
All-organic luminescent nanodots from corannulene and cyclodextrin nano-assembly: continuous-flow synthesis, non-linear optical properties, and bio-imaging applications†
Sivaramapanicker
Sreejith
 a,
Nishanth Venugopal
Menon
b,
Yue
Wang
c,
Hrishikesh
Joshi
a,
Shiying
Liu
b,
Kok Chan
Chong
a,
Yuejun
Kang
d,
Handong
Sun
ce and
Mihaiela C.
Stuparu
*af
a,
Nishanth Venugopal
Menon
b,
Yue
Wang
c,
Hrishikesh
Joshi
a,
Shiying
Liu
b,
Kok Chan
Chong
a,
Yuejun
Kang
d,
Handong
Sun
ce and
Mihaiela C.
Stuparu
*af
aDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang link, 637371, Singapore. E-mail: mstuparu@ntu.edu.sg
bSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 62 Nanyang Drive, 637459, Singapore
cDivision of Physics and Applied Physics, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang link, 637371, Singapore
dFaculty of Materials and Energy, Institute for Clean Energy and Advanced Materials, Southwest University, 2 Tiansheng Road, Beibei, Chongqing, 400715, China
eCentre for Disruptive Photonic Technologies, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, 637371, Singapore
fSchool of Materials Science and Engineering, Nanyang Technological University, 639798, Singapore
First published on 20th April 2017
Abstract
Control of structure and function, at the nanometer scale, remains a formidable challenge in the arena of self-assembled soft materials. Here, we report on the design of a small molecule-based two-component assembly system in which the assembly partners can recognize each other through host–guest interactions. One component is hydrophobic and carries a donor–acceptor type of electronic structure. This is realized by employing a bucky-bowl corannulene derivative. The other component is hydrophilic and hollow. This is achieved by using γ-cyclodextrin, the largest and least studied member of the cyclic oligosaccharide family. In a chemically polar aqueous environment, the two partners come together to form an amphiphilic structure that assembles further into nanosized, quasicrystalline, dot-like, non-toxic, all-organic structures showing two-photon activity and bright green luminescence in water upon excitation at 800 nm. The devised synthesis is achieved by a simple mixing process carried out under continuous-flow conditions. Therefore, in a scalable manner, a constant supply of the assembly components results in continuous fabrication of the nanostructures. Non-linear optical activity and biocompatibility aspects suggest utility of the prepared new class of soft organic nano-dots as contrast agents or labeling tags for visualizing biological specimens. This aspect is examined and demonstrated through two-photon fluorescence imaging of cancer cell lines.
Introduction
Ultra-small luminescent nanostructures capable of non-linear optical activity offer a broad range of applications. The most important is perhaps their use in fluorescence imaging of biological matter.1–6 Interestingly, inorganic and metal-based chemistries allow for fabrication of such small (sub – 20 nm) functional structures in a routine fashion.7–9 Organic chemistry approaches to access such size regimes with functionality remain rather restricted.10 The two known general classes of ultra-small and non-linearly optical active organic materials are dendrimers11,12 and polymer dots13,14 constructed using covalent synthesis methods. Self-assembly approaches operating upon supramolecular interactions2,15,16 offer an alternative and more practical route in which assembly precursors are directly mixed to generate complex functional structures.17–26 If carried out under dynamic (flow) conditions, continuous production can be envisaged leading to the scalability of the materials. Finally, if the assembly components are based on small organic molecules, a high reproducibility on all levels, from synthesis to properties and bio-applications, is expected due to a precise and mono-disperse underlying molecular structure. However, such an approach to access the ultra-small size regime of organic nanostructures capable of specific functions is seldom reported.27Here, we show that a new class of nanosized green luminescent structures of pure organic composition with non-linear optical activity can be obtained using a non-covalent synthetic approach based upon host–guest interactions28–30 between a corannulene derivative and a cyclic oligosaccharide under aqueous conditions.31 The synthesis involves a simple mixing process of the assembly precursors in a microfluidic chamber. Besides simplicity of fabrication, the microfluidic process also ensures access to a large quantity of the material through a continuous feed of the assembly precursors. The new class of organic dots is well dispersible in water, polycrystalline, biocompatible, and readily taken up by cancer cells. These properties, combined with their two-photon activity, render them suitable for bio-imaging applications.
Results and discussion
In the present system, one of the assembly components, corannulene, is a non-planar polycyclic aromatic hydrocarbon (Fig. 1).32–36 It can be considered as the cap region of fullerene C60. Due to this non-planarity of the structure, many interesting properties arise, some of which resonate with the attributes of fullerene C60. For example, corannulene is an electron acceptor.37–40 Unlike C60, however, corannulene can be precisely substituted in different geometric fashions, and these derivatives show high solubility in a range of organic solvents.41–46 This attribute, combined with the recently developed kilogram scale synthesis of corannulene by Siegel and co-workers,47 facilitates application of this unique structural motif in the fabrication of functional materials. In this regard, field effect transistors have been made,48–50 redox-active metal organic frameworks and porous organic networks have been created,40–51 ferroelectric materials have been realized,52 and corannulene-encoded polymers53,54 capable of fullerene C60 recognition55 have been accessed. Recently, discrete conjugated oligomers with two-photon activity have also been reported.56 The present work further extends the repertoire of corannulene chemistry by demonstrating the preparation of soft organic nanomaterials of biological relevance.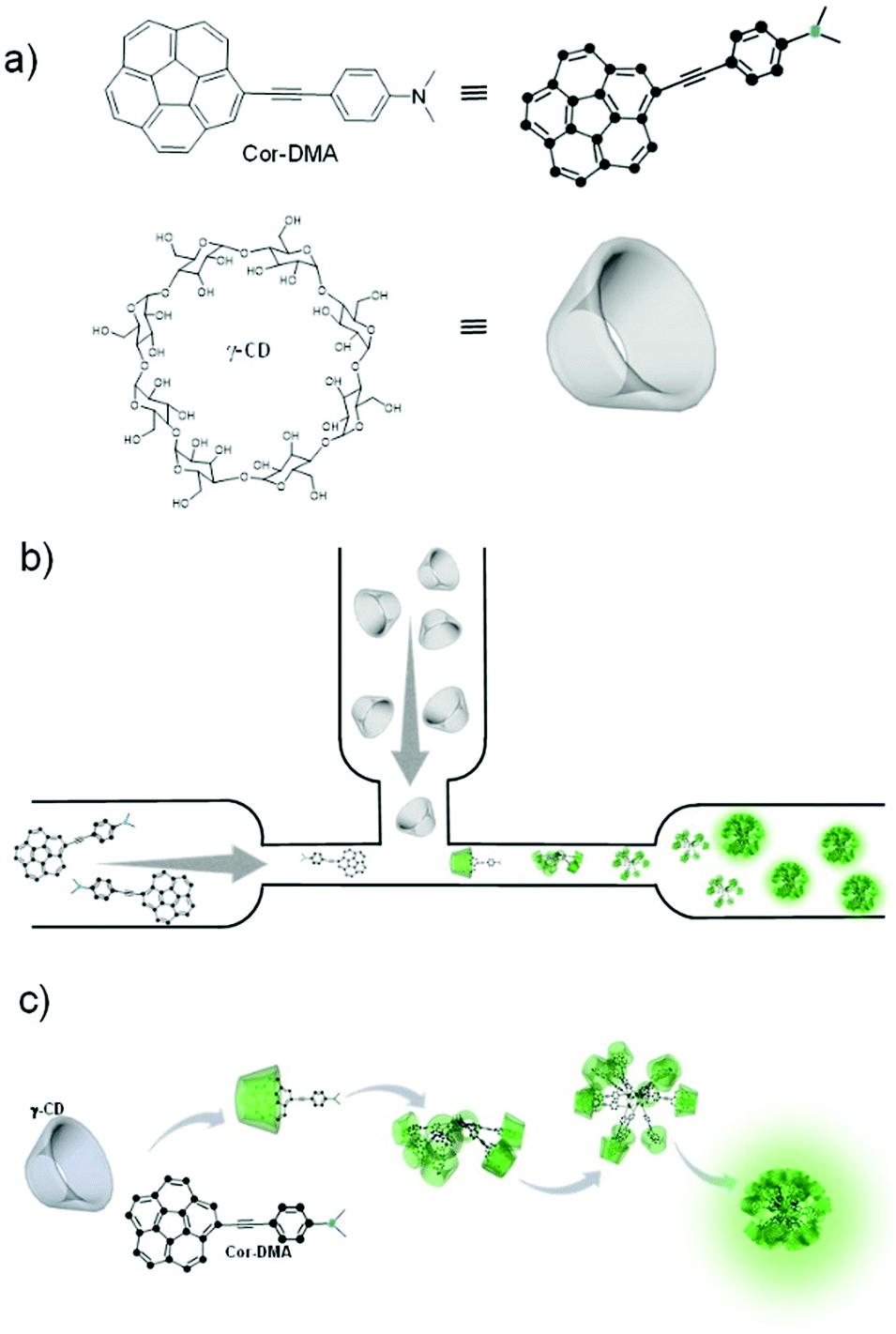 | ||
| Fig. 1 All-organic luminescent nanodot formation. (a) Chemical structures and cartoon representation of the assembly precursors. (b) Their interaction via host–guest complexation in a microfluidic channel. (c) Schematic representation of the formation of a nanodot structure through an assembly process. | ||
To develop such a material platform, bare corannulene cannot be used due to its marginal optical properties. Therefore, appropriate substitution of the corannulene nucleus is necessary. For example, substitution with electron donating units such as a dimethylaniline motif can enhance the intramolecular charge transfer (ICT) capability of the corannulene nucleus.57 However, regarding biological applications, such optically active yet water insoluble structures are of little use. We hypothesized that this limitation could be alleviated if the optically active component can be encapsulated by a hydrophilic molecular sheath.58–60 For this, we decided to employ cyclic oligosaccharides due to their known capability of providing refuge to hydrophobic molecules through the formation of a host–guest type of inclusion complex in water, their low cytotoxicity and biocompatibility aspects.61,62 γ-Cyclodextrin (γ-CD), the largest and least explored oligosaccharide composed of eight cyclic sugar units and with a cavity size of 9.5 Å proved suitable for hosting corannulene63 and paved the way to the fabrication of luminescent nano-dots via an assembly process of the two small molecules under aqueous conditions (Fig. 1).
At the outset, electron deficient corannulene (Cor) is attached to a dimethylaniline (DMA)-based electron rich species through an acetylene linkage (Fig. 1). The acetylene linkage offers rigidity and planarity to the structure and allows electronic communication between the donor (DMA) and the acceptor (Cor) moieties. The UV-Vis spectrum of Cor–DMA in acetonitrile (Fig. S1, ESI†) shows two absorption bands centered at 292 nm (ε = 99857 M−1 cm−1) and 392 nm (ε = 44325 M−1 cm−1). The latter is broad and diagnostic of an intramolecular charge transfer (ICT) process.57 Fluorescence emission upon excitation at 380 nm results in a broad band centered at 562 nm (Φf = 48%) (Fig. S1, ESI†). Depending upon the polarity of the medium, this band shows a significant shift in its position. For example, when going from a non-polar solvent, such as hexane, to a polar solvent, such as DMF, the extent of the solvatochromic shift is observed to be about 116 nm (Fig. S1, ESI†). These characteristics (position of the absorption band, its high molar absorptivity, and solvatochromism) confirmed the ICT process64 in the donor–acceptor, Cor–DMA, assembly precursor.
After studying the preliminary photophysical properties, the interaction between γ-CD and Cor–DMA is considered. For this, aliquots of an aqueous solution of γ-CD were added to an acetonitrile solution of Cor–DMA. Upon increasing the content of γ-CD, a hyperchromic shift in the absorption spectra of Cor–DMA is observed, until the addition reaches a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (Fig. 2a). Further increase in the ratio of Cor–DMA to γ-CD, 1:8 and 1:16, creates a hypochromic shift with further broadening and scattered spectral features. These changes suggest that initially, the two assembly precursors interact with each other at a molecular level and then form larger aggregates.65,66 The use of excess γ-CD helps in forcing the initial complexation event to completion. The surplus γ-CD, being water soluble, can be removed by centrifugation if required. Subsequently, a fluorescence emission study was undertaken. In this study, concomitant changes in the emission profile were observed (Fig. 2b). For example, the emission signal centered at 562 nm initially undergoes a decrease in intensity and then blue-shifts to about 500 nm upon an increase in the content of γ-CD in the Cor–DMA solution. A contour intensity profile visualizing the dynamic evolution of blue-shifted emission (ca. 62–63 nm) indicating the formation of the Cor–DMA–γ-CD inclusion complex is shown in Fig. 2c. These changes in the emission of Cor–DMA could also be visualized by the naked eye (Fig. 2d–f).
:4 (Fig. 2a). Further increase in the ratio of Cor–DMA to γ-CD, 1:8 and 1:16, creates a hypochromic shift with further broadening and scattered spectral features. These changes suggest that initially, the two assembly precursors interact with each other at a molecular level and then form larger aggregates.65,66 The use of excess γ-CD helps in forcing the initial complexation event to completion. The surplus γ-CD, being water soluble, can be removed by centrifugation if required. Subsequently, a fluorescence emission study was undertaken. In this study, concomitant changes in the emission profile were observed (Fig. 2b). For example, the emission signal centered at 562 nm initially undergoes a decrease in intensity and then blue-shifts to about 500 nm upon an increase in the content of γ-CD in the Cor–DMA solution. A contour intensity profile visualizing the dynamic evolution of blue-shifted emission (ca. 62–63 nm) indicating the formation of the Cor–DMA–γ-CD inclusion complex is shown in Fig. 2c. These changes in the emission of Cor–DMA could also be visualized by the naked eye (Fig. 2d–f).
 | ||
| Fig. 2 Interaction of the assembly precursors. (a) UV/Vis absorption changes in Cor–DMA upon addition of γ-CD in varying aliquots. (b) Parallel recordings of the corresponding fluorescence changes in Cor–DMA at 560 nm upon excitation at 380 nm upon addition of various aliquots of γ-CD. (c) Contour map showing changes in fluorescence upon complexation of Cor–DMA with γ-CD. (d–f) Digital pictures showing the solution of Cor–DMA (d), and mixtures of the two assembly precursors (1:2; e, and 1:16; f). Scale bar, 10 mm. | ||
To further investigate the host–guest interaction between the assembly precursors, 1H NMR titration studies using a solvent mixture of DMSO-d6 and water-d2 (D2O) were undertaken. DMSO is a good solvent for both assembly components. D2O, on the other hand, is a bad solvent for Cor–DMA. Therefore, we expected that the complex formation and the subsequent assembly process would be enforced upon increasing the content of D2O in the solvent mixture. As expected, in 100% DMSO, no shift in the proton resonances of corannulene (7.9 to 8.2 ppm) and DMA (6.8 and 7.55 ppm, 2.99 pm) is observed in the presence of γ-CD (Fig. 3 and Fig. S2, S3, ESI†). However, the addition of water resulted in significant differences. For example, the aromatic resonances of the corannulene unit (H1–H3) merged, and a downfield shift is observed for the resonances (H1, O2H, O3H and O6H) of the γ-CD unit.67,68 Remarkably, the resonance from the aromatic and the aliphatic protons of the DMA segment remained unchanged. This study indicated that in water, complex formation occurs between the cavity of γ-CD and the corannulene segment of the Cor–DMA molecule.
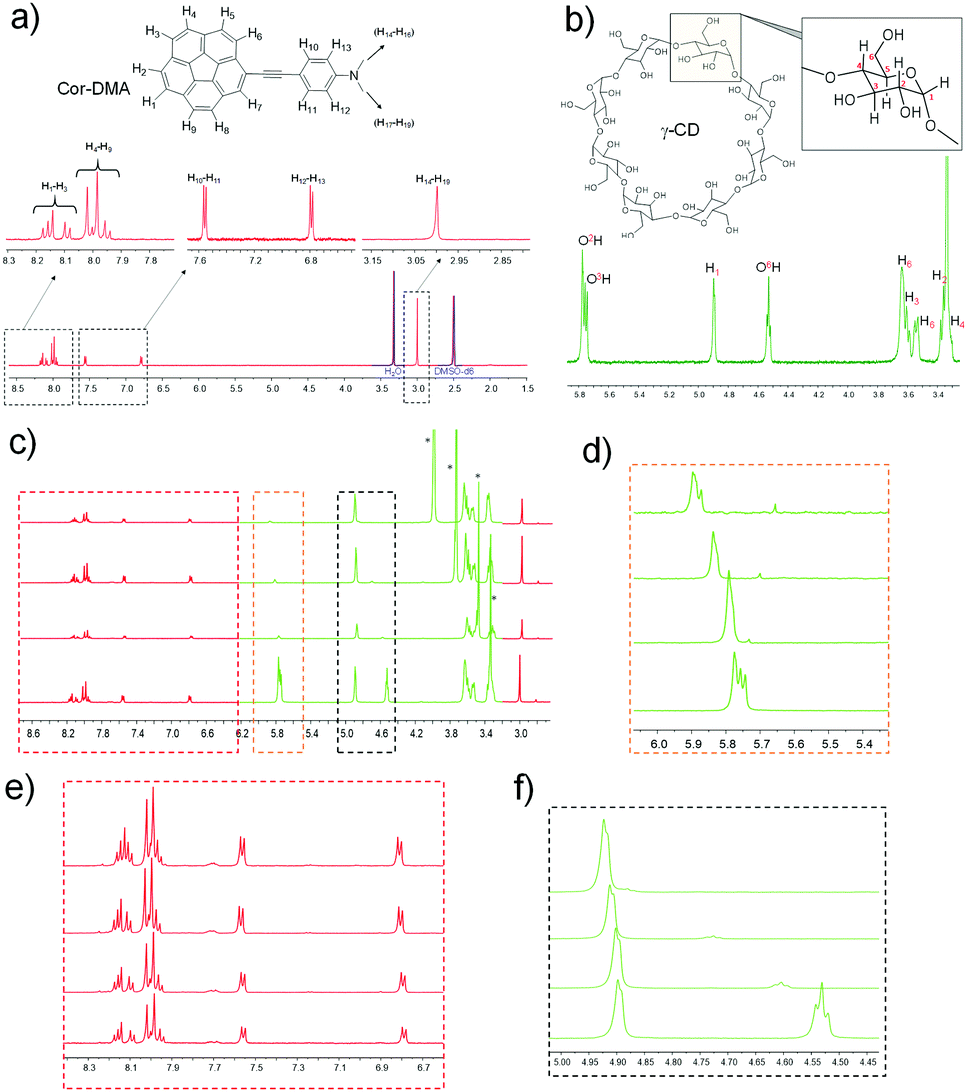 | ||
| Fig. 3 Studying supramolecular interaction using NMR spectroscopy (500 MHz, DMSO-d6). (a) 1H-NMR of Cor–DMA. (b) 1H-NMR of γ-CD. (c) Changes in the chemical shift of protons in 1H-NMR of a mixture of 1:1 Cor–DMA and γ-CD upon addition of aliquots of D2O. (d) Zoomed-in image of the orange dotted stall of 3c showing down field shift of signals of the inner surface proton of γ-CD. (e) Zoomed-in image of the red dotted stall of 3c showing merge of the two resonating signals of Cor–DMA. (f) Zoomed-in image of the black dotted stall of 3c showing down field shift of the outer surface proton of γ-CD. | ||
We observed that after addition of γ-CD, in a few minutes, the aqueous solution turns turbid and a green luminescent precipitate starts to form sediments (Fig. S4, ESI†). Transmission Electron Microscopy (TEM) was employed to examine the nature of the precipitate. This study indicated that the precipitate was composed of spherical nanoparticles with an average size of ∼25 nm (Fig. S5a, ESI†). Longer sedimentation times resulted in observation of larger size aggregates (Fig. S5b, ESI†). Dynamic light scattering (DLS) analysis also showed large aggregates with sizes ranging from 100 to 300 nm (Fig. S6, ESI†). Inspired by these observations, we anticipated that under controlling conditions such as the mixing environment, volume, and time interval for host–guest interactions, a steering of physicochemical properties of aggregates could be achieved. To investigate this possibility of controlling the morphology and size of Cor–DMA–γ-CD aggregates, we conducted inclusion complex formation studies in a microfluidic device. It was anticipated that the hydrodynamic focusing during fluid flow in a microfluidic channel would enable the formation of small sized aggregates. To test this idea, initially, a traditional “Y” junction was used with a flow rate of 2 mL per hour (Fig. S7, ESI†). Since mixing inside straight microfluidic channels relies on diffusion of fluids, it was observed that the “Y” design failed to deliver small size and narrow dispersity aggregates. Therefore, the device configuration was changed to a ‘T’ junction (Fig. 4). This device configuration resulted in the formation of low-dispersity ultra-small dots. The fabrication process also had visual cues. For example, Cor–DMA appears yellowish on its own and γ-CD is colourless. Upon mixing, however, formation of a luminescent material followed by precipitation is observed (Fig. 4).
 | ||
| Fig. 4 Design of the microchip and preparation of Cor–DMA–γ-CD dots under flow conditions. (a) Photographs of the microfluidic chip employed for the formation of nano-dots. Zoomed-in image shows microscopic variation in the channel diameter introduced for effective mixing. (b) Frame of a fluidic device under performance and illuminated with 365 nm UV light. (c) Aggregates resulted from host–guest interaction of Cor–DMA with γ-CD. (d) Transparent solution of Cor–DMA–γ-CD aggregates dispersed in water in a 10 mm cuvette and (e) green luminescence from the dispersed aggregates under 365 nm UV illumination. | ||
The luminescent solution collected from the microfluidic chamber is centrifuged to remove excess cyclodextrin and to obtain pure green luminescent dots. These nanostructures could be dispersed in water through sonication (Fig. 4). The fact that they are dispersible in water suggests that cyclodextrins are indeed creating a hydrophilic shell around the assembled nanostructure. The TEM image from this solution (Fig. 5) shows that the diffusion-mediated microfluidic mixing of Cor–DMA with γ-CD leads to the formation of ultra-small dots with a radius of 2.5–5 nm (Fig. S8, ESI†). The High Resolution-TEM (HR-TEM) image obtained by focusing on a relatively large particle of size >5 nm reveals a quasi-crystalline diffraction pattern with a lattice fringe distance of 0.23 Å (Fig. 5d). The polycrystalline diffraction pattern indicates the presence of carbon segments, corannulene, assembled together in clusters. DLS analysis of the formed dots shows a narrow size distribution with an average diameter of 10 nm (Fig. S8b, ESI†). The surface charges of the Cor–DMA–γ-CD dots were examined using ζ-potential measurements in distilled water. The Cor–DMA–γ-CD dots exhibited a surface potential of −15 ± 3 mV indicative of the presence of –OH functional groups on the surface of dots.
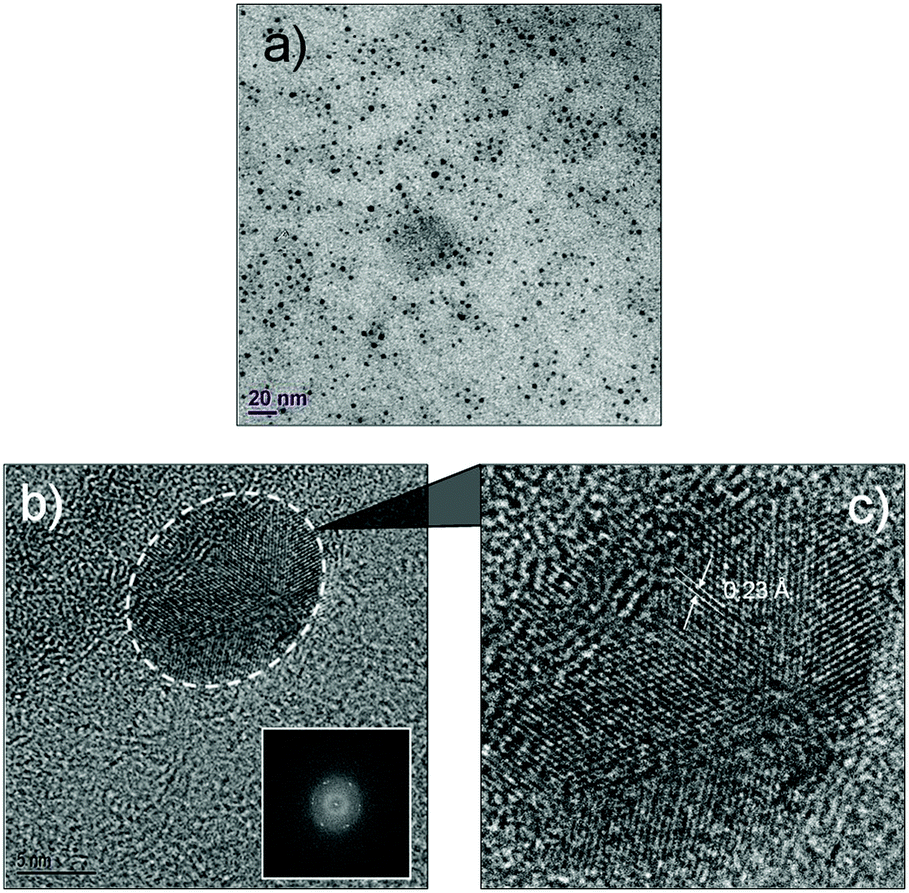 | ||
| Fig. 5 Ultra-small Cor–DMA–γ-CD dots. (a) TEM micrographs showing the uniform spherical morphology of the Cor–DMA–γ-CD dots. (b) HR-TEM images showing the morphology of a single Cor–DMA–γ-CD dot, selected area diffraction pattern (inset) and (c) a high-resolution zoomed-in image of a selected dot. | ||
The UV/Vis absorption spectrum of the Cor–DMA–γ-CD dots (1 mg mL−1 in PBS at pH 7.4) exhibits broad absorption features ranging from 300 to 600 nm with maximum absorbance at 382 nm corresponding to Cor–DMA ICT absorption (Fig. 6a). The scattering in the lower region of the spectrum (i.e. <350 nm) mainly shows the characteristics of dispersed nano-materials under aqueous conditions. The existence of Cor–DMA units in the hydrophobic environment of the nano-assembly was manifested by a ca. 10 nm blue-shifted absorption when compared to Cor–DMA absorption (382 nm) in acetonitrile.69Fig. 6b shows the photoluminescence from the Cor–DMA–γ-CD dots (1 mg mL−1) in PBS at pH 7.4 upon excitation at λexc = 380 nm. An intense and broad green emission could be observed with a maximum centered at 500 nm (Φabsolute = 20%).
 | ||
| Fig. 6 Linear optical properties of Cor–DMA–γ-CD dots. (a) UV/Vis absorption spectrum of the Cor–DMA–γ-CD dots (1 mg mL−1 in PBS buffer at pH 7.4). (b) Photoluminescence spectra of the Cor–DMA–γ-CD dots in PBS buffer at pH 7.4 (λexc = 380 nm). | ||
Isothermal Titration Calorimetry (ITC) measurements were then carried out to explore the kinetics of Cor–DMA and γ-CD interaction and to understand the assembly process (see Section S7 of the ESI† for in-depth discussion along with Fig. S9, S10 and Scheme S1). The information obtained from ITC analysis suggests that the overall reaction shows an association constant (Ka) of 4.15 × 105 M−1 ± 0.78 × 105 M−1 with exothermic and spontaneous (ΔH = −299.85 ± 19.43 kJ mol−1; ΔG = −26.34 ± 0.14 kJ mol−1) characteristics. The relatively high Ka value of Cor–DMA and γ-CD interaction indicates high stability of the assembled dot structure. Next, the solution concentration was lowered by a 10-fold dilution to allow the complexation reaction to achieve equilibrium at the molecular level and without the formation of larger structures. The change in enthalpy of this process (ΔH) was estimated to be −47.32 kJ mol−1, six times lower than the overall assembly process. Thus, it is reasonable to assume that initially a molecular level inclusion complex is formed that further assembles into a nano-dot structure.
The studies suggested that the formation of a higher ordered assembled structure is due to the amphiphilic nature of the host–guest complex formed upon combination of the two assembly precursors. In this complex, corannulene, due to its appropriate size, enters the γ-CD cavity for which the DMA segment is too small to form a stable complex. The net result of this is generation of an amphiphilic structure. In this structure, the DMA segment that remains outside of the γ-CD cavity is hydrophobic and γ-CD, now hosting the corannulene segment, is hydrophilic. Once this structure is formed, under aqueous conditions, it assembles further in such a way that γ-CD forms an outer protective layer and DMA forms an internal core. To further check if these conclusions were true, trifluoroacetic acid (TFA) is added to the solution of nano-dots. TFA is a strong acid and is known to protonate the nitrogen atom of the dialkyl aniline part of the DMA segment. Once protonated, the DMA part becomes cationic and the complex loses its amphiphilic characteristic. In this case, the assembly of well-defined dot-like structures should not occur any longer. Indeed, TEM observation from such samples showed an absence of any well-defined nano-dot structure and only random microscopic aggregation could be seen (Fig. S11, ESI†). This observation implies that the amphiphilic characteristic of the Cor–DMA–γ-CD inclusion complex is vital in ensuring further assembly into well-defined nanostructures. To further confirm this, an alternative experiment was carried out. In this experiment, TFA was introduced during the mixing process. Once again, we failed to obtain well-defined nanostructures. Therefore, based on these control experiments, and the detailed photophysical, 1H-NMR, ITC, ζ-potential and TEM studies discussed earlier, the proposed mechanism of formation of the dots (Fig. 1 and Scheme S2, ESI†) seems reasonable.
The two-photon up-conversion process in Cor–DMA (in acetonitrile) and Cor–DMA–γ-CD dots (in water) was evaluated by measuring input power intensity dependent changes upon excitation wavelength tuned from 720 to 900 nm using a femtosecond laser (Fig. 7). The linear relationship of emission intensities of Cor–DMA and Cor–DMA–γ-CD dots on the square of the corresponding excitation power confirmed the two-photon process.70 In acetonitrile, the maximum two-photon absorption (TPA) cross-section exhibited by Cor–DMA is 33 GM (Fig. 7a and b), while the Cor–DMA–γ-CD dots exhibit a TPA value of 32 GM in water (Fig. 7a and b). Cor–DMA–γ-CD dots also exhibit intense and broad two-photon excited green emission in water (Fig. 7c and d) upon excitation at 800 nm revealing that the photophysical identity of the Cor–DMA fluorophore remains unperturbed during the evolution of Cor–DMA–γ-CD dots.
 | ||
| Fig. 7 Non-linear optical properties of Cor–DMA and Cor–DMA–γ-CD dots. (a) Two-photon action spectra of Cor–DMA in acetonitrile (black) and the Cor–DMA–γ-CD dots in water (red). (b) A logarithmic plot of fluorescence output with excitation power intensity of the Cor–DMA and Cor–DMA–γ-CD dots (λex = 800 nm). Two-photon excited emission of (c) Cor–DMA (1 × 10−4 M) and (d) the Cor–DMA–γ-CD dots in water (λex = 800 nm). | ||
To investigate the bio-applicability of the prepared nano-materials, first, the cytotoxicity of Cor–DMA–γ-CD dots was evaluated using the MTT (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide) viability assay using MCF-7 cancer cell lines (Fig. S12, ESI†). The analysis was carried out by using γ-CD as a positive control and indicated that both Cor–DMA–γ-CD dots and γ-CD show no toxic effects in a low to moderate concentration range. Subsequently, cellular uptake of the Cor–DMA–γ-CD dots was established by flow cytometry using a solution concentration of 100 μg mL−1 (Fig. S13, ESI†). Encouraged by these results, the two-photon in vitro imaging capability of the Cor–DMA–γ-CD dots was examined by incubating MCF-7 cells for a period of 6 h following standardized protocols. Fig. 8 shows two-photon microscopy images and results recorded with 800 nm excitation and emission collected in the 500–600 nm window. This study revealed the good intra-cellular distribution of the Cor–DMA–γ-CD dots in the cytoplasm. The overlay image of Fig. 8a and b (Fig. 8c) and the intensity profile diagram (Fig. 8d) established the capability of the Cor–DMA–γ-CD dots as a new class of two-photon bioimaging agents.
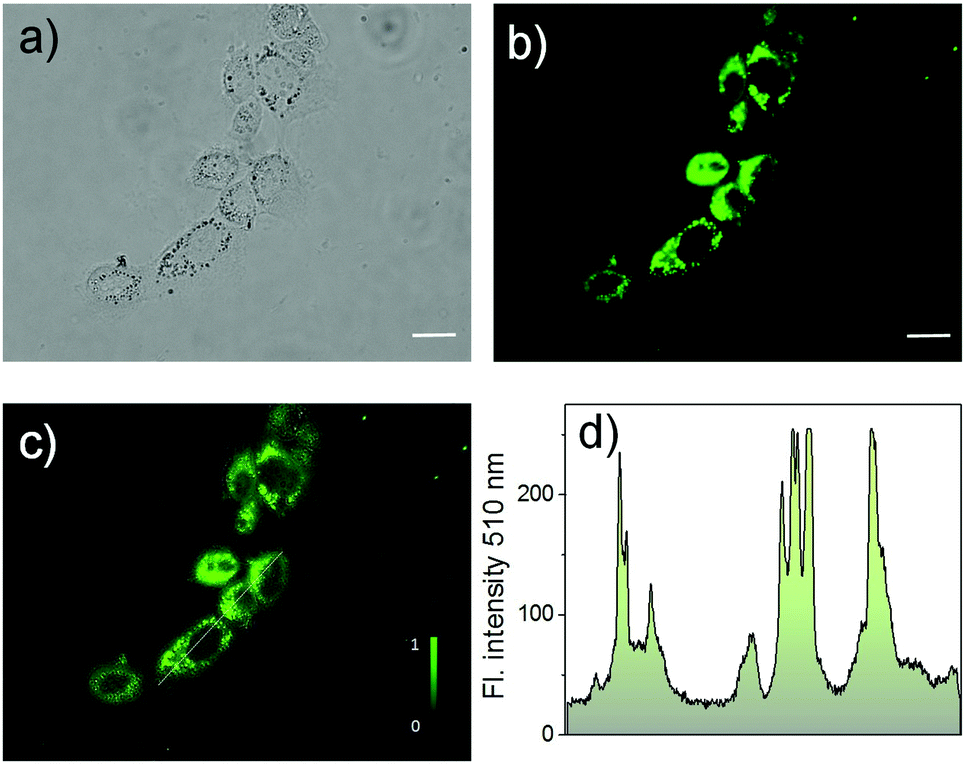 | ||
| Fig. 8 Two-photon microscopy images of the MCF-7 cell line incubated with Cor–DMA–γ-CD dots. (a) Bright field, (b) fluorescence and (c) overlay of images (a) and (b) of cell lines incubated with Cor–DMA–γ-CD dots, and (d) two-photon excited intensity profiles recorded through the lines labelled in image (c). Scale bar – 20 μm. | ||
Conclusions
In summary, two small molecules, an oligosaccharide (γ-CD) and a corannulene derivative (Cor–DMA), can develop a solvophobically driven host–guest type of partnership under chemically polar aqueous conditions resulting in ultra-small particles. The synthesis is carried out in a dynamic process using a microfluidic chamber and is scalable. The prepared nanostructures can be dispersed in water due to the presence of a γ-CD-based periphery, and are electronically active because of the donor–acceptor type of communication in the Cor–DMA molecule. The γ-CD is also the reason that these organic dots are non-toxic and biocompatible. The electronic component renders the particles bright green luminescent (500 nm, Φabsolute = 20%), and multi-photon active (TPA = 32 GM) in water. Combining all the properties, the organic dots prove to be good fluorescence imaging agents of cancer cells upon two-photon-excitation (λex = 800 nm). Clearly, there is large room for modulation of the structure, size, and as a result, properties (for example, color modulation) of the nanostructures in the present concept. It is envisaged that this concept will lead to the development of a new organic dot platform useful for bioimaging applications that is based on a non-planar polycyclic aromatic structural motif.Acknowledgements
MCS would like to thank Nanyang Technological University, Singapore for financial support under Grant No. M4081566. Y. W. and H. S. thank the Singapore Ministry of Education through the Academic Research Fund under Projects Tier 1-RG92/15. Y. K. acknowledges the research funding from the Ministry of Education of Singapore through a Tier 2 Academic Research Fund (ARC 22/13).References
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS PubMed.
- R. Weissleder and M. J. Pittet, Nature, 2008, 452, 580–589 CrossRef CAS PubMed.
- M. Dahan, S. Levi, C. Luccardini, P. Rostaing, B. Riveau and A. Triller, Science, 2003, 302, 442–445 CrossRef CAS PubMed.
- F. Pinaud, S. Clarke, A. Sittner and M. Dahan, Nat. Methods, 2010, 7, 275–285 CrossRef CAS PubMed.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS PubMed.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CAS.
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed.
- B. H. Kim, M. J. Hackett, J. Park and T. Hyeon, Chem. Mater., 2014, 26, 59–71 CrossRef CAS.
- S. Mahesh, A. Gopal, R. Thirumalai and A. Ajayaghosh, J. Am. Chem. Soc., 2012, 134, 7227–7230 CrossRef CAS PubMed.
- O. Mongin, T. R. Krishna, M. H. V. Werts, A.-M. Caminade, J.-P. Majoral and M. Blanchard-Desce, Chem. Commun., 2006, 915–917 RSC.
- O. Mongin, A. Pla-Quintana, F. Terenziani, D. Drouin, C. Le Droumaguet, A.-M. Caminade, J.-P. Majoral and M. Blanchard-Desce, New J. Chem., 2007, 31, 1354–1367 RSC.
- C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086–3109 CrossRef CAS PubMed.
- J. Pecher and S. Mecking, Chem. Rev., 2010, 110, 6260–6279 CrossRef CAS PubMed.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312–1319 CAS.
- L. C. Palmer, Y. S. Velichko, M. Olvera de la Cruz and S. I. Stupp, Philos. Trans. R. Soc., A, 2007, 365, 1417–1433 CrossRef CAS PubMed.
- S. I. Stupp, V. LeBonheur, K. Walker, L. S. Li, K. E. Huggins, M. Keser and A. Amstutz, Science, 1997, 276, 384–389 CrossRef CAS PubMed.
- J. M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS PubMed.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
- C. Tang, E. M. Lennon, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Science, 2008, 322, 429–432 CrossRef CAS PubMed.
- C. Tang, K. Sivanandan, B. C. Stahl, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, ACS Nano, 2010, 4, 285–291 CrossRef CAS PubMed.
- W. Qin, D. Ding, J. Liu, W. Z. Yuan, Y. Hu, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2011, 22, 771–779 CrossRef.
- K. Li, W. Qin, D. Ding, N. Tomczak, J. Geng, R. Liu, J. Liu, X. Zhang, H. Liu, B. Liu and B. Z. Tang, Sci. Rep., 2013, 3, 1150 Search PubMed.
- D. Ding, C. C. Goh, G. Feng, Z. Zhao, J. Liu, R. Liu, N. Tomczak, J. Geng, B. Z. Tang, L. G. Ng and B. Liu, Adv. Mater., 2013, 25, 6083–6088 CrossRef CAS PubMed.
- Y. Yao, X. Chi, Y. Zhou and F. Huang, Chem. Sci., 2014, 5, 2778–2782 RSC.
- D. J. Cram and J. M. Cram, Science, 1974, 183, 803–809 CAS.
- M. Juríček, N. L. Strutt, J. C. Barnes, A. M. Butterfield, E. J. Dale, K. K. Baldridge, J. F. Stoddart and J. S. Siegel, Nat. Chem., 2014, 6, 222–228 CrossRef PubMed.
- B. M. Schmidt, T. Osuga, T. Sawada, M. Hoshino and M. Fujita, Angew. Chem., Int. Ed., 2016, 55, 1561–1564 CrossRef CAS PubMed.
- J. Zhang and P. X. Ma, Nano Today, 2010, 5, 337–350 CrossRef CAS PubMed.
- L. T. Scott, Pure Appl. Chem., 1996, 68, 291–300 CAS.
- Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS PubMed.
- V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868–4884 CrossRef CAS PubMed.
- A. Sygula, Eur. J. Org. Chem., 2011, 1611–1625 CrossRef CAS.
- P. W. Rabideau and A. Sygula, Acc. Chem. Res., 1996, 29, 235–242 CrossRef CAS.
- A. Ayalon, M. Rabinovitz, P. C. Cheng and L. T. Scott, Angew. Chem., Int. Ed. Engl., 1992, 31, 1636–1637 CrossRef.
- D. Eisenberg, J. M. Quimby, E. A. Jackson, L. T. Scott and R. Shenhar, Chem. Commun., 2010, 46, 9010–9013 RSC.
- B. M. Schmidt, B. Topolinski, P. Roesch and D. Lentz, Chem. Commun., 2012, 48, 6520–6523 RSC.
- W. B. Fellows, A. M. Rice, D. E. Williams, E. A. Dolgopolova, A. K. Vannucci, P. J. Pellechia, M. D. Smith, J. A. Krause and N. B. Shustova, Angew. Chem., Int. Ed., 2016, 55, 2195–2199 CrossRef CAS PubMed.
- E. A. Jackson, B. D. Steinberg, M. Bancu, A. Wakamiya and L. T. Scott, J. Am. Chem. Soc., 2007, 129, 484–485 CrossRef CAS PubMed.
- B. D. Steinberg, E. A. Jackson, A. S. Filatov, A. Wakamiya, M. A. Petrukhina and L. T. Scott, J. Am. Chem. Soc., 2009, 131, 10537–10545 CrossRef CAS PubMed.
- Q. Zhang, K. Kawasumi, Y. Segawa, K. Itami and L. T. Scott, J. Am. Chem. Soc., 2012, 134, 15664–15667 CrossRef CAS PubMed.
- T. J. Seiders, K. K. Baldridge, G. H. Grube and J. S. Siegel, J. Am. Chem. Soc., 2001, 123, 517–525 CrossRef CAS PubMed.
- Y.-T. Wu, D. Bandera, R. Maag, A. Linden, K. K. Baldridge and J. S. Siegel, J. Am. Chem. Soc., 2008, 130, 10729–10739 CrossRef CAS PubMed.
- G. H. Grube, E. L. Elliott, R. J. Steffens, C. S. Jones, K. K. Baldridge and J. S. Siegel, Org. Lett., 2003, 5, 713–716 CrossRef CAS PubMed.
- A. M. Butterfield, B. Gilomen and J. S. Siegel, Org. Process Res. Dev., 2012, 16, 664–676 CrossRef CAS.
- K. Shi, T. Lei, X.-Y. Wang, J.-Y. Wang and J. Pei, Chem. Sci., 2014, 5, 1041–1045 RSC.
- R.-Q. Lu, Y.-N. Zhou, X.-Y. Yan, K. Shi, Y.-Q. Zheng, M. Luo, X.-C. Wang, J. Pei, H. Xia, L. Zoppi, K. K. Baldridge, J. S. Siegel and X.-Y. Cao, Chem. Commun., 2015, 51, 1681–1684 RSC.
- R.-Q. Lu, W. Xuan, Y.-Q. Zheng, Y.-N. Zhou, X.-Y. Yan, J.-H. Dou, R. Chen, J. Pei, W. Weng and X.-Y. Cao, RSC Adv., 2014, 4, 56749–56755 RSC.
- A. A. K. Karunathilake, C. M. Thompson, S. Perananthan, J. P. Ferraris and R. A. Smaldone, Chem. Commun., 2016, 52, 12881–12884 RSC.
- D. Miyajima, K. Tashiro, F. Araoka, H. Takezoe, J. Kim, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2009, 131, 44–45 CrossRef CAS PubMed.
- M. C. Stuparu, Angew. Chem., Int. Ed., 2013, 52, 7786–7790 CrossRef CAS PubMed.
- M. C. Stuparu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2641–2649 CrossRef CAS.
- A. Sygula, F. R. Fronczek, R. Sygula, P. W. Rabideau and M. M. Olmstead, J. Am. Chem. Soc., 2007, 129, 3842–3843 CrossRef CAS PubMed.
- J. Li, A. Terec, Y. Wang, H. Joshi, Y. Lu, H. Sun and M. C. Stuparu, J. Am. Chem. Soc., 2016, 139, 3089–3094 CrossRef PubMed.
- Y.-L. Wu, M. C. Stuparu, C. Boudon, J.-P. Gisselbrecht, W. B. Schweizer, K. K. Baldridge, J. S. Siegel and F. Diederich, J. Org. Chem., 2012, 77, 11014–11026 CrossRef CAS PubMed.
- M. T. Stone and H. L. Anderson, Chem. Commun., 2007, 2387–2389 RSC.
- D. Thomsson, R. Camacho, Y. Tian, D. Yadav, G. Sforazzini, H. L. Anderson and I. G. Scheblykin, Small, 2013, 9, 2619–2627 CrossRef CAS PubMed.
- M. J. Frampton and H. L. Anderson, Angew. Chem., Int. Ed., 2007, 46, 1028–1064 CrossRef CAS PubMed.
- J. Szejtli, Chem. Rev., 1998, 98, 1743–1753 CrossRef CAS PubMed.
- M. E. Davis and M. E. Brewster, Nat. Rev. Drug Discovery, 2004, 3, 1023–1035 CrossRef CAS PubMed.
- H. Joshi, S. Sreejith, R. Dey and M. C. Stuparu, RSC Adv., 2016, 6, 110001 RSC.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS PubMed.
- Y. El Ghoul, B. Martel, A. El Achari, C. Campagne, L. Razafimahefa and I. Vroman, Polym. J., 2010, 42, 804–811 CrossRef CAS.
- Y. Sameena and I. V. M. V. Enoch, J. Lumin., 2013, 138, 105–116 CrossRef CAS.
- A. Botsi, K. Yannakopoulou, B. Perly and E. Hadjoudis, J. Org. Chem., 1995, 60, 4017–4023 CrossRef CAS.
- T. Mori, Y. H. Ko, K. Kim and Y. Inoue, J. Org. Chem., 2006, 71, 3232–3247 CrossRef CAS PubMed.
- R. Dondon and S. Fery-Forgues, J. Phys. Chem. B, 2001, 105, 10715–10722 CrossRef CAS.
- Y. Wang, V. D. Ta, Y. Gao, T. C. He, R. Chen, E. Mutlugun, H. V. Demir and H. D. Sun, Adv. Mater., 2014, 26, 2954–2961 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00063d |
| This journal is © the Partner Organisations 2017 |