
Recent advances in the synthesis of Janus nanomaterials of block copolymers
Renhua
Deng
ab,
Fuxin
Liang
a,
Jintao
Zhu
b and
Zhenzhong
Yang
*a
aState Key Laboratory of Polymer Physics and Chemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: yangzz@iccas.ac.cn
bKey Laboratory of Materials and Chemistry for Energy Conversion and Storage of the Ministry of Education, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan 430074, China
First published on 30th August 2016
Abstract
We present a review of the very recent advances in the synthesis of block copolymer (BCP) Janus nanomaterials. Although Janus micelles can form by the self-assembly of BCPs in solution, patchy or core–shell structures are usually dominant. Structural transformation of the core–shell structure or disassembly of the patchy structure is further employed to achieve Janus nanomaterials. Disassembly of ABC tri-block terpolymer supramolecular structures is advantageous in tuning much more easily the shapes of the Janus nanomaterials from spherical to cylindrical and sheet/disc-like. Narrow molecular weight distributions and strict processing conditions are required. Emulsion droplet confined self-assembly of BCPs can directly achieve Janus nanomaterials in a sufficiently small droplet and/or at low polymer concentration. A neutral emulsion interface is required using proper surfactants. Alternatively, a general method has been proposed to prepare Janus nanomaterials by guided self-assembly of BCPs within a confined environment by the strong interaction and the selective crosslinking of one block. The aforementioned Janus nanomaterials are in the form of a polymer cluster. Single chain Janus nanomaterials of BCPs can be prepared by intramolecular crosslinking of one block in a very dilute solution. It remains challenging to synthesize single chain Janus nanomaterials at high solid content for example tens of percent. At the end of this review, a perspective on BCP Janus nanomaterials is provided.
 Renhua Deng | Renhua Deng received a Bachelor’s degree (2010) and PhD (2015) from Huazhong University of Science and Technology (HUST) under the supervision of Prof. Jintao Zhu. From 2012 to 2015, he undertook a PhD program as a visiting student at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS) under the supervision of Prof. Zhenzhong Yang. In 2016, he joined Prof. Steven P. Armes' group as a postdoctoral research associate at the University of Sheffield. His research interests include the synthesis and self-assembly of block copolymers. |
 Fuxin Liang | Fuxin Liang received a Master’s degree from Liaoning University in 2007 and a PhD in polymer physics and chemistry from ICCAS in 2010. He is currently an associate professor at ICCAS. His research interests include the synthesis and applications of Janus materials, and phase change capsules. |
 Jintao Zhu | Jintao Zhu received a Bachelor’s (2000) and Master’s degree (2002) at Hunan University, and a PhD (2005) at Changchun Institute of Applied Chemistry, Chinese Academy of Science. After postdoctoral research at University of Alberta and University of Massachusetts at Amherst, he joined HUST as a full professor (2009). His current research focuses on the confined assembly of polymers and inorganic nanoparticles, and polymer hybrid materials with well-ordered structures. |
 Zhenzhong Yang | Zhenzhong Yang received a Bachelor’s at Tsinghua University (1991), Master’s at Jilin University (1994), and PhD at ICCAS (1997). After one year’s postdoctoral research at the polymer central laboratory BASF, Ludwigshafen, he joined ICCAS as assistant professor, starting his academic career. He was promoted to associate professor and full professor in 1999 and 2003. His research interests include methodology for polymer composites with tunable microstructure and spatial distribution of components and their performances. The composites involve polymer based composite hollow spheres and functional capsules, and mesoporous materials. Currently, he focuses on the synthesis, performance and practical applications of Janus composites. |
1. Introduction
Janus particles (JPs) are asymmetric in composition and/or shape. In particular, their directional interaction can lead to new complex superstructures, which are inaccessible from their symmetric counterparts.1 During the past 25 years, JPs have experienced fast development in their synthesis methodology and the exploration of diverse potential applications such as solid surfactants, building blocks toward superstructures, optical detection and display, and self-propelled nanomotors. In the category of architecture and dimensionality, JPs can be divided into spherical (including snowman-, hamburger-, hemisphere- and asymmetric patchy-), cylindrical, sheet/disc-like and other shapes.2 Many synthetic approaches have been developed to prepare JPs with varied compositions from organic to inorganic and inorganic/organic composites.3 Micrometer sized JPs can be prepared via Pickering emulsion interfacial synthesis,4 microfluidics,5 swelling polymerization and phase separation.6 Nanoscale JPs are mainly generated by polymer self-assembly and selective modification under partial protection.7Block copolymers (BCPs) are composed of different polymer segments, providing a huge family from which to derive Janus nanomaterials. Self-assembly of BCPs is an elegant process to create nanoscale supramolecular structures either in solution or bulk.8 Depending on the interaction strength, the microstructure is tunable ranging from spherical to cylindrical and lamellar. Polymeric nanomaterials with the corresponding shapes are thus derived.9 Sufficiently narrow molecular weight distributions of BCPs and strict processing conditions determine the uniformity of the supramolecular structures.10 The currently attainable architectures of the particles range from spherical micelles, to cylinders and disks (Fig. 1).11 The Janus nanomaterials formed by BCP self-assembly in solution or dis-assembly of BCP particles/bulk are typically hairy, while those obtained in confined spaces are solid.12 Both AB di- and ABC tri-BCPs can be used to derive Janus nanomaterials with varied architectures. BCP Janus nanomaterials with covalent integrity are more stable than polymer mixtures. Since the polymer chains are flexible, the Janus nanomaterials are soft. It is flexible to tune the affinity, specific interaction and responsive performance using the corresponding polymer chains. The BCP Janus nanomaterials display much stronger interfacial activity than their BCP counterparts. They have demonstrated diverse promising applications such as building units toward superstructures,13 and interfacial stabilizers.14
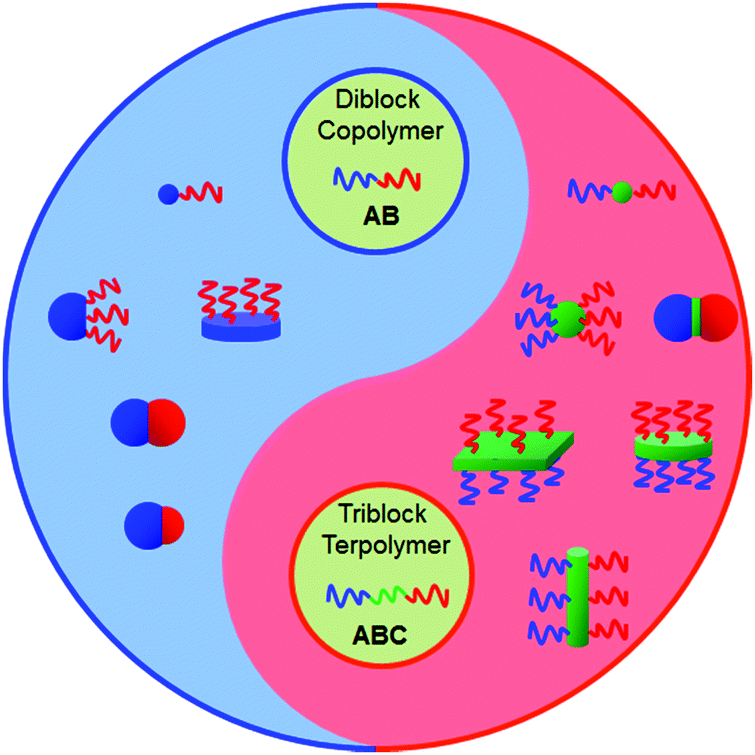 | ||
| Fig. 1 Representative morphologies of BCP Janus nanomaterials. Their architectures are tunable by using either AB di- or ABC tri-BCPs and varied processing methods. | ||
In this Review, we present a comprehensive overview of the very recent progress in the synthesis of BCP Janus nanomaterials and their physical properties. We will focus on Janus nanomaterials from linear AB di- and ABC tri-BCPs. Janus nanomaterials formed by grafting polymer chains onto preformed NPs are not included. The synthesis of Janus micelles by the self-assembly of BCPs in solution is described in Section 2. The disassembly of ABC tri-block terpolymer superstructures (patchy particles and bulk) to derive Janus spherical micelles, cylinders and sheets/discs is described in Section 3. The synthesis of Janus nanomaterials by confined self-assembly of BCPs is described in Section 4. The synthesis of single chain Janus nanomaterials by intramolecular crosslinking is described in Section 5. In Section 6, perspectives on and future concerns about BCP Janus nanomaterials are discussed.
2. Janus micelles by self-assembly of BCPs in solution
2.1 Janus micelles from di-block copolymers
In a selective solvent, di-block copolymers can self-assemble into micelles with controllable morphology ranging from spheres and cylinders, to vesicles etc. The soluble block tends to form a corona at the outer surface while the insoluble block forms the internal core. By proper sequential processing, structural transformation of the core–corona (or core–shell) micelles can be driven to form Janus micelles. In a selective solvent, the soluble poly(ethylene oxide) (PEO) block of poly(2-vinyl pyridine)-block-PEO (P2VP-b-PEO) forms the corona while the insoluble P2VP forms the core above the critical micellization concentration (Fig. 2).15 After the pyridine units be protonated by adding HCl, vesicles are achieved as a result of the structural transformation. The structural transformation is driven by the preferential parallel alignment of the restricted intermolecular conformation after complexation. De-protonation occurs after adding hydrazine monohydrate, which can induce dissociation of the vesicles. Non-centrosymmetric Janus micelles of about 25 nm are derived after re-organization of the aggregates. If HAuCl4 is added instead of HCl, the P2VP blocks are partially crosslinked within the core. After de-protonation with hydrazine monohydrate and thus dissociation of the composite micelles, a P2VP/Au composite dot is obtained on the opposite side of the PEO domain. Janus composite micelles are thus formed. Although the morphological transformation is related to interfacial energy alteration, the formation mechanism of the Janus micelles remains unclear. | ||
| Fig. 2 (a) Schematic of the formation of a P2VP-b-PEO Janus micelle and a composite micelle by morphological transformation; (b) TEM image of the Janus micelles. Reprinted with permission from ref. 15. | ||
Monte Carlo simulation predicts that the morphology of the Janus micelle from an AB/BC di-block copolymer mixture can be tunable, spanning cylinder, lamella, vesicle, and ring morphologies.16 However, co-micelles with mixed A/C corona are always dominant experimentally.17 According to the simulation, the co-micelles are thermodynamically stable when the A and C blocks are less incompatible.18 Sufficiently strong incompatibility between the A and C blocks is necessary to generate Janus micelles. However, a mixture of pure AB and BC core–corona micelles is obtained rather than AB/BC co-micelles if the repulsive interaction is too strong. The incompatibility window between A and C blocks is very narrow for Janus micelles. Alternatively, variation of the block solubility makes it feasible to induce transformation of the co-micelles into Janus micelles. As an example, a micelle with a mixed corona of PEO/PAA (PAA: poly(acrylic acid)) is formed by co-assembly of PEO-b-PAA and poly(2-vinyl naphthalene)-block-PAA (P2VN-b-PAA) via non-covalent crosslinking of the PAA core with 1,2-propanediamine (PDA).19 After substituting the solvent DMF with water at pH = 7, P2VN blocks in the mixed corona become collapsed forming isolated domains. In water, the micelle is surrounded with PEO chains. By decreasing the pH to 3.1, intra-micellar complexation between PEO/PAA occurs. After PDA is quarternized and ejected from the PAA core, an asymmetric Janus nanoparticle is achieved with a hydrophobic P2VN head and a hydrophilic PEO/PAA complex head on the opposite side. On the other hand, increasing the interaction of the B blocks of AB/BC can avoid the formation of a mixture of AB and BC micelles. In the case of the AB/B′C di-block copolymer pair, co-micellization between AB and B′C is greatly promoted by tagging the hydrogen bonding DNA base pair between the B and B′ blocks.20 Janus micelles are formed with PAA occupying one-half of the corona and PS on the other half. However, the Janus micelles are in the minority. Other non-Janus micelles with varied morphologies co-exist. The Monte Carlo simulation indicates that the micelle morphology is highly dependent on the balance between the A/C repulsive interaction and the hydrogen bonding.21 Strong hydrogen bonding favors the formation of Janus micelles.22 Similarly, co-assembly of AB/CD BCP mixtures occurs when there exists a non-covalent attractive interaction e.g., electrostatic interaction or hydrogen bonding between the core-forming blocks.23 Non-centrosymmetric ablated Janus nanoparticles of AB/CD di-block copolymers are prepared by the co-assembly of poly(N-methyl-2-vinylpyridinium iodide)-block-PEO (P2MVP-b-PEO) and PAA-block-poly(acryl amide) (PAA-b-PAAm). The nanoparticles consist of a mixed core of P2MVP/PAA. PEO and PAAm are segregated on opposite sides.24 The electrostatic attraction between B/C blocks ensures co-assembly within the same micelle. The subtle repulsion between water-soluble A/D blocks ensures segregation of the corona into two distinct domains.
2.2 Janus micelles from ABC tri-block terpolymers
Computer simulation predicts that Janus micelles could be made more easily from ABC tri-block terpolymers when incompatibility of the A/C blocks (χAC) is high.25 Experimentally, it is difficult to simultaneously satisfy the requirements of: (1) sufficiently high χAC above a threshold for phase separation, and (2) solubility of the A/C blocks in the same solvent. There is no report on the straightforward formation of ABC Janus micelles in A/C co-solvents. Similarly, proper post-processing can make it possible to achieve Janus micelles from the micelles with a mixed corona. Although Janus micelles are derived after the transformation by annealing, some non-Janus patchy micelles co-exist.26 PEO-block-poly(ε-caprolactone)-block-poly(2-aminoethyl methacrylate) (PEO-b-PCL-b-PAMA) can form a PCL core with a mixed PEO/PAMA corona. A soluble silica precursor tetramethyl orthosilicate (TMOS) can swell the PCL core. During the sol–gel process, the hydrolyzed intermediates of TMOS are anionic in a slightly acidic environment. The cationic PAMA chains are thereby strongly bridged. As a result, the electrostatic interaction favors lateral segregation of the cationic PAMA corona chains. Eventually, the PAMA and PEO chains are separated into two domains at the core surface.27 The poly(styrene)-block-polybutadiene-block-P2VP (PS-b-PB-b-P2VP) micelles with a mixed PS/P2VP corona can be converted into Janus NPs after an intra-micellar crosslinking of the P2VP chains (Fig. 3).28 In the mixed corona, at a PS/P2VP length ratio of 3.9/1, inter-micellar crosslinking of the P2VP chains is effectively prohibited by protection from the long PS block chains. The PB core is large and dynamic, and the crosslinkable P2VP block chains at the corona are too short to form an integrated shell covering the entire PB core surface. At an early stage of the intra-micellar crosslinking, multiple small crosslinked P2VP domains form. Eventually, the crosslinked P2VP domains aggregate into an individual large domain at one side of the PB core. Meanwhile, the PS blocks at the corona are sufficiently large to be excluded toward the other side. A Janus nanoparticle is thus achieved. | ||
| Fig. 3 (a) Schematic of the formation of Janus micelles from PS-b-PB-b-P2VP; (b) TEM image of the Janus micelles. Reprinted with permission from ref. 28. | ||
The other cases where the solvents are selective for only one block to form core–shell–corona or patchy multi-compartment micelles (MCMs) are summarized in recent reviews.29 Although patchy MCMs are not Janus, they can derive Janus nanoparticles by disassembly. This will be discussed in Section 3.1.
3. Janus nanomaterials by disassembly of ABC tri-block terpolymer supramolecular structures
3.1 Janus nanomaterials by disassembly of patchy multi-compartment micelles
Disassembly of MCMs with crosslinked B-patches is effective toward the formation of Janus nanomaterials.30 Simulation results indicate that ABC tri-block terpolymers can form patchy MCMs in C (or A) block selective solvents.31 Experimentally, patchy MCMs are easily achieved by a two-step self-assembly.32 Micelles with a mixed shell are firstly prepared in A/C block co-solvents, which can form patchy ones after the solvent is substituted with a C (or A) block selective solvent.33 Janus nanomaterials are prepared by selectively crosslinking the patches followed by their disassembly.34 In a poor solvent for the B block of poly(tert-butyl acrylate)-block-poly(2-cinnamoyloxyethyl methacrylate)-block-poly(succinated glyceryl monomethacrylate) (PtBA-b-PCEMA-b-PSGMA), complexation occurs between the C block and added sparteine. Hamburger-like micelles are thus achieved. The complexed sparteine/PSGMA blocks form the “patty”, and the PCEMA blocks form the two “buns” with PtBA blocks on their surface. After crosslinking the PCEMA “bun” compartments, the hamburger micelles could be separated along their central “patty” after removal of the sparteine. Janus nanomaterials with PSGMA and PtBA on opposite sides are achieved after ultrasonication. In a poor solvent of the middle B block, ABC tri-block terpolymers can self-assemble into micelles with mixed A/C patchy corona surrounding the B cores. The micelles collapse into MCMs after substitution for another poor solvent for both the A and B blocks. These MCMs are composed of A core and B patches and an emanating C corona. After crosslinking of the B patches, Janus nanomaterials are achieved via dissolution with a good solvent for both the A and C blocks (Fig. 4).35 The method is feasible for a wide range of tri-block terpolymers such as PS-b-PB-b-PMMA (PMMA: poly(methyl methacrylate)), PS-b-PB-b-PtBMA (PtBMA: poly(tert-butyl methacrylate)), PS-b-PB-b-P2VP and PtBA-b-PCEMA-b-PDMAEMA (PDMAEMA: poly(2-(dimethylamino)-ethyl methacrylate)). Similarly, Janus nanomaterials are prepared from PMMA-b-PCEMA-b-PDMAEMA by selective crosslinking and disassembly of the hamburger-like MCMs.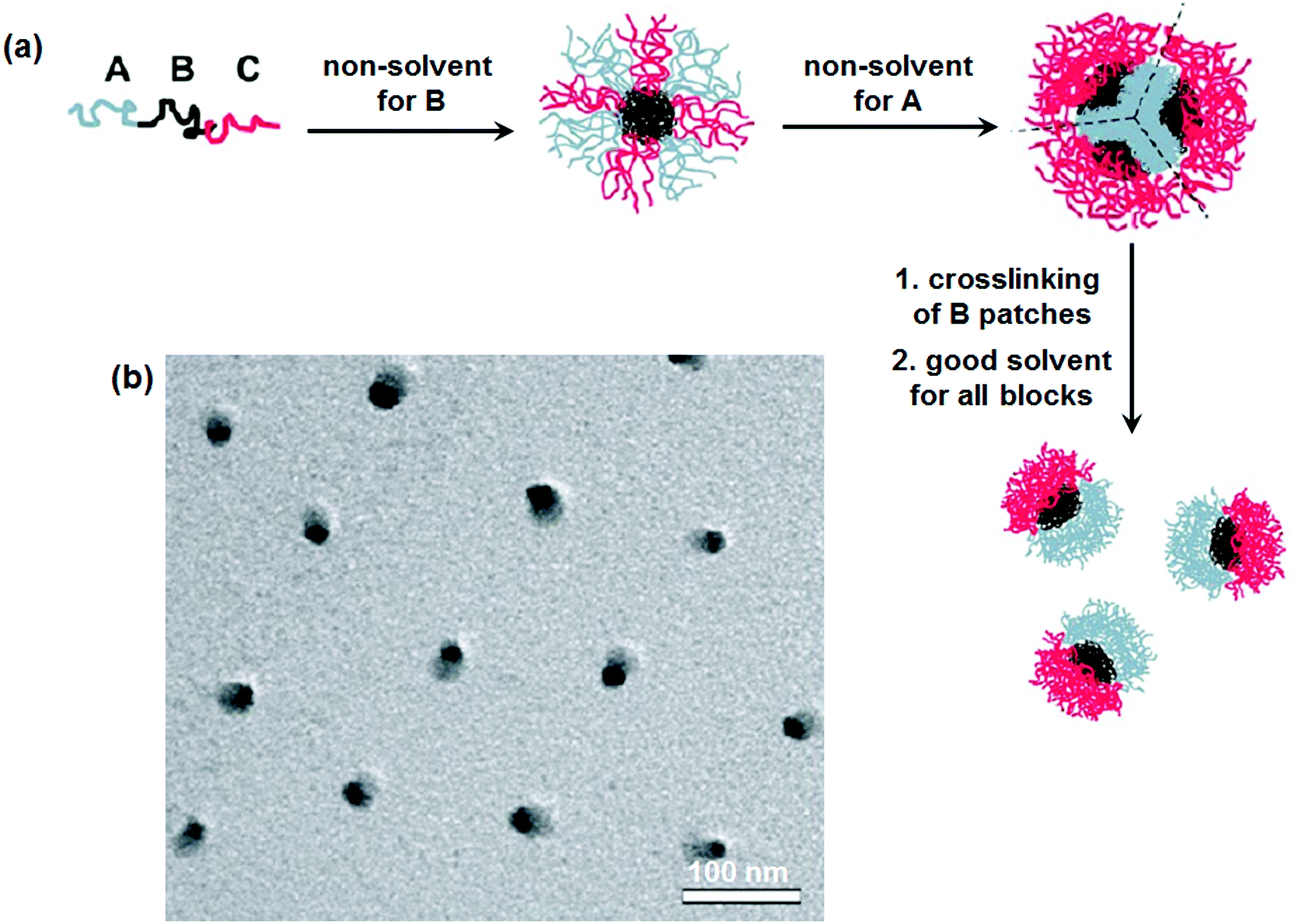 | ||
| Fig. 4 (a) Schematic of the formation of Janus micelles from ABC tri-block terpolymers; (b) TEM image of the PS-b-PB-b-PtBMA Janus micelles. Reprinted with permission from ref. 35. | ||
The Janus nanomaterials can serve as a highly efficient solid compatibilizer for polymer blends. The combined Pickering effect and amphiphilicity of the Janus nanomaterials are responsible for the improved compatibilization performance. Janus nanomaterials can also play the role of nucleation agent during the preparation of poly(2,6-dimethyl-1,4-phenylene ether)/poly-(styrene-co-acrylonitrile) (PPE/SAN) blend foams. Homogenous foams with smaller cells are produced. Partial coverage of the raspberry structure results in stress concentration at the blend interface during the saturation step. The new surfaces formed by cleavage (and maybe microcrazes) result in a higher absorption of the blowing agent, which affects the foaming behavior.36 The Janus nanoparticles were found to assemble in a densely packed monolayer at the PS/PMMA interface, allowing for the stabilization of bicontinuous morphologies in PS/PMMA films.37 The PS-b-PB-b-PMMA Janus nanomaterials can facilitate the dispersion of multi-walled carbon nanotubes (MWNTs) in a variety of solvents. The Janus nanomaterials with a PS hemisphere are preferentially adsorbed onto the MWNTs by π–π stacking, which stabilizes the MWNTs through steric repulsion with the solvophilic PMMA hemisphere. A low Janus nanomaterials/MWNT weight ratio (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 wt/wt) is sufficient for the stabilization.38
:90 wt/wt) is sufficient for the stabilization.38
It is noticed that some conditions are required to achieve Janus nanomaterials from ABC tri-block terpolymers via the disassembly approach. The terpolymers should possess a specific range of block fractions (VA/VB > 1) to ensure the formation of patchy MCMs. Otherwise, linear MCMs (or segmented cylinders) would be obtained.33 The intermediate B block should be crosslinkable to guarantee the integrity of the Janus nanomaterials during disassembly in good solvents. The solubility of the C block should be different from that of the A and B blocks.
3.2 Janus nanomaterials by disassembly of the supramolecular bulk
Selective crosslinking and disassembly of ABC tri-block terpolymer supramolecular bulk was proposed to prepare Janus nanomaterials much earlier than the patchy MCMs route.39 In bulk, ABC tri-block terpolymers can form well defined self-assembled supramolecular structures depending on the polymer–polymer interaction parameter (χ) and the block volume fraction. It is easier to tune the shape of the Janus nanomaterials to shapes spanning spheres, cylinders and platelets.40PS-b-PB-b-PMMA is selected as an example (Fig. 5). After the B block forms the dispersed phase (sphere, cylinder and lamella), an in situ selective crosslinking is performed. Janus nanomaterials can be prepared by disassembling the partially crosslinked bulk with good A/C block solvents.41 Janus cylinders (or sheets/discs) are synthesized when B block dispersed domains are in the shape of cylinders and lamellae. This cylinder/lamella transformation is easily achieved by increasing the B block fraction while keeping the A and C block fractions symmetric. Janus cylinders are synthesized with PS and PMMA hemi-cylinder corona covering the crosslinked PB core.42 The Janus cylinders are long, up to micrometer lengths, which can become shorter, to nanometer lengths, by sonication. Similarly, Janus sheets/discs are synthesized from the PS-b-PB-b-PtBMA lamellar microstructure. The Janus sheets could be crushed to smaller sheets by sonication. Irregular protrusions of the sheets can be preferentially shaved off the edges. Eventually, the sheets appear round in contour shape (e.g., Janus discs). However, the discs are not strictly circular in shape. A hybrid Janus sheet is achieved via gelation of a poly(3-triethoxysilyl propylmethacrylate) (PTEPM) dispersed phase of the P2VP-b-PTEPM-b-PS lamella–lamella bulk. Interestingly, various shapes of Janus nanomaterials such as spheres, cylinders, ribbons and platelets can be achieved from single poly(tert-butoxystyrene)-block-PB-bock-PtBMA (PtBS-b-PB-b-PtBMA) by simply changing the solvent selectivity and crosslinking conditions.43 Non-equilibrium lamellar/spherical morphology is favored when using selective solvent casting. Janus spheres are generated. Interestingly, a Janus ribbon can be obtained by connecting two Janus cylinders along their main axis after trapping a metastable transition state during a phase transition from lamella-cylinder to undulated-lamella. Some other superstructures can derive non-centrosymmetric Janus nanomaterials of other shapes such as tubes and helices.44 It is necessary to strictly control the processing conditions. A sufficiently narrow molecular weight distribution is required to ensure uniformity of the supramolecular structures.45 It remains challenging to adopt this method for AB di-block copolymers.
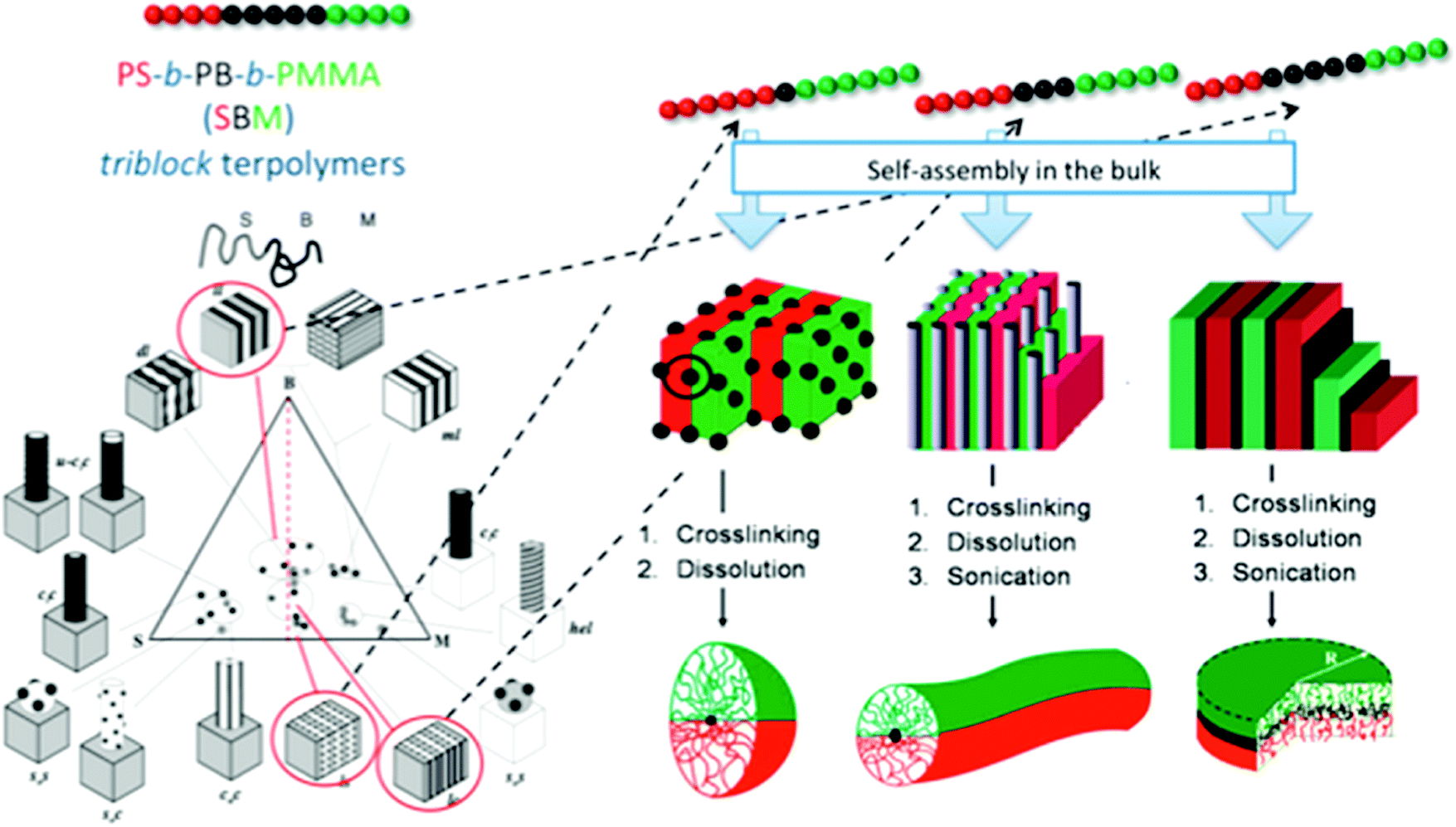 | ||
| Fig. 5 Janus nanomaterials of different architectures prepared via selective crosslinking of the middle block of tri-block terpolymer bulk and subsequent dissolution. Left: Phase diagram of PS-b-PB-b-PMMA; right: synthetic strategy toward Janus micelles, Janus cylinders and Janus discs. Reprinted with permission from ref. 40a, 41 and 42. | ||
4. Janus nanomaterials by the confinement of BCPs
4.1 Janus nanomaterials by the disassembly of BCP confined supramolecular structures
If the aforementioned combined approach is adopted for AB di-block copolymer bulk, a core–corona structure usually results rather than a Janus one. In a selective solvent, AB di-block copolymers tend to self-assemble into core–corona micelles. Different from self-assembly in solution, when confinement is imposed, self-assembly of AB di-block copolymers can form some new structures for example particles with separated A and B domains on the surface.46 Janus nanomaterials can be derived from these new structures. For example, di-block copolymer PS-block-poly(4-vinyl pyridine) (PS-b-P4VP) can self-assemble into a patchy particle with P4VP protuberances on the surface after self-assembly in a confined emulsion droplet (Fig. 6a).47 The patchy structure is attributed to the nearly neutral interface for both PS and P4VP blocks. After selective crosslinking of the P4VP protuberances, Janus nanoparticles are derived after disassembly with a good PS solvent. This route can be extended to AB/BC di-block copolymer mixtures and ABC tri-block terpolymers, since patchy particles are available in the confined space.48 Janus nanodiscs with a perfectly round contour are further synthesized by the route shown in Fig. 6b.49 The ellipsoidal particles are composed of P4VP/PS alternatively stacking lamellae when the emulsion interface is neutral using polyvinyl alcohol (PVA). A sandwiched nanodisc of P4VP/PS/P4VP is achieved after disassembly from the P4VP layer using the selective solvent ethanol. After both the top and bottom P4VP layers are crosslinked, Janus nanodiscs of PS-b-P4VP are achieved by disassembly from the PS layer with a good solvent. This method is valid for ABC tri-block terpolymers. Janus nanodiscs with A and C at the surfaces are obtained by one step disassembly of the crosslinked ellipsoid. In the presence of the PS-b-P4VP Janus nanodisc, a stable paraffin/water emulsion forms. The paraffin droplet surface is armored with a layer of the Janus nanodiscs, whose PS side faces the paraffin core while the P4VP side faces the continuous aqueous phase.49 Further experiments and simulations of Janus nanomaterials at a toluene/water interface indicate that the oil/water interface stability is greatly affected by the geometry of the Janus nanomaterials. As the shape changes from sphere to disc and cylinder, the adsorption kinetics, packing behavior, energy barrier, and equilibrium interfacial tension are varied.50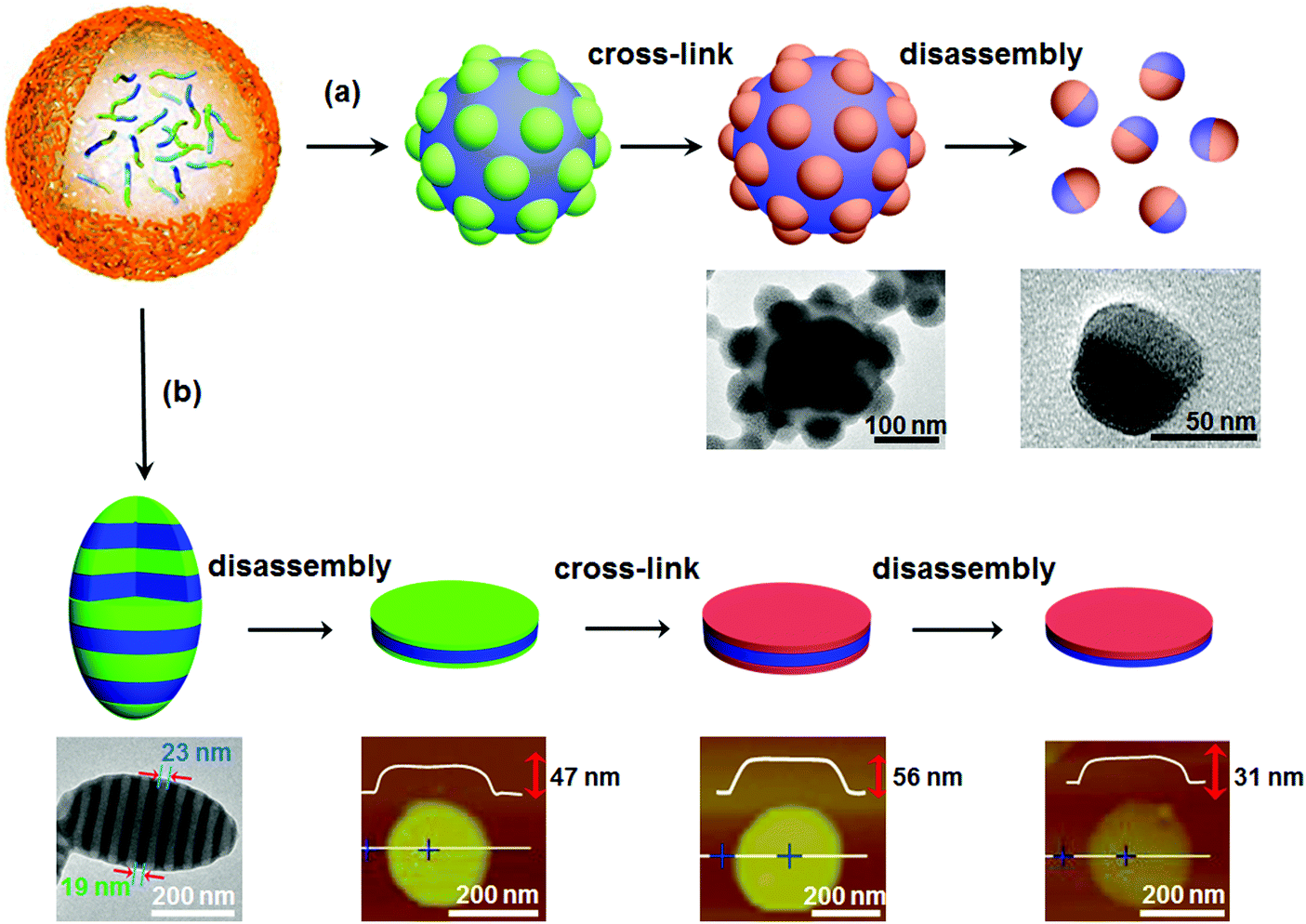 | ||
| Fig. 6 Synthesis of Janus nanomaterials by confined self-assembly, crosslinking and disassembly of di-block copolymers: (a) Janus nanoparticles, and (b) Janus nanodiscs. Reprinted with permission from ref. 47 and 49. | ||
4.2 Direct synthesis of Janus nanomaterials by the confined self-assembly of BCPs
Although disassembly of patchy BCP particles is effective toward the synthesis of Janus nanomaterials, the approach is essentially based on the combined approach. It is rational to expect direct formation of Janus nanoparticles from BCPs when the confinement is sufficiently strong. Computer simulation predicts that self-assembly of AB di-block copolymers at a neutral interface can lead to Janus nanoparticles at low D/L levels below 0.8 (D: confinement diameter; L: BCP periodic length in the bulk).51 Experimentally, PS-b-PI Janus nanoparticles are achieved at D/L ≤ 1.0.52 Some other non-Janus nanoparticles are co-existent at D/L > 1.0. Patchy particles of PS-b-P4VP are prepared by solvent evaporation induced self-assembly within emulsion droplets, which are stabilized with a nearly neutral interface.53 PS-b-P4VP can self-assemble into Janus nanoparticles when the droplets are small enough. Janus nanoparticles appear when D/L ≤ 0.8. Two requirements should be satisfied. A neutral interface is determinative for both blocks to be exposed to the surface to an equal degree. The particles should be small enough, otherwise multiple compartment particles are dominant. Both requirements are easily realized by the emulsion solvent evaporation method. Accordingly, Janus nanomaterials of P4VP based BCPs are synthesized in a straightforward way (Fig. 7).12 P4VP block is more affinitive to the emulsion interface whilst the other block is slightly more affinitive to the solvent. During solvent evaporation, P4VP chains start to aggregate while the other block chains remain soluble. The aggregated P4VP head tends to migrate towards the interface. After the solvent fully evaporates, the other block chains are condensed forming another head at the opposite side. This method is general without requirements for the molecular weight distribution and block fraction of the BCPs. In principle, the method is feasible for lots of P4VP based BCPs with nearly the full range of fP4VP. When two BCPs with varied molecular weights and block fractions are mixed, Janus nanoparticles are always obtained. The Janus balance is thereby tunable.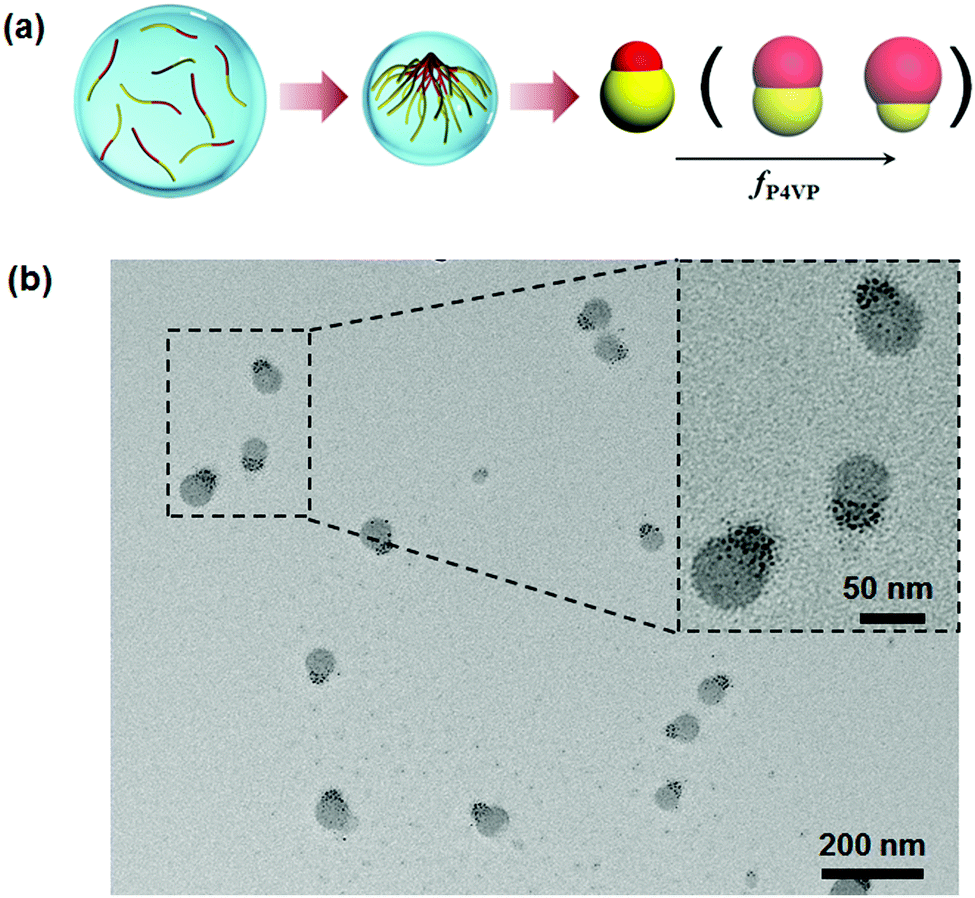 | ||
| Fig. 7 (a) Illustrative synthesis of Janus nanoparticles by the solvent evaporation induced assembly of P4VP based di-block copolymers in emulsion droplets; (b) TEM image of the PS-b-P4VP Janus nanoparticles. Reprinted with permission from ref. 12. | ||
In selective solvents, BCP Janus nanoparticles with asymmetry and directional interaction serve as amphiphilic solid units for new superstructures.47 The Janus nanoparticles are firstly dispersed in a good solvent to form a jellyfish-like structure. When spreading the dilute dispersion onto a hydrophobic substrate such as carbon film, the hydrophobic PS chains preferentially adhere onto the carbon substrate, while the hydrophilic P4VP heads are directed towards the air. Solvent evaporation induces self-assembly of the Janus nanoparticles. The morphology of the aggregates is affected by the Janus balance. For those Janus nanoparticles with a smaller hydrophilic P4VP head and larger hydrophobic PS tail, ordered surface patterns form due to coalescence of the PS chains. In contrast, in the case of the inverted Janus nanoparticles with a larger hydrophilic P4VP head and a smaller hydrophobic PS tail, disordered individual particles instead of an ordered monolayer form after solvent evaporation. Formation of the monolayer is suppressed by the steric hindrance of the larger P4VP heads. A similar ordered surface pattern has been observed for “symmetric” PS-b-P4VP Janus nanoparticles. Adding ethanol can decrease the solubility of the PS chains in chloroform, inducing the formation of a new superstructure after solvent evaporation. Since the mixed solvent remains selective for PS, the P4VP sides of the Janus nanoparticles are aggregated inwardly, while the PS sides face outwardly. ABC tri-block terpolymer Janus nanoparticles with a crosslinked PB core, hydrophobic PS and hydrophilic PMAA chains on two sides exhibit similar self-assembly behavior.35 Upon dialyzing the Janus nanoparticle dispersion in THF against water, hollow spheres (vesicles) form when the hydrophilic P4VP head is small and the hydrophobic PS tail is large.12 The vesicle exterior surface is coarse and covered with the hydrophilic P4VP heads. The interior surface is smooth due to coalescence of the PS chains.
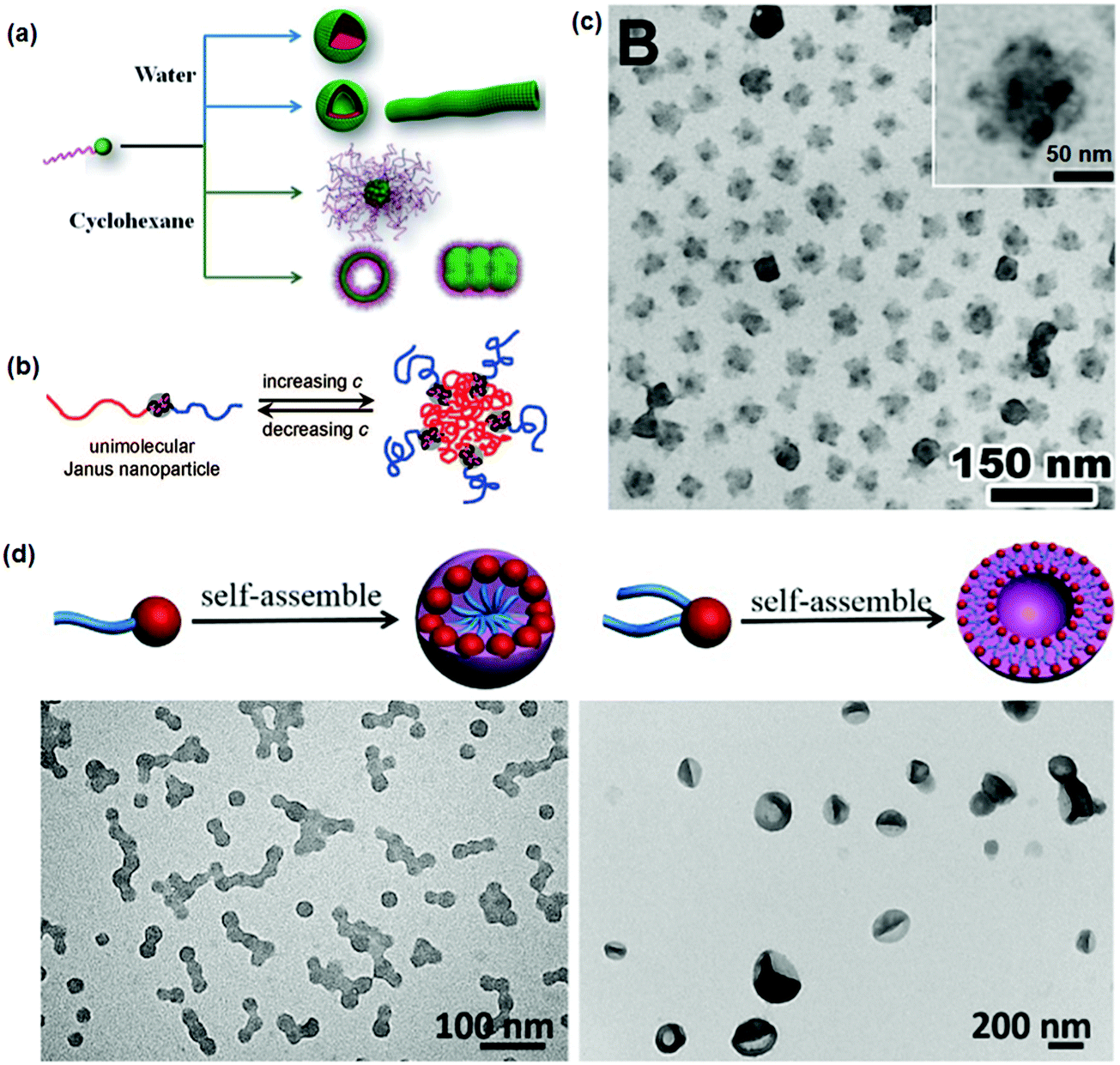
In order to further avoid the requirement of molecular weight distribution, guided self-assembly of BCPs within a confined space is proposed to prepare Janus nanoparticles (Fig. 8).54 In a good solvent, PS-b-PAA di-block copolymers are absorbed within mesoporous silica channels on the surface of an iron oxide (Fe3O4) core. Coordination between PAA and Fe3O4 is responsible for the preferential absorption. After substitution with a PS selective solvent, the PS-b-PAA clusters experience a phase separation. The PAA blocks collapse on the core surface while the PS blocks extend outwardly. After selective crosslinking of the PAA blocks, robust Janus nanoparticles are released from the mesoporous channels after etching the mesoporous silica layer. Using the crosslinked PAA (cPAA) as a nanoreactor, other functional species can thereby favorably grow. As an example, Fe3O4@cPAA–PS composite Janus nanoparticles are derived. After another sequential growth of polymer chains from the opposite side by coordination with carboxyl-terminated PEO, PEO–Fe3O4@cPAA–PS composite Janus nanoparticles are achieved. They serve as a magnetically responsive solid emulsifier.
 | ||
| Fig. 8 (a) Illustrative synthesis of the Janus nanoparticles by guided self-assembly of di-block copolymers in mesoporous silica channels; (b) TEM images of the PS–cPAA and PS–cPAA@Fe3O4–PEO Janus nanoparticles. Reprinted with permission from ref. 54. | ||
5. Single chain Janus nanomaterials of BCPs
Intramolecular crosslinking of an individual polymer chain can give nanoparticles.55 In the case of block copolymers, single chain Janus nanoparticles with a crosslinked colloidal head and soluble block tail were constructed as early as 1997.56 In a sufficiently dilute polymer solution below the overlapping concentration, the polymer chain dimension should be smaller than the average distance between two chains. Polymer coils are isolated without any contact with each other. Intramolecular crosslinking is dominant while intermolecular crosslinking is negligible. Single chain Janus nanoparticles of PS-b-PCEMA are generated by photo-crosslinking of the PCEMA block. In a THF/cyclohexane mixed solvent, at a PS-b-PCEMA concentration of 2.0 mg mL−1, a small amount of unimer (5–25 wt%) is co-existent with the core–corona micelles. The unimers can form single chain Janus nanoparticles after intramolecular crosslinking. Single chain Janus nanoparticles of PEG-block-poly(styrene-co-chloromethylstyrene) (PEG-b-P(S-co-CMS)) are prepared by UV triggered intramolecular Diels–Alder crosslinking.57 The solid concentration remains rather low at 0.017 mg mL−1 in order to avoid intermolecular crosslinking. In order to increase the solid content, a continuous addition strategy is proposed to synthesize single chain Janus nanoparticles of PEG-block-poly(styrene-co-benzocyclobutene) (PEG-b-P(S-co-BCB)).58 In this case, the benzocyclobutene-coupling is thermally activated and quickly accomplished upon adding polymer to the solution. The PEG-b-P(S-co-BCB) concentration is kept at a very low level ensuring intramolecular crosslinking during the whole reaction. Eventually, the solid concentration can reach very high levels. This approach can be regarded as an accumulation of numerous intramolecular crosslinking reactions at an extremely dilute polymer concentration. Single chain Janus nanoparticles are prepared by adding PtBA-b-PCEMA or PtBA-b-P(CEMA-ran-CEMA) solution at a low pumping speed to a solvent mixture under continuous UV irradiation and stirring.59 The UV irradiation ensures that the newly added BCPs can be immediately converted into single chain Janus nanoparticles. The slow addition ensures that the BCP concentration remains low during the whole photo-crosslinking process. Eventually, the concentration of the Janus nanoparticles is 27 times higher than the copolymer critical micellization concentration. Slow addition of crosslinkers into the BCP solution is an alternative method in order to increase the yield of single chain Janus nanoparticles. In the PS-b-PDMAEMA solution (0.4 mg mL−1), alkylamine is added dropwise for quaternization intramolecular crosslinking of the PDMAEMA blocks, resulting in single chain Janus nanoparticles.60 Hybrid single chain Janus nanoparticles with a silica head and PEO tail are prepared via an intramolecular hydrolysis/polycondensation reaction.61 Di-block copolymer PEO-b-PMMA-co-poly(3-(trimethoxysilyl)-propyl methacrylate) (PEO-b-P(MMA-co-TMSPMA)) is dissolved in a solvent at a low concentration of 0.5 mg mL−1. Ammonium hydroxide solution in THF is added dropwise to induce the hydrolysis/polycondensation. Similarly, hybrid single chain Janus nanoparticles composed of a hydrophilic silica head tethered with one or two hydrophobic PS tails, are prepared from PS-b-P(tBA-co-TMSPMA) and PS-b-P(tBA-co-TMSPMA)-b-PS (Fig. 9).62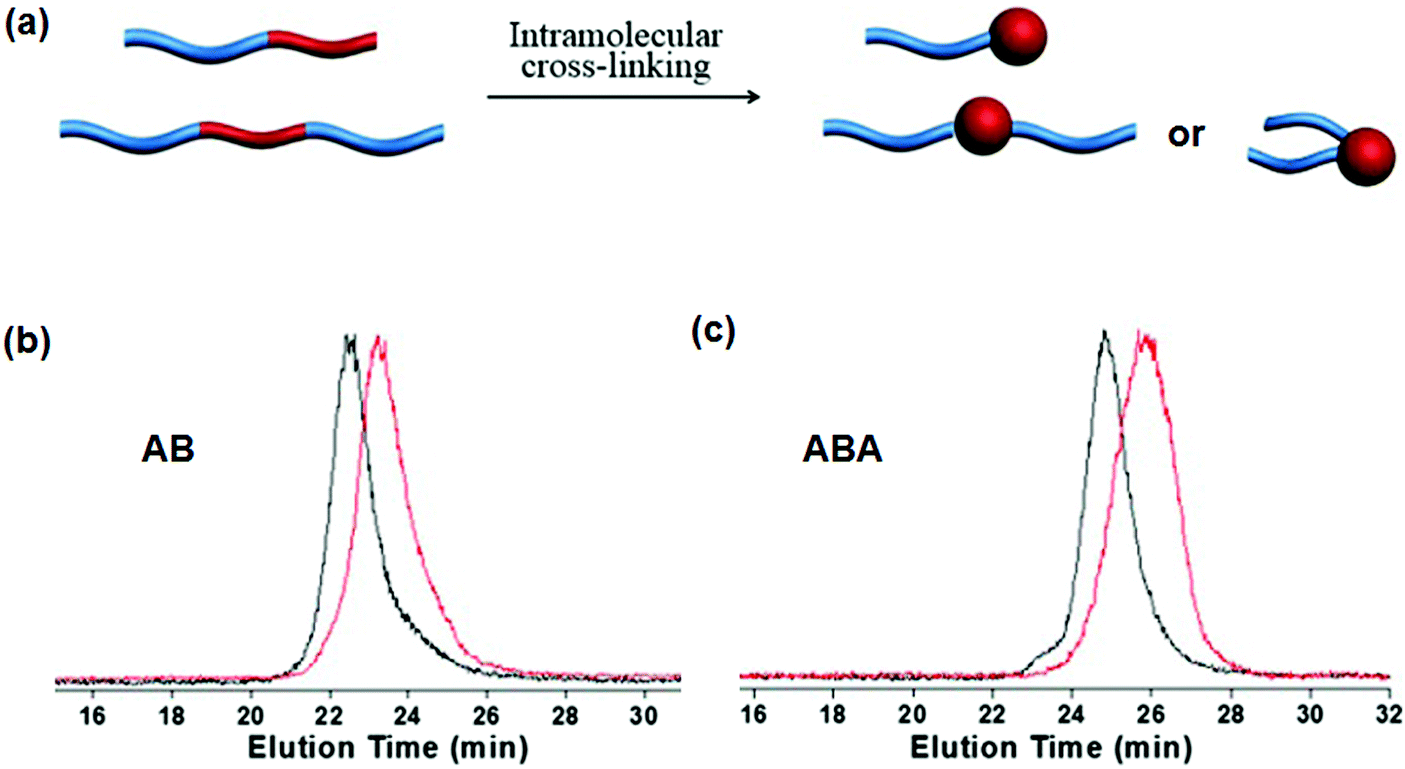 | ||
| Fig. 9 (a) Schematic of single chain Janus nanoparticles prepared from AB and ABA BCPs; GPC curves of (b) PS-b-P(tBA-co-TMSPMA) and (c) PS-b-P(tBA-co-TMSPMA)-b-PS before (black) and after (red) intramolecular crosslinking. Reprinted with permission from ref. 62. | ||
In order to avoid low synthesis efficiency by the continuous slow addition strategy, a fast crosslinking method is proposed to synthesize single chain Janus nanoparticles from PS-b-P2VP-b-PEO tri-block terpolymer (Fig. 10).63 The middle P2VP block is selectively crosslinked via quaternization using 1,4-dibromobutane (DBB). Efficient intramolecular crosslinking of a short P2VP block is performed at a relatively high polymer concentration (20 mg mL−1) and/or with excess DBB/2VP of a molar ratio of 8:1. Janus nanoparticles are prepared with a crosslinked P2VP core and a PS and PEO chain on opposite sides. The intermolecular crosslinking at a high polymer concentration is attributed to a steric shielding of the long PS and PEO end blocks. Single chain Janus nanoparticles with a PEO tail and crosslinked PCEMA head are synthesized by photo crosslinking of the PCEMA block. Intermolecular crosslinking at high BCP concentration (4.0 mg mL−1) can be avoided by slowing down the crosslinking rate.
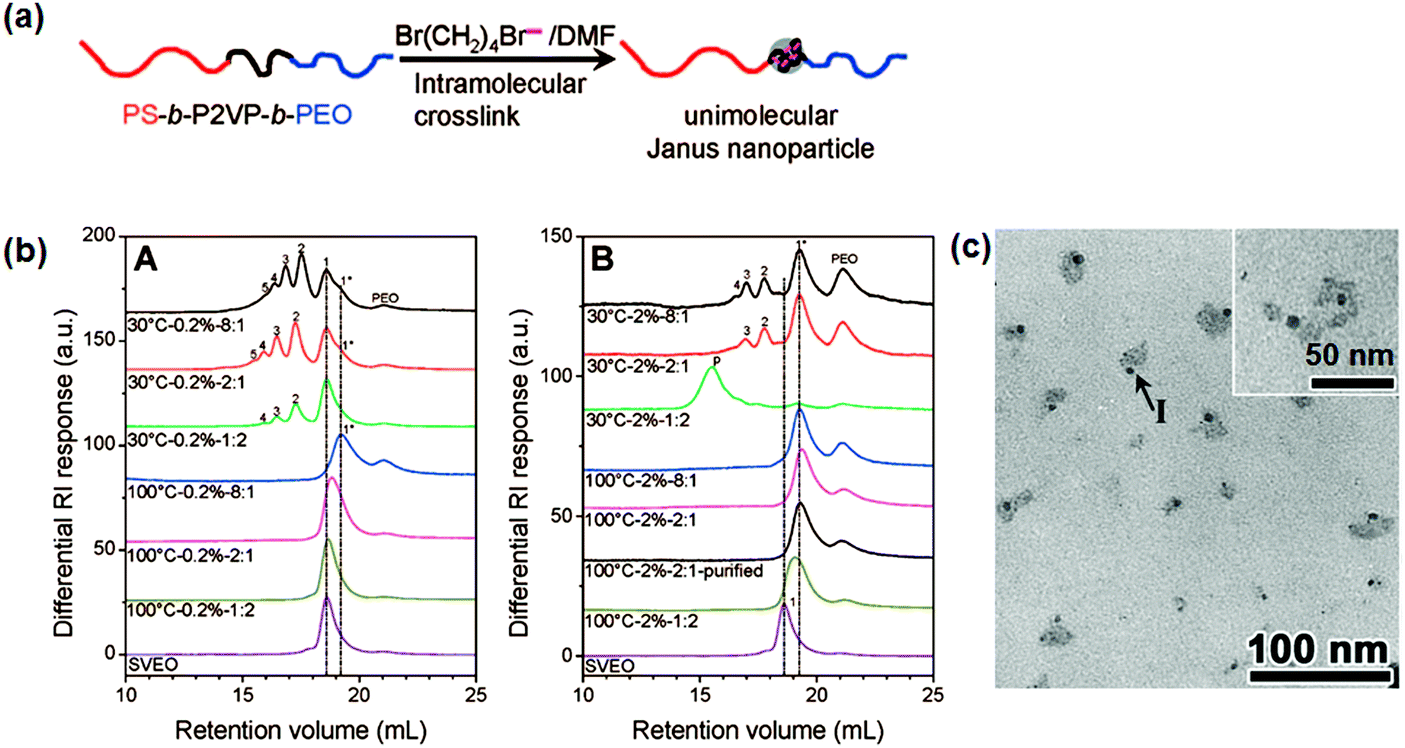 | ||
| Fig. 10 (a) Schematic of single chain Janus nanoparticles prepared from ABC BCPs; (b) differential refractive index SEC curves for the pure PS-b-P2VP-b-PEO and DBB crosslinked PS-b-P2VP-b-PEO products under different reaction conditions, and polymer concentrations of (A) 2.0 mg mL−1 and (B) 20 mg mL−1; (c) TEM image of the single chain Janus nanoparticles. Reprinted with permission from ref. 63. | ||
Single chain Janus nanoparticles of PMMA-b-P4VP are prepared by crosslinking of the P4VP block using propargyl bromide at a polymer concentration of 1.0 mg mL−1.64 After propargyl bromide molecules are grafted onto the P4VP block, the propargyl groups experience intramolecular self-polymerization upon activation. However, intermolecular crosslinking becomes remarkable at higher concentrations e.g., 5 mg mL−1. It is challenging to develop methods to produce single chain Janus nanoparticles at high solid content for example tens of percent.
The single chain Janus nanoparticles can self-assemble into supermicelles in a dilute selective solvent at a concentration above the CMC.59,63 Different micelles are obtained by changing their composition. In the case of PS-b-PDMAEMA single chain Janus nanoparticles, the aggregates change from spherical micelles to vesicles when the PDMAEMA head becomes smaller in “head” selective solvents.60 When using a PS tail selective solvent, bunchy micelles are achieved with the PDMAEMA head forming the core and the PS tails forming the corona. Surface charge density is significant in influencing the morphology and size of the self-assembled micelles (Fig. 11a).65 The spherical micelles of PDMAEMA74-b-PS100 single chain Janus nanoparticles become larger with increasing charge density in PDMAEMA head selective solvents. When increasing the charge density of PDMAEMA30-b-PS169 single chain Janus nanoparticles, the aggregates change from spherical micelles and vesicles to a mixed morphology of worm-like cylinders and vesicles. In the PS tail selective solvent, the aggregates of PDMAEMA74-b-PS297 single chain Janus nanoparticles change from bunchy micelles to a mixture of vesicles and large composite vesicles with increasing charge density. In the PEO tail selective solvent, the PCEMA head and PEO are more overlapped in the supermicelles than in the linear BCPs (Fig. 11b and c).66 When both heads of the single chain Janus nanoparticles are spherical and rigid, their contact area becomes lower. No entanglement among the crosslinked heads occurs in the supermicelles. When PEO-b-P(MMA-co-TMSPMA) single chain Janus nanoparticles are added to a mixed solvent of THF/water, spherical micelles, vesicles, or large compound micelles are obtained depending on the silica head size and the Janus nanoparticle concentration. This morphological evolution is different from that of BCPs.61 Co-assembly of single chain Janus nanoparticles with BCPs gives rise to unprecedented phase-separated bilayer vesicles. The self-assembly of single chain Janus nanoparticles with a silica head and one/two PS tails is further investigated by varying the hydrophilic silica head size (Fig. 11d).62 While BCPs can form spherical micelles, the derivative single chain Janus nanoparticles can self-assemble into vesicles. When increasing the hydrophilic head size, spherical micelles can form rather than vesicles. The conformation of the PS chain tail is critical for the self-assembly, where the bi-tailed single chain Janus nanoparticles with highly stretched PS tails favor the formation of vesicles.
 | ||
| Fig. 11 (a) Schematic of the self-assembly of di-block copolymer single chain Janus nanoparticles into micelles with different morphologies; (b) schematic of the self-assembly of tri-block terpolymer single-chain Janus nanoparticles into MCMs; (c) TEM image of the MCMs; (d) superstructures from the mono-tailed and bi-tailed single chain Janus nanoparticles. Reprinted with permission from ref. 62, 65 and 66. | ||
The example PMMA-b-P4VP single chain Janus nanoparticles can serve as an emulsifier to stabilize water/chlorobenzene emulsions.64a The emulsifying performance is better than that of low molecular surfactants for example Span-80. Using single chain Janus nanoparticles as a stabilizer, suspension polymerization of styrene is carried out.67 Macrocellular polymer foams with an interconnected open-cell structure are prepared using a single chain Janus nanoparticle stabilizer solely with high internal phase emulsions (HIPEs) as templates.64b Besides as an emulsifier for HIPEs, Janus nanoparticles can simultaneously act as a nucleation agent and stabilizer for metal catalytic species. By virtue of the strong interaction of the P4VP “heads” of the Janus NPs embedded in the polyHIPEs and the H2PdCl4 precursor, Pd nanoparticles are in situ synthesized and supported within the produced poly(HIPE) foam without the requirement for functional monomers or post-functionalization.
6. Summary and outlook
Although some approaches have been proposed to synthesize BCP Janus nanomaterials, a challenge remains for their large scale production and practical application. Despite great efforts in BCP self-assembly in solution to generate Janus nanomaterials, it is difficult to achieve Janus micelles in one step using simple solvophobic blocks. The corona chains should undergo sufficient phase separation to yield fully phase-segregated micelles. Additional treatment is required for segment re-arrangement thus breaking symmetry towards Janus micelles. Dis-assembly of ABC tri-block terpolymer bulk superstructures can easily derive Janus nanomaterials with tunable shapes from spheres and cylinders to discs. The bulk superstructures usually contain defects and a lack of sufficient long-range order. Confined self-assembly of BCPs in emulsion droplets following disassembly can produce both Janus nanoparticles and nanodiscs. It is significant to generate a nearly neutral wetting interface using proper surfactants.Single chain Janus nanoparticles can be easily achieved by intramolecular crosslinking of BCPs. They are uniform and able to complex with other functional species to derive functional composite species. The polymer concentration should be extremely low in order to avoid intermolecular crosslinking and polymer chain entanglement. In principle, this intramolecular crosslinking approach is universal in generating the derivative single chain Janus nanoparticles from the parent BCPs. The core issue is to develop methods to perform the intramolecular crosslinking at high polymer concentration.
A breakthrough in synthesis methodology is highly desired to produce single chain Janus nanoparticles on a larger scale. If this occurs, a huge family of Janus composite nanomaterials with responsive polymers and functional inorganic species will be derived for diverse practical applications. Janus nanocomposites can act as units to build adaptive superstructures in solution or at surfaces, which could be manipulated with external fields such as a magnetic field. Janus nanocomposites as functional solid stabilizers for oil/water emulsions or polymer blends can deliver functional species at the interface. In polymer blends, the morphology is easily tunable under external fields. In the bi-continuous morphology, the blend will be highly conductive if the Janus nanoparticles are conductive. Other applications including interfacial catalysis, nano-motors, and medical applications such as controlled drug release or targeted delivery will be also promising in the near future.
Acknowledgements
This work is supported by MOST (2012CB933200), and NSFC (51233007, 51173191).References
- (a) A. Walther and A. H. Müller, Chem. Rev., 2013, 113, 5194–5261 CrossRef CAS PubMed; (b) F. X. Liang, C. L. Zhang and Z. Z. Yang, Adv. Mater., 2014, 26, 6944–6949 CrossRef CAS PubMed; (c) M. Lattuada and T. A. Hatton, Nano Today, 2011, 6, 286–308 CrossRef CAS; (d) J. Z. Du and R. K. O'Reilly, Chem. Soc. Rev., 2011, 40, 2402–2416 RSC; (e) J. Hu, S. X. Zhou, Y. Y. Sun, X. S. Fang and L. M. Wu, Chem. Soc. Rev., 2012, 41, 4356–4378 RSC; (f) S. Jiang, Q. Chen, M. Tripathy, E. Luijten, K. S. Schweizer and S. Granick, Adv. Mater., 2010, 22, 1060–1071 CrossRef CAS PubMed.
- (a) N. Prasad, J. Perumal, C. H. Choi, C. S. Lee and D. P. Kim, Adv. Funct. Mater., 2009, 19, 1656–1662 CrossRef CAS; (b) F. X. Liang, K. Shen, X. Z. Qu, C. L. Zhang, Q. Wang, J. J. Li, J. G. Liu and Z. Z. Yang, Angew. Chem., Int. Ed., 2011, 50, 2379–2382 CrossRef CAS PubMed; (c) H. L. Yang, F. X. Liang, Y. Chen, Q. Wang, X. Z. Qu and Z. Z. Yang, NPG Asia Mater., 2015, 7, e176 CrossRef CAS; (d) H. Zhao, F. X. Liang, X. Z. Qu, Q. Wang and Z. Z. Yang, Macromolecules, 2015, 48, 700–706 CrossRef CAS.
- (a) M. Lattuada and T. A. Hatton, J. Am. Chem. Soc., 2007, 129, 12878–12889 CrossRef CAS PubMed; (b) S. Yin, C. Wang, Z. Yu, J. Wang, S. Liu and S. Chen, Adv. Mater., 2011, 23, 2915–2919 CrossRef CAS PubMed; (c) J. He, M. T. Perez, P. Zhang, Y. Liu, T. Babu, J. Gong and Z. Nie, J. Am. Chem. Soc., 2012, 134, 3639–3642 CrossRef CAS PubMed.
- B. Liu, C. L. Zhang, J. G. Liu, X. Z. Qu and Z. Z. Yang, Chem. Commun., 2009, 3871–3873 RSC.
- (a) T. Nisisako, T. Torii, T. Takahashi and Y. Takizawa, Adv. Mater., 2006, 18, 1152–1156 CrossRef CAS; (b) Z. H. Nie, W. Li, M. Seo, S. Q. Xu and E. Kumacheva, J. Am. Chem. Soc., 2006, 128, 9408–9412 CrossRef CAS PubMed.
- C. L. Zhang, B. Liu, C. Tang, J. G. Liu, X. Z. Qu, J. J. Li and Z. Z. Yang, Chem. Commun., 2010, 46, 4610–4612 RSC.
- (a) F. Wurm, H. M. Konig, S. Hilf and A. F. Kilbinger, J. Am. Chem. Soc., 2008, 130, 5876–5877 CrossRef CAS PubMed; (b) C. Tang, C. L. Zhang, Y. J. Sun, F. X. Liang, Q. Wang, J. J. Li, X. Z. Qu and Z. Z. Yang, Macromolecules, 2013, 46, 188–193 CrossRef CAS; (c) R. H. Deng, S. Liu, F. X. Liang, K. Wang, J. T. Zhu and Z. Z. Yang, Macromolecules, 2014, 47, 3701–3707 CrossRef CAS.
- (a) Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC; (b) A. H. Gröschel and A. H. Müller, Nanoscale, 2015, 7, 11841–11876 RSC.
- Y. Chen, Macromolecules, 2012, 45, 2619–2631 CrossRef CAS.
- A. Walther and A. H. E. Müller, Soft Matter, 2008, 4, 663–668 RSC.
- (a) H. Xu, R. Erhardt, V. Abetz, A. H. E. Müller and W. A. Goedel, Langmuir, 2001, 17, 6787–6793 CrossRef CAS; (b) A. Walther, M. Drechsler and A. Müller, Soft Matter, 2009, 5, 385–390 RSC; (c) A. Walther, M. Drechsler, S. Rosenfeldt, L. Harnau, M. Ballauff, V. Abetz and A. H. E. Müller, J. Am. Chem. Soc., 2009, 131, 4720–4728 CrossRef CAS PubMed.
- R. H. Deng, H. Li, J. T. Zhu, B. Li, F. X. Liang, F. Jia, X. Z. Qu and Z. Z. Yang, Macromolecules, 2016, 49, 1362–1368 CrossRef CAS.
- (a) R. Erhardt, M. Zhang, A. Böker, H. Zettl, C. Abetz, P. Frederik, G. Krausch, V. Abetz and A. H. E. Müller, J. Am. Chem. Soc., 2003, 125, 3260–3267 CrossRef CAS PubMed; (b) T. M. Ruhland, A. H. Gröschel, A. Walther and A. H. E. Müller, Langmuir, 2011, 27, 9807–9814 CrossRef CAS PubMed.
- (a) A. Walther, K. Matussek and A. H. E. Müller, ACS Nano, 2008, 6, 1167–1178 CrossRef PubMed; (b) A. Walther, M. Hoffmann and A. H. Müller, Angew. Chem., Int. Ed., 2008, 47, 711–714 CrossRef CAS PubMed.
- X. Li, H. Yang, L. M. Xu, X. N. Fu, H. W. Guo and X. K. Zhang, Macromol. Chem. Phys., 2010, 211, 297–302 CrossRef CAS.
- Y. P. Sheng, X. P. Yang, N. Yan and Y. T. Zhu, Soft Matter, 2013, 9, 6254–6262 RSC.
- (a) M. Štěpánek, K. Podhájecká, E. Tesařová and K. Procházka, Langmuir, 2001, 17, 4240–4244 CrossRef; (b) T. Hui, D. Chen and M. Jiang, Macromolecules, 2005, 38, 5834–5837 CrossRef CAS; (c) G. Li, L. Shi, R. Ma, Y. An and N. Huang, Angew. Chem., Int. Ed., 2006, 45, 4959–4962 CrossRef CAS PubMed; (d) R. Ma, B. Wang, Y. Xu, Y. An, W. Zhang, G. Li and L. Shi, Macromol. Rapid Commun., 2007, 28, 1062–1069 CrossRef CAS.
- V. V. Palyulin and I. I. Potemkin, Macromolecules, 2008, 41, 4459–4463 CrossRef CAS.
- L. Cheng, G. Zhang, L. Zhu, D. Chen and M. Jiang, Angew. Chem., Int. Ed., 2008, 47, 10171–10174 CrossRef CAS PubMed.
- J. Hu and G. Liu, Macromolecules, 2005, 38, 8058–8065 CrossRef CAS.
- Y. Han and W. Jiang, J. Phys. Chem. B, 2011, 115, 2167–2172 CrossRef CAS PubMed.
- Y. Han, J. Cui and W. Jiang, J. Phys. Chem. B, 2012, 116, 9208–9214 CrossRef CAS PubMed.
- (a) I. K. Voets, A. de Keizer and M. A. Cohen Stuart, Macromolecules, 2006, 39, 5952–5955 CrossRef CAS; (b) S. H. Kim, J. P. K. Tan, F. Nederberg, K. Fukushima, Y. Y. Yang, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2009, 42, 25–29 CrossRef CAS.
- (a) I. K. Voets, A. de Keizer, P. de Waard, P. M. Frederik, P. H. H. Bomans, H. Schmalz, A. Walther, S. M. King, F. A. M. Leermakers and M. A. Cohen Stuart, Angew. Chem., Int. Ed., 2006, 45, 6673–6676 CrossRef CAS PubMed; (b) I. K. Voets, R. Fokkink, T. Hellweg, S. M. King, P. de Waard, A. de Keizer and M. A. Cohen Stuart, Soft Matter, 2009, 5, 999–1005 RSC.
- (a) J. Cui and W. Jiang, Langmuir, 2010, 26, 13672–13676 CrossRef CAS PubMed; (b) O. V. Borisov and E. B. Zhulina, Polymer, 2013, 54, 2043–2048 CrossRef CAS.
- B. Fang, A. Walther, A. Wolf, Y. Xu, J. Yuan and A. H. E. Müller, Angew. Chem., Int. Ed., 2009, 48, 2877–2880 CrossRef CAS PubMed.
- J. Du and S. P. Armes, Soft Matter, 2010, 6, 4851–4857 RSC.
- Z. Zhang, C. Zhou, H. Dong and D. Chen, Angew. Chem., Int. Ed., 2016, 55, 6182–6186 CrossRef CAS PubMed.
- (a) I. W. Wyman and G. Liu, Polymer, 2013, 54, 1950–1978 CrossRef CAS; (b) A. O. Moughton, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2012, 45, 2–19 CrossRef CAS.
- W. Zhang, J. He, H. Baoa and X. Dong, RSC Adv., 2015, 5, 104223 RSC.
- (a) W. Kong, W. Jiang, Y. Zhu and B. Li, Langmuir, 2012, 28, 11714–11724 CrossRef CAS PubMed; (b) Y. Zhou, H. G. Xia, X. P. Long, X. G. Xue and W. Qian, Macromol. Theory Simul., 2014, 24, 85–88 CrossRef; (c) Y. Zhou, X. Long, X. Xue, W. Qian and C. Zhang, RSC Adv., 2015, 5, 7661–7664 RSC.
- A. Walther, C. Barner-Kowollik and A. H. E. Müller, Langmuir, 2010, 26, 12237–12246 CrossRef CAS PubMed.
- A. H. Gröschel, F. H. Schacher, H. Schmalz, O. V. Borisov, E. B. Zhulina, A. Walther and A. H. E. Müller, Nat. Commun., 2012, 3, 710 CrossRef PubMed.
- J. Dupont and G. Liu, Soft Matter, 2010, 6, 3654–3661 RSC.
- A. H. Gröschel, A. Walther, T. I. Löbling, J. Schmelz, A. Hanisch, H. Schmalz and A. H. E. Müller, J. Am. Chem. Soc., 2012, 134, 13850–13860 CrossRef PubMed.
- (a) S. Bärwinkel, R. Bahrami, T. I. Löbling, H. Schmalz, A. H. E. Müller and V. Altstädt, Adv. Eng. Mater., 2016, 18, 814–825 CrossRef; (b) R. Bahrami, T. I. Löbling, A. H. Gröschel, S. Holger, A. H. E. Müller and V. Altstädt, ACS Nano, 2014, 8, 10048–10056 CrossRef CAS PubMed.
- K. C. Bryson, T. I. Lobling, A. H. E. Müller, T. P. Russell and R. C. Hayward, Macromolecules, 2015, 48, 4220–4227 CrossRef CAS.
- A. H. Gröschel, T. I. Löbling, P. D. Petrov, M. Müllner, C. Kuttner, F. Wieberger and A. H. E. Müller, Angew. Chem., Int. Ed., 2013, 52, 3602–3606 CrossRef PubMed.
- R. Saito, A. Fujita, A. Ichimura and K. Ishizu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2091–2097 CrossRef CAS.
- (a) A. Walther, X. André, M. Drechsler, V. Abetz and A. H. E. Müller, J. Am. Chem. Soc., 2007, 129, 6187–6198 CrossRef CAS PubMed; (b) K. Zhang, L. Gao and Y. Chen, Polymer, 2010, 51, 2809–2817 CrossRef CAS.
- R. Erhardt, A. Böker, H. Zettl, H. Kaya, W. Pyckhout-Hintzen, G. Krausch, V. Abetz and A. H. E. Müller, Macromolecules, 2001, 34, 1069–1075 CrossRef CAS.
- Y. F. Liu, V. Abetz and A. Müller, Macromolecules, 2003, 36, 7894–7898 CrossRef CAS.
- A. Wolf, A. Walther and A. H. E. Müller, Macromolecules, 2011, 44, 9221–9229 CrossRef CAS.
- L. Wang, J. Lin and X. Zhang, Polymer, 2013, 54, 3427–3442 CrossRef CAS.
- A. Walther, A. Göldel and A. H. E. Müller, Polymer, 2008, 49, 3217–3227 CrossRef CAS.
- (a) R. Deng, S. Liu, J. Li, Y. Liao, J. Tao and J. Zhu, Adv. Mater., 2012, 24, 1889–1893 CrossRef CAS PubMed; (b) R. Deng, F. Liang, W. Li, Z. Yang and J. Zhu, Macromolecules, 2013, 46, 7012–7017 CrossRef CAS; (c) B. Yu, J. Deng, B. Li and A. Shi, Soft Matter, 2014, 10, 6831–6843 RSC; (d) N. Yan, Y. Zhu and W. Jiang, Soft Matter, 2016, 12, 965–972 RSC; (e) J. Xu, K. Wang, J. Li, H. Zhou, X. Xie and J. Zhu, Macromolecules, 2015, 48, 2628–2636 CrossRef CAS.
- R. H. Deng, F. X. Liang, X. Z. Qu, Q. Wang, J. T. Zhu and Z. Z. Yang, Macromolecules, 2015, 48, 750–755 CrossRef CAS.
- R. H. Zheng, G. J. Liu and X. H. Yan, J. Am. Chem. Soc., 2005, 127, 15358–15359 CrossRef CAS PubMed.
- R. H. Deng, F. X. Liang, P. Zhou, C. L. Zhang, X. Z. Qu, Q. Wang, J. L. Li, J. T. Zhu and Z. Z. Yang, Adv. Mater., 2014, 26, 4469–4472 CrossRef CAS PubMed.
- T. M. Ruhland, A. H. Gröschel, N. Ballard, T. S. Skelhon, A. Walther, A. H. Müller and S. A. Bon, Langmuir, 2013, 29, 1388–1394 CrossRef CAS PubMed.
- (a) S. Li, P. Chen, L. Zhang and H. Liang, Langmuir, 2011, 27, 5081–5089 CrossRef CAS PubMed; (b) P. Chen, H. Liang and A. Shi, Macromolecules, 2008, 41, 8938–8943 CrossRef CAS.
- T. Higuchi, A. Tajima, K. Motoyoshi, H. Yabu and M. Shimomura, Angew. Chem., Int. Ed., 2008, 47, 8044–8046 CrossRef CAS PubMed.
- R. H. Deng, H. Li, F. X. Liang, J. T. Zhu, B. H. Li, X. L. Xie and Z. Z. Yang, Macromolecules, 2015, 48, 5855–5860 CrossRef CAS.
- P. Zhou, F. X. Liang, Y. J. Liu, R. H. Deng, H. L. Yang, Q. Wang, X. Z. Qu and Z. Z. Yang, Sci. China Mater., 2015, 58, 961–968 CrossRef.
- (a) M. Gonzalez-Burgos, A. Latorre-Sanchez and J. A. Pomposo, Chem. Soc. Rev., 2015, 44, 6122–6142 RSC; (b) S. Mavila, O. Eivgi, I. Berkovich and N. G. Lemcoff, Chem. Rev., 2016, 116, 878–961 CrossRef CAS PubMed.
- J. Tao and G. Liu, Macromolecules, 1997, 30, 2408–2411 CrossRef CAS.
- O. Altintas, J. Willenbacher, K. N. R. Wuest, K. K. Oehlenschlaeger, P. Krolla-Sidenstein, H. Gliemann and C. Barner-Kowollik, Macromolecules, 2013, 46, 8092–8101 CrossRef CAS.
- (a) E. Harth, B. V. Horn, V. Y. Lee, D. S. Germack, C. P. Gonzales, R. D. Miller and C. J. Hawker, J. Am. Chem. Soc., 2002, 124, 8653–8660 CrossRef CAS PubMed; (b) Y. Kim, J. Pyun, J. M. F. E. Chet, C. J. Hawker and C. W. Frank, Langmuir, 2005, 21, 10444–10458 CrossRef CAS PubMed.
- G. Njikang, G. Liu and S. A. Curda, Macromolecules, 2008, 41, 5697–5702 CrossRef CAS.
- J. Wen, L. Yuan, Y. Yang, L. Liu and H. Zhao, ACS Macro Lett., 2013, 2, 100–106 CrossRef CAS.
- W. Li, C. Kuo, I. Kanyo, S. Thanneeru and J. He, Macromolecules, 2014, 47, 5932–5941 CrossRef CAS.
- W. Li, S. Thanneeru, I. Kanyo, B. Liu and J. He, ACS Macro Lett., 2015, 4, 736–740 CrossRef CAS.
- L. Cheng, G. Hou, J. Miao, D. Chen, M. Jiang and L. Zhu, Macromolecules, 2008, 41, 8159–8166 CrossRef CAS.
- (a) F. Xu, Z. Fang, D. Yang, Y. Gao, H. Li and D. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 6717–6723 CrossRef CAS PubMed; (b) F. Yi, F. Xu, Y. Gao, H. Li and D. Chen, RSC Adv., 2015, 5, 40227–40235 RSC.
- J. Wen, J. Zhang, Y. Zhang, Y. Yang and H. Zhao, Polym. Chem., 2014, 5, 4032–4038 RSC.
- F. Zhou, M. Xie and D. Chen, Macromolecules, 2013, 47, 365–372 CrossRef.
- Y. Zhang and H. Zhao, Polymer, 2015, 64, 277–284 CrossRef CAS.
| This journal is © the Partner Organisations 2017 |