DOI:
10.1039/C6QM00252H
(Research Article)
Mater. Chem. Front., 2017,
1, 1185-1193
Graphene encapsulated Fe3O4 nanorods assembled into a mesoporous hybrid composite used as a high-performance lithium-ion battery anode material†
Received
3rd October 2016
, Accepted 4th January 2017
First published on 5th January 2017
Abstract
The discovery of new anode materials and engineering their fine structures are the core elements in the development of new-generation lithium ion batteries (LIBs). To this end, we herein report a novel nanostructured composite consisting of approximately 75% Fe3O4 nanorods and 25% reduced graphene oxide (rGO). Microscopy and spectroscopy analyses have identified that the Fe3O4 nanorods are wrapped (or encapsulated) by the rGO nanosheets via covalent bonding, which further self-assemble into a mesoporous hybrid composite networked by the graphene matrix. The composite has an average pore size around 20 nm and exhibits a high surface area of 152 m2 g−1, which is 76 times as high as that of conventional Fe3O4 powder. We have used the composite as an LIB anode material to fabricate coin-type prototype cells with lithium as the cathode. Systematic half-cell testing evaluations show that the electrochemical performance of the present composite material is amongst the best of the transition metal-oxide based LIB anode materials. The performances are characterized by a high reversible capacity of 1053 mA h g−1 subjected to 250 charge–discharge cycles at 500 mA g−1 and an excellent rate capability with the deliverable energy of 788–541 mA h g−1 upon the application of high current densities of 1000–5000 mA g−1. Overall, we have demonstrated that Fe3O4 nanorod–rGO hybrid composite is an interesting and promising material for the fabrication of LIB anodes.
Introduction
Lithium ion batteries (LIBs) have played a dominant role in electrochemical energy conversion and storage systems since their first commercialization by Sony in 1991.1 We have also witnessed extensive research and state-of-the-art progress in the field of LIBs over the past two decades. Driven by the increasing demand from portable electronic devices and electric vehicles, the current research and development (R&D) of LIBs have been strongly motivated to build more powerful, more robust and more durable batteries using cost-effective and environmentally-friendly new electrode materials.2–4
To build more powerful LIBs, one of the major concerns is their anode material. Graphite has mostly been used as the LIB anode material, but its limited capacity (theoretically 372 mA h g−1) has hindered the design and fabrication of more powerful LIBs and the search for new substitutes has become a crucial focus in LIB studies.5–8 Transition metal oxides (TMOs) not only have a high-energy capacity but are also earth-abundant and cost-effective materials in general. Different types of metal oxides, e.g. simple metal oxides (SMOs), single-phased mixed metal oxides (MMOs) and graphene-supported metal oxide, have been explored as LIB anode materials and have shown new promises.7,9 However, some general setbacks of MO based anode materials are notable, including their low electrical conductivity, slow electrochemical kinetics and large-volume expansion (possibly >200%) during the charge–discharge process, which often results in a low-rate performance. As an example, these undesirable disadvantages are clearly seen in Fe3O4 based anode systems.8–14
To circumvent these issues, two main approaches have been attempted to improve the structural integrity and electrical conductivity of the Fe3O4-based materials. One is to design nanostructured Fe3O4, such as nanoparticles,15,16 nanowires,12 microspheres,17,18 and hollow spheres.19 The other is to synthesize Fe3O4/carbon hybrids, including carbon-coated Fe3O4 nanospindles20,21 and nanorods22 and Fe3O4/carbon nanotubes.23 More recently, a number of Fe3O4/graphene (Fe3O4–G) composite systems have been reported.24–36 The integration of graphene nanosheets with Fe3O4 is expected to benefit the hybrid via the unique physicochemical properties of graphene such as large surface area, high electrical conductivity and robust mechanical strength.37,38 However, most studies have focused on engineering the size and morphology of the Fe3O4 nanostructures themselves (Table 1) with much less effort devoted to the engineering of the integral pore structures of the Fe3O4/graphene composites.39–41 Moreover, the overall performance of Fe3O4–G-based LIB anode systems is not as good as expected and needs to be enhanced for their potential practical applications.
Table 1 A comparison of the electrochemical performance of mesoporous Fe3O4/rGO with the previously reported and Fe3O4/rGO related systems
| Material composite |
Synthesis method |
Cycling stability |
Rate capability |
Ref. |
| Current (mA g−1) |
Cycling number |
Capability (mA h g−1) |
Current (mA g−1) |
Capability (mA h g−1) |
| Note: NPs: nanoparticles; NFs: nanoflowers; NCs: nanoclusters. |
| Fe3O4 nanorods/rGO |
Hydrothermal/annealing |
500 |
250 |
1053 |
5000 |
541 |
This work |
| Bare Fe3O4 NPs |
Commercial available |
500 |
250 |
289 |
5000 |
19 |
This work |
| Fe3O4 NPs/rGO |
Hydrothermal |
100 |
10 |
1102 |
1600 |
474 |
24
|
| Fe3O4 NFs/rGO |
Hydrothermal |
92.5 |
50 |
605 |
n/a |
n/a |
25
|
| Fe3O4 NPs/rGO |
Hydrothermal |
100 |
100 |
650 |
n/a |
n/a |
26
|
| Hollow Fe3O4/rGO |
Hydrothermal/electrostatic interaction |
100 |
50 |
900 |
n/a |
n/a |
27
|
| Hollow Fe3O4 NPs graphene/C |
Polyol-media solvothermal method/hydrothermal |
100 |
100 |
870.4 |
5000 |
445 |
28
|
| Fe3O4 NPs/rGO |
Chemical co-precipitation |
46.3 |
20 |
857 |
9260 |
200 |
29
|
| Fe3O4 NCs/rGO |
Ultrasonic assisted co-precipitation |
2000 |
100 |
1000 |
4000 |
860 |
30
|
| Fe3O4 NPs/rGO |
Gas/liquid interface reaction |
100 |
100 |
1048 |
1000 |
410 |
31
|
| Fe3O4–graphene |
Hydrolysis/annealing |
35 |
30 |
1026 |
1750 |
520 |
32
|
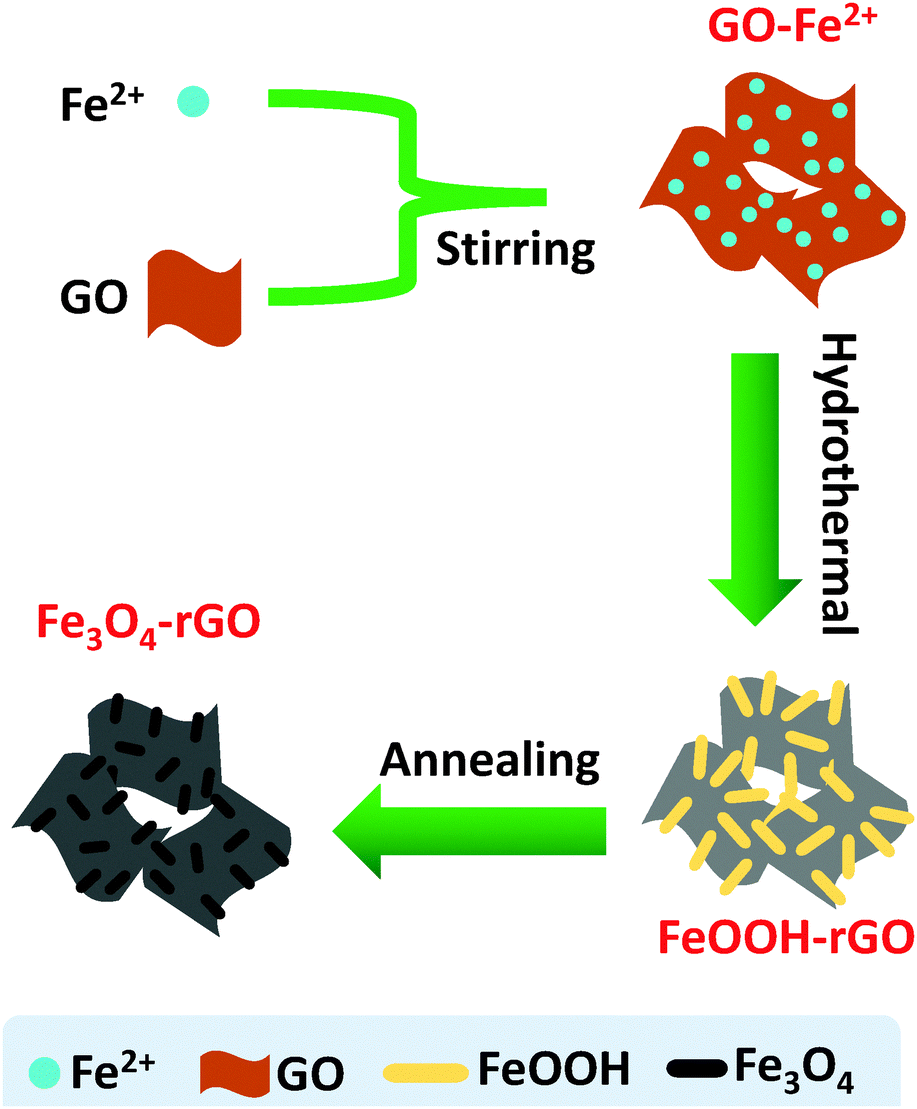
In this study, we are interested in engineering a hybrid composite consisting of high loading Fe3O4 nanorods and rGO nanosheets. We have used a self-constructing approach for the synthesis of the mesoporous hybrid, with the pores formed by networking the Fe3O4 nanorods and rGO sheets. We have shown that in the presence of GO, a hydrothermal reaction allows the conversion of Fe2+ to FeOOH nanorods and the simultaneous reduction of GO to rGO with the FeOOH nanorods wrapped by the rGO nanosheets. Through annealing, the thermal reduction of the FeOOH nanorods enables the formation of a Fe3O4/rGO composite with self-constructed mesopores. The rGO nanosheets provide conductive routes to the Fe3O4 nanorods. Moreover, the mesoporous structure can promote Li ion diffusion and buffer the volume expansion during the charge/discharge process. Consequently, the present hybrid composite has demonstrated high electrochemical performance characterized by a high reversible capacity and notably high cycling stability.
Materials and methods
Chemicals and materials
Expanded graphite (325 mesh) was purchased from Qingdao Jinrilai Graphite Co., Ltd, China. Sodium nitrate (NaNO3, ≥99%), hydrogen peroxide solution (H2O2, 30%), urea (CH4N2O, ≥99%), ammonium fluoride (NH4F, ≥98%) and iron sulphate heptahydrate (FeSO4·7H2O, ≥99%) were purchased from Aladdin, China. Potassium permanganate (KMnO4, ≥99.5%), concentrated sulphuric acid (H2SO4) and concentrated hydrochloric acid (HCl) were obtained from Sinopharm Chemical Reagent Co., Ltd. Iron oxide(II, III) powders (Fe3O4, ≥97%) were obtained from Shanghai Macklin Biochemical Co., Ltd. All chemicals were used as received without further purification. Ultrapure water (>18.25 MΩ cm) was obtained from a UPH-IV purifier manufactured by Chengdu Ultrapure Technology Co., Ltd, China.
Synthesis of graphene oxide
GO was synthesized from expanded graphite according to a modified Hummers’ method.42 First, into an ice-bathed beaker 138 mL of concentrated H2SO4 was added, followed by the addition of 3 g of NaNO3 and expanded graphite, sequentially. This mixture was continuously stirred while 18 g KMnO4 was added slowly. The mixture was stirred for 30 min before being transferred into a refrigerator to maintain a temperature of 4 °C for 24 h. The above-mentioned pre-oxidization step resulted in a black and viscous solution, which was stirred at 35 °C for 30 minutes before further processing. The mixture was then heated to 98 °C and maintained at this temperature for 15 min with the slow addition of 300 mL of water. H2O2 (30 wt% aqueous solution) was slowly added until no bubble production was observed. The resulting mixture was treated with excess 5% HCl solution to remove the residual metal ions. The solid product was separated via centrifugation (8000 rpm) and washed with ultrapure water until the solution became pH neutral (pH around 7.0). The resulting GO solids can be re-dispersed in water when needed.
Synthesis of the nanorod Fe3O4/rGO composite
FeSO4·7H2O (1.39 g), urea (0.9 g), and NH4F (0.37 g) were dissolved together in a 70 mL suspension of GO (1 mg mL−1) with stirring for about 20 min in an ice-water bath. The mixed aqueous solution was then transferred into a Teflon-lined stainless steel autoclave (100 mL), which was sealed and incubated at 80 °C. After 12 h, the precipitate was filtered to remove the loosely agglomerated products and washed with water and ethanol, sequentially, at least three times before being fully vacuum dried at 80 °C. Finally, the resulting powder was annealed at 450 °C for 4 h under an argon gas atmosphere to obtain the Fe3O4/rGO composite. As a control, the same protocol was followed but in the absence of GO.
Materials characterization
The crystal features of the samples were analysed by X-ray diffraction (XRD, Rigaku Dmaxrc diffractometer). The microstructures of the as-prepared materials were characterized via scanning electron microscopy (SEM, HITACHI SU-70) and high-resolution transmission electron microscopy (HRTEM, FEI Titan) instruments fitted with a field-emission electron source and a spherical aberration corrector on the condenser lens system operated at 200 kV. Raman spectra were recorded on a Renishaw InVia Raman spectrometer equipped with a 633 nm excitation laser. X-ray photoelectron spectroscopy (XPS) was performed on a Thermo-Scientific system at RT using Al-Kα radiation (1484.6 eV) as the excitation X-ray source. Fourier transform infrared spectroscopy (FTIR) measurements were performed on a Vector 22 spectrometer (Bruker) for testing the chemical bonding. The specific surface area was estimated via nitrogen adsorption–desorption isotherms at 77 K using an ASAP 2020 Surface Area & Pore Size Analyzer (Micromeritics). For Brunauer–Emmett–Teller analysis, prior to the absorption experiments, the samples were degassed at 140 °C for 8 h. Thermogravimetric analysis (TGA) was performed on a Mettler-Toledo TGA/SDTA851e Thermo Analyzer by increasing temperature from room temperature to 800 °C at a rate of 10 °C min−1 in air.
Fabrication and electrochemical characterization of the LIB anodes
A homogenous slurry, prepared by dispersing the as-prepared active materials or commercial Fe3O4 powders (70 wt%), super P (20 wt%) and polyvinylidene fluoride (PVDF) (10 wt%) in N-methyl-2-pyrrolidone (NMP), was cast onto a Cu foil and dried in a vacuum oven at 80 °C for 12 h, resulting in a mass loading of 1.0 mg cm−2. The battery performance was tested using 2016 coin-type cells containing a mixture of 1 M LiPF6 in ethylene carbonate, dimethyl carbonate and diethyl carbonate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 in volume) as the electrolyte, with a Li sheet and Celgard 2300 as the counter electrode and separator, respectively. Half-type cells were assembled in a glove box at room temperature under argon atmosphere. Galvanostatic charge/discharge in the voltage range of 0.01–3.0 V was carried out using a Neware-CT-3008 test system (Shenzhen, China). The applied current density (in mA g−1) was defined based on the amount of active material. Cyclic voltammetry (CV) between 0.01–3.0 V at a very slow scan rate of 0.1 mV s−1 and electrochemical impedance spectroscopy (EIS) in the frequency range of 0.01 Hz to 100 kHz were conducted using a CHI 660E electrochemical workstation (Shanghai, China).
:1:1 in volume) as the electrolyte, with a Li sheet and Celgard 2300 as the counter electrode and separator, respectively. Half-type cells were assembled in a glove box at room temperature under argon atmosphere. Galvanostatic charge/discharge in the voltage range of 0.01–3.0 V was carried out using a Neware-CT-3008 test system (Shenzhen, China). The applied current density (in mA g−1) was defined based on the amount of active material. Cyclic voltammetry (CV) between 0.01–3.0 V at a very slow scan rate of 0.1 mV s−1 and electrochemical impedance spectroscopy (EIS) in the frequency range of 0.01 Hz to 100 kHz were conducted using a CHI 660E electrochemical workstation (Shanghai, China).
Results and discussion
Composite formation and reaction mechanism
A combination of hydrothermal and annealing processing is an effective approach for the preparation of rGO supported metal oxide composites. The overall chemical process involves several reactions, but the related chemical reactions are yet to be fully clarified to date. Herein, we offer a detailed description of the relevant processes and chemical reactions. In the present case, the overall procedure used to form the Fe3O4/rGO nanorods composite is illustrated in Scheme 1. FeOOH nanorods were produced as a result of the electrostatic trapping of Fe2+ ions on the surface of GO sheets and their following hydrolysis upon heating. In detail, in the presence of air the trapped Fe2+ was partially oxidized into Fe(OH)3 on the GO sheets (eqn (1)), followed by their dehydration into FeOOH (eqn (2)).43 In an alternative pathway, the adsorbed Fe2+ reacted with O2 and OH−generated by the hydrolysis of urea (eqn (3) and (4)),44 to directly form FeOOH (eqn (5)). Therefore, we would expect that there are two types of crystalline phase for the FeOOH nanorods. Indeed, the XRD patterns have firmly proven this expectation (Fig. 1a). In the meantime, GO was subjected to a simultaneous reduction into rGO in the presence of urea (eqn (6)).45,46 The annealing step at 450 °C allowed the FeOOH nanorods to be partially reduced into mixed-valent (Fe(II) and Fe(III)) Fe3O4 with the assistance of rGO (eqn (7)).47 In contrast, the control experiments performed using the same procedure, but in the absence of GO, only produced stacked α-Fe2O3 nanoparticles as the final product (Fig. S1, ESI†). This observation clearly indicates that GO plays an essential role in the generation of Fe3O4 nanorods. In other words, similar Fe3O4 nanorods cannot be produced alone without the presence of GO under the present experimental conditions. In short, based on these experimental observations we have identified the crucial reactions shown below.| | | Fe2+ + O2 + H2O → Fe(OH)3 | (1) |
| | | CO(NH)2 + H2O → NH3 + CO2 | (3) |
| | | NH3 + H2O → NH4+ + OH− | (4) |
| | | Fe2+ + OH− + O2 → FeOOH + H2O | (5) |
| |  | (6) |
| | | FeOOH + rGO → Fe3O4 + rGO + CO2 + H2O | (7) |
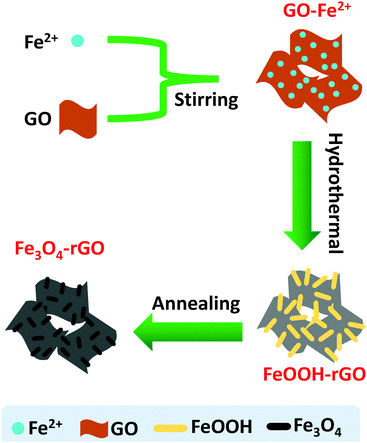 |
| | Scheme 1 A schematic of the synthetic procedure used to prepare the mesoporous Fe3O4/rGO composite. Not drawn to scale. | |
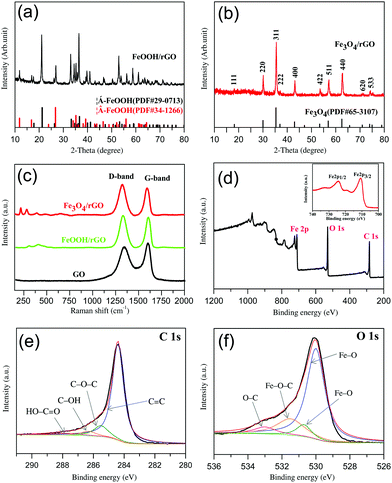 |
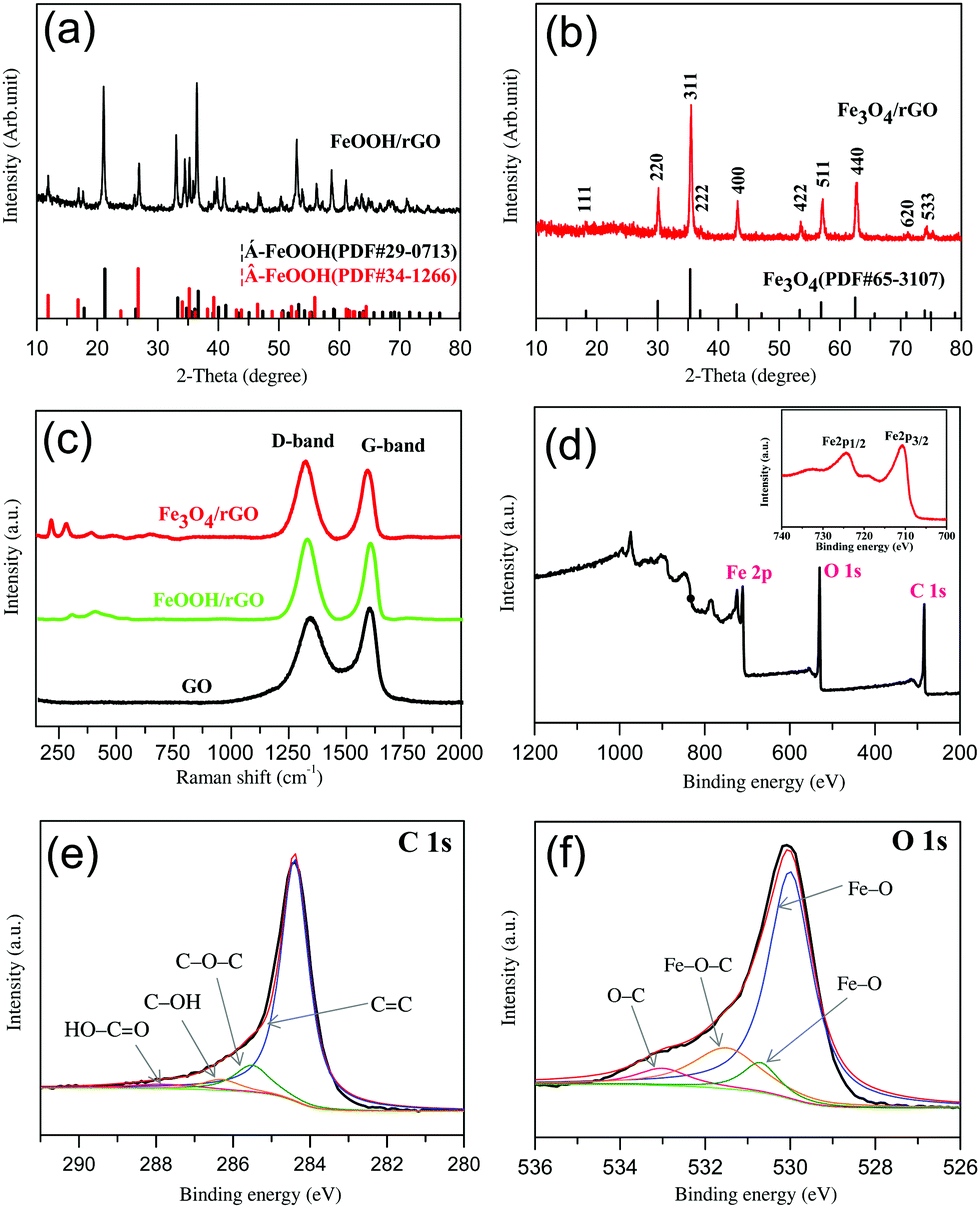
| | Fig. 1 Spectroscopic characterization of the materials: (a) and (b) the XRD patterns of FeOOH/rGO (a) and Fe3O4/rGO (b); (c) the Raman spectra of GO, FeOOH/rGO and Fe3O4/rGO; (d) the XPS survey spectrum of Fe3O4/rGO. Inset: Magnification of the XPS Fe2p spectrum; (e) the XPS C 1s spectrum of Fe3O4/rGO: experimental data (black curve) and an overall fitting (red curve), the spectrum is deconvoluted into various C–O species; (f) the XPS O 1s spectrum of Fe3O4/rGO: experimental data (black curve) and an overall fitting (red curve), the spectrum is deconvoluted into various Fe–O species. | |
Structural and morphological characteristics
As mentioned above, for the intermediate precursor FeOOH/rGO there are two types of crystalline phase observed. As shown in Fig. 1a, these diffraction peaks can be clearly assigned to Goethite (JCPDS no. 29-0713) and Akaganeite-Q (JCPDS no. 34-1266),44,48 respectively. However, the final product Fe3O4/rGO has only one crystalline phase (Fig. 1b). All the diffraction peaks are consistent with those of Fe3O4 (JCPDS no. 65-3107). The relatively low-intensity diffraction peak located at 24°–28° is attributed to stacked graphene sheets.32 The presence of Fe3O4 is further confirmed via Raman spectroscopy (Fig. 1c) with the peaks at 219 and 283 cm−1 referring to its skeletal vibration, the 484 and 653 cm−1 peaks for its T2g and A1g mode, and the 394 cm−1 peak for the Fe–O–Fe vibration.49,50 The D and G bands of Fe3O4/rGO at 1324 and 1593 cm−1, which are red-shifted when compared to pure GO, indicate their hybrid structure. The intensity ratio (r = ID/IG) of Fe3O4/rGO (r = 1.14) is larger than that of FeOOH/rGO (1.05) and GO (r = 0.90), indicative of the improvement in the sp2 hybridized carbon due to the increased degree of GO reduction into rGO.51,52
XPS and FTIR spectroscopy were used to identify the chemical nature of Fe3O4 and rGO in the composite. The XPS survey spectrum confirms the presence of carbon, oxygen, and iron (Fig. 1d). Double peaks at 710.7 and 724.3 eV are observed in the high-resolution Fe 2p spectrum (the inset of Fig. 1d), corresponding to Fe 2p3/2 and Fe 2p1/2 of Fe3O4.53,54 The peak at 284.4 eV in the Fe3O4/rGO C 1s spectrum (Fig. 1e) is consistent with C–C sp255 with the other three peaks at 285.5, 286.3 and 287.8 eV corresponding to C–O–C, C–OH and HO–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.56 The presence of Fe3O4 can be further confirmed by the O 1s spectrum (Fig. 1f) with fitted peaks at 530 eV and 530.7 eV.57 The peak at 533 eV can be assigned to C–O.58 It is known that the O 1s of Fe–O–C bond is in the range of 531–533 eV.59–61 We thus refer the observed peak at 531.5 eV to Fe–O–C formed between Fe3O4 and graphene. To further investigate the existence of chemical bonding between graphene and Fe3O4 nanorods, the FTIR spectra of the GO and Fe3O4/rGO samples were recorded and compared (in the ESI,† Fig. S2). The absorption bands at 1727 & 1621, 1398 and 1057 cm−1 are consistent with oxygen-containing groups on GO including CO stretching, C–O–H bending and C–O stretching vibration, respectively.55,61 These peaks are not observed in the Fe3O4/rGO samples due to the high-degree reduction of GO. The absorption bands observed at 1556 & 1513, 1187, 1134 and 563 cm−1 originate from the aromatic skeletal CC stretching vibration of rGO,62 C–O–C stretching vibration of epoxide,63 Fe–O–C bonding61 and Fe–O stretching in Fe3O4,61 respectively. In short, the XPS and FTIR analyses suggest that a robust interface between Fe3O4 and graphene was established to form a hybrid material, and the nature of the interactions between the two components rests on chemical bonding rather than just physical adsorption.
O.56 The presence of Fe3O4 can be further confirmed by the O 1s spectrum (Fig. 1f) with fitted peaks at 530 eV and 530.7 eV.57 The peak at 533 eV can be assigned to C–O.58 It is known that the O 1s of Fe–O–C bond is in the range of 531–533 eV.59–61 We thus refer the observed peak at 531.5 eV to Fe–O–C formed between Fe3O4 and graphene. To further investigate the existence of chemical bonding between graphene and Fe3O4 nanorods, the FTIR spectra of the GO and Fe3O4/rGO samples were recorded and compared (in the ESI,† Fig. S2). The absorption bands at 1727 & 1621, 1398 and 1057 cm−1 are consistent with oxygen-containing groups on GO including CO stretching, C–O–H bending and C–O stretching vibration, respectively.55,61 These peaks are not observed in the Fe3O4/rGO samples due to the high-degree reduction of GO. The absorption bands observed at 1556 & 1513, 1187, 1134 and 563 cm−1 originate from the aromatic skeletal CC stretching vibration of rGO,62 C–O–C stretching vibration of epoxide,63 Fe–O–C bonding61 and Fe–O stretching in Fe3O4,61 respectively. In short, the XPS and FTIR analyses suggest that a robust interface between Fe3O4 and graphene was established to form a hybrid material, and the nature of the interactions between the two components rests on chemical bonding rather than just physical adsorption.
The morphology of the as-prepared composites was characterized using SEM and TEM. FeOOH nanorods 20–100 nm in diameter and 50–300 nm in length were anchored on the rGO (Fig. 2a). After thermal annealing, the FeOOH nanorods were converted into Fe3O4 nanorods. The Fe3O4 nanorods have a typical diameter of 10–80 nm and are 20–250 nm in length (Fig. 2b and c). Furthermore, the conversion of FeOOH to Fe3O4 nanorods resulted in a volume contraction of the material, leading to a porous structure upon removing the –OH groups and the generation of CO2.47 TEM further confirms the high loading of Fe3O4 nanorods on rGO (Fig. 2d). HRTEM images show the contrast difference between light graphene shells and the dark Fe3O4 nanorods (Fig. 2e and f). The well-defined lattice fringes with an interplanar spacing of 0.48 nm (Fig. 2f) are attributed to the (111) plane of the Fe3O4 nanorods.20,56,64 More importantly, HRTEM images have revealed that the Fe3O4 nanorods are wrapped (or encapsulated) by graphene layers through covalent bonding (Fig. 1f), which is highly desirable and is expected to enhance both the stability and electrical conductivity of metal oxide nanorods.
 |
| | Fig. 2 SEM images of FeOOH/rGO (a) and Fe3O4/rGO (b and c); (d) TEM image of Fe3O4/rGO; (e and f) HRTEM of the Fe3O4/rGO sample. The arrows in (f) confirm that the Fe3O4 nanorods were encapsulated by graphene sheets. | |
In summary, we have identified the key structural and morphological features of the present composite based on microscopy and spectroscopy. These characteristics include (1) Fe3O4 existing in the form of nanorods, (2) the Fe3O4 nanorods attached covalently to the rGO sheets, (3) the rGO sheets wrapping the Fe3O4 nanorods to form loose core–shell structures because of the structural flexibility of the graphene sheets and (4) the rGO sheets further inducing the self-assembly of the rGO wrapped Fe3O4 nanorods into a mesoporous structure via the rGO sheet driven networking. On basis of these features, we have proposed a hierarchical structure (Fig. 3), in which both the top-graphic and side views are schematically illustrated.
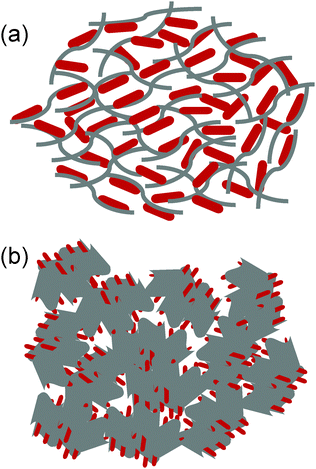 |
| | Fig. 3 A schematic of the Fe3O4/rGO composite structures: (a) top-graphic view showing the wrapping and confinement of Fe3O4 nanorods in the rGO matrix and (b) the side-view showing the further assembly of Fe3O4/rGO into the mesoporous structure. Not drawn to scale. | |
Surface area and component mass ratio
We performed the BET and TGA analyses on the composite to quantify the pore size, surface area and component mass ratio. Fig. 4a shows the typical nitrogen adsorption–desorption isotherms obtained for Fe3O4/rGO and commercial Fe3O4 powder samples. The observed hysteresis loop of Fe3O4/rGO in the relative pressure range of 0.4–1.0 represents a Type IV isotherm, which is indicative of a mesoporous structure (according to IUPAC), while the bare Fe3O4 shows non-porous structural features with a Type II isotherm. The BET specific surface area of Fe3O4/rGO was estimated to be 152 m2 g−1, approximately 76 fold of the surface area of bare Fe3O4 (2 m2 g−1). The SEM image of bare Fe3O4 further supports its non-porous structure (Fig. S3, ESI†). Thus, the specific surface area of Fe3O4/rGO is much higher than that of the previously reported Fe3O4/carbon nanomaterial composites (Table S1, ESI†). The pore-size-distribution histograms (Fig. 4b and Fig. S4, ESI†) show that there are two types of pores in the Fe3O4 nanorods/rGO samples. The small pores are in the size range of 2–5 nm, which is most likely from individual porous Fe3O4 nanorods. The large pores are centered at about 20 nm, which originate from the networking assembly of Fe3O4 nanorods into a hierarchical structure (Fig. 3).
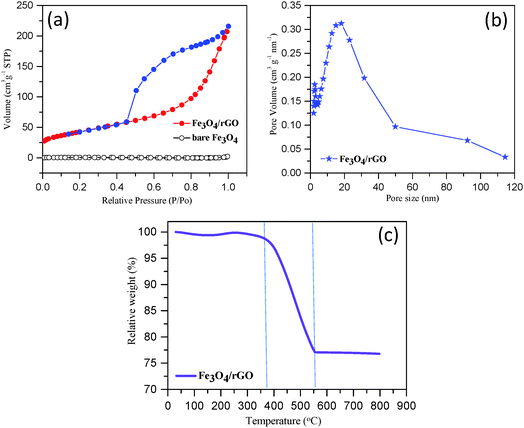 |
| | Fig. 4 Determination of the surface-area, pore-size-distribution and component-ratio: (a) the typical nitrogen adsorption–desorption isotherms of bare Fe3O4 and Fe3O4/rGO samples, (b) the pore-size distribution of Fe3O4/rGO composite and (c) the TGA curve obtained for Fe3O4/rGO. | |
The TGA curve obtained for Fe3O4/rGO is shown in Fig. 4c. A slight weight loss occurred below 150 °C, which was due to the removal of moisture or gaseous contents in the sample.65 A dramatic weight loss occurred in the temperature range from 375 °C to 560 °C. In this temperature range, the carbon component was combusted into CO2. Moreover, Fe3O4 was converted to Fe2O3 resulting in a theoretical weight increase of 3.45% (i.e., 1/2 equivalent of O is combined for each Fe3O4 unit).22 At temperatures higher than 560 °C, the sample weight remained largely unchanged (Fig. 4c). Therefore, we are able to estimate the mass ratio of Fe3O4 nanorods to rGO in the composite as approximately 75:25 (or 3:1, w/w).
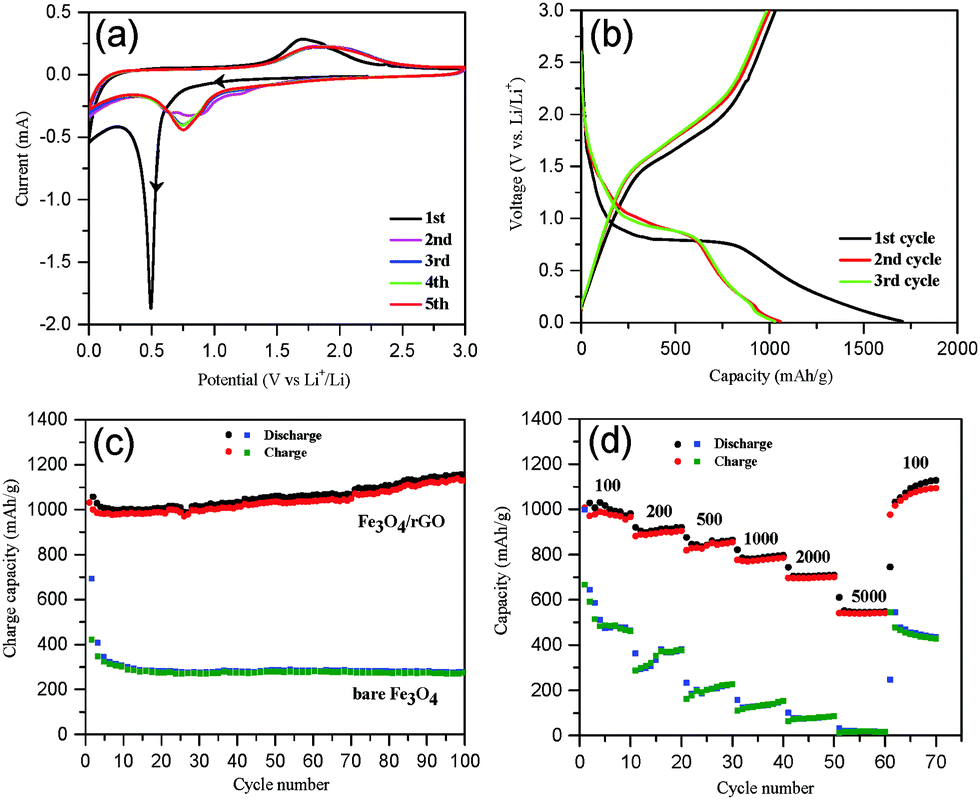
Electrochemical performance
The electrochemical performance of Fe3O4/rGO was tested first by cyclic voltammetry with five successive cycles. The initial potential was set at the open circuit potential (OCP), and the scan was run along the direction of cathodic potentials. As shown in Fig. 5a, a sharp cathodic peak around 0.5 V (vs. Li+/Li) was observed in the first scan, arising from the reduction of Fe3+ and Fe2+ to Fe0, the generation of Li2O (eqn (8) and (9)) and the formation of a solid electrolyte interphase (SEI) layer.66 This was followed by a sloping curve down to 0.01 V, indicating the reversible reaction of lithium and rGO (eqn (10)).67 In the first anodic scan, a broad peak at 1.71 V was due to the re-oxidation of Fe0 to Fe3O4. In the subsequent cycles, however, both the cathodic and anodic peaks shifted to more positive voltages and the currents decreased, which are indicative of a certain degree of irreversible Li insertion from the first cycle. The cyclic voltammograms (CVs) almost overlap each other after the second cycle (Fig. 5a), indicating that the insertion/extraction of lithium ions occurred in the same process. In contrast, the CVs of the bare Fe3O4 sample (Fig. S5, ESI†) exhibit an irreversible decrease in the intensity of the redox peaks.| | | Fe3O4 + xLi + xe− → LixFe3O4 | (8) |
| | | LixFe3O4 + (8 − x)Li+ + (8 − x)e− → 3Fe0 + Li2O | (9) |
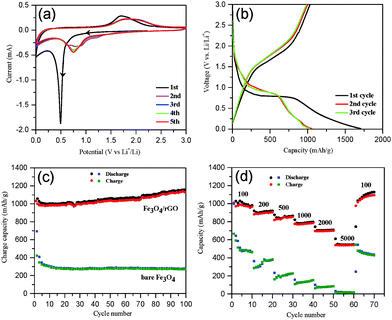 |
| | Fig. 5 (a) Cyclic voltammograms for the first to fifth cycles of Fe3O4/rGO over a voltage range of 0.01–3.0 V at a scan rate of 0.1 mV s−1; (b) the galvanostatic charge/discharge profile obtained for Fe3O4/rGO at a current density of 100 mA g−1; (c) a comparison of the cyclic performance of Fe3O4/rGO and bare Fe3O4 at 100 mA g−1; (d) the rate capability of Fe3O4/rGO and bare Fe3O4 at different current densities. | |
Second, galvanostatic charge–discharge (GCD) tests were performed. The GCD profiles of Fe3O4/rGO at 100 mA g−1 for the first three cycles are shown in Fig. 5b. The initial discharge and charge capacity were 1707 and 1032 mA h g−1 (the black curve in Fig. 5b), respectively. An initial columbic efficiency of ca. 61% was observed, similar to the previous reports.68,69 The relatively low efficiency is generally regarded as a consequence of the irreversible formation of the Li2O and SEI layers and of the irreversible trapping of partial lithium ions in the Fe3O4 lattices.15,66 However, the charge/discharge curves in the subsequent cycles (after the second cycle) overlap well, a proof of the superior capacity after initial activation. A comparison of the cycling performance of the Fe3O4/rGO and bare Fe3O4 at 100 mA g−1 is shown in Fig. 5c. The initial discharge and charge capacities of bare Fe3O4 were 993 and 603 mA h g−1, respectively, with an initial columbic efficiency of 60.7%, whereas the second columbic efficiency was 85%. The capacity of bare Fe3O4 decreased by 62% from 993 to 376 mA h g−1 after 100 cycles. In contrast, the Fe3O4/rGO electrodes permitted highly reversible columbic efficiencies of more than 94% after the second cycle. The discharge capacities of Fe3O4/rGO increased gradually and reached 1149 mA h g−1 at the 100th cycle, which was attributed to the formation of a polymeric gel-like film around the active material.15,32 In addition, we have performed the charge/discharge tests using cells comprised of a pure GO anode (without Fe3O4) as a control experiment. The results are provided in the ESI† (Fig. S6). The capacity of the pure rGO anode was approximately 310 mA h g−1 at the beginning but decreased gradually and stabilized at ca. 280 mA h g−1 after 100 cycles (Fig. S6, ESI†). It is noticed that the capacity of the Fe3O4/rGO composites exceeds the theoretical capacity of Fe3O4 (about 926 mA h g−1). Thus, the extra capacity is contributed by the rGO component and the synergistic effects between the larger electrochemical active surface area of rGO and the grain boundary area of the Fe3O4 nanorods.69,70
To further highlight the performance of the composites, their rate capabilities are compared with bare Fe3O4via testing at various current densities between 100 and 5000 mA g−1 (Fig. 5d). The Fe3O4/rGO displayed a much better rate performance with the reversible discharge capacity being decreased from 1002 to 541 mA h g−1 upon increasing the current density from 100 to 5000 mA g−1. In contrast, bare Fe3O4 showed a dramatic drop in the discharge capacity from 476 to 19 mA h g−1. Moreover, the discharge capacity of Fe3O4/rGO recovered to 1096 mA h g−1 upon resetting to 100 mA g−1 after 60 cycles. The rate capability of the present mesoporous hybrid is significantly better than those previously reported Fe3O4/rGO composites (Table 1). In order to clarify the excellent electrochemical performance of Fe3O4/rGO, the XRD patterns of the Fe3O4/rGO electrode materials were recorded before and after 100 cycles and are compared in Fig. S7 (ESI†). Before cycling, the XRD pattern can be well assigned to Fe3O4 (JCPDS no. 65-3107) and Cu (JCPDS no. 04-0836) as expected. Furthermore, according to the Scherrer equation the average crystal size of Fe3O4 was estimated to be ca. 30 nm. After 100 cycles at 100 mA g−1, however, no relevant peaks for Fe3O4 were observed. This was attributed to the conversion of crystal Fe3O4 into amorphous Fe3O4 during the discharge/charge process.2,3 However, the SEM image indicates that Fe3O4 was stably retained on the anode electrode (Fig. S8a, ESI†). In addition, the structural stability of Fe3O4/rGO was further investigated via SEM imaging of the electrodes after 100 cycles at 100 mA g−1. No disassembly or cracking of the Fe3O4/rGO was observed (Fig. S8a, ESI†), implying the stabilization of Fe3O4 on the electrode during cycling. It could be explained by the buffer function of graphene and the mesoporous structure, which helped to maintain the integrity of the Fe3O4 nanorods. In contrast, cracks and aggregations were clearly found on the bare Fe3O4 electrode (Fig. S8b, ESI†).
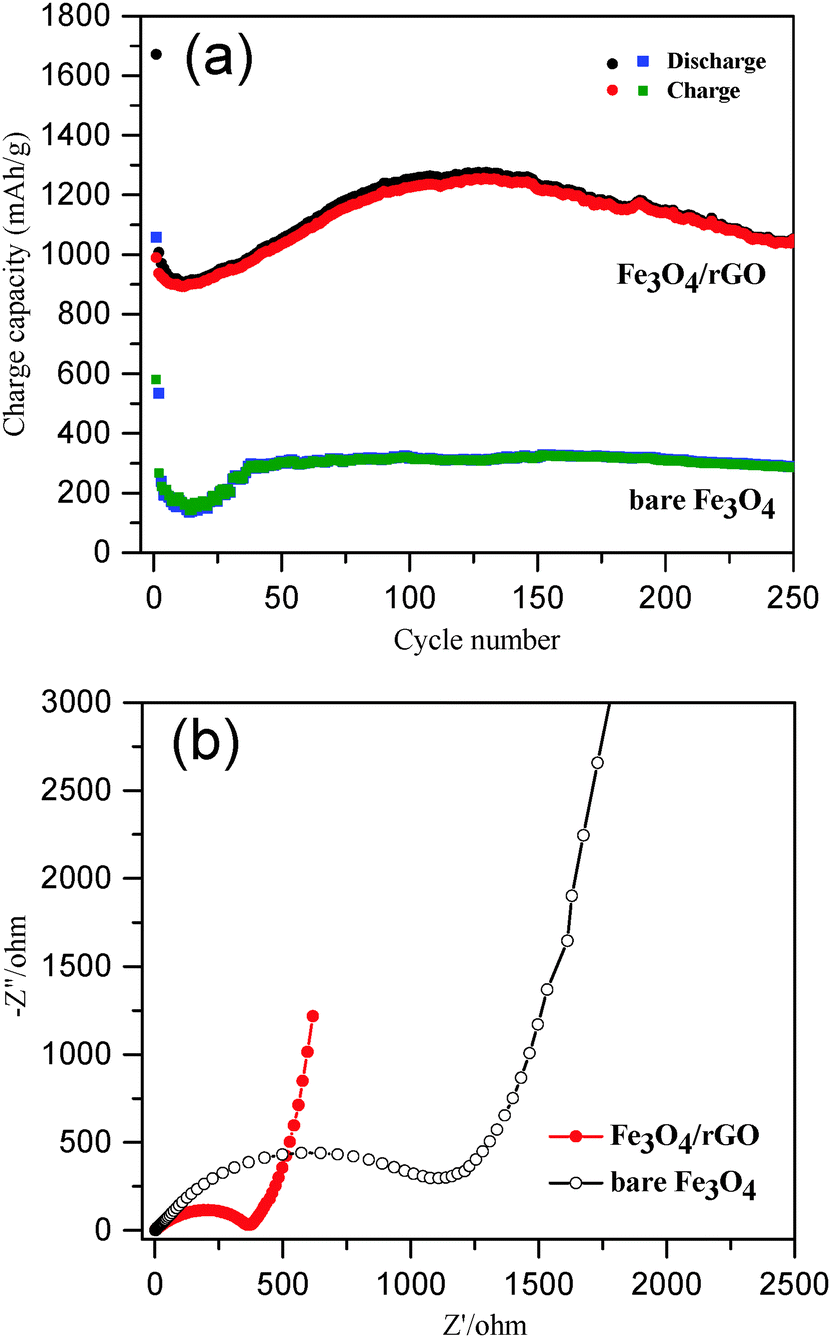
The long-term cycling performance of Fe3O4/rGO and bare Fe3O4 was compared at a high current density of 500 mA g−1 (Fig. 6a). The initial discharge and charge capacities of the Fe3O4/rGO were 1672 and 990 mA h g−1, respectively. The decrease in the capacity at the beginning was attributed to the formation of a SEI layer and the reaction of lithium ions with the residual oxygen-containing functional groups.32,69,70 The reversible discharge capacity of the Fe3O4/rGO increased from the 13th cycle, reaching a maximum value of 1276 mA h g−1 after 130 cycles. This unusual phenomenon is a result of the long-term activation process, so that the active materials were rearranged to provide more active sites for the insertion/extraction of lithium ions during the cycling process.24,31,32,69,71 The Fe3O4/rGO showed a discharge capacity of 1053 mA h g−1 after the 250th cycle and a columbic efficiency consistently more than 99% after 140 cycles. However, the bare Fe3O4 electrode exhibited a much lower reversible discharge capacity of 289 mA h g−1 after the 250th cycle. Table 1 compares the performance of various Fe3O4/rGO composites. For example, Liu et al. demonstrated a Fe3O4 nanoflowers/graphene composite, which exhibited a capacity of 605 mA h g−1 at 92.5 mA g−1 after 50 cycles.25 Wang et al. reported graphene-encapsulated Fe3O4 nanoparticles with a 3D laminated structure, which showed a capacity of 650 mA h g−1 at 100 mA g−1 after 100 cycles.26 The Fe3O4/rGO composite reported here exhibited a much higher specific capacity and better cyclic stability, which was attributed to the integral mesoporous structure of the hybrids of Fe3O4 nanorods/rGO and the robust interfaces between the two components as discussed above.
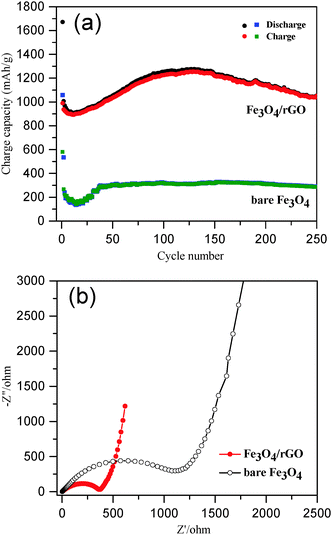 |
| | Fig. 6 (a) A comparison of the cycle performance of Fe3O4/rGO and bare Fe3O4 at a current density of 500 mA g−1; (b) the Nyquist plots of Fe3O4/rGO and bare Fe3O4 electrodes in fresh coin cells over the frequency range from 0.01 Hz to 100 kHz. | |
For better understanding of the improved electrochemical performance of Fe3O4/rGO compared with that of bare Fe3O4, EIS measurements were performed (Fig. 6b). The size of the semicircles in the high frequency region reflects the impedance (or relative kinetics) of charge transfer (Rct) through the electrode/electrolyte interfaces.72,73 The Rct of the Fe3O4/rGO composite based electrodes was determined to be 364 Ω, which was significantly lower than that found for the bare Fe3O4 based electrodes (1118 Ω). These results have further proven the advantage of the self-constructing mesoporous hybrids of Fe3O4 nanorods/rGO as LIB anodes.
Conclusions
We have demonstrated a two-step procedure for the facile synthesis of mesoporous hybrids comprised of 75% Fe3O4 nanorods and 25% reduced graphene oxide. The structural features of this composite are characterized by the covalent bonding between the rGO sheets and metal oxide, wrapping metal oxide nanorods by rGO sheets and hierarchical self-assembly into robust mesoporous ensembles. Such composites can offer advantageous physicochemical properties as a lithium-ion battery anode material, including high stability, enhanced electrical conductivity, appropriate pore size and high-energy capacity. The material has been systematically tested as an LIB anode component. The results show that the electrochemical performance of the present composite are among the best of transition metal-oxide based LIB anode materials. A high reversible capacity of 1053 mA h g−1 subjected to 250 charge–discharge cycles at 500 mA g−1 was achieved, accompanied by an excellent rate capability with the deliverable energy of 788–541 mA h g−1 upon the application of high current densities of 1000–5000 mA g−1. This work points to the conclusion that the combined one-dimensional Fe3O4 nanorod and two-dimensional rGO sheet hybrid composite is a promising LIB anode material for potential practical applications.
Acknowledgements
This study was sponsored by the National Natural Science Foundation of China (NSFC, No. 51301096), Shandong Provincial Natural Science Foundation (ZR2013EMQ012) and the Fundamental Research Funds of Shandong University (No. 2014JC017). The study was also financially supported by the Danish Council for Independent Research Technology and the Production Sciences (DEF-1335-00330) and the Danish Agency for Science, Technology and Innovation for the International Network Programme (4070-00131B). L. C. is grateful to the scientific exchange visit to DTU under the International Network Project, supported by the Danish Agency for Science, Technology and Innovation (Ref. No. 5132-00053B). Q. C. acknowledges the Danish Council for Independent Research DFF-FTP for a graphene-based project (Project No. 12-127447) and the Danish Agency for Science, Technology and Innovation for an International Network project (Ref. No. 5132-00053B). The authors wish to thank Prof. Jingdong Zhang for her support.
References
- T. Nagaura and K. Tozawa, Prog. Batteries Sol. Cells, 1990, 9, 209–217 CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- J. X. Qiu, C. Lai, Y. Wang, S. Li and S. Zhang, Chem. Eng. J., 2014, 256, 247–254 CrossRef CAS.
- B. Wang, X. L. Wu, C. Y. Shu, Y. G. Guo and C. R. Wang, J. Mater. Chem., 2010, 20, 10661–10664 RSC.
- S. Xu, C. M. Hessel, H. Ren, R. Yu, Q. Jin, M. Yang, H. Zhao and D. Wang, Energy Environ. Sci., 2014, 7, 632–637 CAS.
- S. Li, Y. Wang, C. Lai, J. Qiu, M. Ling, W. Martens, H. Zhao and S. Zhang, J. Mater. Chem. A, 2014, 2, 10211–10217 CAS.
- N. A. Kaskhedikar and J. Maier, Adv. Mater., 2009, 21, 2664–2680 CrossRef CAS.
- F. Wu, J. Bai, J. Feng and S. Xiong, Nanoscale, 2015, 7, 17211–17230 RSC.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- P. L. Taberna, S. Mitra, P. Poizot, P. Simon and J. M. Tarascon, Nat. Mater., 2006, 5, 567–573 CrossRef CAS PubMed.
- T. Muraliganth, A. V. Murugan and A. Manthiram, Chem. Commun., 2009, 7360–7362 RSC.
- N. Kang, J. H. Park, J. Choi, J. Jin, J. Chun, I. G. Jung, J. Jeong, J. G. Park, S. M. Lee, H. J. Kim and S. U. Son, Angew. Chem., Int. Ed., 2012, 51, 6626–6630 CrossRef CAS PubMed.
- L. Ji, Z. Lin, M. Alcoutlabi and X. Zhang, Energy Environ. Sci., 2011, 4, 2682 CAS.
- Y. Chen, B. H. Song, X. S. Tang, L. Lu and J. M. Xue, Small, 2014, 10, 1536–1543 CrossRef CAS PubMed.
- S. H. Lee, S. H. Yu, J. E. Lee, A. Jin, D. J. Lee, N. Lee, H. Jo, K. Shin, T. Y. Ahn, Y. W. Kim, H. Choe, Y. E. Sung and T. Hyeon, Nano Lett., 2013, 13, 4249–4256 CrossRef CAS PubMed.
- Q. Q. Xiong, J. P. Tu, Y. Lu, J. Chen, Y. X. Yu, Y. Q. Qiao, X. L. Wang and C. D. Gu, J. Phys. Chem. C, 2012, 116, 6495–6502 CAS.
- B. Wang, H. B. Wu, L. Zhang and X. W. Lou, Angew. Chem., Int. Ed., 2013, 52, 4165–4168 CrossRef CAS PubMed.
- F. X. Ma, H. Hu, H. B. Wu, C. Y. Xu, Z. Xu, L. Zhen and X. W. Lou, Adv. Mater., 2015, 27, 4097–4101 CrossRef CAS PubMed.
- J. Zhang, K. Wang, Q. Xu, Y. Zhou, F. Cheng and S. Guo, ACS Nano, 2015, 9, 3369–3376 CrossRef CAS PubMed.
- W. M. Zhang, X. L. Wu, J. S. Hu, Y. G. Guo and L. J. Wan, Adv. Funct. Mater., 2008, 18, 3941–3946 CrossRef CAS.
- T. Zhu, J. S. Chen and X. W. Lou, J. Phys. Chem. C, 2011, 115, 9814–9820 CAS.
- C. Ban, Z. Wu, D. T. Gillaspie, L. Chen, Y. Yan, J. L. Blackburn and A. C. Dillon, Adv. Mater., 2010, 22, 145–149 CrossRef PubMed.
- J. Su, M. Cao, L. Ren and C. Hu, J. Phys. Chem. C, 2011, 115, 14469–14477 CAS.
- X. Li, X. Huang, D. Liu, X. Wang, S. Song, L. Zhou and H. Zhang, J. Phys. Chem. C, 2011, 115, 21567–21573 CAS.
- J. Z. Wang, C. Zhong, D. Wexler, N. H. Idris, Z. X. Wang, L. Q. Chen and H. K. Liu, Chem. – Eur. J., 2011, 17, 661–667 CrossRef CAS PubMed.
- D. Chen, G. Ji, Y. Ma, J. Y. Lee and J. Lu, ACS Appl. Mater. Interfaces, 2011, 3, 3078–3083 CAS.
- Y. Zuo, G. Wang, J. Peng, G. Li, Y. Ma, F. Yu, B. Dai, X. Guo and C. P. Wong, J. Mater. Chem. A, 2016, 4, 2453–2460 CAS.
- L. Ji, Z. Tan, T. R. Kuykendall, S. Aloni, S. Xun, E. Lin, V. Battaglia and Y. Zhang, Phys. Chem. Chem. Phys., 2011, 13, 7170–7177 RSC.
- S. K. Behera, Chem. Commun., 2011, 47, 10371–10373 RSC.
- P. Lian, X. Zhu, H. Xiang, Z. Li, W. Yang and H. Wang, Electrochim. Acta, 2010, 56, 834–840 CrossRef CAS.
- G. Zhou, D. W. Wang, F. Li, L. Zhang, N. Li, Z. S. Wu, L. Wen, G. Q. Lu and H. M. Cheng, Chem. Mater., 2010, 22, 5306–5313 CrossRef CAS.
- J. Luo, J. Liu, Z. Zeng, C. F. Ng, L. Ma, H. Zhang, J. Lin, Z. Shen and H. J. Fan, Nano Lett., 2013, 13, 6136–6143 CrossRef CAS PubMed.
- Y. Wan, Z. Yang, G. Xiong, R. Guo, Z. Liu and H. Luo, J. Power Sources, 2015, 294, 414–419 CrossRef CAS.
- W. Chen, S. Li, C. Chen and L. Yan, Adv. Mater., 2011, 23, 5679–5683 CrossRef CAS PubMed.
- Y. Gong, S. Yang, Z. Liu, L. Ma, R. Vajtai and P. M. Ajayan, Adv. Mater., 2013, 25, 3979–3984 CrossRef CAS PubMed.
- D. Wu, F. Zhang, H. Liang and X. Feng, Chem. Soc. Rev., 2012, 41, 6160–6177 RSC.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- B. G. Choi, S. J. Chang, Y. B. Lee, J. S. Bae, H. J. Kim and Y. S. Huh, Nanoscale, 2012, 4, 5924–5930 RSC.
- M. Zhang, Z. Sun, T. Zhang, D. Sui, Y. Ma and Y. Chen, Carbon, 2016, 102, 32–38 CrossRef CAS.
- Y. Liu, Z. Cheng, H. Sun, H. Arandiyan, J. Li and M. Ahmad, J. Power Sources, 2015, 273, 878–884 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- Q. X. Low and G. W. Ho, Nano Energy, 2014, 5, 28–35 CrossRef CAS.
- B. Tang, G. Wang, L. Zhuo, J. Ge and L. Cui, Inorg. Chem., 2006, 45, 5196–5200 CrossRef CAS PubMed.
- Z. Lei, L. Lu and X. S. Zhao, Energy Environ. Sci., 2012, 5, 6391–6399 CAS.
- M. Mowry, D. Palaniuk, C. C. Luhrs and S. Osswald, RSC Adv., 2013, 3, 21763 RSC.
- S. M. Yuan, J. X. Li, L. T. Yang, L. W. Su, L. Liu and Z. Zhou, ACS Appl. Mater. Interfaces, 2011, 3, 705–709 CAS.
- X. Mou, B. Zhang, Y. Li, L. Yao, X. Wei, D. S. Su and W. Shen, Angew. Chem., Int. Ed., 2012, 51, 2989–2993 CrossRef CAS PubMed.
- P. R. Graves, C. Johnston and J. J. Campaniello, Mater. Res. Bull., 1988, 23, 1651–1660 CrossRef CAS.
- O. N. Shebanova and P. Lazor, J. Solid State Chem., 2003, 174, 424–430 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51–87 CrossRef CAS.
- P. Marcus and J. M. Grimal, Corros. Sci., 1992, 33, 805–814 CrossRef CAS.
- T. Xia, X. Xu, J. Wang, C. Xu, F. Meng, Z. Shi, J. Lian and J.-M. Bassat, Electrochim. Acta, 2015, 160, 114–122 CrossRef CAS.
- C. J. Powell, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 1–3 CrossRef CAS.
- Y. Chang, J. Li, B. Wang, H. Luo, H. He, Q. Song and L. Zhi, J. Mater. Chem. A, 2013, 1, 14658 CAS.
- S. Bhuvaneswari, P. M. Pratheeksha, S. Anandan, D. Rangappa, R. Gopalan and T. N. Rao, Phys. Chem. Chem. Phys., 2014, 16, 5284–5294 RSC.
- Y. C. Dong, R. G. Ma, M. J. Hu, H. Cheng, C. K. Tsang, Q. D. Yang, Y. Y. Li and J. A. Zapien, J. Solid State Chem., 2013, 201, 330–337 CrossRef CAS.
- G. Kataby, M. Cojocaru, R. Prozorov and A. Gedanken, Langmuir, 1999, 15, 1703–1708 CrossRef CAS.
- C. Combellas, M. Delamar, F. Kanoufi, J. Pinson and F. I. Podvorica, Chem. Mater., 2005, 17, 3968–3975 CrossRef CAS.
- J. Zhou, H. Song, L. Ma and X. Chen, RSC Adv., 2011, 1, 782 RSC.
- C. Nethravathi and M. Rajamathi, Carbon, 2008, 46, 1994–1998 CrossRef CAS.
- M. Acik, G. Lee, C. Mattevi, M. Chhowalla, K. Cho and Y. J. Chabal, Nat. Mater., 2010, 9, 840–845 CrossRef CAS PubMed.
- L. Zhuo, Y. Wu, L. Wang, J. Ming, Y. Yu, X. Zhang and F. Zhao, J. Mater. Chem. A, 2013, 1, 3954 CAS.
- J. S. Chen, Y. L. Tan, C. M. Li, Y. L. Cheah, D. Luan, S. Madhavi, F. Y. C. Boey, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 6124–6130 CrossRef CAS PubMed.
- Y. Piao, H. S. Kim, Y. E. Sung and T. Hyeon, Chem. Commun., 2010, 46, 118–120 RSC.
- P. Lian, X. Zhu, S. Liang, Z. Li, W. Yang and H. Wang, Electrochim. Acta, 2012, 55, 3909–3914 CrossRef.
- Y. Dong, K. C. Yung, R. Ma, X. Yang, Y. S. Chui, J. M. Lee and J. A. Zapien, Carbon, 2015, 86, 310–317 CrossRef CAS.
- Y. Dong, Z. Zhang, Y. Xia, Y. S. Chui, J. M. Lee and J. A. Zapien, J. Mater. Chem. A, 2015, 3, 16206–16212 CAS.
- S. Laruelle, S. Grugeon, P. Poizot, M. Dolle, L. Dupont and J. M. Tarascon, J. Electrochem. Soc., 2002, 149, A627–A634 CrossRef CAS.
- S. H. Choi and Y. C. Kang, Carbon, 2014, 79, 58–66 CrossRef CAS.
- S. A. Needham, G. X. Wang, K. Konstantinov, Y. Tournayre, Z. Lao and H. K. Liu, Electrochem. Solid-State Lett., 2006, 9, A315–A319 CrossRef CAS.
- D. Aurbach, J. Power Sources, 2000, 89, 206–218 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The XRD pattern of α-Fe2O3, FT-IR of GO and Fe3O4/rGO, SEM and CV of commercial Fe3O4, BET surface areas comparison table, pore size distribution of commercial Fe3O4 and Fe3O4/rGO, SEM images of cycled batteries. See DOI: 10.1039/c6qm00252h |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Pengchao
Si
*a and
Qijin
Chi
a,
Pengchao
Si
*a and
Qijin
Chi