
Phase-separation induced hollow/porous carbon nanofibers containing in situ generated ultrafine SnOx as anode materials for lithium-ion batteries†
Yuan
Liu
ab,
Xiaodong
Yan
 c,
Jinle
Lan
b,
Yunhua
Yu
*b,
Xiaoping
Yang
b and
Yuanhua
Lin
*a
c,
Jinle
Lan
b,
Yunhua
Yu
*b,
Xiaoping
Yang
b and
Yuanhua
Lin
*a
aSchool of Materials Science and Engineering, State Key Lab of New Ceramics and Fine Processing, Tsinghua University, Beijing 100084, China. E-mail: linyh@mail.tsinghua.edu.cn
bState Key Laboratory of Organic–Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: yuyh@mail.buct.edu.cn; Fax: +86-10-6442-7698; Tel: +86-10-6442-2084
cDepartment of Chemistry, University of Missouri – Kansas City, Kansas City, Missouri 64110, USA
First published on 7th February 2017
Abstract
Hollow-structured carbon nanofibers embedded with ultrafine SnOx nanoparticles (SnOx/H-CNFs) have been prepared by a simple single-spinneret electrospinning technique assisted by phase separation between polyvinylpyrrolidone and tetraethyl orthosilicate. The nitrogen adsorption–desorption isothermal analysis shows that the SnOx/H-CNFs possess a large specific surface area of 739 m2 g−1 and large amounts of mesopores. The high specific surface area provides a large electrode/electrolyte contact interface for Li+ transport and storage, while the hollow structure shortens the Li+-diffusion pathway and thus favors the rate capability. Moreover, the hollow and porous carbon matrix can act as a buffer zone to accommodate the volume change of the highly active SnOx during the lithiation/delithiation processes. Consequently, the SnOx/H-CNF electrode delivers a high reversible capacity of 732 mA h g−1 at the rate of 500 mA g−1 upon the 100th cycle, good rate performance (254 mA h g−1 at 4 A g−1), and long-term cycling stability (513 mA h g−1 at 1 A g−1 after 500 cycles).
Introduction
Over the past decade, considerable efforts have been devoted to the research of high-performance and long-lifespan lithium-ion batteries (LIBs), aiming to meet the high criteria of their applications in large-scale power systems.1–3 As one of the critical components in LIBs, the anode plays an important role in determining the overall electrochemical performance. So far, graphite is still the most successfully commercialized anode material. However, its limited practical capacity (theoretical capacity: 372 mA h g−1) and low power capability lag far behind the expectation of the desired next-generation LIBs.4,5 Therefore, alternative anode materials with high theoretical capacities have been intensively investigated, including Si (4212 mA h g−1), Ge (1623 mA h g−1), Sn (990 mA h g−1), and nanosized SnO2 (1494 mA h g−1).6,7 However, the practical applications of these high-capacity anode materials are hampered by their huge volume expansion, which can lead to loss of electrical contact between the electrode material and the current collector during the lithiation/delithiation processes and thus cause fast capacity decay.6–9 To overcome this issue, carbon-based composites have been developed, in which the high-capacity materials (HCMs) are dispersed in a carbon matrix. The carbon matrix can accommodate the volume change, confine the HCMs in the carbon matrix, and thus maintain the electrical contact during cycling; meanwhile, the HCMs can still efficiently play their role in offering high lithium-storage capacity.10,11 Nevertheless, the bulk carbon matrix may increase the Li+-diffusion distance and Li+-transport resistance toward the deeply embedded HCM particles, which is apparently detrimental to the capacity output of the composite electrode at fast charge–discharge rates.12Recently, the nanoscale carbon coating technique has been proposed as an effective method to partially solve this issue with a shortened Li+ transport distance from the electrolyte to the HCMs. Among all the nanoHCMs/C composites, an electrospinning-derived one-dimensional (1D) carbon-coated nanostructure has attracted much attention considering its easy synthesis and scalability.13–18 More importantly, the 1D morphology feature facilitates the electron-transport efficiency along the axial direction and simultaneously reduces the Li+-diffusion distance in the radial direction.19 However, the Li+ diffusion distance from the surface to the core of a CNF is still at least equal to its radius. Therefore, it is still imperative to further shorten the Li+ diffusion pathway and make full use of the electrode materials, especially the interior HCM particles. The introduction of a porous structure into the CNF matrix has been proved to be an effective strategy. Generally, the porous structure, which benefits ion transport and storage, is highly dependent on its size, shape and electrolyte wettability.20–24 For instance, Zhi's group demonstrated that multiple open hollow channels in CNFs are more beneficial for improving the ion-transport capability than closed pores.20 This is because hollow channels with a large inner diameter (50–60 nm) act as an ion-buffering reservoir, which can offer fast ion-transport pathways similar to those in the bulk electrolyte. Later, Shen et al. also found that more mesopores could bring in a much better rate capability for an Sn-loaded CNF electrode (349.2 vs. 55.7 mA h g−1 at a high current density of 4 A g−1).22 These studies demonstrate that large open pores may act as Li+-carrying electrolyte reservoirs, favoring the Li+ transport and storage processes. Therefore, controlling the pore structure offers a promising way to tune the electrochemical properties of the HCM/CNF composites.
Template methods have been used intensively to control the porous structure of electrospinning-derived CNFs or HCM/CNF composites for LIBs. For example, Kang's group synthesized porous CNFs by etching off the silica template in CNFs obtained from pyrolysis of electrospun polyamic acid/tetraethoxysilane (TEOS) nanofibers.23 Later, Kim and co-workers prepared Co3O4/porous CNFs25 and Si-encapsulated porous CNFs26 using polymethyl methacrylate and Fe3C as the templates, respectively. However, the porous structures were only controlled by the templates. Herein, we demonstrated that the porous structure of SnOx-containing CNFs could be simply tuned by controlling the TEOS/polyvinylpyrrolidone (PVP) ratio. SnOx-containing hollow-structured CNFs (SnOx/H-CNFs) and wormhole-like porous structures (SnOx/W-CNFs) were fabricated by single-spinneret electrospinning of tin(II) 2-ethylhexanoate-containing TEOS/polyvinylpyrrolidone/ethanol solution, assisted by phase separation between TEOS and PVP. The structure–property relationship was investigated comparatively. Benefiting from the synergistic effect of the ultrafine SnOx nanoparticles, the high specific surface area and the hollow structure, the SnOx/H-CNF electrode delivered a high reversible capacity of 732 mA h g−1 at the rate of 500 mA g−1 upon the 100th cycle, and good rate performance (254 mA h g−1 at 4 A g−1), both of which exceeded those of SnOx-containing CNFs in SnOx/W-CNF and pure SnOx/CNF electrodes.
Experimental
Materials synthesis
In a typical synthesis, 1 g of polyvinylpyrrolidone (PVP, Mw = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000 g mol−1) was dissolved in 10 mL of ethanol at room temperature for 2 h under magnetic stirring. 2 mmol of tin(II) 2-ethylhexanoate (TET) was added stepwise to the PVP solution. Then, a certain amount of TEOS (4 or 12 mmol) was added to the mixed solution, and kept stirring for 0.5 h to obtain a precursor solution for electrospinning. During electrospinning, the precursor solution was delivered to a stainless steel needle by a syringe pump with a flow rate of 0.5 mL h−1 and electrospun at a voltage of 14 kV by a high voltage supply. The electrospun fibers were collected as a thin web on a roller wrapped in aluminium foil. The rotation speed of the roller was 500 rpm, and the distance between the needle and the roller was 15 cm. The collected precursor fiber webs were kept in air at room temperature for 24 h, and then carbonized at 600 °C for 1 h with a heating rate of 3 °C min−1 under a nitrogen atmosphere to prepare SnOx/SiO2/CNF nanocomposites. Finally, SnOx/W-CNF and SnOx/H-CNF nanocomposites were, respectively, obtained after using NaOH solution to remove the SiO2 template. For comparison, SnOx/CNFs were prepared following similar procedures without TEOS in the precursor solution and the post-etching process.
300000 g mol−1) was dissolved in 10 mL of ethanol at room temperature for 2 h under magnetic stirring. 2 mmol of tin(II) 2-ethylhexanoate (TET) was added stepwise to the PVP solution. Then, a certain amount of TEOS (4 or 12 mmol) was added to the mixed solution, and kept stirring for 0.5 h to obtain a precursor solution for electrospinning. During electrospinning, the precursor solution was delivered to a stainless steel needle by a syringe pump with a flow rate of 0.5 mL h−1 and electrospun at a voltage of 14 kV by a high voltage supply. The electrospun fibers were collected as a thin web on a roller wrapped in aluminium foil. The rotation speed of the roller was 500 rpm, and the distance between the needle and the roller was 15 cm. The collected precursor fiber webs were kept in air at room temperature for 24 h, and then carbonized at 600 °C for 1 h with a heating rate of 3 °C min−1 under a nitrogen atmosphere to prepare SnOx/SiO2/CNF nanocomposites. Finally, SnOx/W-CNF and SnOx/H-CNF nanocomposites were, respectively, obtained after using NaOH solution to remove the SiO2 template. For comparison, SnOx/CNFs were prepared following similar procedures without TEOS in the precursor solution and the post-etching process.
Characterization
The morphology and mircrostructure of these samples were observed using a field emission scanning electron microscope (FE-SEM, Supra55, Carl Zeiss) and a high-resolution transmission electron microscope (HR-TEM, Tecnai G2 F30 S-TWIN) equipped with a scanning transmission electron microscope (STEM). The crystal structure and composition of these SnOx/PCNF samples were investigated by wide angle X-ray diffraction (XRD) (WAXD, D8 Advance, Bruker, Cu Kα, λ = 0.154 nm). Thermogravimetric analysis (TGA) was conducted on a TGA instrument (TA-Q50, America) at a heating rate of 10 °C min−1 from 25 to 800 °C in air. Nitrogen adsorption and desorption isotherms were recorded with a Quantachrome SI instrument at 77 K and the pore-size distribution was calculated using the non-local density functional theory (NLDFT) method.Electrochemical measurements
Electrochemical measurements were investigated using 2025-type coin cells which were assembled in an Ar-filled glove box (OMNI-LAB, [O2] < 0.02 ppm, [H2O] < 0.02 ppm). All the samples were first mixed with carbon black and poly(vinylidene difluoride) (PVDF) to form a slurry at a weight ratio of 7:2:1 in N-methyl pyrrolidone (NMP). Subsequently, the working electrodes were prepared by coating the slurry onto nickel foams through a doctor blade method, and then dried in a vacuum oven at 120 °C overnight. Lithium metal foil was used as a counter electrode and Celgard 2300 membrane was used as a separator. The electrolyte for the tests was 1 M LiPF6 in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1:1 v/v). The charge/discharge curves were obtained between 0.005 and 3.0 V on a Land CT2001A (China). Cyclic voltammetry (CV) measurements were performed between 0.005 and 3 V using an Autolab PGSTAT 302 N (Metrohm) workstation with a scan rate of 0.1 mV s−1. Electrochemical impedance spectra (EIS) measurements were also conducted at the same electrochemical workstation with an amplitude of 10 mV and a frequency in the range of 10 kHz to 0.1 Hz. The mass loading of the active material (1.2 mg cm−2), including both SnOx and CNFs, was taken into account when the capacities were calculated.
Results and discussion
The effect of varying the amount of TEOS on the morphology of the porous composite nanofibers is shown by the SEM images shown in Fig. 1. Fig. 1a and b show that both SnOx/W-CNFs and SnOx/H-CNFs possess a well-defined fibrous morphology without any clear cross-linking, and their length can reach up to hundreds of micrometers. The magnified SEM images (insets in Fig. 1a and b) display that these composite nanofibers have small diameters of 200–300 nm and a rough/porous surface. The surface of the SnOx/H-CNFs is more rough than that of the SnOx/W-CNFs. This can be explained by the different content of TEOS. The more TEOS there is in the precursor, the larger the pores created by removing the SiO2 will be. Furthermore, by careful observation of the cross-section of a single nanofiber from the SnOx/W-CNFs and SnOx/H-CNFs, a dramatic difference was revealed. Obviously, the SnOx/H-CNFs have a hollow structure. The inner porous structures of the SnOx/W-CNFs and SnOx/H-CNFs were further revealed by TEM. As shown in Fig. 1c–f, the SnOx/W-CNFs only have wormhole-like closed pores, while the SnOx/H-CNFs present a hollow structure. Again, the TEM images demonstrate the porous nature of the surface of the SnOx/W-CNFs and SnOx/H-CNFs. To explore the reason that leads to the formation of the hollow structure in the SnOx/H-CNFs, we carefully characterized its carbonized precursor nanofibers (SnOx/SiO2/CNFs) without the post-etching treatment. Interestingly, the SnOx/SiO2/CNF nanocomposite also possesses a clear hollow structure as demonstrated by its SEM and TEM images (Fig. S1, ESI†). For comparison, the SnOx/CNF composite only exhibits a solid nanofibrous structure as evidenced by the TEM image (Fig. S2, ESI†). These results imply that TEOS not only acts as a simple sacrificial template precursor, but also can induce a hollow structure by phase-separation dynamics during the solvent evaporation.27 Dayal et al. even used theoretical analysis and simulations to explain the formation of different fiber morphologies in a polymer–solvent system.28 They demonstrated that the fiber morphologies depended on the competition between the phase separation dynamics and the evaporation rate of the solvent.28 According to their assumption, hollow fibers can be obtained only when the solvent evaporation rate is so high that it can quickly induce sharp phase separation. Herein, we believe that the volatilization-induced phase separation between TEOS and PVP plays an important role in promoting the formation of a hollow structure. More specifically, as shown by Fig. 2, once the precursor solution was jetted from the needle tip to form the nanofibers under the high-voltage electrostatic field, ethanol evaporated rapidly. This led to a gradient distribution of ethanol along the radial direction with the highest concentration in the center of the nanofibers. TEOS and TET are soluble in ethanol, and thus they would diffuse along the concentration gradient of ethanol and gather at the center of the nanofibers. Meanwhile, PVP would move toward the opposite direction due to its incompatibility with TEOS and TET, thus promoting the formation of TEOS/TET–PVP core–shell nanofibers. Finally, the hollow structure was formed in the electrospun nanofibers after the inner TEOS completely volatilized.27 In addition, it should be noted that the volatile TEOS itself is also hydrolysable.5 The hydrolyzed TEOS will stay in the outer surface layer and on the inner and exterior walls of the nanofibers, and subsequently be converted into silicon oxide during the carbonization process. Therefore, a porous carbon wall can be generated in these tubular nanofibers after the removal of these silicon oxides through the etching treatment. | ||
| Fig. 1 SEM images of (a) SnOx/W-CNFs and (b) SnOx/H-CNFs. Insets in (a and b) show the magnified SEM images of the cross-section of the composite nanofibers. TEM images of (c and e) SnOx/W-CNFs and (d and f) SnOx/H-CNFs. | ||
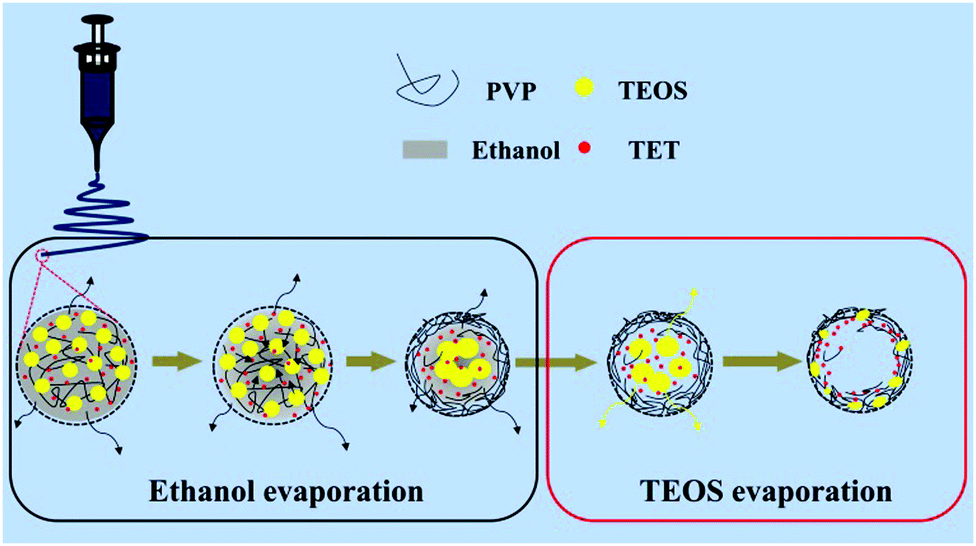 | ||
| Fig. 2 Schematic illustration of the formation mechanism of the hollow nanofibers from a cross-sectional view. | ||
Fig. 3a displays a HR-TEM image of SnOx/H-CNFs. It is obvious that large amounts of SnOx nanoparticles are uniformly dispersed in the tube-like nanofibers. Furthermore, the magnified HR-TEM image (the inset in Fig. 3a) demonstrates that the size of the incorporated SnOx nanoparticles is very small (less than 5 nm). This is beneficial for obtaining good cycling performance because the fracture and pulverization of the tin-based nanoparticles will become less severe during cycling with decreasing particle size.7 The nanocrystals have d-spacing values of 0.33 and 0.26 nm, corresponding to the (110) and (101) planes of SnO2, respectively. The presence and distribution of elements are further confirmed by energy dispersive spectrometry (EDS) elemental mapping analysis. As shown in Fig. 3b, the tin and oxygen are uniformly distributed within the hollow nanofibers, in agreement with the HR-TEM analysis. The phase structure and composition of the SnOx/H-CNFs were also characterized using XRD measurement. The XRD pattern of the SnOx/H-CNFs shows a wide peak at about 24° (Fig. 3c). This indicates the amorphous nature of the PVP-derived carbon. According to the JCPDS card, the diffraction peaks located at 2θ = 26.6, 33.8, 37.9, 51.7, 54.7, 64.7 and 65.9° correspond to the (110), (101), (200), (211), (220), (112) and (301) diffraction peaks of SnO2, respectively; the diffraction peaks at 2θ = 30.6, 32.0, 43.8 and 44.9° correspond to the (200), (101), (220) and (211) diffraction peaks of Sn, respectively. This suggests that tin oxide was partially reduced during the carbonization process because of the carbon thermal reduction. Fig. 3d shows the TGA curve of the SnOx/H-CNFs. The SnOx/H-CNFs experience two typical weight loss stages: one is explained by the desorption of water vapor and the removal of the adsorbed volatile organics in the temperature range of 25–200 °C; another is caused by the oxidation or burning of the carbon matrix from 200 to 550 °C.15 Assuming the complete burn-off of the carbon matrix and the complete conversion of SnOx to SnO2, the weight percentage of SnOx in the SnOx/H-CNFs is calculated to be about 33 wt%.
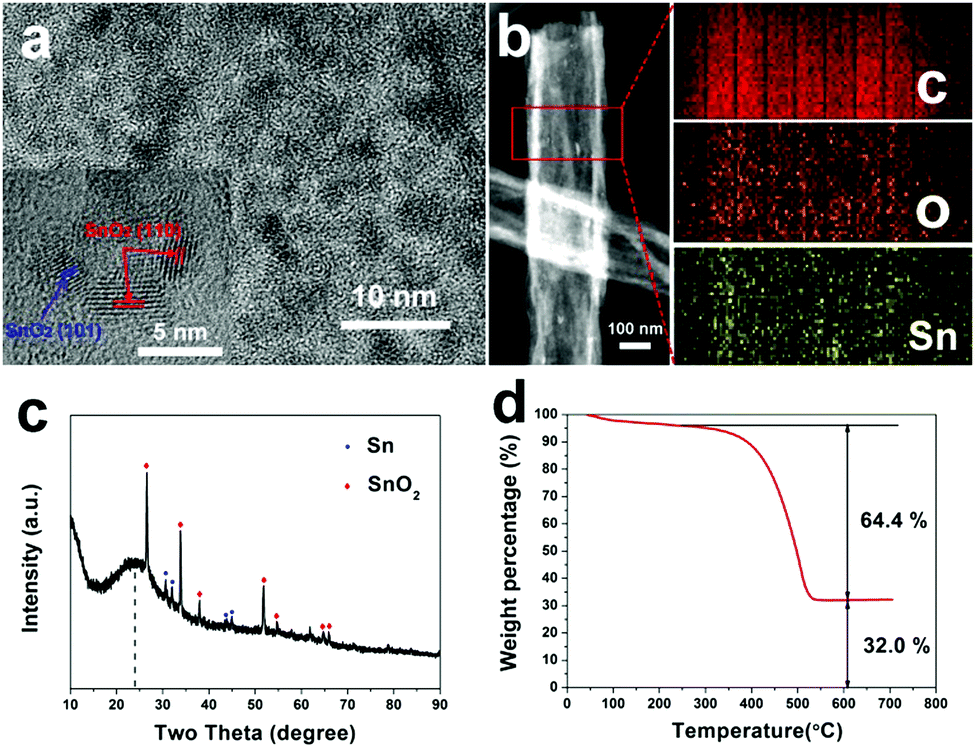 | ||
| Fig. 3 (a) HR-TEM images of SnOx/H-CNFs. The inset shows the SnO2 nanocrystals. (b) STEM image of SnOx/H-CNFs and the corresponding elemental mapping images of carbon, oxygen, and tin. (c) XRD pattern and (d) TG curve of SnOx/H-CNFs. | ||
The specific surface area and pore structure of the SnOx/W-CNFs and SnOx/H-CNFs were determined by nitrogen adsorption–desorption isothermal analysis. Fig. 4a shows the nitrogen adsorption–desorption isotherms and the pore size distribution of the SnOx/W-CNFs and SnOx/H-CNFs. Both samples demonstrated a type-IV isotherm with nitrogen uptake mainly occurring at a relative pressure of above 0.2. This implies that both samples are rich in mesopores. The H1-type hysteresis loop further indicates their mesoporous structure. Pore size distribution analyses by the DFT method confirm that pores with diameters of greater than 2.0 nm are the dominating component (Fig. 4b), which agrees well with the nitrogen sorption/desorption isotherms. The specific surface areas of the SnOx/W-CNFs and SnOx/H-CNFs, derived from the Brunauer–Emmett–Teller (BET) method, are calculated to be 44 and 739 m2 g−1, respectively. The total pore volume of the SnOx/W-CNFs and SnOx/H-CNFs is 0.0544 and 0.7257 cm3 g−1, respectively. Apparently, SnOx/H-CNFs exhibit a larger specific surface area and more open pores, which could be highly suitable for the easy permeation of the Li+-carrying electrolyte into the inner part of these composite nanofibers to reach the embedded nanoparticles. Thus, good rate capability and high capacity are expected.22,29
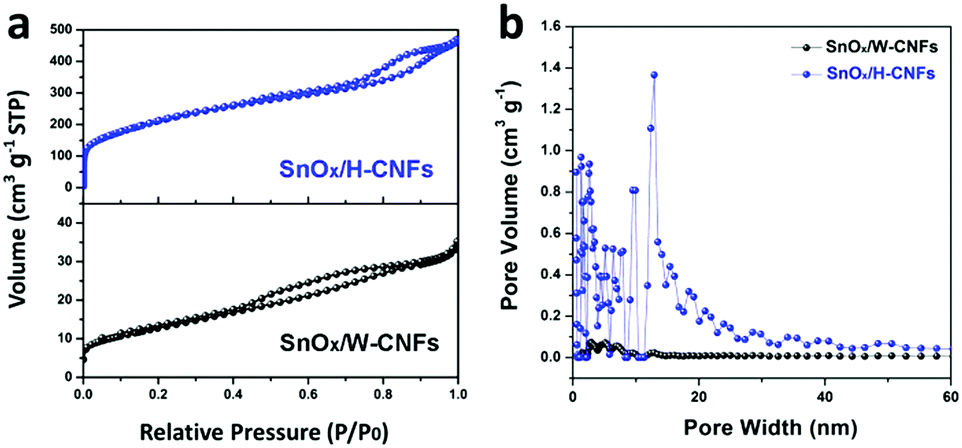 | ||
| Fig. 4 (a) Nitrogen adsorption–desorption isotherms and (b) the pore size distribution of SnOx/W-CNFs and SnOx/H-CNFs. | ||
Fig. 5a shows the discharge (lithiation) and charge (delithiation) voltage profiles of the SnOx/H-CNF electrode from the 1st, 3rd, 5th and 10th cycles at a current density of 200 mA g−1 between 0.005 and 3 V. The initial charge and discharge capacities of the SnOx/H-CNF electrode are 1006 and 1507 mA h g−1, respectively. It should be noted that the low initial Coulombic efficiency of 67% is mainly attributed to the irreversible formation of the solid electrolyte interphase (SEI) layer on the large electrode/electrolyte interface. After the 1st discharge/charge process, the Coulombic efficiency gradually climbed to 97% at the 3rd cycle, and was maintained at almost 100% from the 5th cycle. This shows its excellent cycling stability. In addition, CV analysis was employed to further understand the lithium-storage behavior of the SnOx/H-CNF electrode. Fig. S3 (ESI†) presents the first three CV curves of the SnOx/H-CNF electrode at 0.1 mV s−1. The cathodic peak observed in the first cycle at about 0.8 V is mainly attributed to the formation of the SEI layer, and this peak disappears from the second cycle. Another cathodic peak from 0.75 to 0.005 V is assigned to the alloying process of LixSn and the intercalation of Li+ storage in the hollow CNFs. In the following anodic sweep, two humps located at 0.55 and 1.25 V correspond to the de-alloying process of Li–Sn and the conversion reaction from Sn and Li2O to SnOx, respectively. Fig. 5b compares the cycling performance of the SnOx/CNF, SnOx/W-CNF, and SnOx/H-CNF electrodes at 500 mA g−1. After 100 cycles, both the SnOx/CNF and SnOx/W-CNF electrodes show low capacities of 406 and 468 mA h g−1, respectively. Their capacity loss during cycling might be derived from the gradual partial irreversible insertion of Li+ into the vacancies of the carbon matrices.29 In contrast, the SnOx/H-CNF electrode shows a high charge capacity of 721 mA h g−1, and more importantly undergoes moderate capacity fading upon the 100th cycle. The improved Li-storage capacity exhibited by the SnOx/H-CNF electrode can be attributed to the open channels of the 1D carbon matrix that not only shorten the transport length of Li+ ions by providing a large electrode/electrolyte contact area but also provide adequate buffer area for releasing the mechanical stress induced by the volume change of the embedded high-capacity SnOx particles during cycling.30 The rate capabilities of these electrodes are compared in Fig. 5c. The SnOx/H-CNF electrode shows the best rate capability among all the electrodes, presenting a reversible capacity of 254 mA h g−1 at 4 A g−1. This is about 46% and 200% larger than that of the SnOx/W-CNF (173 mA h g−1) and SnOx/CNF (86 mA h g−1) electrodes at 4 A g−1, respectively. The excellent high-rate performance of the SnOx/H-CNFs electrode further demonstrates that the unique hollow/porous architecture can highly improve the Li+-diffusion capability along the 1D CNF matrix. As compared with the all-closed or semi-closed worm-like pores, the hollow-like voids have larger amounts of open ends and continuous channels, and a larger pore size for the fast and total penetration of the Li+-carrying electrolyte into the inner part of the electrode, thus decreasing the Li+-diffusion length and simultaneously making full use of the embedded SnOx nanoparticles at high-rate current. On the other hand, the reversible capacity of the SnOx/H-CNF electrode returns to its initial value at a small rate of 200 mA g−1 after the high-rate charge–discharge processes. This indicates the robust stability of the SnOx/H-CNF electrode. Fig. S4 (ESI†) shows the SEM images of the SnOx/CNF, SnOx/W-CNF and SnOx/H-CNF electrodes after 100 cycles. It can be observed that all the composite nanofibers maintain a well-defined fibrous morphology, demonstrating that the as-synthesized CNFs are a good and stable matrix that can adequately buffer the volume change of SnOx nanoparticles and keep the structural stability of the electrodes. EIS impedance spectroscopy was employed to reveal the ion transport behavior throughout the electrodes. The impedance spectra of the SnOx/CNF, SnOx/W-CNF and SnOx/H-CNF electrodes after 100 cycles are shown in Fig. S5 (ESI†). All the Nyquist plots consist of one semicircle in the high frequency range and an inclined line in the low frequency region. The semicircle corresponds to the resistance of the electrodes and the ion-transport resistance.20,31,32 The smaller the diameter, the lower the overall resistance. The diameter of the semicircle for the SnOx/H-CNF electrode is smaller than that for the SnOx/CNF and SnOx/W-CNF electrodes. This evidences the improved electrolyte accessibility arising from the open hollow structure of the SnOx/H-CNFs. The outstanding high-rate performance of the SnOx/H-CNF electrode could be contributed to the improved Li+ transport/storage capability as schematically illustrated in Fig. 6. The Li+ ion diffusion will be simplified here. The existence of some inaccessible small pores can interrupt and/or prolong the diffusion of Li+, thus increasing the Li+ transport resistance. Compared with the SnOx/W-CNFs, the hollow channel in the SnOx/H-CNFs can provide fast ion transport pathways like those in the bulk electrolyte. When all hollow channels are filled with the electrolyte, Li+ can migrate from both the outer surface and the inner surface of the tube-like nanofiber, thus greatly decreasing the Li+-diffusion distance and the Li+-transport resistance.20
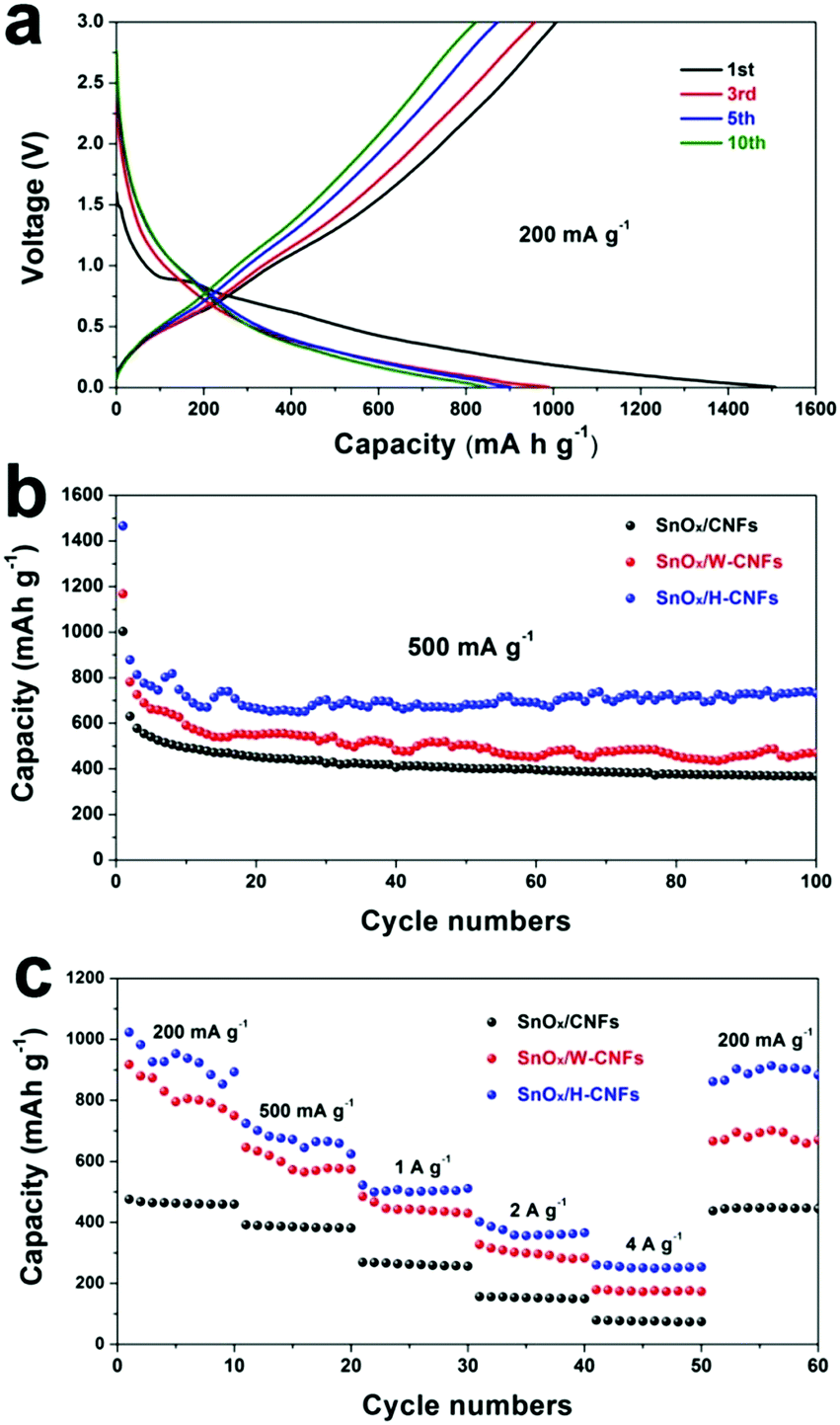 | ||
| Fig. 5 (a) Galvanostatic discharge/charge profiles of the SnOx/H-CNF electrode at the rate of 200 mA g−1. (b) Cycling performance of the SnOx/CNF, SnOx/W-CNF, and SnOx/H-CNF electrodes at the rate of 500 mA g−1 within a voltage window of 0.005–3.0 V vs. Li+/Li. (c) Rate performance of the SnOx/CNF, SnOx/W-CNF, and SnOx/H-CNF electrodes at various rates from 200 to 4000 mA g−1. | ||
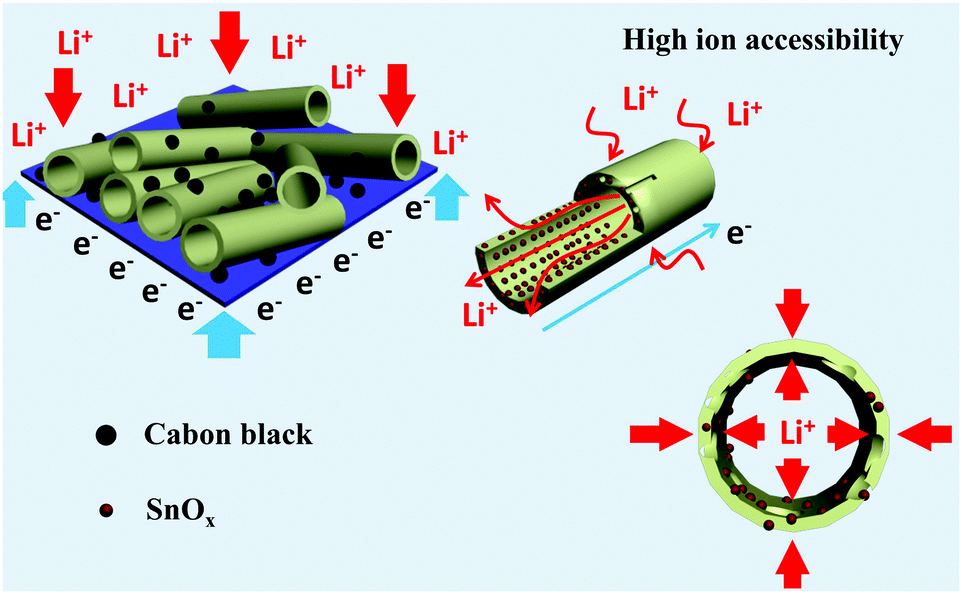 | ||
| Fig. 6 Schematic illustration showing the structure and ion transport in a hollow/porous structure. | ||
Fig. 7 shows the long-term cycling performance of the SnOx/H-CNF electrode at a high rate of 1 A g−1. It can be seen that the SnOx/H-CNF electrode demonstrates an excellent charge–discharge reversibility and superior cycling stability, with a high reversible capacity of 513 mA h g−1 and a remarkably high Coulombic efficiency of ∼100% during the 500 cycles. In addition, it should be noted that the specific capacities of the SnOx/H-CNFs gradually increase with cycling after about 50 cycles, as shown by Fig. 7. This phenomenon is also observed in previous tin oxide/carbon nanocomposites, which contributed to the gradual activation of active materials or the reversible formation of an organic polymeric/gel-like film that delivers excess lithium-storage capacity.8,9 To further verify the good durability of the hollow structure, the morphology and structure of the SnOx/H-CNF electrode after 500 cycles were explored by HR-TEM measurements (inset in Fig. 7). The composite nanofibers after the cycling test still have a well-maintained fibrous morphology. This demonstrates that the hollow CNF substrate can effectively buffer the volume change in the SnOx particles during cycling, thus maintaining the integrity of the overall electrode. This is very beneficial for the cycling performance of the SnOx/H-CNF electrode.
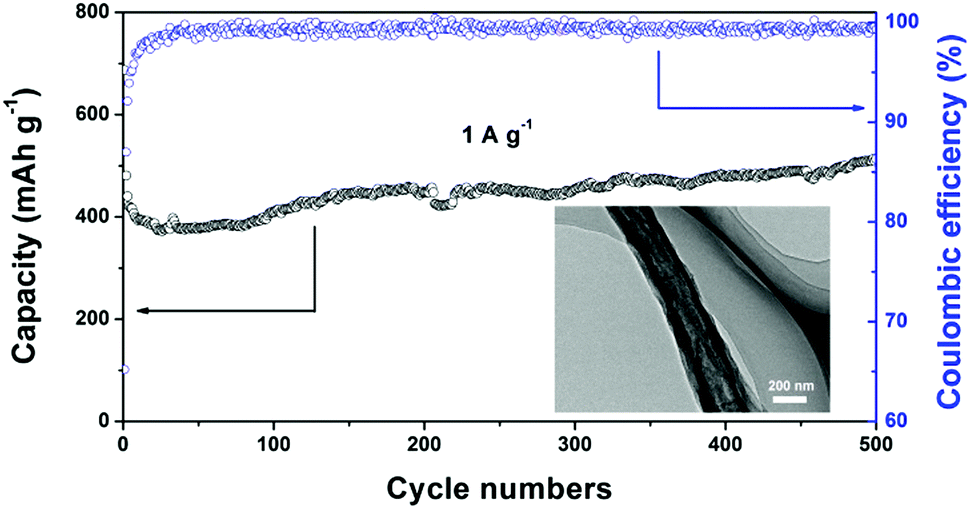 | ||
| Fig. 7 Long-term cycling performance of the SnOx/H-CNF electrode at 1 A g−1. The inset shows the HR-TEM image of the cycled SnOx/H-CNFs obtained after 500 cycles. | ||
Based on these results, the excellent performance of the SnOx/H-CNF electrode may be ascribed to its distinct structure and morphology that offer the following benefits. First, the ultrafine SnOx nanoparticles can provide a large number of active sites for lithium storage, contributing to high reversible capacity. Second, the hollow CNF substrate not only offers sufficient 1D conductive pathways for electron transport, but also buffers the volume expansion and prevents the aggregation of the nanosized SnOx during cycling. Third, the hollow nature of the SnOx/H-CNFs with large amounts of mesopores in the tube walls can ensure a high contact area between the electrolyte and the active material and a shortened Li+ ion diffusion length, thus facilitating the transport and diffusion of lithium ions toward the inner part of the active material, especially the embedded SnOx nanoparticles, during the charge/discharge process.
Conclusion
In summary, hollow carbon nanofibers containing SnOx (SnOx/H-CNFs) have been prepared by a single-spinneret electrospinning technique, followed by carbonization and the removal of silicon oxide. The hollow structure of the SnOx/H-CNFs mainly stems from ethanol/TEOS evaporation-induced phase separation. The removal of the retained silicon oxide produces large amounts of mesopores or channels in the tube walls of the SnOx/H-CNF, resulting in a large specific surface area of 739 m2 g−1. The high specific surface area provides a large electrode/electrolyte contact interface and facilitates the penetration of the Li+-carrying electrolyte into the electrode for fast charge–discharge processes. The unique hollow structure releases the strain caused by the volume expansion of the high-capacity SnOx particles and shortens the Li+ ion diffusion length during cycling. Therefore, the as-prepared SnOx/H-CNF electrode shows high reversible capacity, stable cycling performance and good rate capability. Moreover, this simple single-spinneret electrospinning method can be extended to fabricate porous electrode materials far and beyond the lithium-ion batteries.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 51072013, 51272021, 51142004 and 51402010) and the Natural Science Foundation of Jiangsu Province (No. BK20131147 and BK20140270).Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacín, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS PubMed.
- C.-M. Park, J.-H. Kim, H. Kim and H.-J. Sohn, Chem. Soc. Rev., 2010, 39, 3115 RSC.
- N. Nitta and G. Yushin, Part. Part. Syst. Charact., 2014, 31, 317 CrossRef CAS.
- X. Yan, D. Teng, X. Jia, Y. Yu and X. Yang, Electrochim. Acta, 2013, 108, 196 CrossRef CAS.
- F.-H. Du, K.-X. Wang and J.-S. Chen, J. Mater. Chem. A, 2016, 4, 32 CAS.
- X. Zhou, Z. Dai, S. Liu, J. Bao and Y.-G. Guo, Adv. Mater., 2014, 26, 3943 CrossRef CAS PubMed.
- Y. Liu, J.-L. Lan, Q. Cai, Y.-H. Yu, Y.-H. Lin and X.-P. Yang, Part. Part. Syst. Charact., 2015, 32, 952 CrossRef CAS.
- X. Zhou, L.-J. Wan and Y.-G. Guo, Adv. Mater., 2013, 25, 2152 CrossRef CAS PubMed.
- J. Zhu, G. Zhang, X. Yu, Q. Li, B. Lu and Z. Xu, Nano Energy, 2014, 3, 80 CrossRef CAS.
- Y. Xu, Q. Liu, Y. Zhu, Y. Liu, A. Langrock, M. R. Zachariah and C. Wang, Nano Lett., 2013, 13, 470 CrossRef CAS PubMed.
- B. Zhao, S. Jiang, C. Su, R. Cai, R. Ran, M. O. Tade and Z. Shao, J. Mater. Chem. A, 2013, 1, 12310 CAS.
- B. Zhang, Y. Yu, Z. Huang, Y.-B. He, D. Jang, W.-S. Yoon, Y.-W. Mai, F. Kang and J.-K. Kim, Energy Environ. Sci., 2012, 5, 9895 CAS.
- Y. Liu, X. Yan, Y. Yu and X. Yang, ACS Sustainable Chem. Eng., 2016, 4, 2951 CrossRef CAS.
- Y. Liu, X. Yan, J.-L. Lan, D. Teng, Y. Yu and X. Yang, Electrochim. Acta, 2014, 137, 9 CrossRef CAS.
- J. Zhu, T. Wang, F. Fan, L. Mei and B. Lu, ACS Nano, 2016, 10, 8243 CrossRef CAS PubMed.
- L. Mei, M. Mao, S. Chou, H. Liu, S. Dou, D. H. L. Ng and J. Ma, J. Mater. Chem. A, 2015, 3, 21699 CAS.
- L. Xue, K. Fu, Y. Li, G. Xu, Y. Lu, S. Zhang, O. Toprakci and X. Zhang, Nano Energy, 2013, 2, 361 CrossRef CAS.
- J. Shin, W.-H. Ryu, K.-S. Park and I.-D. Kim, ACS Nano, 2013, 7, 7330 CrossRef CAS PubMed.
- H. He, L. Shi, Y. Fang, X. Li, Q. Song and L. Zhi, Small, 2014, 10, 4671 CrossRef CAS PubMed.
- A. Vu, Y. Qian and A. Stein, Adv. Energy Mater., 2012, 2, 1056 CrossRef CAS.
- Z. Shen, Y. Hu, Y. Chen, X. Zhang, K. Wang and R. Chen, J. Power Sources, 2015, 278, 660 CrossRef CAS.
- D. Nan, J.-G. Wang, Z.-H. Huang, L. Wang, W. Shen and F. Kang, Electrochem. Commun., 2013, 34, 52 CrossRef CAS.
- Z. Jin, X. Yan, Y. Yu and G. Zhao, J. Mater. Chem. A, 2014, 2, 117060 Search PubMed.
- S. Abouali, M. A. Garakani, B. Zhang, H. Luo, Z.-L. Xu, J.-Q. Huang, J. Huang and J.-K. Kim, J. Mater. Chem. A, 2014, 2, 16939 CAS.
- Z.-L. Xu, B. Zhang, S. Abouali, M. A. Garakani, J. Huang, J.-Q. Huang, E. K. Heidari and J.-K. Kim, J. Mater. Chem. A, 2014, 2, 17944 CAS.
- X. H. Li, C. L. Shao and Y. C. Liu, Langmuir, 2007, 23, 10920 CrossRef CAS PubMed.
- P. Dayal and T. Kyu, J. Appl. Phys., 2006, 100, 43512 CrossRef.
- Y. Liu, X. Yan, Y. Yu and X. Yang, J. Mater. Chem. A, 2015, 3, 20880 CAS.
- J. Zhu, Y. Shan, T. Wang, H. Sun, Z. Zhao, L. Mei, Z. Fan, Z. Xu, I. Shakir, Y. Huang, B. Lu and X. Duan, Nat. Commun., 2016 DOI:10.1038/ncomms13432.
- Y. Shi, L. Wen, F. Li and H.-M. Cheng, J. Power Sources, 2011, 196, 8610 CrossRef CAS.
- S. Yoon, J. Lee, T. Hyeon and S. M. Oh, J. Electrochem. Soc., 2000, 147, 2507 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00377j |
| This journal is © the Partner Organisations 2017 |