
A sulfur–eugenol allyl ether copolymer: a material synthesized via inverse vulcanization from renewable resources and its application in Li–S batteries†
Alexander
Hoefling‡
a,
Dan Thien
Nguyen‡
b,
Young Joo
Lee
c,
Seung-Wan
Song
b and
Patrick
Theato
 *a
*a
aInstitute for Technical and Macromolecular Chemistry, University of Hamburg, Bundesstr. 45, 20146 Hamburg, Germany. E-mail: theato@chemie.uni-hamburg.de
bDepartment of Chemical Engineering and Applied Chemistry, Chungnam National University, Daejeon, 305-764, Republic of Korea
cInstitute of Inorganic and Applied Chemistry, University of Hamburg, Martin-Luther-King-Platz 6, 20146 Hamburg, Germany
First published on 24th April 2017
Abstract
The demand for eco-friendly and renewable resources has dramatically increased in scientific and technological areas including energy storage systems and production of new functional materials. Sulfur copolymers with high sulfur contents of up to 90 wt% were prepared via inverse vulcanization from environment-friendly, sustainable raw materials: cost-effective waste-product elemental sulfur and eugenol allyl ether (EAE), which is obtained from clove oil. The thermal properties and electrochemical activities of the resulting poly(S-co-EAE) materials can be tuned by controlling the EAE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S feed ratio. Employed as a cathode material in Li–S batteries, the copolymer with 90 wt% sulfur content provides good cycling stability at a capacity of ∼650 mA h g−1 and high Coulombic efficiencies (>99%) over 100 cycles.
:S feed ratio. Employed as a cathode material in Li–S batteries, the copolymer with 90 wt% sulfur content provides good cycling stability at a capacity of ∼650 mA h g−1 and high Coulombic efficiencies (>99%) over 100 cycles.
Introduction
Despite its long-known use in the vulcanization of polydienes, further utilization of elemental sulfur in materials science has rarely been reported. Nowadays, some 70 million tons of elemental sulfur are produced annually, mostly as a byproduct from oil refinery, natural-gas processing or coking via the Claus process.1 Recent developments including increased demand for fuels and exploitation of sulfur-rich resources led to a surplus production of roughly 7 million tons of elemental sulfur per year.2 Thus, the vast availability of sulfur at low costs motivates the use of elemental sulfur as a feedstock for sustainable, cost-effective and functional materials.Sulfur-based copolymeric materials have emerged in the past three years, pioneered by the group of Pyun.3 Initially, Pyun and coworkers converted elemental sulfur with 1,3-diisopropenylbenzene (DIB) by simple mixing at temperatures up to 185 °C, yielding hyperbranched copolymers with sulfur contents of up to 95 wt%.3,4 The conversion of elemental sulfur with unsaturated organic comonomers leading to chemically stable polymeric materials with high sulfur contents (>50 wt%) was termed “inverse vulcanization”. Consequently, this simple, economic and solvent-free synthesis protocol was successfully extended to further comonomers such as diethynylbenzene (DEB),5 1,4-diphenylbutadiyne6 or benzoxazines.7,8 The obtained sulfur copolymers found uses as cathode materials in lithium sulfur (Li–S) batteries,6,9 as self-healable optical lenses,10 as electrolytes in dye-sensitized solar cells11 or as mercury absorbers.12
Li–S batteries are of great interest in meeting the growing demand for high capacity energy storage systems owing to their low toxicity, low cost and vast abundance of active materials. In addition, compared to conventional Li-ion batteries with heavy metal oxides as cathode materials, Li–S batteries provide ten times higher theoretical capacity (1670 mA h g−1) and five times higher specific energy density (∼2570 W h kg−1).13,14 However, Li–S batteries still suffer from short cycle lifetimes, self-discharge and limited rate capability.13–15 Various mechanisms responsible for these obstacles have been suggested including dissolution of long-chain polysulfides and the low conductivity of sulfur. Among the vast approaches to tackle these problems, sulfur copolymers have drawn significant attention recently due to their simple synthetic methods, demonstrating improved cycling stability and high specific capacities of Li–S batteries.16–19 Presumably, this enhanced cyclability results from the enhanced compatibility with conductive carbon and better mechanical stability of the resulting cathode materials; however, the exact mechanism contributing to the improved cycling performance is not well understood.20
Despite the advantage of facile synthesis, preparation of sulfur copolymers still requires comonomers often derived from the petrochemical industry. Thus, aiming for the employment of abundant organic substances from sustainable resources as comonomers in the inverse vulcanization method, we encountered earlier that elemental sulfur and vegetable oils formed composite materials with enhanced performance as cathode active materials in Li–S batteries.21 Various vegetable oils consisting of different types and amounts of unsaturated fatty acids have been investigated, providing an initial capacity of 880 mA h g−1 and a capacity retention of 63% after 100 cycles. However, copolymers with a sulfur content exceeding 80 wt% could not be achieved due to the insolubility of vegetable oils in molten sulfur. In contrast, phenylpropanoids like eugenol provide better compatibility with liquid sulfur due to their aryl moieties, allowing for facile control of homogeneity and viscosity. Moreover, the presence of oxygen can offer an additional functionality, such as chemical adsorption sites which can help in immobilizing polysulfide and preventing dissolution of polysulfide.22,23 Eugenol is non-toxic and widely available in nature, majorly found in clove oil from Syzygium aromaticum and in different essential oils.24 It is well-known as a spice, and a precursor for synthetic vanillin production and is used in dental medicine since the 19th century due to its antimicrobial activity. In polymer science, it has found application as polyeugenol25 or methacrylate.26
Herein, we present a novel sulfur copolymeric material from entirely sustainable resources prepared by inverse vulcanization of abundant, low cost by-product elemental sulfur and eugenol allyl ether obtained from renewable resources. The resulting copolymeric materials, poly(S-co-EAE), are then successfully employed as cathode active materials in Li–S batteries.
Experimental
Synthesis of poly(S-co-EAE)
Elemental sulfur and eugenol allyl ether were combined in a 20 mL glass vial equipped with a magnetic stirring bar on a 2 g scale. Under vigorous stirring, the mixture was heated at 150 °C in an oil bath for one hour, while the color changed from yellow to dark red. In addition, the viscosity of the mixture increased and stirring stopped. After the gel point passed, the glass vial was transferred into a second oil bath heated to 180 °C and kept there for 10 minutes in order to ensure complete conversion. The resulting dark red glassy materials with EAE contents ranging from 10 to 50 wt% were insoluble in common organic solvents.Electrochemical characterization
Poly(S-co-EAE) cathodes were prepared by casting a slurry, composed of poly(S-co-EAE) active material (70 wt%), carbon black (super-P, 15 wt%), carbon fibers (CF, Aldrich, 5%) and a binder (10 wt%), onto aluminum foil. The binder consisted of sodium carboxymethyl cellulose (CMC, Mw 250000 g mol−1, Sigma-Aldrich) and poly(acrylic acid) (PAA, Mw 450000 g mol−1, Sigma-Aldrich) in a 1 to 1 weight ratio. The cathodes with a sulfur loading of roughly 0.75 mg cm−2 were vacuum-dried at 45 °C for 24 h. In an argon-filled glove box (M.O. Tek) the cathodes were assembled into 3-electrode cells or CR 2032 coin cells against lithium metal, a separator (Celgard C210) and a solution of 2 M lithium bis(trifluoromethane)sulfonamide (LiTFSI, Sigma-Aldrich) and 0.31 M lithium nitrate (LiNO3, Sigma-Aldrich) in 1,3-dioxolane (DOL, Sigma-Aldrich) and 1,2-dimethoxy ethane (DME, Sigma-Aldrich) in a 1 to 1 volume ratio, as an electrolyte. The optimized electrolyte/sulfur ratio of 25 μL mg−1 was used. Cyclic voltammetry (CV) was carried out with 3-electrode cells that consisted of poly(S-co-EAE) as a working electrode and lithium metal as reference and counter electrodes, at a scan rate of 20 μV s−1 between 1.5 and 2.8 V vs. Li/Li+, using a potentiostat (VPS SP-150, Bio-Logic). The current density of CVs was normalized to the weight of sulfur. The electrochemical charge–discharge cycling ability of poly(S-co-EAE) cathodes was tested in CR 2032 coin cells at a constant current of 0.1 C (167 mA g−1) in the voltage window of 1.5–2.6 V. The specific gravimetric capacities of all cathodes were calculated based on the weight of sulfur.
Results and discussion
First of all, the conversion of elemental sulfur and eugenol in an inverse vulcanization approach was investigated. The starting materials were simply mixed and heated at 175 °C for 20 min, gradually yielding a single red-colored, transparent and homogenous phase (detailed synthesis is described in the ESI†). Successful conversion of the terminal double bonds was verified by NMR, FT-IR (ESI,† Fig. S1 and S2) and TGA measurements (ESI,† Fig. S3), suggesting quantitative conversion of eugenol. However, elemental sulfur started to recrystallize from the cooled melt after 24 h storage for the composites with a low eugenol content (relative weight percentage of eugenol ≤ 30%), which can be confirmed by sharp melting peaks between 100 and 120 °C in DSC measurements (ESI,† Fig. S4). Additionally, SEC experiments confirmed that only low-molecular mass products were obtained (ESI,† Fig. S5), accounting for the good solubility of the reaction products in common organic solvents (ESI,† Fig. S6). In conclusion, it appears that the lack of a second functionality for the addition of polymeric sulfur chains prevents the formation of a hyperbranched copolymeric material and is unable to permanently stabilize the polysulfide chains.Thus, eugenol allyl ether (EAE) exhibiting two unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds was designed for the inverse vulcanization with elemental sulfur to obtain stable sulfur copolymers with a high sulfur content. EAE was prepared from sustainable eugenol in a scalable, one-step Williamson ether synthesis that yielded a colorless oil in 58% yield. The conversion of EAE with elemental sulfur resulted in red to dark colored, polymeric materials that were insoluble in common organic solvents (Fig. 1). Copolymers containing various ratios of sulfur to EAE, with sulfur contents ranging from 50 wt% to 90 wt% and EAE contents from 50 wt% to 10 wt% correspondingly, were prepared, and the materials are denoted as S–EAE-X, with X corresponding to the relative amount of EAE in weight percentage. The contents of sulfur and EAE were verified by elemental analysis (ESI,† Table S1).
C double bonds was designed for the inverse vulcanization with elemental sulfur to obtain stable sulfur copolymers with a high sulfur content. EAE was prepared from sustainable eugenol in a scalable, one-step Williamson ether synthesis that yielded a colorless oil in 58% yield. The conversion of EAE with elemental sulfur resulted in red to dark colored, polymeric materials that were insoluble in common organic solvents (Fig. 1). Copolymers containing various ratios of sulfur to EAE, with sulfur contents ranging from 50 wt% to 90 wt% and EAE contents from 50 wt% to 10 wt% correspondingly, were prepared, and the materials are denoted as S–EAE-X, with X corresponding to the relative amount of EAE in weight percentage. The contents of sulfur and EAE were verified by elemental analysis (ESI,† Table S1).
 | ||
| Fig. 1 Top: Schematic representation of the synthesis of EAE and poly(S-co-EAE); bottom: photographs of poly(S-co-EAE) with the EAE content denoted as X in wt%. | ||
For all the materials, conversion of the allylic CC double bonds was confirmed by Fourier transform infrared spectroscopy (FT-IR). In comparison to EAE, the signal at 1638 cm−1 representing stretching vibrations of allylic CC double bonds completely disappeared in FT-IR spectra of poly(S-co-EAE), thus indicating successful consumption of the CC double bonds during the inverse vulcanization step (Fig. 2a). In addition, a dramatic decrease of the signals at 3079 cm−1, 993 cm−1 and 914 cm−1, attributed to the stretching and deformation vibrations of vinylic C–H bonds, was observed. Furthermore, changes in the spectral region between 2800 and 3000 cm−1 indicate alterations in the chemical environment of C–H bonds. Solid state 13C-NMR experiments of poly(S-co-EAE) revealed additional signals with a shift from 30 to 60 ppm compared to EAE, which can be attributed to carbon atoms bearing polysulfide chains, confirming successful conversion and formation of C–S linkage (ESI,† Fig. S7). The presence of another signal at 20 ppm suggests the formation of methyl groups in the molecular structure of poly(S-co-EAE), probably due to hydrogen abstraction by intermediately formed EAE radicals and the concomitant formation of methyl end groups.27
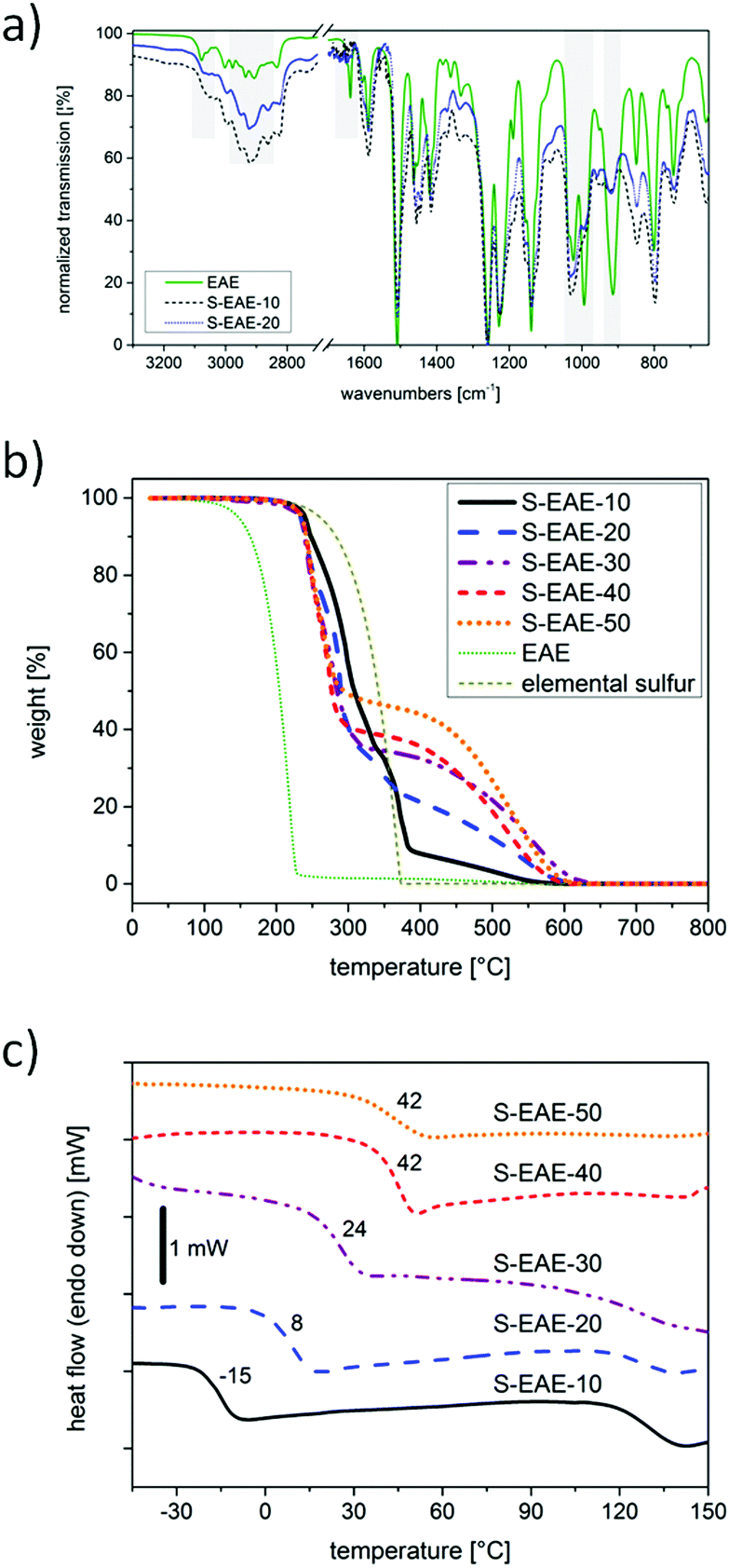 | ||
| Fig. 2 (a) FTIR spectra of pure EAE and poly(S-co-EAE); (b) thermogravimetric analysis of poly(S-co-EAE) and starting materials, measured under an air flow of 20 mL min−1 at a scan rate of 10 K min−1; and (c) DSC thermograms of poly(S-co-EAE) at a scan rate of 10 K min−1. The samples are denoted as S–EAE-X, with X = wt% of EAE. Tg (°C) of each material is denoted in the DSC figure. | ||
Thermogravimetric analysis of poly(S-co-EAE) materials showed that decomposition was characterized by two steps, the first weight loss at ∼230 °C and the second gradual weight loss between 400 and 600 °C (Fig. 2b). The weight loss in the first step is well correlated with the sulfur content and that of the second step with the employed amount of EAE, indicating that the feed ratios were retained in the final copolymer. An influence of the EAE content on the thermal properties of poly(S-co-EAE) could also be observed in DSC thermograms (Fig. 2c). The amorphous copolymers exhibited glass transition temperatures (Tgs) ranging from −15 °C to 42 °C, that increase linearly with an increase of the EAE feed ratio in the range of 10 to 40 wt% (ESI,† Fig. S8). This demonstrates that the thermomechanical properties of poly(S-co-EAE) materials can be precisely tuned within the aforementioned limits. In contrast to the composite materials, resulting from inverse vulcanization of elemental sulfur and eugenol (S–Eg, ESI,† Fig. S4), no transition corresponding to the sharp melting transition of sulfur was observed, indicating complete conversion and stabilization of sulfur in poly(S-co-EAE) materials. Another broad phase transition was observed around 125 °C, which became more pronounced with increasing sulfur content. Recently, it has been reported that self-healing of sulfur copolymer materials can be induced by thermal treatment above 90 °C through S–S bond rearrangement.10 Thus, this broad transition at approximately 125 °C can be attributed to the enhanced dynamic bond character of S–S bonds. In particular, for sulfur copolymers with high sulfur contents, this dynamic bond character appears to be more pronounced due to the large number of polysulfide bonds. Further study of the self-healing properties and their effect on battery design will be addressed in the future.28
Further structural analysis was necessary to obtain a thorough understanding of the materials. Even though poly(S-co-EAE) with high sulfur contents are insoluble due to the high amount of polysulfide chains, a soluble sample could be prepared with a low sulfur content of 24 wt% (S–EAE-76, molar ratio of EAE:elemental sulfur = 1:2), enabling characterization with solution-based techniques. The 1H-NMR spectrum verified the conversion of the CC double bonds and formation of C–S bonds via the appearance of different broad signals in the aliphatic region (2.5–4.4 ppm) and the decrease of the signals representing vinylic protons (5.9 and 6.0 ppm) (ESI,† Fig. S9a). SEC revealed that the prepared polymers had a molecular weight of Mw = 12000 g mol−1 (ESI,† Fig. S10). This is in contrast to the soluble products that were formed by the reaction of elemental sulfur with eugenol, which exhibited only low molecular weight components with Mw < 1000 g mol−1 (ESI,† Fig. S5). Very recently, it was reported that only low molecular weight polysulfides can be synthesized from renewable resources, a sulfur–limonene mixture.29 Our results show that high-molecular weight copolymeric materials with high sulfur contents up to 90 wt% can be prepared via inverse vulcanization of waste-product elemental sulfur and EAE from renewable resources. The molecular weight and the number of sulfur atoms in the polysulfide chains depend on the relative amount of sulfur and the organic linker group. Likely, low EAE feed ratios (high sulfur) result in macrocyclic structures containing high-order sulfur chains with only a few branches. An image of the microstructure of poly(S-co-EAE) correlated with the sulfur content is depicted in ESI,† Fig. S11.
The applicability of poly(S-co-EAE) as a cathode in Li–S cells has been evaluated in preliminary studies. After quick testing of the poly(S-co-EAE) copolymers with various ratios of EAE to sulfur (ESI,† Fig. S12), it was determined that the S–EAE-10 cathode yielded higher capacities and a more stable cycling ability than others. To investigate the electrochemical activity of the S–EAE-10 cathode, cyclic voltammetry (CV) was conducted, as shown in Fig. 3a. During the 1st cycle, two cathodic peaks at 2.27 and 2.05 V were observed, which are typical for the stepwise reduction of elemental sulfur in liquid electrolyte Li–S batteries.14 The peak current at 2.27 V was attributed to the reduction of S–EAE-10, yielding high-order inorganic polysulfides (Li2Sx, x = 6–8) and high-order organic polysulfides which are bonded to EAE (C–Sx–Li, x = 6–8) (ESI,† Fig. S13). At around 2.05 V the high-order polysulfides were further reduced to low-order inorganic polysulfides (Li2Sx, x = 1–3) and low order organic polysulfides (C–Sx–Li, x = 1–3).9,30–33 In the reverse process, the broad anodic peak near 2.43 V was attributed to the oxidation of the low-order lithium polysulfides to high-order lithium polysulfides and eventually to sulfur,34,35 as suggested by the reverse reactions in the ESI,† Fig. S13. Well maintained cathodic and anodic peaks throughout the three cycles indicated thorough reversibility of the electrochemical reaction. The potential difference between the cathodic and anodic peaks decreased with cycling, indicating an improvement of the reaction reversibility. The electrochemical discharge–charge cycling performance of the S–EAE-10 cathode was studied as shown in Fig. 3b. Starting with a high specific capacity of 870 mA h g−1, quite a steep loss was observed during the first 20 cycles. This initial loss was attributable to the irreversible loss of soluble polysulfide species, and the deposition of polysulfides and decomposed electrolyte components on the lithium metal anode and/or S–EAE-10 cathode, as often observed on elemental sulfur.13,15,36 However, after 20 cycles, the discharge capacity was well retained in the range between 700 and 620 mA h g−1. The overall capacity retention during 100 cycles was 64%. After the 2nd cycle, the Coulombic efficiency reached 99% and was maintained over 98 cycles. Challenges for improving the cycling performance through the optimization of several critical factors such as coating conditions, electrolyte, separator, etc., and mechanistic studies remain and would be reported in the forthcoming paper.
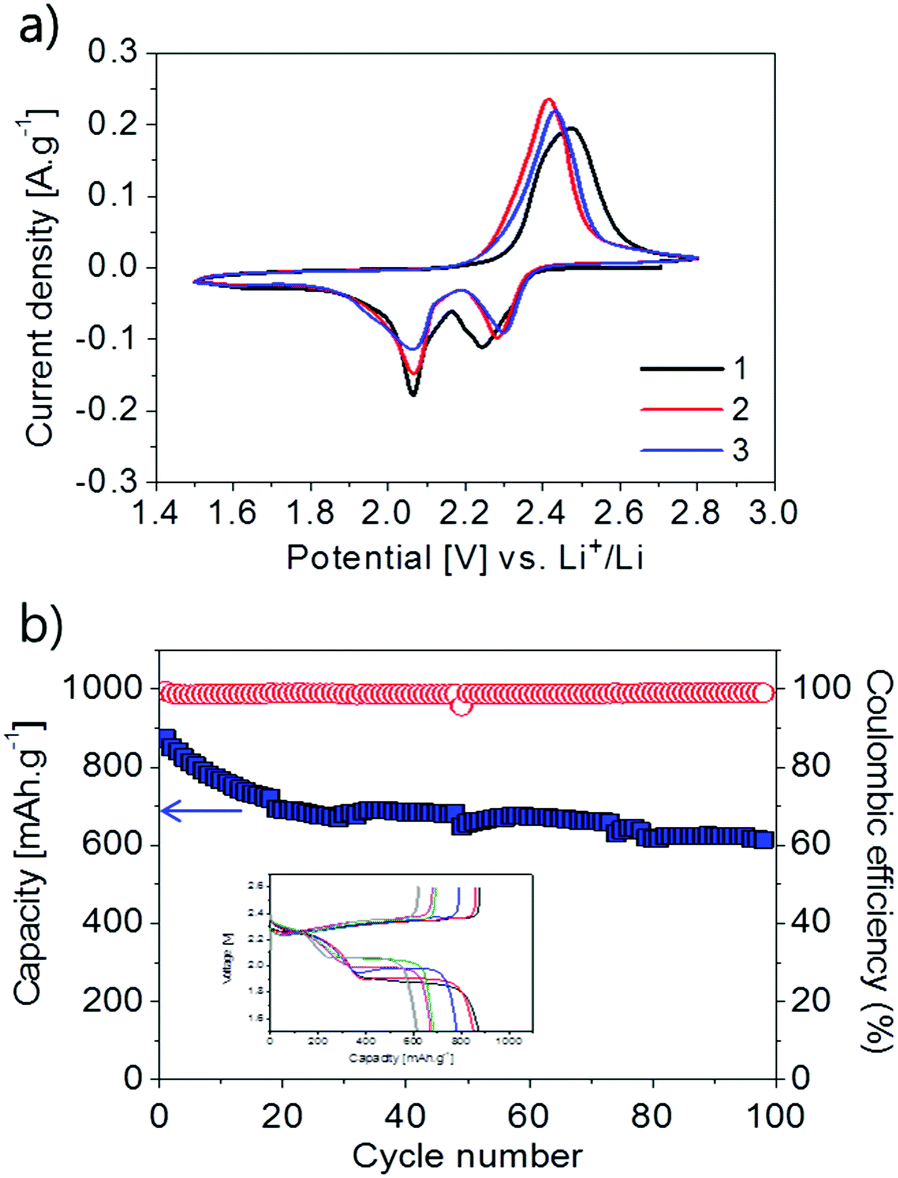 | ||
| Fig. 3 (a) Cyclic voltammograms of the S–EAE-10 cathode at a scan rate of 20 μV s−1; and (b) cycling performance of the S–EAE-10 cathode at 0.1C. | ||
Conclusions
Our study demonstrates that sulfur copolymeric materials can be synthesized with different molecular weights and tunable thermal properties entirely from renewable resources through a simple inverse vulcanization protocol. These materials are electrochemically active in Li–S cells, which can contribute to the development of sustainable, cost-effective, high capacity active materials for Li–S batteries. Furthermore, due to their processabilities, high sulfur content and low costs, these sulfur-copolymers can be used in versatile applications.Acknowledgements
The work was supported under the framework of international cooperation program managed by the National Research Foundation of Korea (2015K2A5A3000068) and the German Academic Exchange Service (57141898).References
- A. F. Hollemann, E. Wiberg and N. Wiberg, Lehrbuch der Anorganischen Chemie, de Gruyter, Berlin, 102nd edn, 2007 Search PubMed.
- J. Peacock, Sulphur Market Outlook, London, 2009 Search PubMed.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, Á. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518 CrossRef CAS PubMed.
- J. J. Griebel, R. S. Glass, K. Char and J. Pyun, Prog. Polym. Sci., 2016, 58, 90 CrossRef CAS.
- Z. Sun, M. Xiao, S. Wang, D. Han, S. Song, G. Chen and Y. Meng, J. Mater. Chem. A, 2014, 2, 9280 CAS.
- P. T. Dirlam, A. G. Simmonds, T. S. Kleine, N. a. Nguyen, L. E. Anderson, A. O. Klever, A. Florian, P. J. Costanzo, P. Theato, M. E. Mackay, R. S. Glass, K. Char and J. Pyun, RSC Adv., 2015, 5, 24718 RSC.
- M. Arslan, B. Kiskan and Y. Yagci, Macromolecules, 2016, 49, 767 CrossRef CAS.
- S. Shukla, A. Ghosh, U. K. Sen, P. K. Roy, S. Mitra and B. Lochab, ChemistrySelect, 2016, 1, 594 CrossRef CAS.
- A. G. Simmonds, J. J. Griebel, J. Park, K. R. Kim, W. J. Chung, V. P. Oleshko, J. Kim, E. T. Kim, R. S. Glass, C. L. Soles, Y.-E. Sung, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 229 CrossRef CAS.
- J. J. Griebel, N. A. Nguyen, S. Namnabat, L. E. Anderson, R. S. Glass, R. A. Norwood, M. E. Mackay, K. Char and J. Pyun, ACS Macro Lett., 2015, 4, 862 CrossRef CAS.
- P. Liu, J. M. Gardner and L. Kloo, Chem. Commun., 2015, 51, 14660 RSC.
- M. Thielke, L. Bultema, D. Brauer, B. Richter, M. Fischer and P. Theato, Polymers, 2016, 8, 266 CrossRef.
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186 CrossRef CAS PubMed.
- S. S. Zhang, J. Power Sources, 2013, 231, 153 CrossRef CAS.
- A. Manthiram, Y. Fu, S. Chung, C. Zu and Y. Su, Chem. Rev., 2014, 114, 11751 CrossRef CAS PubMed.
- A. Ghosh, S. Shukla, G. S. Khosla, B. Lochab and S. Mitra, Sci. Rep., 2016, 6, 25207 CrossRef CAS PubMed.
- H. Kim, J. Lee, H. Ahn, O. Kim and M. J. Park, Nat. Commun., 2015, 6, 7278 CrossRef CAS PubMed.
- I. Gomez, D. Mecerreyes, J. A. Blazquez, O. Leonet, H. Ben Youcef, C. Li, J. L. Gómez-Cámer, O. Bundarchuk and L. Rodriguez-Martinez, J. Power Sources, 2016, 329, 72 CrossRef CAS.
- B. Ding, Z. Chang, G. Xu, P. Nie, J. Wang, J. Pan, H. Dou and X. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 11165 CAS.
- V. P. Oleshko, J. Kim, J. L. Schaefer, S. D. Hudson, C. L. Soles, A. G. Simmonds, J. J. Griebel, R. S. Glass, K. Char and J. Pyun, MRS Commun., 2015, 5, 353 CrossRef CAS.
- A. Hoefling, Y. J. Lee and P. Theato, Macromol. Chem. Phys., 2017, 218, 1600303 CrossRef.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500 CrossRef CAS PubMed.
- A. Gorkovenko, T. A. Skotheim, L. Boguslavsky, Z. Deng and S. P. Mukherjee, WO9933131, 1999.
- M. S. Brewer, Compr. Rev. Food Sci. Food Saf., 2011, 10, 221 CrossRef CAS.
- M. C. Djunaidi, J. Jumina, D. Siswanta and M. Ulbricht, Asian J. Chem., 2015, 27, 4553 CrossRef CAS.
- L. Rojo, B. Vazquez, J. Parra, A. L. Bravo, S. Deb and J. San Roman, Biomacromolecules, 2006, 7, 2751 CrossRef CAS PubMed.
- L. B. Blight, B. R. Currell, B. J. Nash, T. M. Scott and C. Stillo, Br. Polym. J., 1980, 12, 5 CrossRef CAS.
- Y. Zhao, Y. Zhang, H. Sun, X. Dong, J. Cao, L. Wang, Y. Xu, J. Ren, Y. Hwang, I. H. Son, X. Huang, Y. Wang and H. Peng, Angew. Chem., Int. Ed., 2016, 55, 14384 CrossRef CAS PubMed.
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Angew. Chem., Int. Ed., 2016, 55, 1714 CrossRef CAS PubMed.
- C. Barchasz, F. Molton, C. Duboc, J. C. Leprêtre, S. Patoux and F. Alloin, Anal. Chem., 2012, 84, 3973 CrossRef CAS PubMed.
- H. Yamin, A. Gorenshtein, J. Penciner, Y. Sternberg and E. Peled, J. Electrochem. Soc., 1988, 135, 1045 CrossRef CAS.
- E. Peled, Y. Sternberg, A. Gorenshtein and Y. Lavi, J. Electrochem. Soc., 1989, 136, 1621 CrossRef CAS.
- Y. S. Su, Y. Fu, T. Cochell and A. Manthiram, Nat. Commun., 2013, 4, 2985 Search PubMed.
- C. Zu, M. Klein and A. Manthiram, J. Phys. Chem. Lett., 2014, 5, 3986 CrossRef CAS PubMed.
- H. L. Wu, L. A. Huff and A. A. Gewirth, ACS Appl. Mater. Interfaces, 2015, 7, 1709 CAS.
- R. Xu, J. Lu and K. Amine, Adv. Energy Mater., 2015, 5, 1 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00083a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2017 |