
Nanofiber-supported CuS nanoplatelets as high efficiency counter electrodes for quantum dot-based photoelectrochemical hydrogen production†
F.
Navarro-Pardo‡
 a,
L.
Jin‡
a,
R.
Adhikari
a,
X.
Tong
ab,
D.
Benetti
a,
K.
Basu
a,
S.
Vanka
c,
H. G.
Zhao
*a,
Z. T.
Mi
c,
S. H.
Sun
a,
V. M.
Castano
d,
A.
Vomiero
*e and
F.
Rosei
*af
a,
L.
Jin‡
a,
R.
Adhikari
a,
X.
Tong
ab,
D.
Benetti
a,
K.
Basu
a,
S.
Vanka
c,
H. G.
Zhao
*a,
Z. T.
Mi
c,
S. H.
Sun
a,
V. M.
Castano
d,
A.
Vomiero
*e and
F.
Rosei
*af
aCentre for Energy, Materials and Telecommunications, Institut National de la Recherche Scientifique, 1650 Boul. Lionel-Boulet, Varennes, QC, Canada J3X 1S2. E-mail: zhaoh@emt.inrs.ca; rosei@emt.inrs.ca
bSchool of Chemistry and Material Science, Guizhou Normal University, Guiyang 550001, China
cDepartment of Electrical and Computer Eng., McGill University, 3480 Univ. Str. W, Montreal, QC, Canada H3A 0E9
dCentre of Applied Physics and Advanced Technology, National Autonomous University of Mexico, 3001 Boul. Juriquilla, Juriquilla, Santiago de Queretaro 76230, Mexico
eDivision of Engineering Sciences and Mathematics, Luleå University of Technology, 971 98 Luleå, Sweden. E-mail: alberto.vomiero@ltu.se
fInstitute for Fundamental and Frontier Science, University of Electronic Science and Technology of China, Chengdu, 610054, P. R. China
First published on 24th October 2016
Abstract
We developed a hierarchically assembled hybrid counter electrode (CE) based on copper sulfide (CuS) nanoplatelets grown on polymer nanofibers. The resulting CE was used in a quantum dot (QD)-based photoelectrochemical (PEC) system for H2 generation in the presence of sacrificial agents (S2−/SO32−). The concept is to increase the specific surface area of the CE, aiming at maximizing charge exchange at the electrode, which boosts efficient generation of H2 and to obtain a stable structure for long-term operation of the device. Structural and morphological characterization indicated the presence of a covellite crystalline phase (CuS). PEC tests showed that the CuS nanoplatelets grown in the CEs could replace Pt CEs in either visible-active or near infrared (NIR)-active QD-based PEC systems. Specifically, saturation of the photocurrent density (∼7.5 mA cm−2) occurred at ∼0.6 V versus the RHE, when using a NIR QD-based TiO2 photoanode and a nanofiber-supported CuS as the CE. Stability tests of the nanofiber-supported CuS CE showed that 85% of the initial photocurrent density was maintained after ∼1 h, which is similar to that obtained with the Pt foil CE (86%). In contrast, CuS nanostructures directly deposited on FTO glass without nanofibers (CuS/FTO CE) exhibited poor stability. CuS/FTO CE degraded quickly, showing a 90% drop in the initial photocurrent within 200 s testing whereas a 14% drop in the initial photocurrent was observed for the CuxS on brass within 10 min of testing. Our new nanofiber supported-CuS CE stands out due to its higher performance compared to brass and its similar stability compared to Pt during long term PEC operation. Additionally, our hybrid CE showed a better catalytic performance than the Pt CE and good stability in cyclic voltammetry tests. These results demonstrate that the nanofiber-supported CuS is a promising cost effective alternative to Pt as a highly efficient CE for PEC H2 generation.
Introduction
Hydrogen (H2) as a clean fuel combines the advantages of high energy storage density, ease of transportation, cost-effectiveness and the generation of water as the only byproduct of its use for power generation.1 Photoelectrochemical (PEC) H2 production represents a clean and environmentally sustainable approach to provide abundant energy. Generally, a PEC cell presents reduction–oxidation (redox) reactions driven by electron–hole pairs created by incident photons, namely, the holes oxidize the water/hole scavenger at the surface of an anode, and electrons migrate to the counter electrode (CE) to reduce water and produce H2.2–5 An integrated PEC system has three main components: a photoanode, an electrolyte and a CE.6 Quantum dot (QD)-sensitized photoanodes have recently attracted much attention, because of their excellent optical activity in a broad spectral range as well as the fast exciton dissociation and charge injection from the photoexcited QDs to the wide band gap semiconductor, when proper electronic band alignment is achieved.5–8 In the QD-based PEC system, the electrolyte composed of sulfide, S2−, and sulfite, SO32−, acts as a sacrificial agent since it is a very efficient hole acceptor, enabling the effective separation of charge carriers and minimizing the photocorrosion of QDs.9,10 Currently, Pt is the material of choice as the electrocatalyst for the CE.6 However, there are several disadvantages, which limit its potential applications in PEC H2 production: (i) Pt is a rare and precious metal and (ii) Pt is easily poisoned by the electrolyte and its conductivity and surface activity decreases due to the adsorption of sulfur-containing compounds on its surface.11–14 Therefore, it is highly desirable to develop robust, active and inexpensive non-precious metal-based CEs for cost-effective PEC devices.Metal sulfides have been known for decades as efficient electrocatalysts in PEC cells.15 Copper sulfide (CuxS) has been widely studied in QD-sensitized solar cells (QDSCs),12,16–20 whereas little attention has been given to its application in PEC hydrogen generation. Recently, Cu2S nanostructures grown on brass via acidic activation were found to provide an equivalent performance to that of Pt nanoparticles mixed with carbon black deposited on a carbon cloth electrode, when tested in a PEC H2 production device.21 This procedure has been followed in a few QD-based PEC systems,22,23 though it is well known that the brass substrate suffers from continuous corrosion by the electrolyte and lacks long-term stability.15,24 In previous work on QD-based PEC H2 generation, we fabricated a CE by sputtering a thin layer of Cu subsequently treated with polysulfide solution. The as-prepared CE based on CuS hexagonal nanoplatelets provided similar electrical performances compared to a Pt CE.25 Nevertheless, the nanoplatelets are detached from a fluorine-doped tin oxide (FTO) substrate gradually during long PEC operation times. Fabricating an electrocatalyst/CE, which presents both high PEC performance and long-term stability, using a time effective and cost-efficient process is still challenging.
Other commonly used methods to grow CuxS nanostructures are solvothermal synthesis26 and chemical bath deposition.27 In both approaches, a large quantity of chemical solution is needed, which requires an enormous cost for chemicals, disposal of liquid waste, and additional steps for the integration of these nanostructures in the CE. The development of hybrid CuxS/polymer materials is promising because of the resulting electrical properties of the composite, while the polymer itself allows the preservation of the mechanical robustness of the composite.28,29 Such composite materials are relevant for solar energy applications and can be typically assembled in two ways in a thin film form: (i) by treating a polymer containing an absorbed sulfuring agent with a solution of a metal salt and (ii) by sulfurization of metal compounds absorbed in a polymer.27,30 Regarding the latter approach, carbon-containing oxygen (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and carbon-containing nitrogen (C–N) functionalities at polymer interfaces can act as preferred sites for physical interactions during metal deposition.27,31 Polyamide 6 is characterized for having both C–N and CO functional groups in its structure.31,32 For these reasons, a hybrid material based on CuS/polyamide 6 may be a promising candidate for the development of a CE with enhanced functional properties arising from the electrocatalyst's affinity for the electrolyte used in the QD-based PEC system and from the improvement in the CE mechanical robustness due to the support of the nanoplatelets on the polymer.
O) and carbon-containing nitrogen (C–N) functionalities at polymer interfaces can act as preferred sites for physical interactions during metal deposition.27,31 Polyamide 6 is characterized for having both C–N and CO functional groups in its structure.31,32 For these reasons, a hybrid material based on CuS/polyamide 6 may be a promising candidate for the development of a CE with enhanced functional properties arising from the electrocatalyst's affinity for the electrolyte used in the QD-based PEC system and from the improvement in the CE mechanical robustness due to the support of the nanoplatelets on the polymer.
In this work, we used an electrospinning technique to produce large aspect ratio nanofibers assembled onto FTO glass, which act as supports for magnetron-sputtered Cu films. Transformation from Cu to CuS was achieved through simple and rapid treatment with a polysulfide solution at room temperature. The application of the nanofiber-supported CuS represents an advantage against the commonly used brass treated CE, which further reacts with the sacrificial agent as corroborated by analyzing the morphologies after PEC tests. More importantly, supporting the CuS nanoplatelets on the nanofibers was found to provide similar stability to that of the Pt CE for a testing period of 1 h, in contrast with the poor performance of the flat CuS CE. Additionally, under identical measurement conditions, the nanofiber-supported CuS showed the best catalytic performance and good stability to repeated cyclic voltammetry (CV) tests, performing better than the Pt CE. Our results demonstrate the potential of the nanofiber-supported CuS CE for QD based PEC H2 production and other applications, such as in QDSCs.
Experimental
Materials
Lead acetate trihydrate, trioctylphosphine (TOP, 90%), bis (trimethylsilyl) sulfide (TMS)2S (technical grade, 70%), cadmium oxide (CdO, 99%), oleic acid (OA), oleylamine (OLA), 1-octadecene (ODE), selenium pellets (≥99.999%), trioctyl phosphine oxide (TOPO), hydrochloric acid (HCl), cadmium nitrate tetrahydrate (Cd(NO3)2·4H2O, 98%), zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 98%), sodium sulfide nonahydrate (Na2S·9H2O), sodium sulfite (Na2SO3), toluene, methanol, acetone, ethanol, isopropanol (IPA), polyamide 6 pellets (PA6), and formic acid (FA, 95%) were purchased from Sigma-Aldrich Inc. Ti-Nanoxide BL/SC was bought from Solaronix. Titania paste consisting of ∼20 nm diameter nanoparticles (18 NR-T, paste A) and a blend of active anatase particles (∼20 nm) and larger anatase scatter particle (up to 450 nm) paste (18 NR-AO, paste B) were supplied by Dyesol. Brass substrates containing 64 wt% Cu and 36 wt% Zn were obtained from McMaster-Carr. Cu target (99.99%) was purchased from Ted Pella. All chemicals were used as received.QD synthesis
PbS QDs with diameter ∼3.0 nm were synthesized using a hot injection method, using OA as a ligand.33 In a three-neck reaction flask, a mixture of lead acetate trihydrate (1 mmol), OA (1.2 mL), TOP (1 mL), and ODE (15 mL) were heated to 150 °C for 1 h. After the system was cooled down to ∼100 °C under a vacuum for 15 min, 4.8 mL of a sulphur precursor solution prepared by mixing (TMS)2S (0.5 mmol) with 0.2 mL of TOP was quickly injected into the reaction flask at 130 °C. Subsequently, the reaction was quenched with cold water. The obtained PbS QDs were precipitated with ethanol, centrifuged to remove any unreacted lead oleate and free OA molecules and then re-dispersed in toluene.PbS@CdS QDs were synthesized via a cation exchange method.34 Typically, CdO (2.3 mmol), OA (2 mL) and ODE (10 mL) were heated to 255 °C under N2 for 20 min. The clear solution was cooled down to 155 °C under a vacuum for 15 min. The flask was then reopened and the N2 flux was restored. A PbS QD suspension in toluene (1 mL, absorbance = 3 at the first exciton peak) was diluted in 10 mL toluene, bubbled with N2 for 30 min and then immediately heated to 100 °C. The Cd/OA mixture was added via a syringe. The solution was maintained at 100 °C for 5 minutes and then cooled down to room temperature with cold water. Then PbS@CdS was washed with ethanol and re-dispersed in toluene. The re-dispersion–precipitation procedure was repeated twice.
CdSe QDs with a diameter of 1.65 nm were synthesized by using the hot injection approach.35 Typically, TOPO (1 g) and Cd-oleate (0.38 mmol, 1 mL) in 8 mL of ODE were purged by N2 at room temperature for 30 min. The reaction system was evacuated for 30 min at 100 °C and then the temperature was raised to 300 °C. The mixture of TOP-Se (4 mmol, 4 mL), 3 mL of OLA, and 1 mL of ODE at room temperature was quickly injected into the Cd-oleate suspension under vigorous stirring. The reaction cell was quenched with cold water after injection. 20 mL of ethanol were added, then the suspension was centrifuged, the supernatant was removed and, finally, the QDs were dispersed in toluene.
CdSe@CdS core@shell QDs were obtained using a successive ionic layer adsorption and reaction (SILAR) approach, similar to the procedure described by Ghosh et al.36 Typically, in a 100 mL round-bottom flask, OLA (5 mL), ODE (5 mL) and CdSe QDs (∼2 × 10−7 mol in hexane) were degassed at 110 °C for 30 min. The reaction flask was re-stored with N2 and the temperature was further raised to 240 °C with stirring. Cd(OA)2 dispersed in ODE (0.25 mL, 0.2 M) was added dropwise and the mixture was allowed to react for 2.5 h, followed by dropwise addition of 0.2 M sulfur in ODE with the same volume. All subsequent shells were annealed at 240 °C for ∼10 min following the injection of sulfur and ∼2.5 h following dropwise addition of Cd(OA)2 in ODE. Sulfur/Cd(OA)2 addition volumes for shell addition cycles 1–13 were as follows: 0.25, 0.36, 0.49, 0.63, 0.8, 0.98, 1.18, 1.41, 1.66, 1.92, 2.2, 2.51 and 2.8 mL, respectively. The reaction was cooled to room temperature using cold water. Ethanol was added, then the suspension was centrifuged and the supernatant was removed. The QDs were then dispersed in toluene for further characterization.
TiO2 film preparation
FTO coated glass substrates with sheet resistance 8 Ω sq−1 were cleaned with acetone, methanol, IPA, thoroughly rinsed with deionized water and dried in a N2 stream. A thin and compact TiO2 layer was deposited on the FTO substrates by spin coating a Ti-Nanoxide solution at 2000 rotations per minute for 60 s. Subsequently the films were annealed in air at 500 °C for 30 min after drying and cooled down to room temperature. An active layer was deposited on top of the compact TiO2 layer by tape casting (paste A) and the film was left to stand in air for 15 min. The anodes were then kept at 120 °C for 6 min in a hot plate. A commercial blend of scattering nanoparticles (paste B) was then deposited on top of the active layer, following the previous tape casting procedure. The electrodes were subsequently sintered following temperature profiles at 325 °C/5 min, at 375 °C/5 min, at 450 °C/15 min and at 500 °C/30 min, forming films with thicknesses ∼14 μm, as measured using contact profilometry.Electrophoretic deposition (EPD) of the QDs on the TiO2 film
QDs (PbS/CdS, CdSe or CdSe/CdS) were dispersed in toluene, with a pair of TiO2 deposited FTO slides vertically immersed in the QD solution and facing each other. The distance between them was adjusted to 1 cm. A voltage of 200 V was applied for 120 min.37 To wash off unbound QDs after the EPD process, the samples were rinsed several times with toluene and dried with N2 at room temperature. In a typical SILAR deposition cycle for PbS/CdS QDs,7 Cd2+ ions were deposited from an ethanoic 0.05 M solution of Cd(NO3)2. Similarly, a 0.1 M aqueous Zn(NO3)2 was used as a Zn2+ source. The sulfide sources were 0.05 and 0.1 M solutions of Na2S in methanol/water (50/50 V/V) for the Cd2+ ions, and in water for the Zn2+ ions, respectively. A single SILAR cycle consisted of 1 min of dip-coating the TiO2 working electrode into the metal precursors (Cd2+ or Zn2+) and subsequently into the sulfide solutions. After each bath, the anode was thoroughly rinsed by immersion in the corresponding solvent to remove the chemical residuals from the surface and then drying with a N2 gun. The SILAR cycle was done 4 times for CdS and 2 times for ZnS for each sample for the anode containing PbS/CdS QDs (denoted as TiO2/PbS/CdS/(CdS)4/(ZnS)2). For the CdSe or CdSe/CdS QD-based anode, only 2 cycles of ZnS were further coated via SILAR after depositing the QDs into a mesoporous TiO2 film (denoted as TiO2/CdSe/(ZnS)2 or TiO2/CdSe/CdS/(ZnS)2).CuxS cathode fabrication
Brass foil was immersed into 37% HCl at 70 °C for 5 min, subsequently rinsed with water and dried in air. Ultrasonically cleaned FTO substrates were additionally cleaned for 20 min in an UV/Ozone Ossila cleaning system. Polymer nanofibers were electrospun onto FTO glasses from a 23 wt% solution of PA6 in FA which was kept under stirring for 4 hours. Nanofibers were obtained in a Spraybase, Profector Life Sciences electrospinning unit at 20 kV with a distance of 15 cm and a flow rate of 0.06 mL h−1 with a 26G needle. Subsequently, the nanofibers were sputter coated using a Cu target in a Cressington 208HR high resolution sputter coater, equipped with a quartz crystal microbalance to control the desired thickness. The process was conducted under a 0.01 mbar argon atmosphere, at 80 mA using a rotating disk at a determined angle; the chosen thickness was achieved via two-step Cu coating, 20 nm were deposited first, followed by 15 nm more. The samples were labeled as 40NF, 80NF and 120NF according to the electrospinning deposition time (40, 80 and 120 s) and the flat CE without nanofiber was designated as 00NF. A similar procedure was conducted for nanofiber-supported CuxS in brass (120NF brass). The etched brass foil and the sputter coated Cu samples were immersed in an aqueous polysulfide electrolyte38 (1 M Na2S, 1 M S and 0.1 M NaOH) for 10 min, resulting in the formation of black CuxS. Afterwards the polysulfide excess was removed and washed with distilled water. The samples were dried under the fume cupboard for ∼10 min.Characterization
The morphologies of the CEs were characterized using a field emission scanning electron microscope (FESEM) in a JEOL JSM7401F FE-SEM equipped with an energy-dispersive X-ray spectrometer (EDS). Transmission electron microscopy (TEM) and selected area electron diffraction (SAED) measurements were obtained using a JEOL JEM-2010 TEM. The X-ray diffraction (XRD) pattern was acquired using a Bruker D8 Advance X-ray diffractometer equipped with Cu Kα radiation. X-ray photoelectron spectroscopy (XPS) was performed in a VG Escalab 220i-XL equipped with a hemispherical analyzer recorded for a Twin Anode X-Ray Source. The spectra acquisition parameters (channel exposition, number of scans, analyzer parameters, etc.) were selected so as to provide the best energy resolution and signal/noise ratio. Cu 2p and S 2p photoelectron lines were acquired during the experiment. The C 1s peak (BE = 284.6 eV) was used as an internal reference line to accurately determine the positions of other spectral lines. The fine structure of the photoelectron lines was treated using Casa XPS software (2.3.15 Version).The PEC performance of the photoelectrodes was evaluated in a three-electrode configuration, consisting of a QD-TiO2 photoanode working electrode, a Pt CE or CuS/nanofiber or CuxS supported by brass, and a KCl saturated Ag/AgCl reference electrode. Copper wire was used to connect the photoanode with the outer circuit by using silver paste on the FTO substrate. An insulating epoxy resin was used to cover the sample surface except the active area to avoid any direct contact between the electrolyte and the conducting back-contact and/or connecting wire. Then the sample was fully immersed in the electrolyte with pH = 13, containing 0.25 M Na2S and 0.35 M Na2SO3. All potentials were measured with respect to Ag/AgCl during the PEC test and were converted to the reversible hydrogen electrode (RHE) scale with the following expression VRHE = VAg/AgCl + 0.197 + pH × (0.059).6,39 The photoresponse was measured using an Oriel LCS-100 solar simulator (AM1.5 G, 94011A, S/N: 244, MFD:09/15). The sample was placed 15 cm far from the window of the light source and the light intensity measured using a thermopile was ∼100 mW cm−2. The working area of the photoanode was ∼0.16 cm2. All the current versus potential measurements were carried out at a 20 mV s−1 sweep rate. Current density as a function of time was measured at 0.6 V vs. RHE under continuous AM1.5 G illumination. H2 evolution was measured during the PEC experiment. The produced H2 gas was detected using a ShimadzuGC-8A gas chromatography (GC) device equipped with a thermal conductivity detector. Argon was used as the carrier gas for GC analysis. An air-tight syringe was used for sampling from the vacuum sealed chamber. CV curves for different electrodes were scanned from −1.4 to 0.1 V vs. Ag/AgCl on a CHI760D electrochemical workstation at a scan rate of 100 mV s−1. Additional measurements were carried out at different scan rates (20, 40 and 100 mV s−1) from −1.2 to 0.4 V vs. Ag/AgCl using a 10 times diluted electrolyte aqueous solution.
Results and discussion
SEM images in Fig. 1 reveal the morphologies of the different CuxS CEs. The top three SEM images (Fig. 1a–c) refer to samples before the polysulfide treatment. Fig. 1a shows the flat surface of brass after acidic activation, while large CuxS nanoplatelets are formed after polysulfide treatment (Fig. 1d). Thin Cu coating, sputter deposited on FTO (Fig. 1b), yields small sized CuxS hexagonal nanoplatelets after polysulfide treatment, homogeneously covering the whole surface of this flat CE (Fig. 1e). The smooth surface of the nanofibers after Cu sputter coating is displayed in Fig. 1c. The thickness of the Cu film is ∼35 nm (Fig. S1, ESI†). Incorporation of the nanofiber onto the substrate provides additional Cu sites for the growth of CuxS compared to to those of the Cu film on bare FTO. The thickness of the composite film is between 500 and 950 nm, whereas it is in the range of 300–500 nm in the case of a flat CE (Fig. S2, ESI†). | ||
| Fig. 1 Top view SEM images. (a)–(c) Samples before polysulfide treatment. (a) Acid treated-brass surface, (b) sputter deposited Cu film on the FTO substrate and (c) sputter deposited Cu film on the nanofiber/FTO substrate. (d)–(f) Morphologies obtained after polysulfide treatment: (d) brass CE; (e) 00NF flat CE and (f) 120NF CE. Scale bar equals 4 μm in low magnification images and 1 μm in insets. | ||
The morphology of the ultrasonically detached CuxS nanoplatelets on the nanofiber was further investigated using combined TEM/EDS/SAED analysis (Fig. 2a and b). TEM images reveal the formation of large hexagonal CuxS nanoplatelets. The presence of Cu and S was detected via EDS measurements (Fig. S3, ESI†). Hexagonal nanoplatelets can be obtained from the self-organization of Cu2S and CuS nanocrystals.26,40 A Cu2−xS crystal has five stable phases at room temperature i.e. chalcocite (Cu2S), djurleite, (Cu1.94S), digenite (Cu1.8S), anilite (Cu1.75S) and covellite (CuS) with different crystal structures from hexagonal to rhombohedral.41 HRTEM and SAED show the d-spacing of 3.29 Å and 1.90 Å in the obtained nanocrystal, which correspond to the {100} and {110} d spacing, respectively, of hexagonal CuS (covellite).42 This conclusion was further confirmed by the XRD patterns (Fig. 2c). The crystalline phase of the FTO substrate (JCPDS No. 00-041-1445) is the major contribution to the signal detected in the XRD patterns. However, two weak peaks at 31.2° and 47.2° are clearly detected, assigned to the (103) and (110) reflections of covellite from the nanoplatelet (JCPDS 06-0464).30,42 These results indicate that the obtained nanoplatelets developed on the surface of the nanofibers are mainly composed of hexagonal CuS nanocrystals.
 | ||
| Fig. 2 (a) TEM image, (b) SAED pattern and (c) XRD pattern of the CuxS nanoplatelets. Scale bar equals 5 nm in the high resolution image and 200 nm in the inset. | ||
The as-prepared CEs were further used in a QD-based PEC device. The PEC investigation was performed in the dark and under illumination using a conventional three-electrode configuration with an Ag/AgCl (saturated KCl) reference electrode. A scheme of the system is shown in Fig. 3a. QD-sensitized TiO2 works as a photoelectrode. Tuning the compositions of the QDs is one of the strategies used to tailor their electronic band structure and optical absorption, which contributes significantly to the improvement of PEC performances. In this regard, a standard mesoporous TiO2 film sensitized with CdSe@CdS QDs optically active in the visible region25 or by PbS@CdS QDs optically active in the near infrared (NIR)8 region was used as a photoanode. An aqueous solution containing 0.25 M Na2S and 0.35 M Na2SO3 (pH = 13) served as a sacrificial hole scavenger. Comparison of CEs under the same PEC configuration (using the same anode, electrolyte, and identical measurement conditions) was evaluated using linear voltammetry scan (LVS), containing Pt foil or CuxS CEs. In the visible optically active QD-based PEC system (photoanode sensitized with CdSe/(ZnS)2 or CdSe@CdS/(ZnS)2) a J–V behavior comparable to that of Pt foil was found, by using the CuxS/FTO substrate as a CE (Fig. S4, ESI†), in which the saturated photocurrent density was as high as 10 mA cm−2.
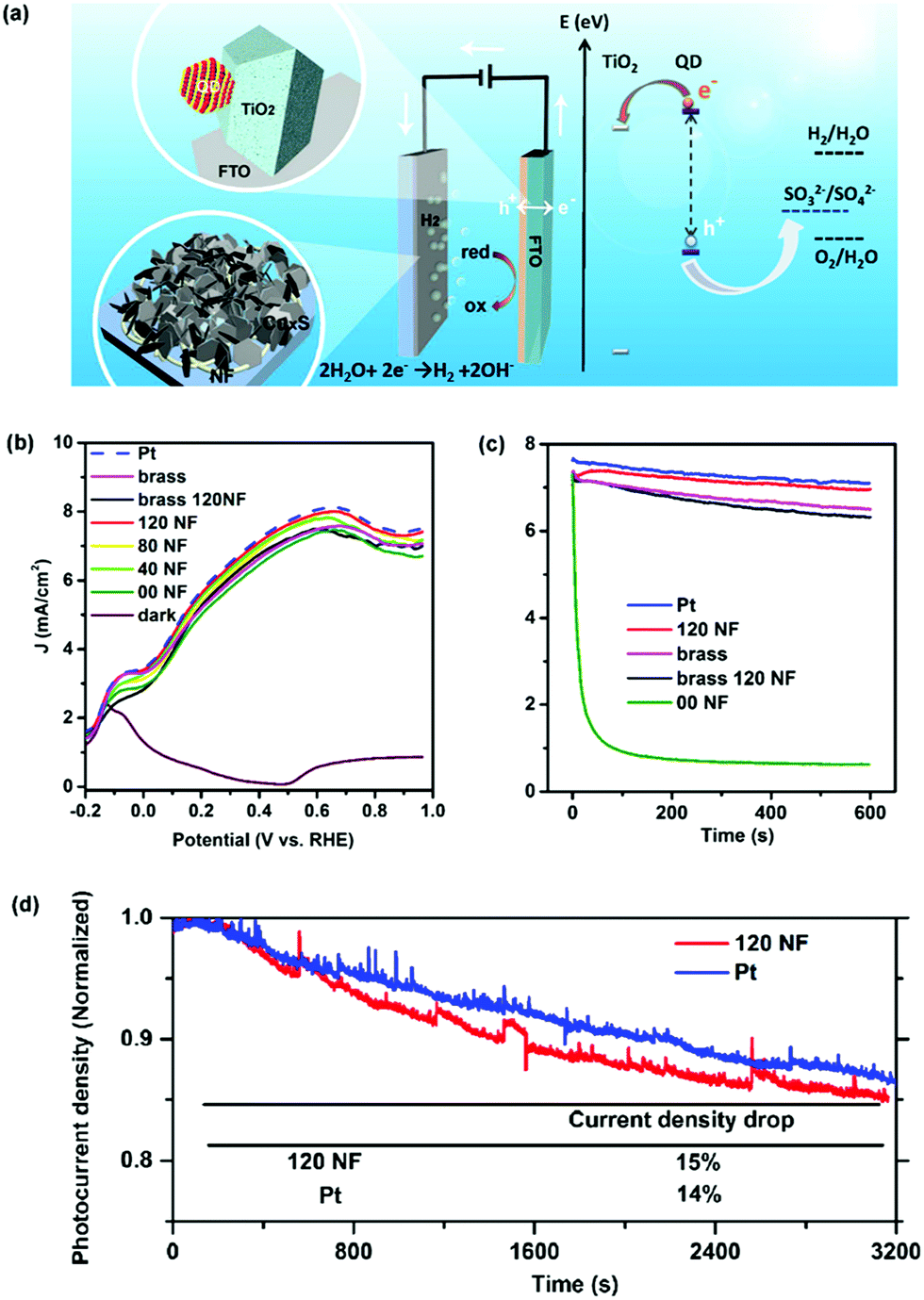 | ||
| Fig. 3 (a) Scheme of the representative PEC device; (b) current density (J) versus the applied voltage of a PEC system based on a TiO2/PbS/CdS/(CdS)4/(ZnS)2 anode, employing different CEs; (c) measured current density as a function of time at 0.6 V vs. RHE under 100 mW cm−2 illumination with an AM 1.5G filter and (d) photocurrent density as a function of time by using the CEs of the nanofiber-supported CuS and the Pt. | ||
Additionally, the NIR optically active QD-based PEC system was further studied with different CuxS CEs, including CuxS/brass, nanofiber-supported CuxS/brass (120NF) and nanofiber-supported CuS/FTO substrate with different electrospinning deposition times (40 s, 80 s and 120 s) (Fig. 3b). In this photoanode comprising TiO2/PbS@CdS/(CdS)4/(ZnS)2 the saturation of the photocurrent density (∼7.5 mA cm−2) occurs at ∼0.6 V versus the RHE, with the conventional Pt foil as a CE. Under identical measurement conditions, all the above-mentioned CuxS- or CuS-based CEs show a very similar J–V response to the Pt cathode within an experimental error. These results clearly show that the low-cost CuxS or CuS-based CEs developed in this work can substitute the expensive Pt to construct a Pt-free high-efficiency PEC apparatus for H2 generation when using QDs as sensitizers.
The stability of the PEC cells was evaluated using potentiostatic (current vs. time) measurements (Fig. 3c), with the same photoanode and identical measurement conditions. For CuxS/brass, the photocurrent densities of the PEC systems had a 13% drop compared to the initial value after 10 min. For the CuxS/FTO CE (00NF), the photocurrent density degraded very fast during the first 200 s, probably due to detachment of the thin CuxS film from the FTO substrate related to corrosion by the electrolyte.43 On the other hand, the photocurrent densities of the PEC systems based on the nanofiber-supported CuS CE (120NF) only showed an 8% decrease compared to its original value after 10 min, which is similar to the Pt electrode, and also resulted in a better performance than that of the CE of the CuxS/brass or nanofiber-supported CuxS/brass (Fig. 3c). It is noteworthy to mention that the poor stability of the CuxS/brass CEs has been related to deposits of CuxS nanoparticles on the photoanode via an electric field-assistant migration; therefore there is competitive light absorption from both the QDs and CuxS nanoparticles, which in turn cannot inject electrons into the QDs or the TiO2 photoanode due to an energy band mismatch.20
The samples using Pt or nanofiber-supported CuS as the CE were further tested for longer operation times. The current density of the sample using the nanofiber-supported CuS as the CE decreased only 15% within ∼1 h which is analogous to the response obtained from the Pt foil (14% drop). These results suggest that the nanofiber-supported CuS on FTO glass can be used as a substitute CE of Pt for both high efficiency and stable PEC H2 production. Additionally, H2 evolution was measured in a PEC system composed of a photoanode sensitized with CdSe@CdS/(ZnS)2 QDs, Pt as the CE and an electrolyte solution containing 0.25 M Na2S and 0.35 M Na2SO3 (pH ∼13).25 The measurements exhibited an almost linear increase over time, getting around 80% of the theoretical value for the evolved H2, as displayed in Fig. S5 (ESI†). The same trend was found in our PEC system composed of PbS@CdS QDs as the photoanode and the nanofiber-supported CuS CE (120NF), as displayed by the theoretically calculated H2 evolution. After ∼1 h, the evolved amount of H2 corresponds to 0.43 mL h−1 (2.66 mL cm−2 h−1, considering the photoanode area).
The morphology of the CEs after the PEC test was analysed and the SEM images are included in Fig. 4. The CuxS/brass-based CE displayed further growth of the CuxS nanoplatelets, which can be an indication of electrolyte induced corrosion of the brass substrate during operation of the PEC system (Fig. 4a). This additional chemical reaction may lead to the variation in the photoactivity of the CuxS nanoplatelets. A noticeable damage by detachment of the CuxS nanoplatelets was found in the CuxS/FTO CE (00NF) (Fig. 4b), which leads to a rapid decrease in photocurrent density during PEC operation (Fig. 3c). In contrast, the surface structure and morphology of the nanofiber-supported CuS/FTO (120NF) CE is less damaged (Fig. 4c), suggesting the good structural stability of the CE, benefiting from the presence of the polymer nanofiber.
 | ||
| Fig. 4 SEM images of the CE surfaces after the PEC test. (a) CuxS/brass based CE, (b) CuxS/FTO (00NF) CE and (c) nanofiber-supported CuS/FTO (120NF) CE. Scale bar equals 4 μm in low magnification images and 1 μm in insets. | ||
Adhesion between the metals and polymer films is governed by the development of physicochemical bonds at the interface.32 Depending on the technique used for metal deposition, polymers with functional groups analogous to PA6 have shown to present new interfacial bonds with Cu; at the interface of sputter deposited Cu/PA6, the adhesion mechanism has been associated with new Cu–O–C bonds.31,32 Additionally, before electrospinning nanofiber deposition, the FTO substrates were UV-Ozone cleaned to remove any remaining organic contaminants. This treatment also improves adhesion within PA6/FTO by rendering a hydrophilic surface in the substrate, which favours affinity for the polar groups of PA6.
XPS analysis was carried out to investigate the chemical binding states of CuS in the CE of the nanofiber-supported CuS/FTO (120NF) before and after PEC testing (Fig. 5). As displayed in Fig. 5a, for the nanofiber-supported CuS/FTO before the PEC test, the XPS spectrum has two main peaks located at 951.8 eV and 931.9 eV assigned to Cu 2p1/2 and Cu 2p3/2, respectively. The latter appears to be consistent with a formal oxidation state of Cu2+, which has binding energies within the range 930–932 eV.44,45 This conclusion was further confirmed by the presence of the weak shake-up satellite peaks at around 943 eV.46,47 After a 10 min PEC test, XPS peaks of Cu 2p1/2 and Cu 2p3/2 were observed at 951.5 and 931.7 eV, respectively. These two peaks almost maintain similar positions as the sample before the PEC test, indicating that there is no detectable chemical variation in Cu2+. S 2p peaks before and after PEC are also included in Fig. 5b, displaying S 2p1/2 and S 2p3/2 peaks appearing at binding energies of 161.5 and 163.1 eV, respectively. These two main doublets are characteristic of S2−.48–50 These results prove the presence of CuS in the CE and are consistent with the conclusions based on the XRD and SAED measurements. In addition, the as-prepared nanofiber-supported CuS is very stable during the PEC measurements.
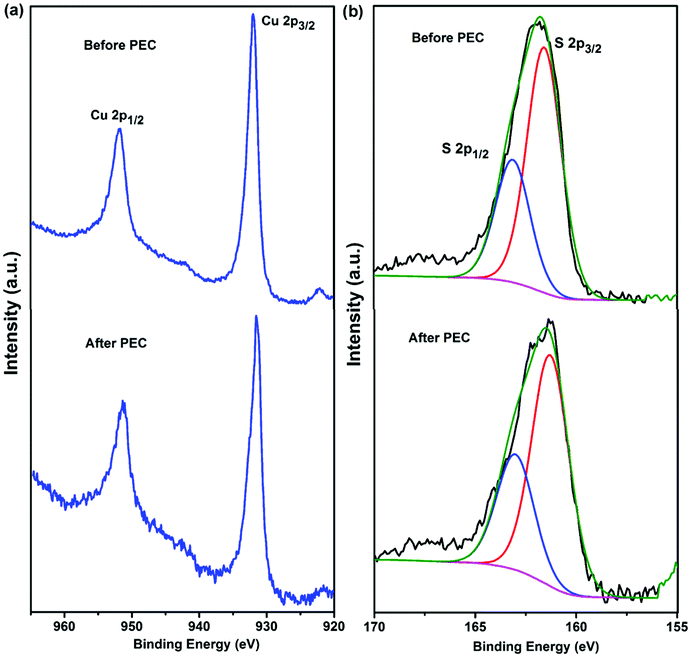 | ||
| Fig. 5 High resolution XPS spectra of Cu 2p (a) and S 2p (b) for the nanofiber-supported CuS (120NF) sample before and after the PEC test. | ||
In the case of the CE based on the CuxS/brass (Fig. S6a, ESI†), there is an accompanying Auger line (Cu LMM) located at 568.6 eV (inset in Fig. S6a, ESI†), indicating the presence of a Cu+ state. After PEC, the satellite peaks at around 943 eV become stronger (highlighted in the red square), implying that the Cu ion was transformed into the chemical state of Cu2+. This feature is due to the oxidation of the Cu film surface.49 As shown in Fig. S6b (ESI†), S presented similar XPS peaks to the nanofiber-supported CuS (120NF), while an additional XPS signal near the S 2p peak was observed at 168.15 eV (highlighted in the red square), which was assigned to the presence of the sulfate species (SO42−) overlayer on the surface of the film.49 SO42− formation has been attributed to the direct oxidation of SO32− at the photoanode.10,51 This behavior indicates that copper in brass could interact with these SO42− species. These findings demonstrate that composite structures obtained by using polyamide 6 nanofibers as a support of the CuS hexagonal nanoplatelets are very stable from the viewpoint of both the chemical structure and morphologies, holding great potential as a highly efficient and stable electrocatalyst CE for PEC hydrogen generation.
Electrochemical stability of the nanofiber-supported CuS CE was compared with a conventional Pt foil CE using repeated CV measurements. Comparison of the behavior obtained under CV measurements of the Pt CE and nanofiber-supported CuS/FTO CE was carried out in a three-electrode cell consisting of Pt foil or 120NF CEs as working electrodes, another Pt foil was used as the counter electrode, and saturated Ag/AgCl was used as the reference electrode. Fig. 6 shows the CV curves obtained with the commonly used Na2S/Na2SO3 electrolyte concentration (0.25 M Na2S and 0.35 M Na2SO3) for PEC H2 generation. Typically, the positive current of CV represent an oxidation of S2− in the electrolyte, while the negative current corresponds to a reduction.16,52–54 The relative catalytic activity CEs can be related to the intensity of the current density peak in the CV curves.55 As shown in Fig. 6, the nanofiber-supported CuS/FTO (120NF) presents remarkably high current density, displaying a 28-fold increase compared to the Pt CE. For the typically used concentration of the electrolyte, 96% of this current density was maintained after 150 cycles. We further investigated this process at different scan speeds and using a 10 times diluted electrolyte (Fig. S7, ESI†). At the commonly used sweep rates (20 mV s−1) during J–V PEC tests the current density in the nanofiber-supported CuS CE is maintained after a few cycles. The stabilized current density of our CE corresponds to a 9-fold increase compared to the one in the Pt CE. These enhancements in the current densities can be interpreted as a better electrocatalytic performance in the CE.55
 | ||
| Fig. 6 Electrochemical CV measurements of the Pt and nanofiber-supported CuS/FTO (120NF) based CEs; the inset is the magnified CV of the Pt CE. | ||
Conclusions and perspectives
Nanofiber-supported CuS nanoplatelets were synthesized for highly efficient and stable PEC H2 production. The nanofibers obtained via electrospinning offered enhanced surface areas for Cu deposited via sputtering and also served as a template to guide the growth of CuS. The nanofiber-supported CuS CE showed a comparable PEC performance to that of Pt CE. This hybrid CE demonstrated a remarkable stability compared to the analogous CuxS/FTO CE and a slightly better performance than that of the typically obtained CuxS from the acidic reaction of brass. The affinity of the tailored CE represents an advantage in the presence of sacrificial agents (S2−/SO32−) and stability tests demonstrated that 86% of the current density was maintained during PEC operation for ∼1 h, similar to the Pt CE, which showed a 15% drop. Additionally, the application of the nanofiber-supported CuS represents an attractive approach compared to the brass treated CEs, which were found to further react with the electrolyte. These results demonstrate that this alternative nanofiber-supported CuS CE has two advantages, namely avoiding the costs of using precious Pt and also obtaining a highly stable and effective QD-based PEC system for H2 generation. Adaptation of this approach can be further explored by changing the composition and/or coating thickness of the catalyst or opting for an inorganic template allowing it to further expand its potential application in other energy conversion devices.Acknowledgements
F. R. acknowledges NSERC for funding through the Discovery Grants program and for an EWR Steacie Memorial Fellowship for funding and partial salary support. F. R. is grateful to the Alexander von Humboldt Foundation for a F. W. Bessel Award. A. V. acknowledges Kempe Foundation and LTU Lab fund program for funding. F. N. P. is grateful for a postdoctoral fellowship from Fonds de recherche du Québec – Nature et technologies (FRQNT) and CONACYT (grant 236094). L. J. is thankful to FRQNT for a PhD Merit Scholarship, R. A. acknowledges NSERC for personal postdoctoral fellowship.References
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- A. Fujishima, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Hagfeldt and M. Graetzel, Chem. Rev., 1995, 95, 49–68 CrossRef CAS.
- J. Hensel, G. Wang, Y. Li and J. Z. Zhang, Nano Lett., 2010, 10, 478–483 CrossRef CAS PubMed.
- R. Van de Krol and M. Grätzel, Photoelectrochemical hydrogen production, Springer, 2012 Search PubMed.
- Y.-L. Lee, C.-F. Chi and S.-Y. Liau, Chem. Mater., 2009, 22, 922–927 CrossRef.
- L. Jin, B. AlOtaibi, D. Benetti, S. Li, H. Zhao, Z. Mi, A. Vomiero and F. Rosei, Adv. Sci., 2016, 3, 1500345 CrossRef PubMed.
- J. F. Reber and K. Meier, J. Phys. Chem., 1984, 88, 5903–5913 CrossRef CAS.
- J. Schneider and D. W. Bahnemann, J. Phys. Chem. Lett., 2013, 4, 3479–3483 CrossRef CAS.
- T. Loučka, J. Electroanal. Chem. Interfacial Electrochem., 1972, 36, 355–367 CrossRef.
- M. Wu, X. Lin, Y. Wang and T. Ma, J. Mater. Chem. A, 2015, 3, 19638–19656 CAS.
- I. Mora-Seró, S. Giménez, T. Moehl, F. Fabregat-Santiago, T. Lana-Villareal, R. Gómez and J. Bisquert, Nanotechnology, 2008, 19, 424007 CrossRef PubMed.
- I. Mora-Seró and J. Bisquert, J. Phys. Chem. Lett., 2010, 1, 3046–3052 CrossRef.
- G. Hodes, J. Manassen and D. Cahen, J. Electrochem. Soc., 1980, 127, 544–549 CrossRef CAS.
- J. G. Radich, R. Dwyer and P. V. Kamat, J. Phys. Chem. Lett., 2011, 2, 2453–2460 CrossRef CAS.
- C. V. Gopi, M. Venkata-Haritha, S.-K. Kim, S. S. Rao, D. Punnoose and H.-J. Kim, RSC Adv., 2015, 5, 2963–2967 RSC.
- R. Milan, M. Hassan, G. S. Selopal, L. Borgese, M. M. Natile, L. E. Depero, G. Sberveglieri and I. Concina, ACS Appl. Mater. Interfaces, 2016, 8, 7766–7776 CAS.
- G. S. Selopal, I. Concina, R. Milan, M. M. Natile, G. Sberveglieri and A. Vomiero, Nano Energy, 2014, 6, 200–210 CrossRef CAS.
- F. Wang, H. Dong, J. Pan, J. Li, Q. Li and D. Xu, J. Phys. Chem. C, 2014, 118, 19589–19598 CAS.
- M. Antoniadou, S. Sfaelou, V. Dracopoulos and P. Lianos, Catal. Commun., 2014, 43, 72–74 CrossRef CAS.
- L.-C. Pop, L. Sygellou, V. Dracopoulos, K. S. Andrikopoulos, S. Sfaelou and P. Lianos, Catal. Today, 2015, 252, 157–161 CrossRef CAS.
- M. Antoniadou, S. Sfaelou and P. Lianos, Chem. Eng. J., 2014, 254, 245–251 CrossRef CAS.
- K. Zhao, H. Yu, H. Zhang and X. Zhong, J. Phys. Chem. C, 2014, 118, 5683–5690 CAS.
- R. Adhikari, L. Jin, F. Navarro-Pardo, D. Benetti, B. AlOtaibi, S. Vanka, H. Zhao, Z. Mi, A. Vomiero and F. Rosei, Nano Energy, 2016, 27, 265–274 CrossRef CAS.
- G. Mondal, P. Bera, A. Santra, S. Jana, T. N. Mandal, A. Mondal, S. I. Seok and P. Bera, New J. Chem., 2014, 38, 4774–4782 RSC.
- V. Janickis, R. Maciulevičiusė, R. Ivanauskas and I. Ancutienė, Colloid Polym. Sci., 2003, 281, 84–89 CAS.
- S. He, G.-S. Wang, C. Lu, J. Liu, B. Wen, H. Liu, L. Guo and M.-S. Cao, J. Mater. Chem. A, 2013, 1, 4685–4692 CAS.
- Y.-Z. Wei, G.-S. Wang, Y. Wu, Y.-H. Yue, J.-T. Wu, C. Lu and L. Guo, J. Mater. Chem. A, 2014, 2, 5516–5524 CAS.
- J. Cardoso, O. GomezDaza, L. Ixtlilco, M. Nair and P. Nair, Semicond. Sci. Technol., 2001, 16, 123–127 CrossRef CAS.
- A. J. Wagner, G. M. Wolfe and D. H. Fairbrother, Appl. Surf. Sci., 2003, 219, 317–328 CrossRef CAS.
- V. Legois, M. Aucouturier, E. Ollivier, E. Darque-Darque and P. Macheto, Surf. Eng., 1998, 14, 259–264 CrossRef CAS.
- T. Zhang, H. Zhao, D. Riabinina, M. Chaker and D. Ma, J. Phys. Chem. C, 2010, 114, 10153–10159 CAS.
- H. Zhao, M. Chaker, N. Wu and D. Ma, J. Mater. Chem., 2011, 21, 8898–8904 RSC.
- B. Dabbousi, J. Rodriguez-Viejo, F. V. Mikulec, J. Heine, H. Mattoussi, R. Ober, K. Jensen and M. Bawendi, J. Phys. Chem. B, 1997, 101, 9463–9475 CrossRef CAS.
- Y. Ghosh, B. D. Mangum, J. L. Casson, D. J. Williams, H. Htoon and J. A. Hollingsworth, J. Am. Chem. Soc., 2012, 134, 9634–9643 CrossRef CAS PubMed.
- L. Jin, H. Zhao, D. Ma, A. Vomiero and F. Rosei, J. Mater. Chem. A, 2015, 3, 847–856 CAS.
- H. McDaniel, N. Fuke, N. S. Makarov, J. M. Pietryga and V. I. Klimov, Nat. Commun., 2013, 4, 2887 Search PubMed.
- R. Trevisan, P. Rodenas, V. Gonzalez-Pedro, C. Sima, R. S. Sanchez, E. M. Barea, I. Mora-Sero, F. Fabregat-Santiago and S. Gimenez, J. Phys. Chem. Lett., 2012, 4, 141–146 CrossRef PubMed.
- Z. Song, H. Lei, B. Li, H. Wang, J. Wen, S. Li and G. Fang, Phys. Chem. Chem. Phys., 2015, 17, 11790–11795 RSC.
- L. Liu, H. Zhong, Z. Bai, T. Zhang, W. Fu, L. Shi, H. Xie, L. Deng and B. Zou, Chem. Mater., 2013, 25, 4828–4834 CrossRef CAS.
- W. Du, X. Qian, X. Ma, Q. Gong, H. Cao and J. Yin, Chem. – Eur. J., 2007, 13, 3241–3247 CrossRef CAS PubMed.
- V. González-Pedro, X. Xu, I. Mora-Sero and J. Bisquert, ACS Nano, 2010, 4, 5783–5790 CrossRef PubMed.
- E. J. Silvester, F. Grieser, B. A. Sexton and T. W. Healy, Langmuir, 1991, 7, 2917–2922 CrossRef CAS.
- S. W. Goh, A. N. Buckley and R. N. Lamb, Miner. Eng., 2006, 19, 204–208 CrossRef CAS.
- T.-L. Li, Y.-L. Lee and H. Teng, Energy Environ. Sci., 2012, 5, 5315–5324 CAS.
- M. Lee and K. Yong, Nanotechnology, 2012, 23, 194014 CrossRef PubMed.
- Y. Xie, A. Riedinger, M. Prato, A. Casu, A. Genovese, P. Guardia, S. Sottini, C. Sangregorio, K. Miszta and S. Ghosh, J. Am. Chem. Soc., 2013, 135, 17630–17637 CrossRef CAS PubMed.
- M. Kundu, T. Hasegawa, K. Terabe, K. Yamamoto and M. Aono, Sci. Technol. Adv. Mater., 2016, 9, 035011 CrossRef.
- R. Pattrick, J. Mosselmans, J. Charnock, K. England, G. Helz, C. Garner and D. Vaughan, Geochim. Cosmochim. Acta, 1997, 61, 2023–2036 CrossRef CAS.
- N. Buehler, K. Meier and J. F. Reber, J. Phys. Chem., 1984, 88, 3261–3268 CrossRef CAS.
- L.-W. Chong, H.-T. Chien and Y.-L. Lee, J. Power Sources, 2010, 195, 5109–5113 CrossRef CAS.
- S. J. Yuan, Z. J. Zhou, Z. L. Hou, W. H. Zhou, R. Y. Yao, Y. Zhao and S. X. Wu, Chem. – Eur. J., 2013, 19, 10107–10110 CrossRef CAS PubMed.
- R.-Y. Yao, Z.-J. Zhou, Z.-L. Hou, X. Wang, W.-H. Zhou and S.-X. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 3143–3148 CAS.
- I. Hwang and K. Yong, ChemElectroChem, 2015, 2, 634–653 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c6qm00144k |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2017 |