
Ultrafine Pt–Ru bimetallic nanoparticles anchored on reduced graphene oxide sheets as highly active electrocatalysts for methanol oxidation
G. Vishwakshan
Reddy
a,
P.
Raghavendra
a,
B.
Ankamwar
b,
P.
Sri Chandana
c,
S. M.
Senthil Kumar
d and
L. Subramanyam
Sarma
*a
aNanoelectrochemistry Laboratory, Dept. of Chemistry, Yogi Vemana University, Kadapa, Andhra Pradesh, India. E-mail: sarma7@yogivemanauniversity.ac.in
bBio-inspired Materials Chemistry Laboratory, Dept. of Chemistry, University of Pune, Pune, Maharashtra, India
cDepartment of Civil and Environmental Engineering, Annamacharya Institute of Technology & Sciences, Kadapa, Andhra Pradesh, India
dElectrochemical Materials Science Division, CSIR-Central Electrochemical Research Institute, Karaikudi, Tamil Nadu, India
First published on 2nd November 2016
Abstract
The controlled fabrication of bimetallic nanocatalysts with a suitable size, structure and morphology is extremely important to realize direct methanol fuel cells (DMFCs) as promising energy-generating sources. In this paper, a facile approach for depositing platinum–ruthenium (Pt–Ru) bimetallic nanoparticles on a reduced graphene oxide (RGO) support has been demonstrated. Two electrocatalysts denoted as Pt–Ru/RGO-AA and Pt–Ru/RGO-AB were synthesized by synchronously reducing H2PtCl6 and RuCl3 on the graphene oxide (GO) support with reducing agents ascorbic acid and methyl ammonia borane, respectively. For comparison of methanol electrooxidation activity, monometallic Pt nanoparticles supported on RGO sheets (Pt/RGO) were also synthesized. All the catalysts were conveniently synthesized under ambient conditions without using any surfactants. The Pt/RGO, Pt–Ru/RGO-AA and Pt–Ru/RGO-AB electrocatalysts were characterized using X-ray diffraction (XRD), high-resolution transmission electron microscopy (HR-TEM), selected area electron diffraction (SAED) and energy dispersive X-ray spectroscopy (EDS). Among all the catalysts, the Pt–Ru/RGO-AB catalyst with an average particle size of 2.8 nm possessed remarkable uniformity on the RGO sheets. The electrochemical performance of RGO-supported Pt and Pt–Ru catalysts towards methanol oxidation was systematically studied using cyclic voltammetry and chronoamperometry. The results demonstrate that the Pt–Ru/RGO-AB catalyst with a high electrochemical active surface area (ECSA) of 130.46 m2 g−1 exhibits a higher mass and ECSA normalized activities towards methanol oxidation. Furthermore, the Pt–Ru/RGO-AB catalyst has a better tolerance towards accumulated CO-like species as realized from its higher ratio of forward peak current density to reverse peak current density (If/Ib) of 3.21. The facile fabrication strategy described here is convenient and could be used for the fabrication of other multi-component nanostructured electrocatalysts for fuel cell reactions.
Introduction
Direct methanol fuel cells (DMFCs) have become a subject of extensive research due to their ability to offer environmentally friendly energy conversion with a high efficiency and promising durability.1–3 In these electrochemical cells, methanol and water molecules are simultaneously oxidized at the anode producing CO2, electrons and protons (known as the methanol oxidation reaction, MOR). Protons released at the anode diffuse through the proton exchange membrane to the cathode and react with electrons and the oxidant air simultaneously and are reduced to water (known as the oxygen reduction reaction, ORR). The direct oxidation of methanol fuel simplifies the fuel-cell technology and attracts applications in various power-hungry portable devices like cellular phones, notebooks and computers. With more recent developments in this field, fuel cells are promising to provide power to automobiles and in large scale electricity-generating set-ups.4 The key to realize the widespread use of DMFCs for mobile and stationary applications is the use of more efficient and stable electrocatalysts which can effectively catalyze both MOR and ORR half-cell reactions. In particular, the development of sufficiently active and selective catalysts to accelerate the kinetics of MOR is a prerequisite for further progress of this technology. So far, platinum is the only metal available for the dissociative adsorption of methanol. However, methanol oxidation produces carbon monoxide and subsequently poisons the active platinum surface required for the continuous adsorption of methanol in real applications.5–7 The oxidative removal of CO on a Pt-only surface requires OH formation which will occur only at a high potential. This situation limits the application of Pt as an MOR catalyst. Several Pt-based alloys including metals such as Fe, Ni, Co, Mo, Sn, Pd and Ru etc. were explored intensively as CO-tolerant MOR electrocatalysts rather than Pt-alone.8–19 According to the literature, Pt-based bimetallic surfaces offer enhanced catalytic activities and stabilities over Pt-alone catalysts due to a “bifunctional mechanism”20 and “ligand effects”.21,22 Among the various bimetallic Pt-based electrocatalysts explored for MOR, Pt–Ru/C catalysts are viewed as highly efficient anode catalysts.23–25 However, in practical fuel cell applications, Pt–Ru/C loadings should be maintained at a high level in order to achieve better device performances. The high cost associated with the use of noble catalyst loading remains one of the major impediments to DMFC commercialization. To circumvent these problems, the reduction of catalyst loading through maximum utilization of the catalysts (like developing catalysts with an extremely smaller size and hence a higher surface area) and catalyst activity optimization by exploring high performance electron-conducting supports is highly sought.An ideal carbon support for an electrocatalyst should possess a high surface area and optimum electronic conductivity to aid electron transport. In addition, the supporting material also has to be sufficiently porous to facilitate the transport of reactants and products during the electrode reactions.26,27 Impressive progress has been made in the research and development of novel carbon supports with good stability and greater corrosion resistance. Novel carbon-based support materials such as carbon black,28 carbon nanotubes (CNTs),29–37 mesoporous carbon,38 amorphous supermicroporous carbons (ASCs),39 graphite nanofibres (GNFs),40 graphene etc.,41–48 and 3D-graphene aerogel have been explored for use as fuel cell catalysts.49 In a recent development, graphene quantum dots (GQDs) have been explored as viable supporting substrates for fuel cell catalysts.50–52 Very recently, we have fabricated uniform Pt–Pd bimetallic nanoparticles on graphene supports as methanol electrooxidation catalysts by introducing halide ions during the synthesis process.53 Most of these established studies revealed that the electrocatalytic activity and durability of the catalysts are greatly improved because of the high electrical conductivities, large surface areas and versatilities in pore sizes and distributions of nanostructured support materials. Among the various carbon nanostructures explored so far, specifically graphene and its derivatives have been identified as the most promising support materials for fuel cell electrocatalysts due to their large surface area (theoretical surface area of 2620 m2 g−1), good dispersion stability, chemical inertness and high electrical conductivity.54,55
In the present study, a simple method was proposed for the controlled deposition of Pt–Ru bimetallic nanoparticles on graphene supports by using methyl ammonia borane (MeAB) as a reducing agent under ambient conditions. MeAB simultaneously reduces graphene oxide (GO), platinum and ruthenium precursors to form Pt–Ru bimetallic nanoparticles on a reduced graphene oxide (RGO) support (denoted as Pt–Ru/RGO-AB). For comparison, Pt–Ru/RGO prepared by using ascorbic acid as a reducing agent (denoted as Pt–Ru/RGO-AA) and Pt/RGO were also fabricated. The activities of Pt–Ru/RGO-AB, Pt–Ru/RGO-AA, Pt/RGO and a commercial E-TEK Pt–Ru/C towards the methanol oxidation reaction (MOR) were investigated in N2–saturated 0.5 M H2SO4 containing 1 M CH3OH. The controlled reducing ability of MeAB yields a homogeneous distribution of ultrafine Pt–Ru bimetallic nanoparticles on RGO sheets at room temperature. As a result, Pt–Ru/RGO-AB as an electrocatalyst exhibits a superior electrocatalytic activity towards MOR when compared to the commercial PtRu/C and home-made Pt–Ru/RGO-AA and Pt/RGO electrocatalysts.
Experimental
Chemicals
Natural graphite powder, H2PtCl6·6H2O, RuCl3, methylamine hydrochloride, sulphuric acid and methanol were purchased from Aldrich and used as-received. Tetra hydro furan, diethyl ether, KMnO4, NaNO3, and ascorbic acid were purchased from Merck India. 20 wt% Pt/C and 30 wt% Pt–Ru/C commercial catalysts were purchased from Alfa-Aesar. All the chemicals used in the present study are of analytical grade and used without further purification.Preparation of graphene oxide (GO)
GO was prepared using a modified Hummer's method as elaborated in our previous report.53 The prepared GO powder was thoroughly washed with distilled water several times until the pH of the washings was neutral. GO powder was dried overnight in a hot-air oven at 60 °C.Preparation of methyl ammonia borane (MeAB)
Methyl ammonia borane was prepared utilizing a procedure reported elsewhere.56 In a typical procedure, appropriate amounts of sodium borohydride and methylamine hydrochloride with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio were added to a 250 mL two-neck round-bottom flask with a neck connected to a condenser. Later, 100 mL of tetrahydrofuran (THF) was added to the flask and stirred vigorously at room temperature under a nitrogen atmosphere for 12 h. The resultant solution was filtered and the filtrate was concentrated under a vacuum at room temperature. Finally, the product was purified using diethyl ether and then utilized as a reducing agent.
:1 molar ratio were added to a 250 mL two-neck round-bottom flask with a neck connected to a condenser. Later, 100 mL of tetrahydrofuran (THF) was added to the flask and stirred vigorously at room temperature under a nitrogen atmosphere for 12 h. The resultant solution was filtered and the filtrate was concentrated under a vacuum at room temperature. Finally, the product was purified using diethyl ether and then utilized as a reducing agent.
Preparation of reduced graphene oxide (RGO)-supported Pt–Ru bimetallic nanocomposites
In a typical synthesis process, 70 mg of GO was added to 80 mL of double distilled water in a 250 mL beaker. The water and GO mixture were first ultrasonically treated with a Labman ultrasonicator operated at an ultrasonic wattage of 300 W for 1 h to ensure the uniform mixing of GO in water. To this, 2.026 mL of 0.05 M aqueous solution of H2PtCl6·6H2O and 15 mg KI dissolved in 6 mL water was added and ultrasonication was continued. In our previous study, it was found that the addition of KI to metal precursor solutions enhances the final dispersion of metal nanoparticles onto a GO support. Later, 2.027 mL of 0.05 M aqueous solution of RuCl3 was added. Precursor amounts of Pt and Ru were taken to get the final weight of 20 wt% Pt and 10 wt% Ruon RGO (Total Pt + Ru is 30 wt%) with a Pt-to-Ru atomic ratio of 1:1. The total mixture was stirred overnight in a beaker at room temperature. This step was deliberately followed to achieve better anchoring of the metal ions to the GO support. The suspension containing metal halides and GO was completely transferred into a 250 mL round bottomed flask and the pH of the entire reaction mixture was maintained at a value of 12 by adding the appropriate amount of NaOH under continuous stirring. The pH-maintained reaction mixture was slowly stirred at room temperature for 6 h. To this, 200 mg of freshly prepared methyl amino borane (MeAB) dissolved in 10 mL distilled water was added slowly under stirring at room temperature. Here, MeAB acts as a reducing agent to simultaneously reduce the GO, platinum and ruthenium precursors to form Pt–Ru on the RGO support. The solid product was allowed to settle down within 5 min. The final Pt–Ru on the RGO product labelled as Pt–Ru/RGO-AB was washed copiously with distilled water and dried in a hot-air oven at 60 °C for 12 h. Separately, for comparison, Pt–Ru/RGO with the same metal loading (20 wt% Pt + 10 wt% Ru) was prepared with L-ascorbic acid as a reducing agent. A similar procedure as the one used for the fabrication of Pt–Ru/RGO-AB was adopted, except that in the last step 200 mg of L-ascorbic acid dissolved in 10 mL of distilled water was added to the reaction mixture instead of MeAB. The product obtained was labelled as Pt–Ru/RGO-AA. Monometallic 20 wt% Pt nanoparticles were deposited on RGO sheets by reducing H2PtCl6 using methyl ammonia borane as a reducing agent.
Physical characterization of the electrocatalysts
X-ray diffraction (XRD) patterns of the synthesized samples were collected using a PANalytical Xpert PRO X-ray powder diffractometer using Cu Kα-radiation (1.5418 Å). XRD scans were recorded with a scan rate of 5° min−1 for 2θ values between 5 and 90°. Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) characterizations were carried out using a JEOL JEM-2100 operated at 200 kV. The samples for the TEM and HRTEM analyses were prepared by ultrasonically dispersing a small amount of solid powders in ethanol and placing a drop of the diluted sample on a carbon-coated copper grid. An Oxford Instruments' energy-dispersive X-ray detector (EDX) equipped with TEM was used to analyse the atomic composition of the Pt–Ru/RGO and Pt/RGO catalysts. Tapping-mode atomic force microscopy (AFM) imaging of the GO sample was performed on an Ntegra Prima Atomic Force Microscope (NTMDT, Russia). Fourier transform infrared spectroscopy (FT-IR) measurements of the catalyst samples were performed on a Perkin Elmer Spectrum Two™ FT-IR spectrometer in the range of 500–4000 cm−1. Inductively coupled plasma-optical emission spectroscopy (ICP-OES) analysis was carried out with an Optima 5300 DV ICP-OES spectrometer (Perkin Elmer, USA) to determine the metal loadings.Electrochemical characterization and methanol electrooxidation investigations
Cyclic voltammograms and chronoamperometry were performed at room temperature in a standard three-electrode electrochemical cell connected to a CHI potentiostat (Model CHI 6002 E, USA). A silver/silver chloride (saturated KCl) (E° = −0.197 V vs. NHE) and a Pt wire were used as the reference and auxiliary electrodes, respectively. The working electrode was fabricated on a glassy carbon disk (3 mm in diameter with a geometric area of 0.0706 cm2). At first, the glassy carbon electrode (GCE) was polished carefully with 1.0, 0.5, and 0.05 μm alumina powder, followed by ultrasonication in ethanol and deionized water several times. The catalyst slurry was prepared by mixing 2 mg of catalyst powder in a mixture of water (1000 μL), ethanol (500 μL) and 0.005% Nafion solution (100 μL) under ultrasonication for 30 min. A 5 μL droplet of well-dispersed catalyst ink was deposited on GCE and air-dried before performing the electrochemical measurements. Cyclic voltammetry experiments were performed in freshly prepared 0.5 M H2SO4 solution saturated with high purity nitrogen by sweeping the electrode potential from −0.2 to 1.2 V vs. Ag/AgCl (saturated KCl) at a scan rate of 50 mV s−1 for 20 runs. Electrochemical surface areas (ECSAs) of the catalyst samples were calculated from the stable voltammograms. Methanol oxidation reaction (MOR) measurements were performed in a solution of 0.5 M H2SO4 containing 1 M CH3OH at a scan rate of 50 mV s−1. Chronoamperometry (CA) measurements were carried out in a solution of 0.5 M H2SO4 containing 1 M CH3OH for 1000 s at 0.65 V vs. Ag/AgCl (saturated KCl). The specific activity of methanol oxidation for the electrocatalysts was measured by normalizing the current with the ECSA values. Furthermore, the mass activity of methanol oxidation for the electrocatalysts was obtained by normalizing the observed current with the Pt-mass loading on the working electrode.Results and discussion
Graphene oxide (GO)
Fig. 1(a) shows the representative TEM micrographs of the GO layers. As seen in Fig. 1(a), GO is transparent with a puff-like morphology. The literature studies show that the transparent nature is the result of an extremely thin monolayer formation of GO with a high graphitic degree formed during the synthesis process.57 The selected area electron diffraction (SAED) pattern of the GO layer shown in Fig. 1(b) reveals a typical six-fold symmetry pattern related to the ordered structure of the high crystalline graphene layers.58,59 The high resolution TEM image shown in Fig. 1(c) shows the continuous well-defined lattice fringes of graphene, and these occur in packets of 4 to 5 sheets. HRTEM analysis further shows that different layers of graphene are formed during chemical exfoliation. Atomic force microscopy (AFM) is also used to characterize the morphology of the GO layer. As seen in Fig. 1(d)–(g), uniform layers of GO sheets with various sizes with an apparent thickness of ∼3 to 5 nm corresponding to few layers of GO were observed.60,61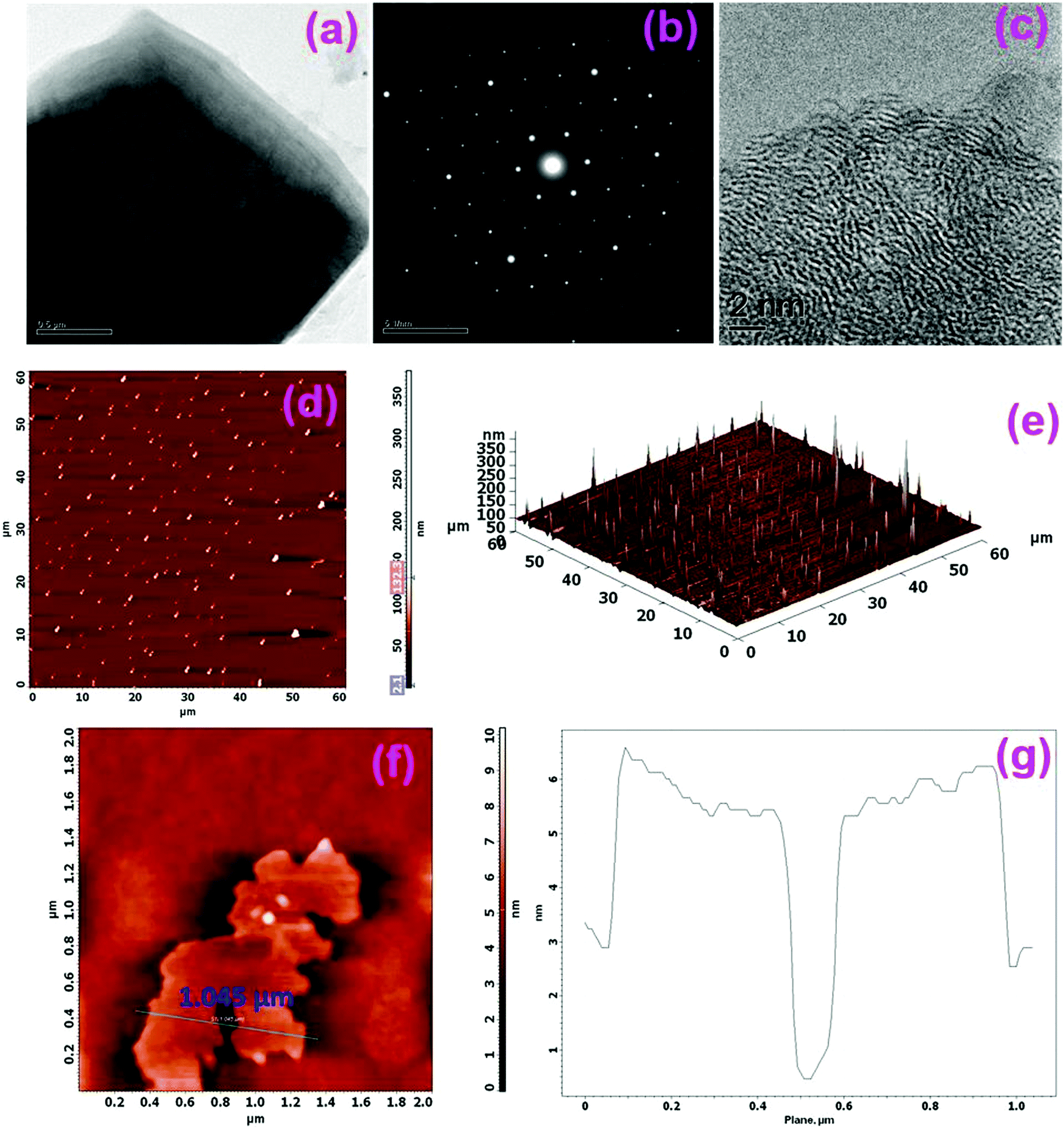 | ||
| Fig. 1 Structural characterization of graphene oxide (GO). (a) TEM image (scale bar, 0.5 μm); (b) corresponding selective area electron diffraction (SAED) pattern; (c) HRTEM image; (d) Tapping-mode 2D-atomic force microscopy (AFM) image; (e) 3D-AFM image; (f) 2D-AFM image of selected GO layers and (g) line-scan profile of the GO layers shown in panel (f). | ||
Morphology studies of Pt/RGO and Pt–Ru/RGO catalysts
TEM was used to examine the surface morphology of the synthesized RGO-supported Pt and Pt–Ru catalysts. As can be seen in the case of the Pt/RGO (Fig. 2A) and Pt–Ru/RGO-AB (Fig. 2G) catalysts ultrafine Pt and Pt–Ru, respectively, are well-dispersed and densely covered on the RGO support without any significant agglomeration. In contrast, in the case of the synthesized Pt–Ru/RGO-AA catalysts, severely agglomerated Pt–Ru particles are deposited on the RGO layer (Fig. 2D). This result demonstrates that ascorbic acid (AA) is inferior to methyl ammonia borane towards the stabilization and growth of the particles. The particle size distribution was estimated statistically by measuring 100 particles in the selected TEM images. As shown in the corresponding particle size histograms, both Pt/RGO (Fig. 2B) and Pt–Ru/RGO-AB (Fig. 2H) exhibited narrow particle size distributions with an average diameter of 4.3 ± 2.2 nm and 2.8 ± 1.1 nm, respectively. Studies have proven that oxygen-containing groups on GO provide anchoring sites for the interactions with metal ions, thereby facilitating a controlled nucleation and growth which results in the higher dispersion and immobilization of metal particles on the reduced GO surface.62–65 However, Pt–Ru/RGO-AA catalysts with severe aggregation show a broader size distribution with an average diameter centred around 52.5 ± 3.4 nm (Fig. 2E).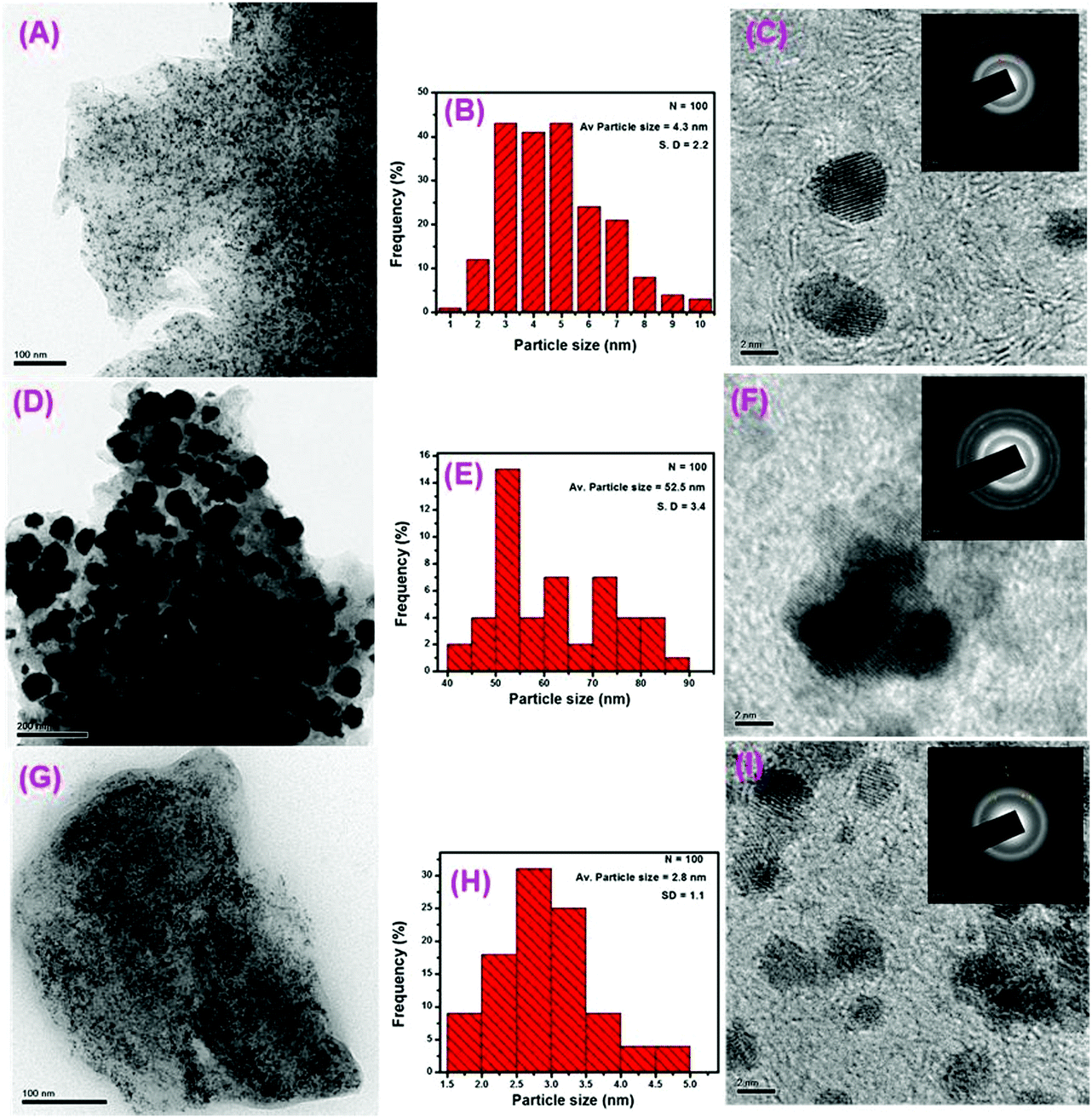 | ||
| Fig. 2 TEM images (A, D and G) of Pt/RGO, Pt–Ru/RGO-AA, and Pt–Ru/RGO-AB, respectively. Particle size histograms (B, E and H) of Pt/RGO, Pt–Ru/RGO-AA, and Pt–Ru/RGO-AB, respectively. HR-TEM images (C, F and I) of Pt/RGO, Pt–Ru/RGO-AA, and Pt–Ru/RGO-AB, respectively. Corresponding SAED patterns are shown as insets. | ||
In addition to the attributes offered by GO, here the use of methyl ammonia borane as a reducing agent plays a key role in achieving the controlled reduction of either Pt or PtRu, and may contribute to the improvement of Pt and PtRu particle dispersion on the RGO support. The higher extent of uniform dispersion and extremely small size observed in the case of the PtRu/RGO-AB nanoparticles suggest that a higher electrocatalytic performance towards methanol oxidation can be expected for this catalyst.
Additional structural features of the Pt/RGO and Pt–Ru/RGO catalysts were evaluated using high-resolution TEM (HR-TEM). As shown in the HR-TEM images (Fig. 2C, F, and I), all the catalysts exhibit highly ordered lattice fringes which correspond to a Pt face centre cubic (fcc) lattice structure. The interplanar spacing of the lattice fringes calculated for the Pt–Ru/RGO-AA and Pt–Ru/RGO-AB catalysts are ∼0.217 and 0.221 nm, respectively, corresponding to the (111) crystal plane of a Pt–Ru alloy consistent with the literature reports.66 Furthermore, the SAED patterns shown as insets in Fig. 2C, F, and I verify the good crystalline features of all the catalysts.
EDS analysis
The EDS scans of Pt/RGO and Pt–Ru/RGO nanocomposites are presented in Fig. 3. The EDS results show the presence of Pt and Ru in the catalyst particles. The Pt and Ru compositions for the Pt–Ru/RGO catalysts are very close to the nominal values (Pt:Ru for Pt–Ru/RGO-AA is 50.4:49.6 and for Pt–Ru/RGO-AB is 50.2:49.8). The Pt and Ru metal loadings on the RGO support calculated from the ICP-OES analyses are found to be 19.3 wt% Pt + 9.1 wt% Ru for the Pt–Ru/RGO-AA catalyst and 19.5 wt% Pt + 9.3 wt% Ru for the Pt–Ru/RGO-AB catalyst. The Pt metal loading on Pt/RGO is found to be 19.3 wt% on the RGO support.
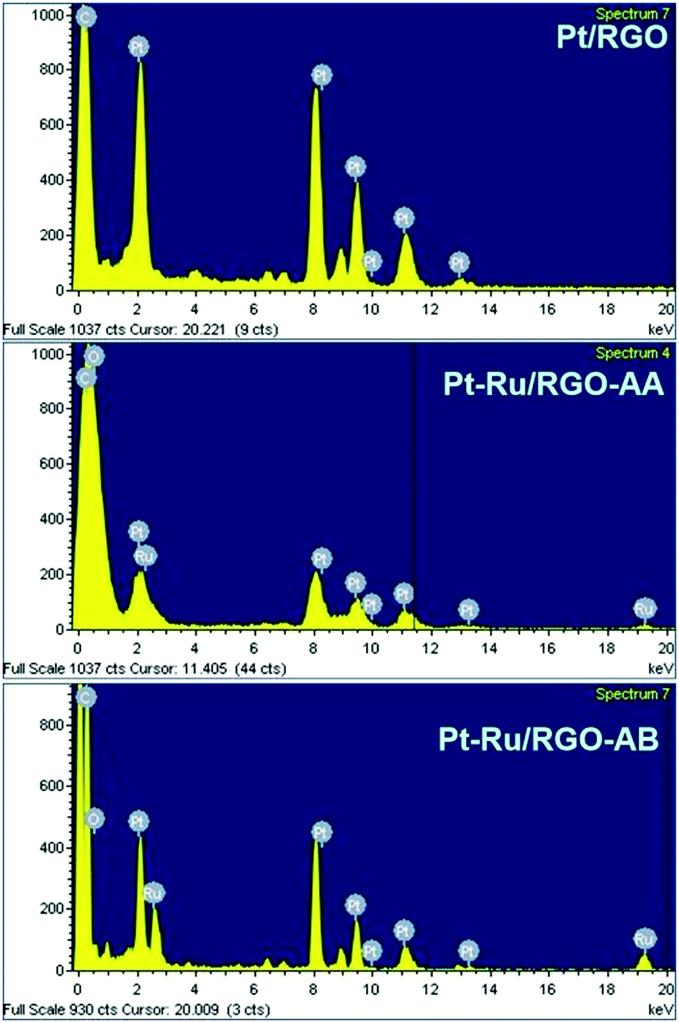 | ||
| Fig. 3 Energy dispersive X-ray spectroscopy (EDS) scans of the Pt/RGO, Pt–Ru/RGO-AA and Pt–Ru/RGO-AB nanocomposites. | ||
XRD analysis
Fig. 4 shows the XRD patterns of the as-synthesized GO and the RGO-supported Pt and Pt–Ru catalysts. The as-synthesized GO exhibits an intense peak at 2θ ∼ 10° related to a C(002) reflection. Literature studies have shown that for pristine graphite the C(002) peak appears at 2θ ∼ 26.5°. The peak shift in the case of GO to 10° is ascribed to the expanded interlayer distance of the graphene layers due to the presence of oxygen-containing groups like carboxyl, epoxide and water molecules between the GO layers formed as a result of chemical exfoliation of pristine graphite.67–69 The XRD for Pt/RGO, Pt–Ru/RGO-AA and Pt–Ru/RGO-AB exhibits diffraction peaks at 2θ ∼ 39.7°, 46.0°, 67.4°, and 80.1° which are conveniently ascribed to the (111), (200), (220) and (311) crystal planes of platinum with a face-centred-cubic (fcc) phase (JCPDF 04-0802). Close inspection of the XRD scans of Pt–Ru/RGO catalysts show that 2θ values shift to higher values compared to bulk platinum. Furthermore, no peaks related to the hexagonal close packed (hcp) structured Ru(0) (JCPDF-06-0663) are found indicating that Ru might be incorporated into the Pt-fcc lattice forming alloy structured Pt–Ru/RGO catalysts.70–72 The absence of peaks relating to the Pt or Ru oxides demonstrates that precursors are mainly converted into metallic nanoparticles. Interestingly, the XRD peaks of the Pt–Ru/RGO-AB catalysts are weak and much broader compared to the peaks for Pt/RGO and Pt–Ru/RGO-AA, indicating the extremely nanoscopic nature of the catalyst. The average crystallite size of the catalysts is calculated using the Scherrer's equation from line broadening analysis by selecting a Pt(220) peak.73,74 The average crystallite sizes of the Pt and Pt–Ru nanoparticles in Pt/RGO, Pt–Ru/RGO-AA and Pt–Ru/RGO-AB are calculated to be about 4.02, 52.13 and 2.48 nm, respectively. The estimated average crystallite sizes of the catalyst particles are in good agreement with the TEM analysis results.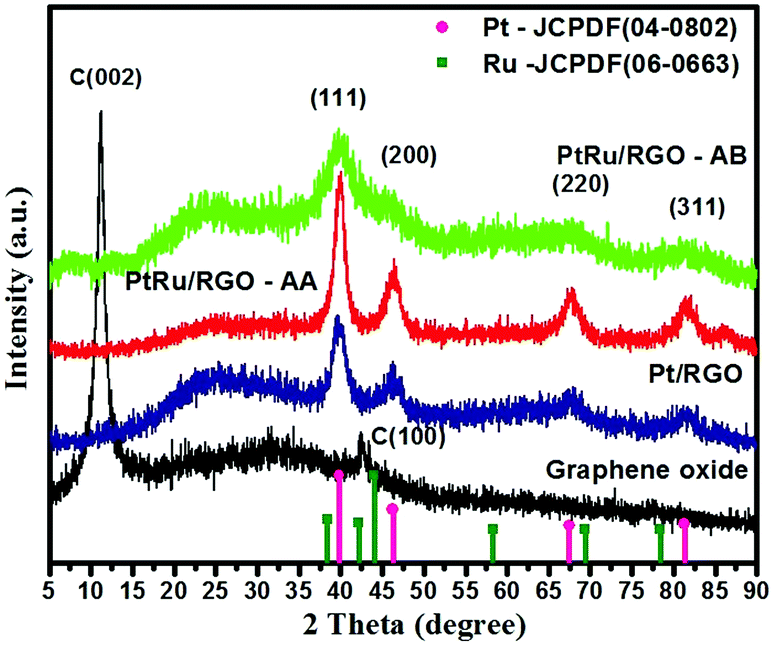 | ||
| Fig. 4 XRD scans of GO and the in-house synthesized Pt/RGO and Pt–Ru/RGO catalysts. | ||
FT-IR analysis
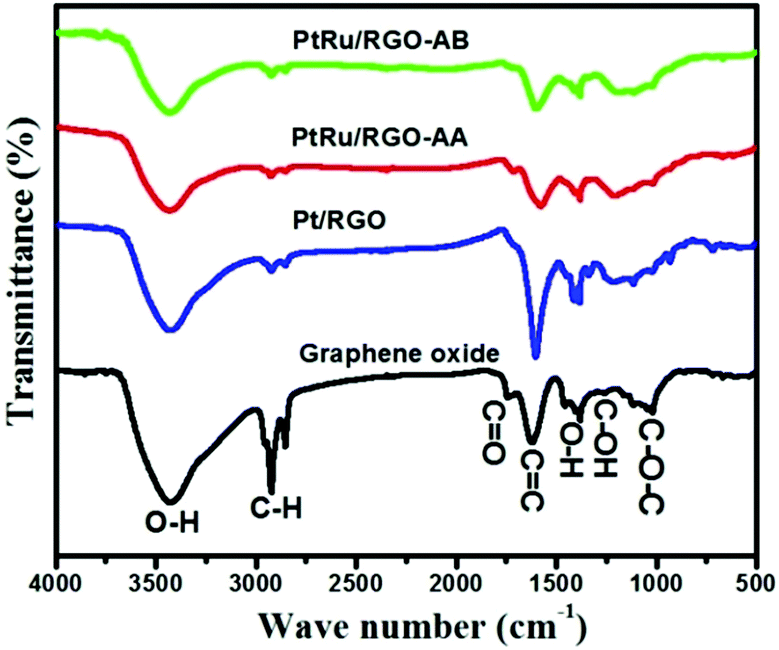
FT-IR spectroscopy provides information about the functional groups present on the GO surfaces and the extent of reduction during the synthesis process. As shown in Fig. 5, the as-synthesized GO shows absorption bands that can be assigned to a stretching vibration of O–H functionalities appearing at 3250–3500 cm−1,75,76 and a characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O absorption band at 1736 cm−1, which is indicative of the presence of carbonyl and carboxyl species in GO.
O absorption band at 1736 cm−1, which is indicative of the presence of carbonyl and carboxyl species in GO.
 | ||
| Fig. 5 FT-IR spectral scans of GO and the in-house synthesized Pt/RGO and Pt–Ru/RGO catalysts. | ||
In addition, the spectrum of GO shows absorption bands at 1405 cm−1 and 1052 cm−1 relating to the C–OH and C–O–C functional groups respectively, indicating the presence of rich-oxygen containing functional groups on GO. After treatment with methyl ammonia borane in the case of the Pt/RGO and Pt–Ru/RGO-AB catalysts, and ascorbic acid (AA) in the case of the Pt–Ru/RGO-AA catalyst, the intensity of these peaks are considerably decreased and some of the peaks totally disappeared indicating the partial removal of oxygen-containing groups upon reduction.
CV analysis
Surface structural characteristics of in-house synthesized RGO-supported Pt and PtRu catalysts and commercial Pt/C and Pt–Ru/C catalysts were assessed by performing cyclic voltammetry measurements on catalysts in N2–saturated H2SO4 (0.5 M) solution in the potential range −0.2 to 1.2 V vs. Ag/AgCl (satd. KCl) at a scan rate of 50 mV s−1. As shown in Fig. 6, peaks associated with hydrogen adsorption/desorption were observed between −0.15 V and 0.1 V. A higher extent of the double layer region observed in the case of the as-synthesized RGO-supported Pt and Pt–Ru catalysts signifies the high surface area of the RGO support. In addition, the irregular current peaks appearing in the double layer region are associated with the residual oxygen-containing functional groups on graphene consistent with the literature.77,78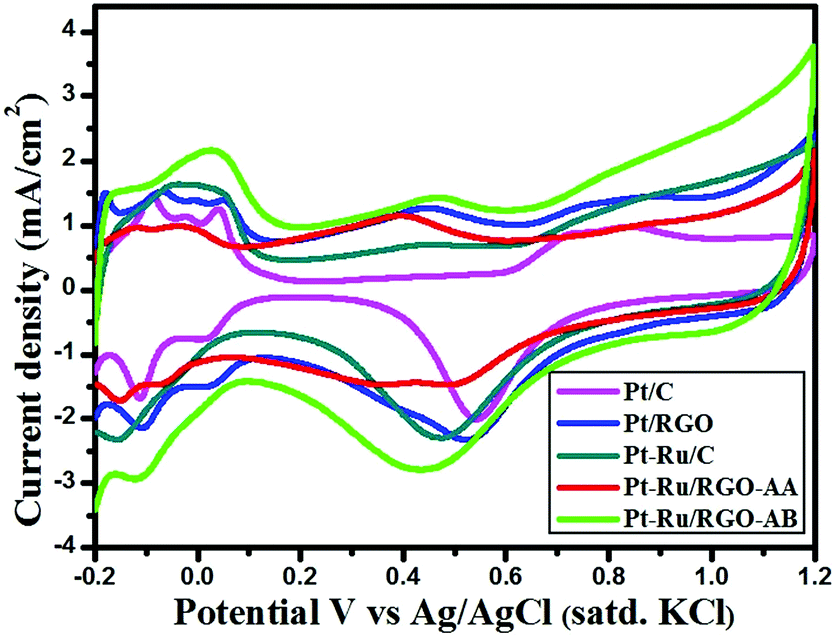 | ||
| Fig. 6 Cyclic voltammograms of the in-house synthesized Pt/RGO, Pt–Ru/RGO and commercial Pt and PtRu/C catalysts in N2–saturated 0.5 M H2SO4 solution at a scan rate of 50 mV s−1. | ||
The calculated ECSA values of commercial and as-synthesized catalysts varied in the following order: Pt–Ru/RGO-AB (130.46 m2 g−1) > Pt–Ru/C (80.76 m2 g−1) > Pt/RGO (63.23 m2 g−1) > Pt/C (59.05 m2 g−1) > Pt–Ru/RGO-AA (58.6 m2 g−1). The higher ECSA value observed for Pt–Ru/RGO-AB might be due to its extremely small size and uniform distribution on the GO layer when compared to the other catalysts, as demonstrated by the TEM analysis. The CV result demonstrates that better methanol oxidation activity can be anticipated for the Pt–Ru/RGO-AB catalyst.
Electrooxidation of methanol
The electrocatalytic activities of the as-synthesized Pt and Pt–Ru/RGO catalysts and commercial Pt/C and Pt–Ru/C catalysts are evaluated for electro-oxidation of methanol by performing cyclic voltammetry in 0.5 M H2SO4 containing 1 M CH3OH solution. The voltammogram was repeated until stable and reproducible CV was obtained. The methanol oxidation current shown in the CVs of Fig. 7A and B represent weight normalized, in terms of Pt-weight (mass activity), and ECSA normalized currents (specific activity), respectively.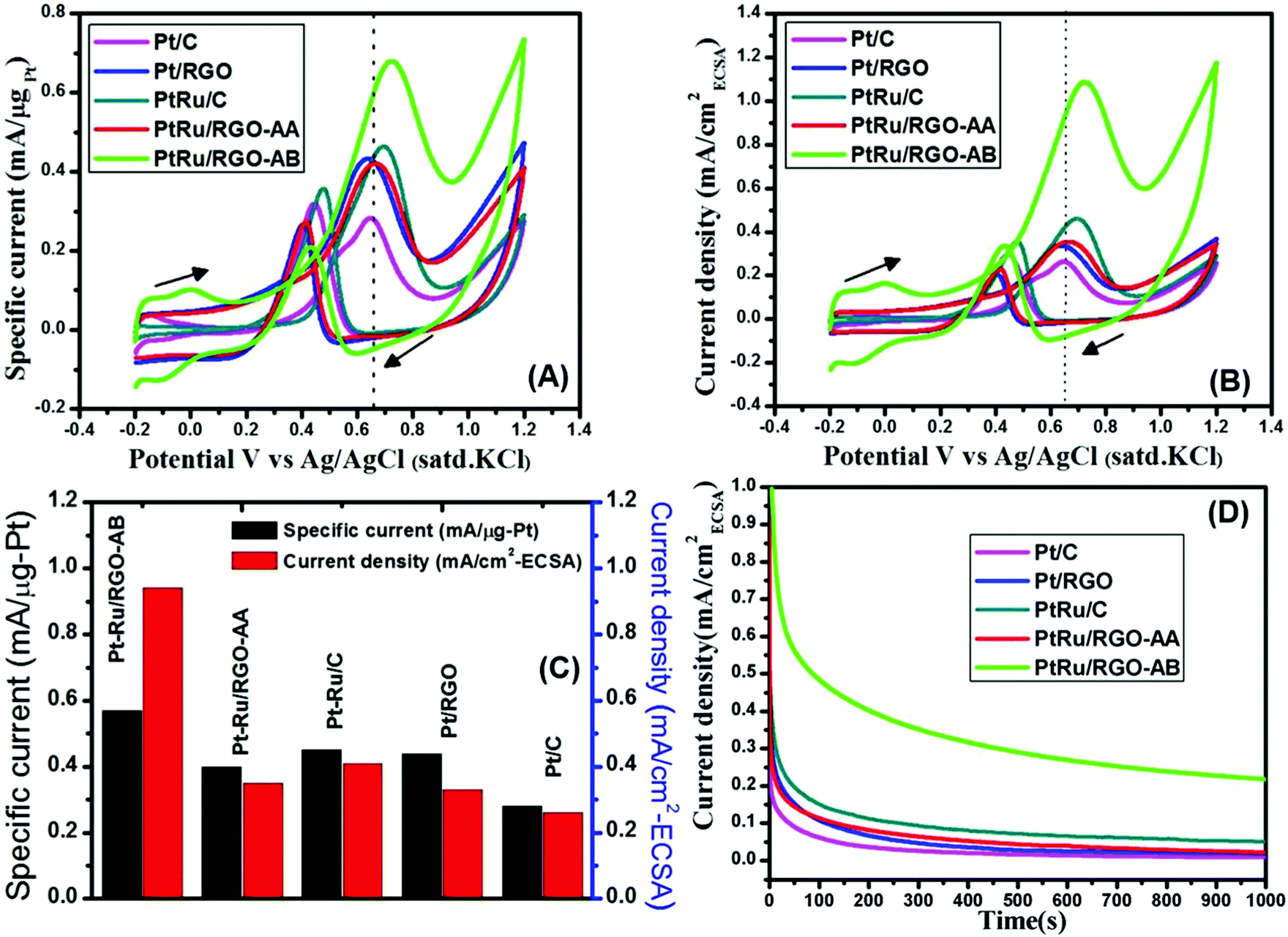 | ||
| Fig. 7 (A) Cyclic voltammograms for methanol oxidation (Pt-mass normalized current) on Pt/RGO, Pt–Ru/RGO, and commercial Pt/C and Pt–Ru/C electrocatalysts; (B) cyclic voltammograms for methanol oxidation (ECSA normalized current) on Pt/RGO, Pt–Ru/RGO, and commercial Pt/C and Pt–Ru/C electrocatalysts; (C) comparison of the specific current and ECSA-normalized current of Pt/RGO, Pt–Ru/RGO, and commercial Pt/C and Pt–Ru/C electrocatalysts; (D) chronoamperometric curves for methanol oxidation on Pt/RGO, Pt–Ru/RGO, and commercial Pt/C and Pt–Ru/C electrocatalysts at a fixed potential of 0.65 V for 1000 s. | ||
As shown in both Fig. 7A and B for all the catalysts, the CVs show a peak ascribed to methanol oxidation in the forward scan and a peak related to the oxidation of carbonaceous motifs in the reverse scan. Established studies show that intermediates formed during methanol oxidation such as CO and other carbonaceous species will be oxidized in the reverse scan. The magnitude of the forward peak current can be taken as a measure of methanol oxidation degree. The forward peak current values of all the catalysts both in terms of mass normalized and ECSA normalized is graphically represented in Fig. 7C. The as-synthesized Pt–Ru/RGO-AB catalyst with its specific current (0.57 mA μg−1-Pt) exhibits a higher methanol oxidation activity than that of the Pt/RGO (0.42 mA μg−1-Pt), Pt–Ru/RGO-AA (0.41 mA μg−1-Pt), commercial Pt/C (0.27 mA μg−1-Pt) and Pt–Ru/C (0.43 mA μg−1-Pt) catalysts at 0.65 V. Comparison of the ECSA normalized current density values at 0.65 V show that the as-synthesized Pt–Ru/RGO-AB catalyst exhibits more than two times higher activity than the commercial Pt–Ru/C catalyst. In addition, the onset potential of methanol electrooxidation in the case of Pt–Ru/RGO-AB is more negative when compared to Pt/RGO, Pt–Ru/RGO-AA, commercial Pt/C, Pt–Ru/C and is centered at about 0.3 V. Furthermore, the ratio of the forward peak current density (If) to backward peak current density (Ib) is generally viewed as an important index to understand the extent of catalyst ability towards CO-like carbonaceous intermediate tolerance. It has been well reported that a higher If/Ib value can be taken as a measure for the higher extent of oxidation of carbonaceous intermediates to carbon dioxide on the catalyst surface.62,79 The calculated If/Ib values follow the order: Pt–Ru/RGO-AB (3.21) > Pt/RGO (1.64) > Pt–Ru/RGO-AA (1.59) > Pt–Ru/C (1.31) > Pt/C (0.89). The better MOR activity both in terms of mass-specific and area-specific activity in acidic medium in the case of as-synthesized Pt–Ru/RGO-AB catalysts is mainly attributed to the combined advantages offered by the extremely small size of the Pt–Ru particles (2.8 nm) and graphene oxide supports. It has been well reported that higher kinetics of methanol electrooxidation reaction could be achieved on Pt–Ru surfaces because Ru is good at providing sites for the adsorption of oxygenated species (OH)ads relatively at more negative potentials than Pt and primarily contributes in the oxidation of adsorbed CO to CO2.80,81 This situation leads to the removal of adsorbed CO intermediates and cleans the surface of Pt for further adsorption of methanol for continuous electrooxidation. Furthermore, incorporation of Ru into the Pt lattice could alter the electronic structure in a positive way by decreasing the strength of the Pt–CO bond since Ru can withdraw more electron density from Pt. The decreased electron density on Pt weakens its ability in back donating the electrons to the CO antibonding molecular orbitals and hence results in a weak Pt–CO bond.82,83 Additionally, GO as a support could also provide higher additional sites for the adsorption of oxygenated species required for the oxidation of CO-like species to CO2. The methanol electrooxidation catalytic activities of the home-made Pt–Ru/RGO catalysts in the present work are compared with the RGO-supported Pt–Ru catalysts reported in the literature.63,84–88 As shown in Table 1, the peak specific current of the methanol electrooxidation of the home-made Pt–Ru/RGO-AB catalysts measured at 25 °C is 0.57 mA μg−1-Pt (=570 mA mg−1-Pt vs. Ag/AgCl) and 2.8 times higher than that of PtRu/FGS (205.7 mg−1-Pt vs. Ag/AgCl).63The electrochemical stabilities of the home-made Pt/RGO and Pt–Ru/RGO catalysts and commercial Pt/C and PtRu/C are studied by performing chromoamperometry (CA) experiments in 0.5 M H2SO4 containing 1 M CH3OH at 0.65 V.
| Catalyst | Metal loading (wt%) | ECSA (m2 g−1) | MOR activity as peak current density | Ref. |
|---|---|---|---|---|
| Pt–Ru/GS-CNTS | 30 | 118.6 | 137 mA mg−1 | 84 |
| Pt–Ru/FGS | 30 | 40.5 | 205.7 mA mg−1 | 63 |
| Pt–Ru/RGO-PVP | 31 | 55.4 | 146 mA mg−1 | 85 |
| Pt–Ru/RGO | 30 | 68 | 20.8 mA cm−2-ECSA | 86 |
| Pt–Ru/RGO | 30 | 10.3 | 24.4 mA cm−2-ECSA | 87 |
| Pt–Ru/GS | 30 | 77.23 | 78.7 mA mg−1 | 84 |
| Pt–Ru–G | 30 | 59.6 | 19 mA cm−2-ECSA | 88 |
| Pt–Ru–RGO | 30 | 130.5 | 570 mA mg−1 | This work |
As shown in Fig. 7D, initially higher current densities can be seen on all the catalysts with the highest being on Pt–Ru/RGO-AB. The initial decay in current density observed in all the samples can be attributed to the formation of CO-like species formed on the electrode surfaces during methanol oxidation.58,89 A closer inspection of the CA curves reveals that the initial decay in current density for carbon black-based Pt, Pt/RGO and Pt–Ru/RGO-AA catalysts is more rapid compared to Pt–Ru/RGO-AB, signifying the higher CO-tolerance ability of the Pt–Ru/RGO-AB catalyst. In the entire time frame, Pt–Ru/RGO-AB retains the highest current density compared to all the other catalysts demonstrating its superior electrocatalytic activity and stability for methanol oxidation. The significantly improved electrocatalytic activity of Pt–Ru/RGO-AB for methanol oxidation indicates the greater number of active sites available for the unit surface area which is in good agreement with the CVs shown in Fig. 6. The enhanced catalytic performance of the Pt–Ru/RGO-AB catalyst can be ascribed to the combined role played by Pt, Ru and the GO support.
Conclusions
In summary, a facile approach for achieving uniform and extremely small sized Pt–Ru bimetallic nanoparticles on RGO supports using amino borane as a reducing agent under ambient conditions is demonstrated. The resulting Pt–Ru/RGO-AB catalysts possess a higher electrochemical active surface area (ECSA) and thus superior methanol electrooxidation activities compared to commercial Pt/C and in-house Pt/RGO and Pt–Ru/RGO-AA catalysts. The surfactant-less synthesis strategy demonstrated here is convenient and could be extended to the fabrication of other multi-metallic nanoparticles on graphene supports for DMFC applications.Acknowledgements
Financial support from the Science and Engineering Research Board (SERB) of the Department of Science and Technology (DST), New Delhi (Project No. SB/S1/PC-98/2012), is greatly acknowledged. One of the authors (GVR) is thankful to the University Grants Commission (UGC), New Delhi, for providing a Senior Research Fellowship.References
- B. D. McNicol, D. A. J. Rand and K. R. Williams, J. Power Sources, 1999, 83, 15–31 CrossRef CAS.
- A. S. Arico, S. Srinivasan and V. Antonucci, Fuel Cells, 2001, 2, 133–161 CrossRef.
- C. Lamy, A. Lima, V. LeRhun, F. Delima, C. Coutanceau and J. M. Leger, J. Power Sources, 2002, 105, 283 CrossRef CAS.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed.
- H. Uchida, K. Izumi, K. Aoki and M. Watanabe, Phys. Chem. Chem. Phys., 2009, 11, 1771–1779 RSC.
- G. Kim and S. H. Jhi, ACS Nano, 2011, 5, 805–810 CrossRef CAS PubMed.
- H. Wang, Z. Jusys and R. J. Behm, J. Phys. Chem. B, 2004, 108, 19413–19424 CrossRef CAS.
- Y. Lu and W. Chen, Chem. Commun., 2011, 47, 2541–2543 RSC.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- Z. Peng and H. Yang, J. Am. Chem. Soc., 2009, 131, 7542–7543 CrossRef CAS PubMed; L. L. He, J. N. Zheng, P. Song, S. X. Zhong, A. J. Wang, Z. Chen and J. J. Feng, J. Power Sources, 2015, 276, 357–364 CrossRef.
- H. You, F. Zhang, Z. Liu and J. Fang, ACS Catal., 2014, 4, 2829–2835 CrossRef CAS.
- F. Colmati, E. Antolini and E. R. Gonzalez, Electrochim. Acta, 2005, 50, 5496–5503 CrossRef CAS.
- A. O. Neto, R. R. Dias, M. M. Tusi, M. Linardi and E. V. Spinacé, J. Power Sources, 2007, 166, 87–91 CrossRef.
- C. T. Habe and M. S. Wrighton, Langmuir, 1993, 9, 3284 CrossRef.
- N. R. Mathe, M. R. Scriba and N. J. Coville, Int. J. Hydrogen Energy, 2014, 39, 18871–18881 CrossRef CAS.
- F. Taufany, C. J. Pan, H. L. Chou, L. S. Sarma, J. Rick, J. M. Lin, J. F. Lee, M. T. Tang and B. J. Hwang, Chem. – Eur. J., 2013, 19, 905–915 CrossRef CAS PubMed.
- B. J. Hwang, L. S. Sarma, G. R. Wang, C. H. Chen, D. G. Liu, H. S. Sheu and J. F. Lee, Chem. – Eur. J., 2007, 13, 6255–6264 CrossRef CAS PubMed.
- L. S. Sarma, T. D. Lin, Y. W. Tsai, J. M. Chen and B. J. Hwang, J. Power Sources, 2005, 139, 44–54 CrossRef CAS.
- C. H. Chen, L. S. Sarma, D. Y. Wang, F. L. Lai, C. C. Al Andra, S. H. Chang, D. G. Liu, C. C. Chen, J. F. Lee and B. J. Hwang, ChemCatChem, 2010, 1, 1–9 Search PubMed.
- M. Watanabe and S. Motoo, J. Electroanal. Chem., 1975, 60, 267–273 CrossRef CAS.
- J. A. Rodriguez and D. W. Goodman, J. Phys. Chem., 1991, 95, 4196 CrossRef CAS.
- J. A. R. Van Veen, T. Frelink and W. Visscher, Surf. Sci., 1995, 335, 353–360 CrossRef.
- A. E. Batista, H. Hoster and T. J. Iwasita, J. Electroanal. Chem., 2003, 554–555, 265–271 CrossRef.
- H. W. Lei, S. Suh, B. Gurau, B. Workie, R. X. Liu and E. S. Smotkin, Electrochim. Acta, 2002, 47, 2913–2919 CrossRef CAS.
- Y. Y. Tong, H. S. Kim, P. K. Babu, P. Waszczuk, A. Wieckowski and E. J. Oldfield, J. Am. Chem. Soc., 2002, 124, 468–473 CrossRef CAS PubMed.
- C. Arbizzani, S. Beninati, E. Manferrari, F. Soavi and M. J. Mastragostino, J. Power Sources, 2007, 172, 578–586 CrossRef CAS.
- V. Rao, P. A. Simonov, E. R. Savinova, G. V. Plaskin, S. V. Cherapanova, G. N. Kryukova and U. Stimming, J. Power Sources, 2005, 145, 178–187 CrossRef CAS.
- B. Fang, N. K. Chaudhari, M. S. Kim, J. H. Kim and J. S. Yu, J. Am. Chem. Soc., 2009, 131, 15330–15338 CrossRef CAS PubMed.
- S. Guo, S. Dong and E. Wang, Adv. Mater., 2010, 22, 1269–1272 CrossRef CAS PubMed.
- X. Yu and S. Ye, J. Power Sources, 2007, 172, 145–154 CrossRef CAS.
- G. Wu, Y. S. Chen and B. Q. Xu, Electrochem. Commun., 2005, 7, 1237–1243 CrossRef CAS.
- G. Che, B. B. Lakshmi, C. R. Martin and E. R. Fischer, Langmuir, 1999, 15, 750–758 CrossRef CAS.
- W. Li, C. Liang, W. Zhou, J. Qiu, Z. Zhou, G. Sun and V. Xin, J. Phys. Chem. B, 2003, 107, 6292–6299 CrossRef CAS.
- D. Villers, S. H. Sun, A. M. Serventi and J. P. Dodelet, J. Phys. Chem. B, 2006, 110, 25916–25925 CrossRef CAS PubMed.
- M. S. Saha, R. Li and X. Sun, J. Power Sources, 2008, 177, 314–322 CrossRef CAS.
- T. Matsumoto, T. Komatsu, H. Nakanoa, K. Arai, K. Nagashima, Y. Yoo, E. Yamazaki, T. Kijima, M. Shimizu, H. Takasawa and Y. Nakamura, Catal. Today, 2004, 90, 277–281 CrossRef CAS.
- G. Girishkumar, K. Vindogopal and P. V. Kamat, J. Phys. Chem. B, 2004, 108, 19960–19966 CrossRef CAS.
- F. Su, J. Zeng, X. Bao, Y. Yu, J. Y. Lee and X. Zhao, Chem. Mater., 2005, 17, 3960–3967 CrossRef CAS.
- Z. R. Ismagilov, M. A. Kerzhenstev, N. V. Shikina, A. S. Lisitsyn, L. B. Okhlopkova, C. N. Barnakov, M. Sakashita, T. Iijima and K. Tadokoro, Catal. Today, 2005, 102–103, 58–66 CrossRef CAS.
- H. Tang, J. H. Chen, L. H. Nie, D. Y. Li, W. Deng, Y. F. Kuang and S. Z. Yao, J. Colloid Interface Sci., 2004, 269, 26–31 CrossRef CAS PubMed.
- B. Xia, Y. Yan, X. Wang and X. W. Lou, Mater. Horiz., 2014, 1, 379–399 RSC.
- P. F. Xu, L. Dong, M. Neek-Amal, M. L. Ackerman, J. H. Yu and S. D. Barber, ACS Nano, 2014, 8, 2697–2703 CrossRef CAS PubMed.
- Y. M. Tan, C. F. Xu, G. X. Chen, N. F. Zheng and Q. J. Xie, Energy Environ. Sci., 2012, 5, 6923–6927 CAS.
- B. Seger and P. V. Kamat, J. Phys. Chem. C, 2009, 113, 7990–7995 CAS.
- Y. Si and T. E. Samulski, Chem. Mater., 2008, 20, 6792–6797 CrossRef CAS.
- P. Kundu, C. Nethravathi, P. A. Deshpande, M. Rajamathi, G. Madras and N. Ravishankar, Chem. Mater., 2011, 23, 2772–2780 CrossRef CAS.
- Y. Xia, J. G. Liua, Y. Zhou, W. Liu, J. Gao, Y. Xie, Y. Yin and Z. Zou, J. Power Sources, 2011, 196, 1012 CrossRef.
- S. M. Choi, M. H. Seo, H. J. Kim and W. B. Kim, Carbon, 2011, 49, 904–909 CrossRef CAS.
- H. Yin, C. Zhang, F. Liu and Y. Hou, Adv. Funct. Mater., 2014, 24, 2930–2937 CrossRef CAS.
- Y. Song and S. W. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 14050–14060 CAS.
- G. Q. He, Y. Song, K. Liu, A. Walter, S. Chen and S. W. Chen, ACS Catal., 2013, 3, 831–838 CrossRef CAS.
- C. P. Deming, R. Mercado, V. Gadiraju, S. W. Sweeney, M. Khan and S. Chen, ACS Sustainable Chem. Eng., 2015, 3, 3315–3323 CrossRef CAS.
- G. Vishwakshan Reddy, P. Raghavendra, P. Srichandana and L. S. Sarma, RSC Adv., 2015, 5, 100522–100530 RSC.
- M. H. Seo, S. M. Choi, H. J. Kim and W. B. Kim, Electrochem. Commun., 2011, 13, 182–185 CrossRef CAS.
- B. P. Vinayan, R. Nagar, N. Rajalakshmi and S. Ramaprabhu, Adv. Funct. Mater., 2012, 22, 3519–3526 CrossRef CAS.
- N. Cao, J. Su, W. Luo and G. Cheng, Int. J. Hydrogen Energy, 2014, 39, 426 CrossRef CAS.
- J. Qi, L. Jiang, Q. Tang, S. Zhu, S. Wang, B. Yi and G. Sun, Carbon, 2012, 50, 2824–2831 CrossRef CAS.
- J. Zhao, H. Li, Z. Liu, W. Hu, C. Zhao and D. Shi, Carbon, 2015, 87, 116–127 CrossRef CAS.
- C. T. Pan, J. A. Hinks, Q. M. Ramasse, G. Greaves, U. Bangert, S. E. Donnelly and S. T. Haigh, Sci. Rep., 2014, 4, 6334 CrossRef CAS PubMed.
- H. C. Schniepp, J. L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. J. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- C. Nethravathi, E. A. Anumol, M. Rajamathi and N. Ravishankar, Nanoscale, 2011, 3, 569–571 RSC.
- J. Zhao, L. Zhang, H. Xue, Z. Wang and H. Hu, RSC Adv., 2012, 2, 9651–9659 RSC.
- J. N. Zheng, S. S. Li, F. Y. Chen, N. Bao, A. J. Wang, J. R. Chen and J. J. Feng, J. Power Sources, 2014, 266, 259–267 CrossRef CAS.
- P. Song, S. S. Li, L. L. He, J. J. Feng, L. Wu, S. X. Zhong and A. J. Wang, RSC Adv., 2015, 5, 87061–87068 RSC.
- M. Xiao, L. Feng, J. Zhu, C. Liu and W. Xing, Nanoscale, 2015, 7, 9467–9471 RSC.
- J. D. Qiu, G. C. Wang, R. P. Liang, X. H. Xia and H. W. Yu, J. Phys. Chem. C, 2011, 115, 15639–15645 CAS.
- G. Wang, J. Yang, J. Park, X. Gou, B. Wang, H. Liu and J. Yao, J. Phys. Chem. C, 2008, 112, 8192 CAS.
- H. K. Jeong, M. H. Jin, K. H. An and Y. H. Lee, J. Phys. Chem. C, 2009, 113, 13060 CAS.
- J. Zhao, H. Xue, L. Zhang, J. Yu and H. Hu, J. Nanopart. Res., 2012, 14, 935 CrossRef.
- G. An, P. Yu, L. Mao, Z. Sun, Z. Liu, S. Miao, Z. Miao and K. Ding, Carbon, 2007, 45, 536–542 CrossRef CAS.
- W. Lin, C. Liang, W. Zhou, J. Qiu, G. Sun, Z. Zhou and Q. Xin, J. Phys. Chem. B, 2003, 107, 6292–6299 CrossRef.
- W. Z. Li, W. Z. Zhou, H. Q. Li, Z. H. Zhou, B. Zhou, J. Q. Sun and Q. Xin, Electrochim. Acta, 2004, 49, 1045–1055 CrossRef CAS.
- Z. Hou, B. Yi, H. Yu, Z. Liu and H. J. Zhang, J. Power Sources, 2003, 123, 116–125 CrossRef CAS.
- J. Guo, R. Wang, W. W. Tjiu, J. Pan and T. Liu, J. Hazard. Mater., 2012, 63, 225–226 Search PubMed.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS PubMed.
- T. Y. Zhang, D. Zhang and M. Shen, Mater. Lett., 2009, 63, 2051–2054 CrossRef CAS.
- W. He, H. Jiang, Y. Zhou, S. Yang, X. Xue, Z. Zou, X. Zhang, D. L. Akins and H. Yang, Carbon, 2012, 50, 265–274 CrossRef CAS.
- Z. Liu, X. Y. Ling, X. Su and J. Y. Lee, J. Phys. Chem. B, 2004, 108, 8234–8240 CrossRef CAS.
- S. Y. Huang, P. Ganesan and B. N. Popov, ACS Catal., 2012, 2, 825–831 CrossRef CAS.
- Y. Choi, J. H. Choi, L. Liu, B. K. Oh and S. Park, Chem. Mater., 2013, 25, 919–926 CrossRef CAS.
- I. Mintsoul, J. Georgieva, S. Armyanov, E. Valova, G. Avdeev, A. Hubin, O. Steenhaut, J. Dille, D. Tsiplakides, S. Balomenou and S. Sotiropoulos, Appl. Catal., B, 2013, 136–137, 160–167 CrossRef.
- S. Papadimitriou, S. Armyanov, E. Valova, A. Hubin, O. Steenhaut, E. Pavlidou, G. Kokkinidis and S. Sotiropoulos, J. Phys. Chem. C, 2010, 114, 5217–5223 CAS.
- Y. S. Wang, S. Y. Yang, S. M. Li, H. W. Tien, S. T. Hsiao, W. H. Liao, C. H. Liu, K. H. Chang, C. C. M. Ma and C. C. Hu, Electrochim. Acta, 2013, 87, 261–269 CrossRef CAS.
- D. Bin, F. F. Ren, H. Wang, K. Zhang, B. Yang, C. Zhai, M. Zhu, P. Yanga and Y. Du, RSC Adv., 2014, 4, 39612–39618 RSC.
- S. H. Lee, N. Kakati, S. H. Jee, J. Maiti and Y. S. Yoon, Mater. Lett., 2011, 65, 3281–3284 CrossRef CAS.
- H. Wang, J. Du, Z. Yao, R. yuea, C. Zhaia, F. Jianga, Y. Du, C. Wang and P. Yanga, Colloids Surf., A, 2013, 436, 57–61 CrossRef CAS.
- L. Dong, R. R. S. Gari, Z. Li, M. M. Craig and S. Hou, Carbon, 2010, 48, 781–787 CrossRef CAS.
- H. Xu, L. X. Ding, J. X. Feng and G. R. Li, Chem. Sci., 2015, 6, 6991–6998 RSC.
| This journal is © the Partner Organisations 2017 |