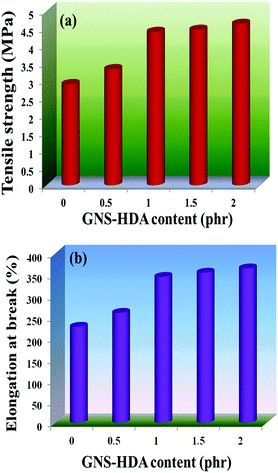 |
| | Fig. 11 (a) Tensile strength and (b) elongation at break of neat XNBR and various GNS–HDA/XNBR composites. | |
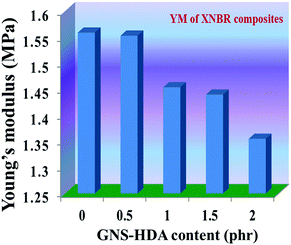 |
| | Fig. 12 Young's modulus of neat XNBR and various GNS–HDA/XNBR composites. | |
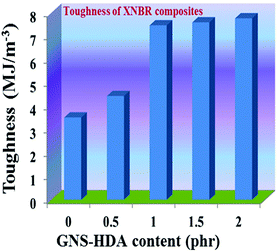 |
| | Fig. 13 Toughness of neat XNBR and various GNS–HDA/XNBR composites. | |
 |
| | Fig. 14 FESEM images of the tensile fracture surface of (a) neat XNBR, (b) 1 phr, (c) 1.5 phr and (d) 2 phr GNS–HDA loading. | |
3.6 Dielectric properties
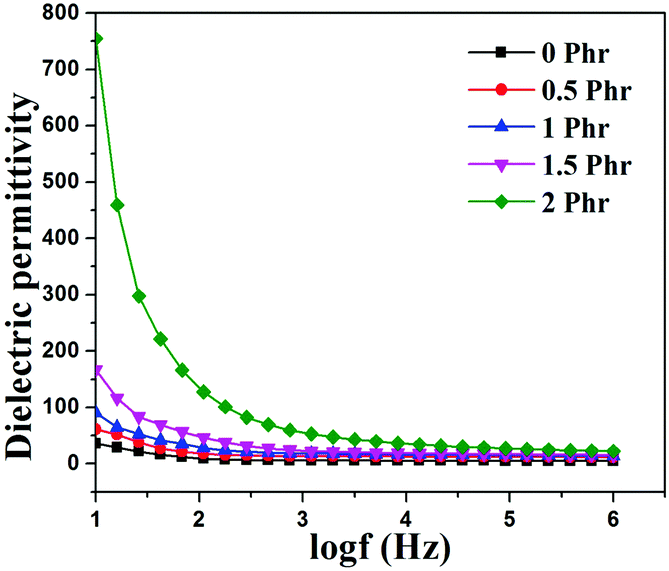
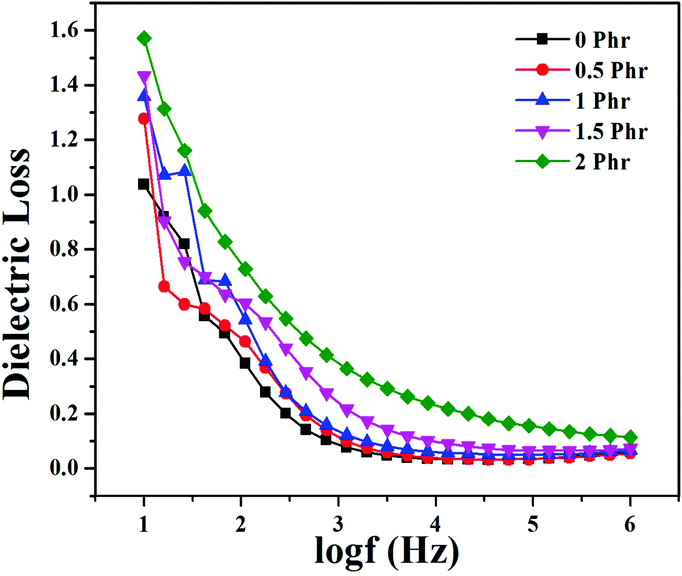
Fig. 15 and 16 show variation in dielectric permittivity (k) and dielectric loss of neat XNBR and its GNS–HDA composites as a function of frequency (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) f). It is inevitable that dielectric permittivity decreases with increasing frequency for XNBR and its GNS–HDA composites. Such strong frequency dependence of dielectric permittivity can be attributed to the interfacial polarization effect between the GNS–HDA and XNBR (Maxwell–Wagner–Sillars effect).39 The findings based on dielectric constants and dielectric loss data of XNBR and its GNS–HDA composites at 100 Hz along with the values reported on other elastomers are presented in Table 1. HDA filled XNBR nanocomposites increase nearly 15 fold, whereas dielectric loss increases only 2 fold compared to neat XNBR. It is also evident from Fig. 15 that the dielectric permittivity of GNS–HDA/XNBR composites is relatively higher in the low frequency range (logf: 1–3) followed by a significant decrease at the higher frequency region. In all probability, accumulation of charge between GNS–HDA and XNBR at the interfaces in the low frequency region accounts for the higher dielectric permittivity values of the composites. At a higher frequency, interfacial polarization lags behind the frequency of electrical field. The dielectric permittivity of the composites increases with the increase in filler loading. Furthermore, it may be interesting to mention that the presence of conductive GNS–HDA fillers (electrodes) in XNBR (insulating) allows forming the microcapacitor network.14 However, successful functionalization also plays a crucial role in the enhancement of its dielectric permittivity. Alternatively, there is a possibility that successful functionalization of graphene sheets can reduce their restacking tendency.35 This, in turn, is likely to enhance a number of microcapacitors in the polymer matrix and ultimately accounts for their higher dielectric permittivity values.14 In polymer composites, the dielectric loss generally occurs due to dipolar loss, conduction loss, and interfacial polarization loss. In graphene polymer composites, conduction loss may occur due to leakage current through direct current conduction through graphene. Fig. 16 shows a variation in dielectric loss as a function of the frequency of XNBR and its nanocomposites; the corresponding data at 100 Hz are recorded in Table 1. It is noted that the dielectric loss of XNBR (0.38) increases with GNS–HDA loading and attains a maximum value of 0.73 in 2 phr loaded composites at 100 Hz. Such negligible enhancement in dielectric loss can arise due to the interruption of π-conjugation in GNS–HDA, which seems to be due to covalent functionalization. This leads to a reduction in the direct current conductance in graphene sheets and hinders the enhancement, if any, in dielectric loss in the case of its XNBR nanocomposites. It may be noted that GNS–HDA/XNBR achieved dielectric permittivity (127 at 100 Hz) compared to that reported in TRG/XNBR (2211 at 100 Hz),39 PDA-GO/XNBR (250 at 100 Hz),37 PEDOT:PSS-RGO@PU (350 at 1000 Hz)36 and GO@CNS/XNBR (650).38 However, the dielectric loss values of GNS–HDA/XNBR (0.8 at 100 Hz) were found to be comparable with the TRG/XNBR (0.6 at 100 Hz),39 PDA-GO/XNBR (1.2 at 100 Hz),37 PEDOT:PSS-RGO@PU (∼0.2 at 1 kHz)45 and GO@CNS/XNBR (1.7 at 100 Hz)46 systems.
f). It is inevitable that dielectric permittivity decreases with increasing frequency for XNBR and its GNS–HDA composites. Such strong frequency dependence of dielectric permittivity can be attributed to the interfacial polarization effect between the GNS–HDA and XNBR (Maxwell–Wagner–Sillars effect).39 The findings based on dielectric constants and dielectric loss data of XNBR and its GNS–HDA composites at 100 Hz along with the values reported on other elastomers are presented in Table 1. HDA filled XNBR nanocomposites increase nearly 15 fold, whereas dielectric loss increases only 2 fold compared to neat XNBR. It is also evident from Fig. 15 that the dielectric permittivity of GNS–HDA/XNBR composites is relatively higher in the low frequency range (logf: 1–3) followed by a significant decrease at the higher frequency region. In all probability, accumulation of charge between GNS–HDA and XNBR at the interfaces in the low frequency region accounts for the higher dielectric permittivity values of the composites. At a higher frequency, interfacial polarization lags behind the frequency of electrical field. The dielectric permittivity of the composites increases with the increase in filler loading. Furthermore, it may be interesting to mention that the presence of conductive GNS–HDA fillers (electrodes) in XNBR (insulating) allows forming the microcapacitor network.14 However, successful functionalization also plays a crucial role in the enhancement of its dielectric permittivity. Alternatively, there is a possibility that successful functionalization of graphene sheets can reduce their restacking tendency.35 This, in turn, is likely to enhance a number of microcapacitors in the polymer matrix and ultimately accounts for their higher dielectric permittivity values.14 In polymer composites, the dielectric loss generally occurs due to dipolar loss, conduction loss, and interfacial polarization loss. In graphene polymer composites, conduction loss may occur due to leakage current through direct current conduction through graphene. Fig. 16 shows a variation in dielectric loss as a function of the frequency of XNBR and its nanocomposites; the corresponding data at 100 Hz are recorded in Table 1. It is noted that the dielectric loss of XNBR (0.38) increases with GNS–HDA loading and attains a maximum value of 0.73 in 2 phr loaded composites at 100 Hz. Such negligible enhancement in dielectric loss can arise due to the interruption of π-conjugation in GNS–HDA, which seems to be due to covalent functionalization. This leads to a reduction in the direct current conductance in graphene sheets and hinders the enhancement, if any, in dielectric loss in the case of its XNBR nanocomposites. It may be noted that GNS–HDA/XNBR achieved dielectric permittivity (127 at 100 Hz) compared to that reported in TRG/XNBR (2211 at 100 Hz),39 PDA-GO/XNBR (250 at 100 Hz),37 PEDOT:PSS-RGO@PU (350 at 1000 Hz)36 and GO@CNS/XNBR (650).38 However, the dielectric loss values of GNS–HDA/XNBR (0.8 at 100 Hz) were found to be comparable with the TRG/XNBR (0.6 at 100 Hz),39 PDA-GO/XNBR (1.2 at 100 Hz),37 PEDOT:PSS-RGO@PU (∼0.2 at 1 kHz)45 and GO@CNS/XNBR (1.7 at 100 Hz)46 systems.
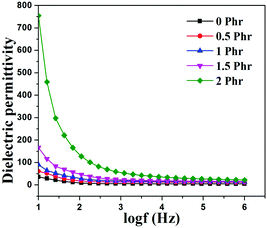 |
| | Fig. 15 Dielectric constant vs. log(frequency) for neat XNBR and various GNS–HDA/XNBR composites. | |
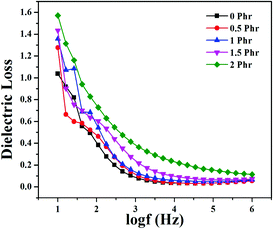 |
| | Fig. 16 Dielectric loss vs. log(frequency) for neat XNBR and various GNS–HDA/XNBR composites. | |
Table 1 Dielectric properties of XNBR and composites
| GNS–HDA (phr) |
Dielectric permittivity @ 100 Hz |
Dielectric loss @ 100 Hz |
| 0 |
8.9 |
0.38 |
| 0.5 |
17.8 |
0.46 |
| 1 |
28 |
0.54 |
| 1.5 |
46.3 |
0.60 |
| 2 |
127.6 |
0.73 |
4. Conclusion
A simple facile solution mixing method has successfully been used in developing highly dispersed HDA functionalized graphene nanocomposites of XNBR and their characterization. The mechanical properties at various loadings of HDA functionalized graphene showed efficient load transfer. As a consequence, XNBR/GNS–HDA-2 exhibited a significant improvement in tensile strength (60%) and elongation at break (62%). Furthermore, measurements of the dielectric properties indicated a maximum enhancement in the dielectric constant by 15 times (100 Hz), whereas the dielectric loss was increased only 2 fold. The thermal study showed that reduction in the glass transition temperature (Tg) of XNBR/GNS–HDA-2 is responsible for the reduction in elastic modulus. This is in all probability due to the lower elastic modulus of the wrinkled and single sheet graphene produced by hexadecylamine functionalization compared to XNBR. Such reduction in elastic modulus including enhanced tensile strength/dielectric permittivity and very low dielectric loss makes these composites useful for applications as flexible dielectric materials in various electronic devices.
Acknowledgements
S. K. Srivastava and Rakesh Manna gratefully acknowledge DRDO, India, and IIT Kharagpur for grants to R. Manna for perusing this research.
Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197 CrossRef CAS PubMed.
- Y. Zhang, Y. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201 CrossRef CAS PubMed.
- L. J. Romasanta, M. Hernandez, M. A. Lopez-Manchado and R. Verdejo, Nanoscale Res. Lett., 2011, 6, 205 CrossRef PubMed.
- Y. Luo, P. Zhao, Q. Yang, D. He, L. Kong and Z. Peng, Compos. Sci. Technol., 2014, 100, 143 CrossRef CAS.
- S. Mo, L. Peng, C. Yuan, C. Zhao, W. Tang, C. Ma, J. Shen, W. Yang, Y. Yu, Y. Min and A. J. Epstein, RSC Adv., 2015, 5, 97738 RSC.
- Y. Song, Y. Shen, H. Liu, Y. Lin, M. Li and C. Nan, J. Mater. Chem., 2012, 22, 16491 RSC.
- R. Shankar, T. K. Ghosh and R. J. Spontak, Soft Matter, 2007, 3, 1116 RSC.
- Y. Jang and T. Hirai, Soft Matter, 2011, 7, 10818 RSC.
- R. Palakodeti and M. R. Kessler, Mater. Lett., 2006, 60, 3437 CrossRef CAS.
- Y. Rao and C. P. Wong, J. Appl. Polym. Sci., 2004, 92, 2228 CrossRef CAS.
- S. M. Ha, W. Yuan, Q. Pei, R. Pelrine and S. Stanford, Adv. Mater., 2006, 18, 887 CrossRef CAS.
- R. Pelrine, R. Kornbluh, Q. Pei and J. Joseph, Science, 2000, 287, 836 CrossRef CAS PubMed.
- C. Wu, X. Huang, G. Wang, X. Wu, K. Yang, S. Lib and P. Jiang, J. Mater. Chem., 2012, 22, 7010 RSC.
- X. Zhang, G. Wang, W. Cao, Y. Wei, M. Caob and L. Guo, RSC Adv., 2014, 4, 19594 RSC.
- J. Shang, Y. Zhang, L. Yu, B. Shen, F. Lv and P. K. Chu, Mater. Chem. Phys., 2012, 134, 867 CrossRef CAS.
- Z. M. Dang, L. Wang, Y. Yin, Q. Zhang and Q. Q. Lei, Adv. Mater., 2007, 19, 852 CrossRef CAS.
- J. Shang, Y. Zhang, L. Yu, B. Shen, F. Lv and P. K. Chu, Mater. Chem. Phys., 2012, 134, 867 CrossRef CAS.
- M. Cano, U. Khan, T. Sainsbury, A. ONeill, Z. Wang, I. T. Mcgovern, W. K. Maser, A. M. Benito and J. N. Coleman, Carbon, 2013, 52, 363 CrossRef CAS.
- M. Fang, Z. Zhang, J. Li, H. Zhang, H. Lu and Y. Yang, J. Mater. Chem., 2010, 20, 9635 RSC.
- K. Manna, S. K. Srivastava and V. Mittal, J. Phys. Chem. C, 2016, 120, 17011 CAS.
- Z. Li, R. Wang, R. J. Young, L. Deng, F. Yang, L. Hao, W. Jiao and W. Liu, Polymer, 2013, 54, 6437 CrossRef CAS.
- S. Hou, S. Su, M. L. Kasner, P. Shah, K. Patel and C. J. Madarang, Chem. Phys. Lett., 2010, 501, 68 CrossRef CAS.
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856 CrossRef CAS PubMed.
- H. Bai, Y. Xu, L. Zhao, C. Li and G. Shi, Chem. Commun., 2009, 1667 RSC.
- R. Liao, Z. Tang, Y. Lei and B. Guo, J. Phys. Chem. C, 2011, 115, 20740 CAS.
- Y. Lei, Z. Tang, R. Liao and B. Guo, Green Chem., 2011, 13, 1655 RSC.
- Z. Tang, C. Zeng, Y. Lei, B. Guo, L. Zhang and D. Jia, J. Mater. Chem., 2011, 21, 17111 RSC.
- Z. Tang, Y. Lei, B. Guo, L. Zhang and D. Jia, Polymer, 2012, 53, 673 CrossRef CAS.
- M. Hernandez, M. d. M. Bernal, R. Verdejo, T. A. Ezquerra and M. A. Lopez-Manchado, Compos. Sci. Technol., 2012, 73, 40 CrossRef CAS.
- J. R. Potts, O. Shankar, S. Murali, L. Du and R. S. Ruoff, Compos. Sci. Technol., 2013, 74, 166 CrossRef CAS.
- S. K. Kumar, M. Castro, A. Saiter, L. Delbreilh, J. F. Feller, S. Thomas and Y. Grohens, Mater. Lett., 2013, 96, 109 CrossRef CAS.
- O. A. Al-Hartomy, A. A. Al-Ghamdi, F. Al-Salamy, N. Dishovsky, R. Shtarkova, V. Iliev and F. El-Tantawy, Int. J. Mater. Chem., 2012, 2, 116 CrossRef.
- J. R. Potts, O. Shankar, S. Murali, L. Du and R. S. Ruoff, Compos. Sci. Technol., 2013, 74(13), 166 CrossRef CAS.
- X. Liu, D. Sun, L. Wang and B. Guo, Ind. Eng. Chem. Res., 2013, 52, 14592 CrossRef CAS.
- T. Chen, J. Qiu, K. Zhu, J. Li, J. Wang, S. Lib and X. Wang, RSC Adv., 2014, 4, 64061 RSC.
- N. Ning, Q. Ma, S. Liu, M. Tian, L. Zhang and T. Nishi, ACS Appl. Mater. Interfaces, 2015, 7, 10755 CAS.
- M. Tian, Q. Ma, X. Li, L. Zhang, T. Nishic and N. Ning, J. Mater. Chem. A, 2014, 2, 11144 CAS.
- M. Tian, J. Zhang, L. Zhang, S. Liu, X. Zan, T. Nishi and N. Ning, J. Colloid Interface Sci., 2014, 430, 249 CrossRef CAS PubMed.
- H. M. Ghazaly and P. P. La, J. Rubber Res., 2001, 4(2), 88 CAS.
- O. H. Nautiyal, Al Ameen J. Med. Sci., 2012, 2(2), 14 Search PubMed.
- L. Shahriary and A. A. Athawale, IJREEE, 2014, 2, 2348 Search PubMed.
- J. Jang, V. H. Pham, S. H. Hur and J. S. Chung, J. Colloid Interface Sci., 2014, 424, 62 CrossRef CAS PubMed.
- G. Wang, X. Shen, B. Wang, J. Yao and J. Park, Carbon, 2009, 47, 1359 CrossRef CAS.
- Z. Tang, Y. Lei, B. Guo, L. Zhang and D. Jia, Polymer, 2012, 53, 673 CrossRef CAS.
- Y. Wan, L. Tang, L. Gong, D. Yan, Y. Li, L. Wu, J. Jiang and G. Lai, Carbon, 2014, 69, 467 CrossRef CAS.
- T. Kuila, S. Bose, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Polym. Test., 2012, 31, 31 CrossRef CAS.
- D. R. Paul and L. M. Robeson, Polymer, 2008, 49, 3187 CrossRef CAS.
- T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin, M. H. Alonso, R. D. Piner, D. H. Adamson, H. C. Schniepp, X. Chen, R. S. Ruoff, S. T. Nguyen, I. A. Aksay, R. K. P. Homme and L. C. Brinson, Nat. Nanotechnol., 2008, 3, 327 CrossRef CAS PubMed.
- R. D. Priestley, C. J. Ellison, L. J. Broadbelt and J. M. Torkelson, Science, 2005, 309, 456 CrossRef CAS PubMed.
- J. R. Pottsa, D. R. Dreyerb, C. W. Bielawskib and R. S. Ruoffa, Polymer, 2011, 52, 5 CrossRef.
- C. J. Ellison and J. M. Torkelson, Nat. Mater., 2003, 2, 695 CrossRef CAS PubMed.
- M. Pluta, J. K. Jeszkaa and G. Boiteux, Eur. Polym. J., 2007, 43, 2819 CrossRef CAS.
- P. Rittigstein and J. M. Torkelson, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 2935 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Stress–strain plots and comparative table of Young's modulus. See DOI: 10.1039/c6qm00025h |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.