
Control of the electrochemical and photophysical properties of N-substituted benzo[ghi]perylene derivatives†
Kokichi
Tokuo
a,
Hayato
Sakai
a,
Tomo
Sakanoue
b,
Taishi
Takenobu
b,
Yasuyuki
Araki
 c,
Takehiko
Wada
c and
Taku
Hasobe
*a
c,
Takehiko
Wada
c and
Taku
Hasobe
*a
aDepartment of Chemistry, Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Yokohama, Kanagawa 223-8522, Japan. E-mail: hasobe@chem.keio.ac.jp
bDepartment of Applied Physics, Nagoya University, Chikusa, Nagoya 464-8603, Japan
cInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan
First published on 15th August 2017
Abstract
In this work, we report the synthesis, and electrochemical and photophysical properties of a series of N-substituted benzo[ghi]perylene (BP) derivatives. The orientation of the lone-pair of electrons toward the aromatic core is expected to have a great effect on the electronic structures and energy levels. Namely, the introduction of electron-withdrawing nitrogen atoms and imide groups stabilized the energy levels of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) in 7,8-diazabenzo[ghi]perylene (DABP) and 7,8-diazabenzo[ghi]peryleneimide (DABPIm) because of the parallel orientation to the aromatic ring plane. This is in sharp contrast with the decreased trend of the HOMO in 1,2-diazonia-7,8-diazabenzo[ghi]peryleneimide (TABPIm). Namely, the nitrogen lone-pair of electrons in the 7 and 8 positions of TABPIm, which are perpendicular to the ring plane (i.e., part of π-system), induced an increased HOMO level. These results are successfully explained by DFT calculations and agree well with the spectroscopic and electrochemical results. With regard to the excited-state dynamics of these derivatives, the introduction of nitrogen atoms and/or an imide unit onto the BP core enables control of the rate constants of both the fluorescence and intersystem crossing (ISC) pathways, which significantly affects the corresponding quantum yields. The quantum yields of fluorescence (ΦFL) decreased with the introduction of nitrogen atoms, whereas an increasing trend of ΦFL was observed with substitution of an imide unit.
Introduction
In recent years, increasing attention has been paid to disk-shaped organic molecules such as polycyclic aromatic hydrocarbons (PAHs) [e.g., perylene, benzo[ghi]perylene (BP) and coronene] because of their characteristic structural and electronic properties for extended π-conjugation.1–10 Since these molecules possess a variety of potential properties, such as efficient absorption and emission in the UV/visible region and high electron density, they are promising semiconductor materials for organic electronics.11–23 Moreover, by introducing new functional substituents onto the peripheral positions of an aromatic core, which induces additional properties (spectroscopic and electrochemical properties) and intermolecular interactions (van der Waals interaction, hydrogen and coordination bonds), highly organized superstructures with specific functionalities can be designed and constructed.4,24–30Although the syntheses and properties of p-type PAHs have been widely reported, development of n-type PAHs has lagged behind that of p-type molecules regardless of recent excellent progress. A general strategy for the synthesis of n-type organic semiconductors is to introduce electron-accepting substituents onto the peripheral positions of typical p-type molecules. The introduction of strong electron-accepting substituents such as imide and cyano groups onto the peripheral positions of a PAH aromatic core typically enables us to control the electrochemical property and excited-state dynamics.31–37 For example, perylenediimides (PDIs) are good electron acceptors with extremely high fluorescence quantum yields. Therefore, they are widely used as electron acceptors and electron transporting materials in the reaction centre models of artificial photosynthesis15,38–46 and organic electronic devices.15,39,47,48 However, as compared to PDI derivatives, the number of examples on the photophysical and electrochemical properties of BP derivatives is extremely limited.49,50
Along with this concept, the heterocyclic PAHs that contain nitrogen,51–53 sulfur11,54,55 and oxygen56 atoms in their aromatic skeletons also have received increasing attention because they combine the unique electronic structure of the smallest benzenoid unit and the outstanding electronic properties of heteroatoms. In particular, the orientation of the lone-pair of electrons toward the aromatic core may play an important role in the electronic structures and energy levels.57,58 However, the systematic substituent effects of BP derivatives including nitrogen-containing heteroring and electron-accepting substituents on the electrochemical properties and excited-state dynamics have yet to be explored.
The photophysical and photochemical properties of phenanthrene, 1,10-phenanthroline and related compounds have been previously reported to examine the nitrogen-substitution effects of the aromatic cores on the excited-state dynamics.59,60 In contrast to the fluorescence quantum yield (ΦFL) of phenanthrene (0.13),61 the ΦFL of 1,10-phenanthroline is close to zero after introducing nitrogen atoms onto the phenanthrene skeleton.60 However, little attention has been paid toward the systematic syntheses and evaluations of the photophysical and electrochemical properties of N- and imide-substituted PAH derivatives. Scheme 1 shows the reported chemical structures of perylene derivatives, especially focusing on the orientation of the lone-pair of electrons of the nitrogen atom toward the aromatic core. The first one-electron oxidation potentials (Eox1) of DAPery,62 Pery62 and Pery-NH57 are reported to be 1.55 V, 1.00 V and 0.93 V vs. SCE, respectively. These results clearly indicate that the orientation of the lone-pair of electrons of the nitrogen atom has an effect on the energies and molecular orbitals of the HOMO and LUMO states.
 | ||
| Scheme 1 Reported chemical structures of N-substituted perylene derivatives. | ||
Based on the above considerations, herein we demonstrate the synthesis, and electrochemical and photophysical properties of N-substituted benzo[ghi]perylene: 7,8-diazabenzo[ghi]perylene (DABP) derivatives. It should be noted that these compound names, especially associated with nitrogen positions onto a BP core, are simply defined for clear understanding considering chemical structures of all compounds in this study. By systematically introducing nitrogen-containing heteroring and imide groups onto benzo[ghi]perylene (BP) cores, we have successfully controlled the electrochemical properties and excited-state dynamics of these derivatives (Scheme 2). In particular, we employed 7,8-diazabenzo[ghi]peryleneimide (DABPIm) and 1,2-diazonia-7,8-diazabenzo[ghi]peryleneimide (TABPIm) to discuss the orientation effects of the nitrogen lone-pair of electrons toward the π-conjugated aromatic core on the electronic structures and energy levels. Actually, the nitrogen lone-pair of electrons in the 1 and 2 positions of TABPIm should be perpendicular to the ring plane (i.e., part of π-system), whereas the nitrogen lone-pair of electrons in the 7 and 8 positions do not participate in the π-systems because of the parallel orientation to the ring plane. As a result, the former case induces strong electron donating behaviour, which is in sharp contrast with the latter electron accepting feature.
 | ||
| Scheme 2 Chemical structures of N-substituted BP derivatives in this study. | ||
Results and discussion
Synthesis
1,12-Diazaperylene (DAP) and DABP were synthesized according to the reported methods.63–65 BPIm was also synthesized according to our reported procedure.49 The syntheses of DABPIm and TABPIm were performed by modifying our reported procedure: benzo[ghi]perylene monoimide by the Diels–Alder reaction of perylene and maleic anhydride.49 Based on this synthetic strategy, the synthetic schemes of DABPIm and TABPIm from DAP were designed as described in Scheme 3. To synthesize DABPIm, first, DAP underwent a Diels–Alder reaction with maleic anhydride to give compound 1. Then, by maleimidation using 4-heptylamine, we obtained DABPIm in 14% yield. TABPIm was obtained via the Diels–Alder reaction of DAP and 4-phenyl-1,2,4-triazoline-3,5-dione in 40% yield. The synthesized N-substituted PAHs were well-characterized by 1H NMR, 13C NMR, high resolution MALDI-TOF mass, and FT-IR spectroscopies [see: Experimental section and Fig. S1–S7 in the ESI†].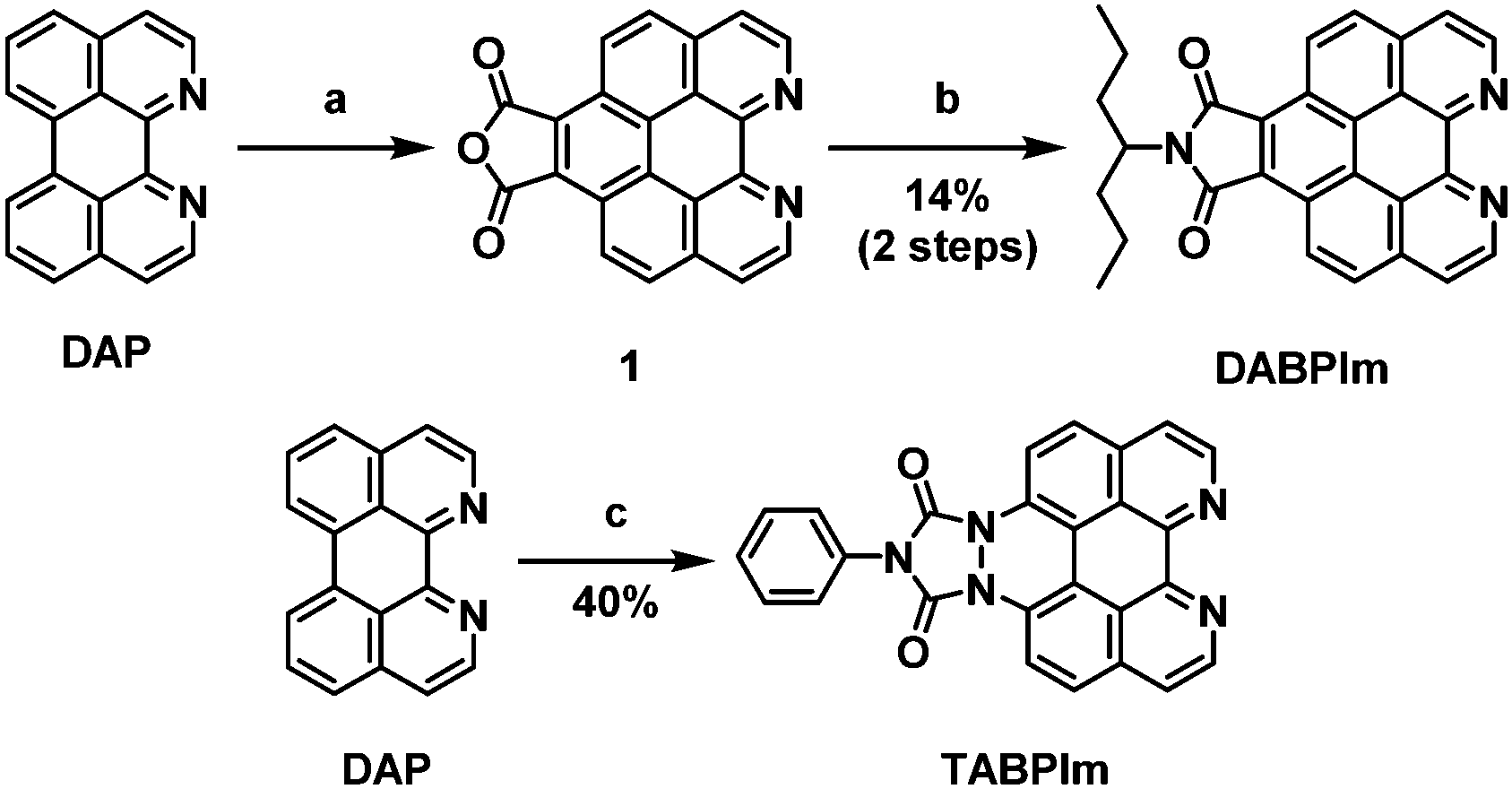 | ||
| Scheme 3 Synthetic schemes of DABPIm and TABPIm. Reagents and conditions: (a) maleic anhydride, p-chloranil, 220 °C, (b) 4-heptylamine, 150 °C, (c) 4-phenyl-1,2,4-triazoline-3,5-dione, p-chloranil, 150 °C. | ||
Steady-state spectroscopic measurements
Steady-state absorption and fluorescence spectra were recorded to examine the electronic structures of the N-substituted BP and reference compounds as shown in Fig. 1. Fig. 1A shows the absorption spectra of BP (spectrum a), DABP (spectrum b), BPIm (spectrum c), DABPIm (spectrum d) and TABPIm (spectrum e) in CH2Cl2. First, to discuss the nitrogen-substituted effects, we compared the spectra between BP and DABP. In the spectrum of reference BP (spectrum a), the characteristic strong peaks were observed at 289, 300, 329, 346, 364, and 385 nm, whereas the absorption peak at 406 nm is assigned to a symmetry-forbidden transition (see: ESI,† Fig. S9).50 The spectrum of DABP (spectrum b) has a broad absorption with maxima at 335, 354, 371, and 392 nm in the UV region, which indicates a small red-shifted trend of DABP as compared to BP. Considering the similar C2v symmetry to BP and fluorescence excitation spectral measurement (ESI,† Fig. S10), a small shoulder absorption at 407 nm may be attributable to a symmetry-forbidden transition. In addition, the spectrum of BPIm (spectrum c) significantly became red-shifted and broadened as compared to BP because of the introduction of the imide unit (vide infra), which is similar to the relationship between DABP and DABPIm (spectrum d). Moreover, TABPIm (spectrum e) demonstrates a significantly broadened and red-shifted spectrum in the ca. 450 to 600 nm region as compared to DABP and DABPIm (vide infra).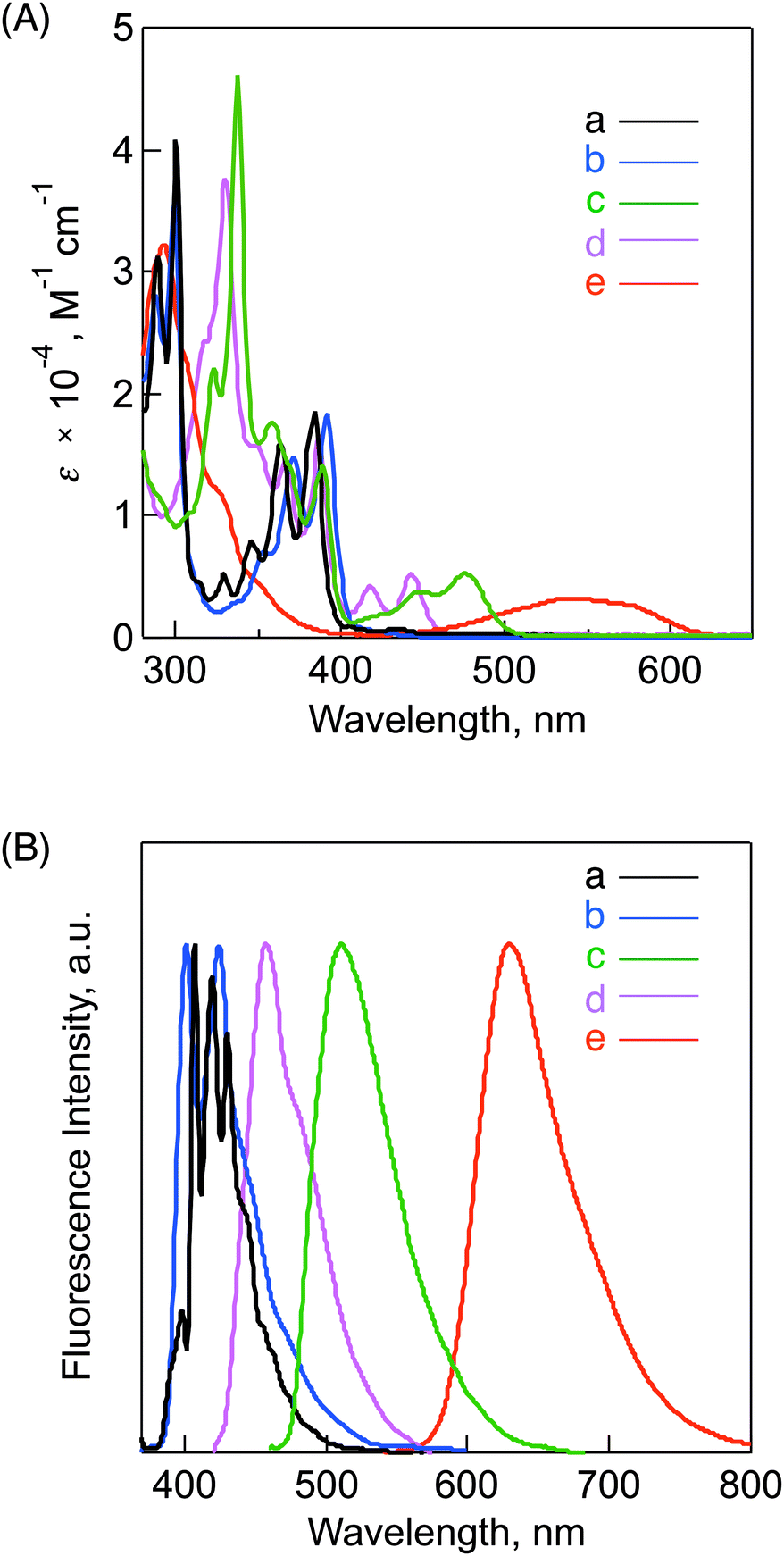 | ||
| Fig. 1 (A) Absorption and (B) fluorescence emission spectra of (a) BP, (b) DABP, (c) BPIm, (d) DABPIm and (e) TABPIm in CH2Cl2. Excitation wavelengths: 365 nm for BP, 300 nm for DABP, 404 nm for DABPIm and 580 nm for TABPIm. | ||
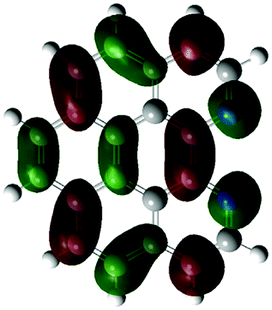
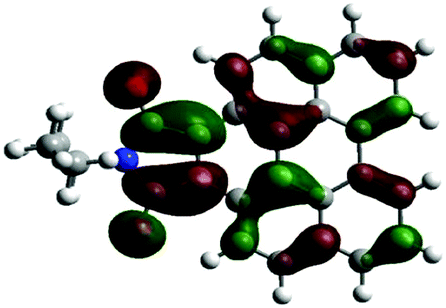
According to the DFT results (Tables 1 and 2), the introduction of nitrogen atoms onto the BP skeleton stabilizes the energies for both the lowest unoccupied molecular orbital: LUMO (−2.20 eV) and highest occupied molecular orbital: HOMO (−5.82 eV) of DABP by ∼0.6 eV as compared to those of BP (LUMO: −1.66 eV and HOMO: −5.20 eV). Consequently, similar spectral features between DABP and BP were mainly explained by the corresponding HOMO–LUMO gaps. In the case of DABPIm, the introduction of an electron-withdrawing imide unit onto the DABP core further stabilizes the LUMO level (−2.63 eV) as compared to the corresponding HOMO level (−6.08 eV), which leads to the decrease in the HOMO–LUMO gap. This is also similar to the relationship between BP and BPIm. It should be noted that both energies of the HOMO and LUMO states of DABPIm are largely stabilized as compared to those of BPIm (LUMO level: −2.35 eV and HOMO level: −5.54 eV) because of the introduction of nitrogen units, while the HOMO–LUMO gap of DABPIm (3.45 eV) became slightly larger than that of BPIm (3.19 eV) as shown in Table 2. This is highly relevant to the blue-shifted trend of DABPIm relative to BPIm (Fig. 1A). In addition, the LUMO level of TABPIm (−2.73 eV) became stabilized by ∼0.1 eV, whereas the HOMO level (−5.36 eV) was increased by ∼0.7 eV as compared to DABPIm. These results significantly contribute to the large red-shifted feature in the region from ca. 450 to 600 nm (vide supra). As mentioned earlier, the nitrogen lone-pair of electrons in the 1 and 2 positions of TABPIm are perpendicular to the aromatic plane, which indicates that the lone-pair of electrons were involved in the π-system, and they behaved like an electron donor. Consequently, this trend significantly induced the increased HOMO level, while the LUMO level became relatively decreased as compared to DABPIm. The molecular orbitals for both the HOMO and LUMO in TABPIm, which are totally different from those of DABPIm, also supported these spectral features (Table 1).
| HOMO | LUMO | |
|---|---|---|
| (a) |

|
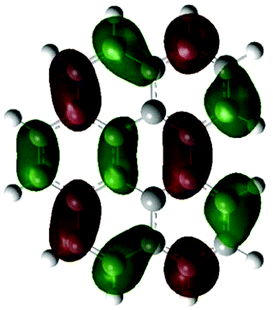
|
| (b) |
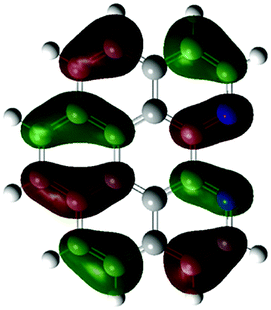
|
|
| (c) |

|
|
| (d) |

|
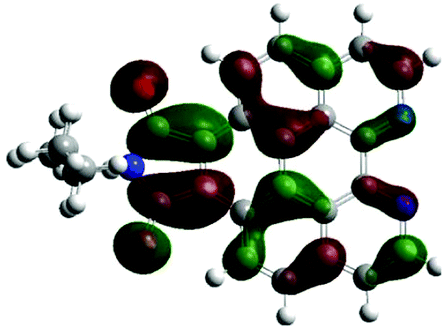
|
| (e) |
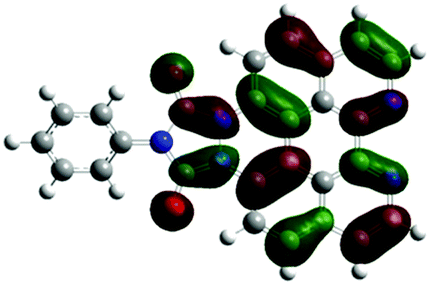
|
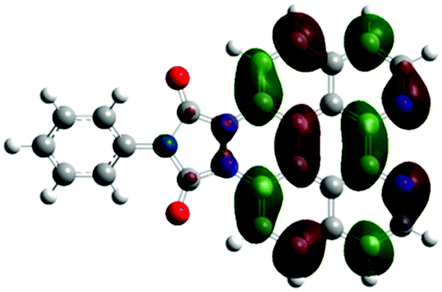
|
| Compound | E ox1,a V | E red1,a V | E red2,a V | HOMO,c eV | LUMO,c eV | HOMO–LUMO gapc, eV |
|---|---|---|---|---|---|---|
| a V vs. SCE. b Determined by DPV. c Calculated at the B3LYP/6-31G(d) level of theory. | ||||||
| BP | 1.12 | −2.20b | — | −5.20 | −1.66 | 3.54 |
| DABP | — | −1.43 | — | −5.82 | −2.20 | 3.62 |
| BPIm | — | −1.33 | — | −5.54 | −2.35 | 3.19 |
| DABPIm | — | −1.11 | −1.63 | −6.08 | −2.63 | 3.45 |
| TABPIm | 1.13 | −1.12 | −1.56 | −5.36 | −2.73 | 2.63 |
The absorption molar coefficients (ε0–0) of DABP (950 M−1 cm−1 at 407 nm), DABPIm (5200 M−1 cm−1 at 440 nm) and TABPIm (2500 M−1 cm−1 at 580 nm) were calculated from the 0–0 absorption bands. Whereas the ε0–0 of DABPIm is quite similar to that of the reference BPIm (5230 M−1 cm−1 at 475 nm), the ε0–0 of DABP (950 M−1 cm−1 at 407 nm) is somewhat larger than that of the reference BP (600 M−1 cm−1 at 406 nm). These values may provide useful discussion on the excited-state dynamics (vide infra).
In the fluorescence spectra of the BP derivatives and reference compounds, similar trends were also observed (Fig. 1B). The spectrum of BP (spectrum a) shows sharp and definite peaks at 399, 408, 420, 431, and 439 nm, whereas a broader spectrum with two strong absorption peaks at 401 and 424 nm was observed in DABP (spectrum b). The spectrum of DABPIm (spectrum d) also became much broader and red-shifted as compared to that of DABP, which is very similar to the relationship between BP and BPIm (spectrum c). TABPIm (spectrum e) demonstrates a significantly red-shifted spectrum in the range from ca. 600 to 800 nm. As discussed above, the HOMO of DABPIm is relatively localized on the DABP unit, which leads to the lower spin densities on the imide unit, whereas the corresponding LUMO extends up to the imide unit (Table 1). Therefore, this trend is probably ascribed to the increase of charge-transfer character by the introduction of the monosubstituted imide unit,49 which suggests that large spectral shifts were observed by varying the solvent polarities (ESI,† Fig. S11). Although this trend is also similar to TABPIm (ESI,† Fig. S12), the electronic configurations in the HOMO and LUMO of TABPIm are in sharp contrast with those of DABPIm (vide supra).
In addition, phosphorescence emission spectra of DABP and DABPIm were also observed in the range of ca. 600–900 nm (Fig. 2). These results indicate the presence of intersystem crossing (ISC) pathways. The estimated energies of T1–S0 are shown in Table 3. It should be noted that the T1–S0 energy of TABPIm was estimated using a TD-DFT method because we could not measure the phosphorescence spectrum due to the very limited solubility.
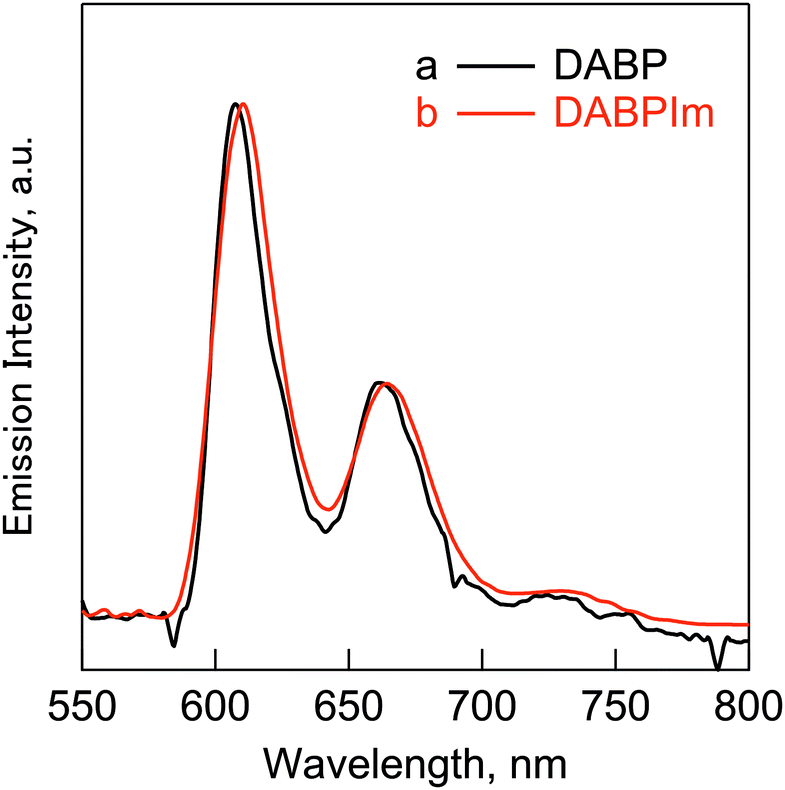 | ||
| Fig. 2 Phosphorescence emission spectra of (a) DABP (black line) and (b) DABPIm (red line) in frozen mixed solvent [toluene/MeOH/diethyl ether/ethyl iodide = 4/2/2/1 (v/v)] (77 K). The excitation wavelengths: 390 nm for DABP and 335 nm for DABPIm. | ||
| Compound | τ FL, ns | Φ FL | Φ ISC | Φ IC | k FL, ×107 s−1 | k ISC, ×107 s−1 | k IC, ×107 s−1 | S1–S0, eV | T1–S0, eV | S1–T1, eV | ε 0–0, M−1 cm−1 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| τ FL: fluorescence lifetime; ΦFL: fluorescence emission quantum yield; ΦISC: intersystem crossing quantum yield; ΦIC: internal conversion quantum yield; kFL: fluorescence emission rate constant; kISC: intersystem crossing rate constant; kIC: internal conversion rate constant. ΦIC = 1 – ΦFL − ΦISC, kFL = ΦFL·τFL−1, kISC = ΦISC·τFL−1, kIC = ΦIC·τFL−1.a Reported value in EtOH.70b Reported value in EtOH.71c Reported value in PMMA.70d ΦISC of DABP was determined by nanosecond transient absorption measurements using triplet–triplet energy transfer to DABP to β-carotene following the reference paper.72e Reported value in toluene.49f The estimated value calculated by using a TD-DFT method at the level of B3LYP/6-31G(d,p) in a suite of Gaussian 09 programs. | |||||||||||
| BP | 188a | 0.25b | 0.53c | 0.22 | 0.13 | 0.28 | 0.12 | 3.05 | 2.02 | 1.03 | 600 |
| DABP | 2.9 | 0.083 | 0.88d | 0.037 | 2.9 | 30 | 1.3 | 3.23 | 2.03 | 1.20 | 950 |
| BPIm | 9.2e | 0.36e | 0.68 ± 0.17e | 0 | 3.9 | 7.4 | — | 2.52 | 2.01 | 0.51 | 5200 |
| DABPIm | 7.2 | 0.33 | 0.51 ± 0.10 | 0.16 | 4.6 | 7.1 | 2.2 | 2.84 | 2.07 | 0.77 | 5200 |
| TABPIm | 4.6 | 0.11 | 0.27 ± 0.040 | 0.62 | 2.4 | 5.9 | 13 | 2.11 | (1.69)f | (0.42) | 2500 |
Electrochemical measurements
The electrochemical properties of the N-substituted BP derivatives were measured by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) to examine the substitution effects on the reduction and oxidation potentials. In CV and DPV, CH2Cl2 was employed as a solvent considering the solubility of these compounds. The electrochemical measurements were performed in CH2Cl2 containing 0.1 M n-Bu4NPF6 with a sweep rate of 0.1 V s−1 (CV) or 0.01 V s−1 (DPV). The successive redox couples are clearly observed in Fig. 3 and ESI,† Fig. S13. The measured half-wave potentials (E1/2) of N-substituted BP derivatives together with reference compounds are summarized in Table 2. The first one-electron reduction (Ered1) and oxidation potentials (Eox) of BP were −2.20 and 1.12 V vs. SCE, respectively (ESI,† Fig. S13). Fig. 3A and B show the voltammograms of BPIm and DABP, respectively. The Ered1 of BPIm and DABP were also assigned to be −1.33 V and −1.43 V vs. SCE, whereas the corresponding Eox values were not determined because of the limited electrochemical window. By introducing nitrogen atoms onto the BP unit, the successive large positive shifts of reduction potentials are observed by ∼0.8–0.9 V. The Ered1 of DABPIm (−1.11 V vs. SCE) also became the more positively shifted by ∼0.3 V as compared to DABP because of the substituted electron-withdrawing imide unit (Fig. 3C). The positive shift trend of Ered1 values in DABP derivatives agrees well with that in DFT calculations (Table 2). Moreover, the Ered1 of TABPIm (−1.12 V vs. SCE) is approximately similar to that of DABPIm, and the corresponding Eox (1.13 V vs. SCE) was successfully assigned within the electrochemical window (Fig. 3D). Consequently, these reduction and oxidation potentials of TABPIm result in the significantly decreased HOMO–LUMO gap, which is in good agreement with the DFT calculations and red-shifted trend in spectroscopic measurements (spectrum e in Fig. 1A and B).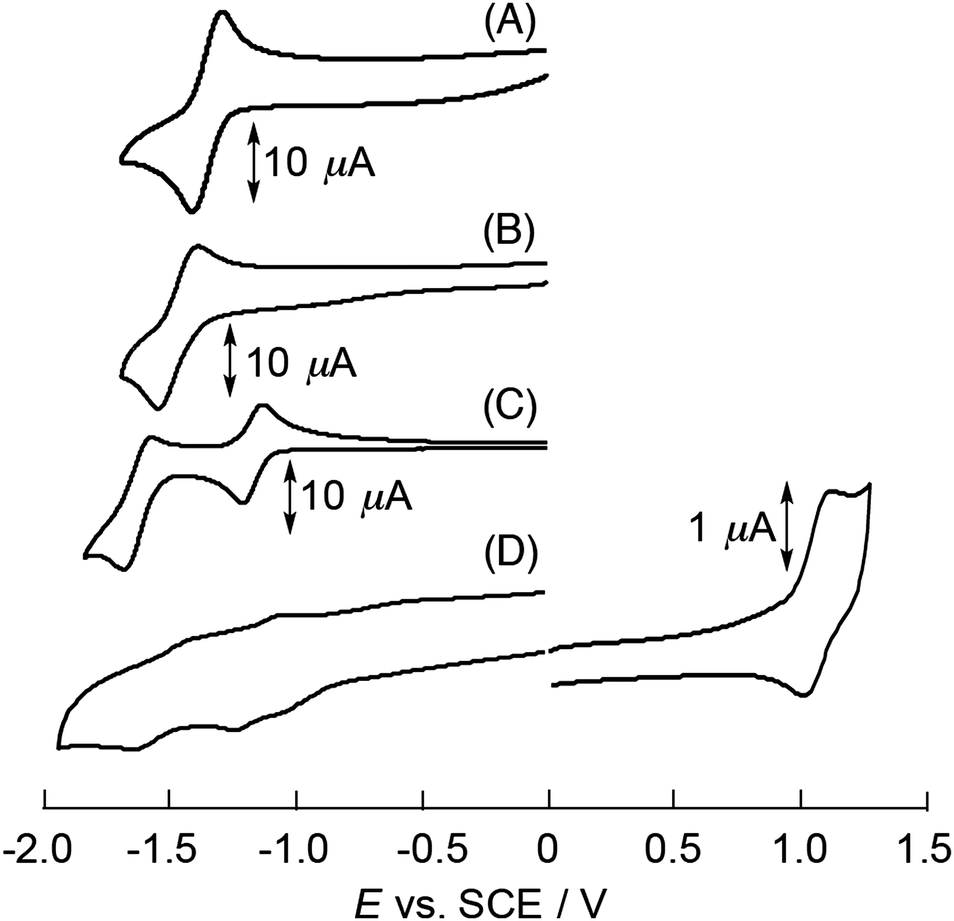 | ||
| Fig. 3 Cyclic voltammograms of (A) BPIm, (B) DABP, (C) DABPIm and (D) TABPIm in CH2Cl2 with 0.1 M n-Bu4NPF6 as the supporting electrolyte. Scan rate: 100 mV s−1. | ||
Spectroelectrochemical measurements of reduced DABP, DABPIm and oxidized TABPIm
The radical anion and cation species of N-substituted BP derivatives were produced using the electrochemical method by applying the negative and positive potentials. Fig. 4A–C show the absorption spectra of DABP˙−, DABPIm˙− and TDABPIm˙+, respectively. In all cases, a few new broad bands with isosbestic points appear corresponding to its radical anion and cation species under the reduction and oxidation process at room temperature. For example, the molar absorption coefficients of DABP˙−, DABPIm˙− and TDABPIm˙+ in the long wavelength region were estimated to be 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 M−1 cm−1 (at 446 nm), 10090 M−1 cm−1 (at 449 nm) and 2470 M−1 cm−1 (at 667 nm), respectively.
900 M−1 cm−1 (at 446 nm), 10090 M−1 cm−1 (at 449 nm) and 2470 M−1 cm−1 (at 667 nm), respectively.
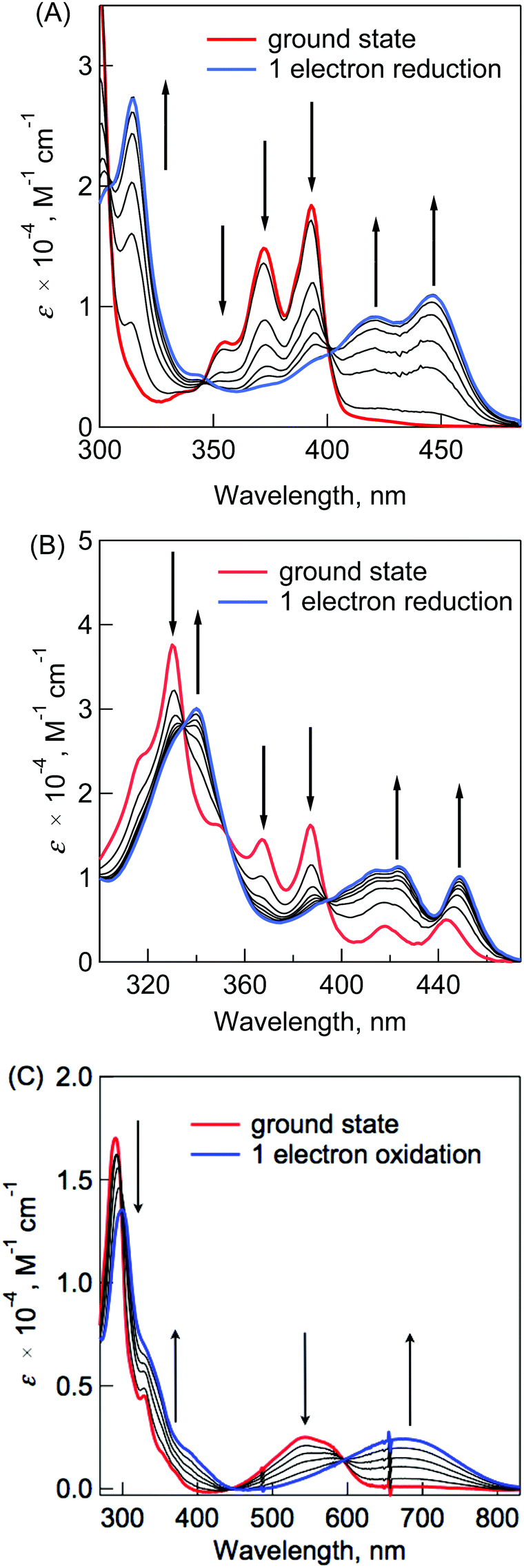 | ||
| Fig. 4 UV-vis spectral changes observed in one-electron transfer reduction of (A) DABP and (B) DABPIm by reducing the electrochemical voltage in CH2Cl2. (C) UV-vis spectral changes observed in one-electron transfer oxidation of TABPIm by increasing the electrochemical voltage in CH2Cl2. | ||
Measurements of fluorescence lifetimes and evaluation of excited-state dynamics
To clarify the detail excited-state dynamics of the N-substituted BP derivatives and reference compounds, first, fluorescence lifetime measurements were performed. The fluorescence decays for these molecules were examined in toluene solution using the pulsed 404 nm laser light, which excited these moieties as shown in Fig. 5. These fluorescence lifetimes (τFL), which were evaluated from a monoexponential fitting, are summarized in Table 3. The τFL values of DABP (2.9 ns), DABPIm (7.2 ns) and TABPIm (4.6 ns) are much shorter than that of BP (188 ns). This indicates that nitrogen-substitution onto the BP unit plays an important role in the decrease of τFL values.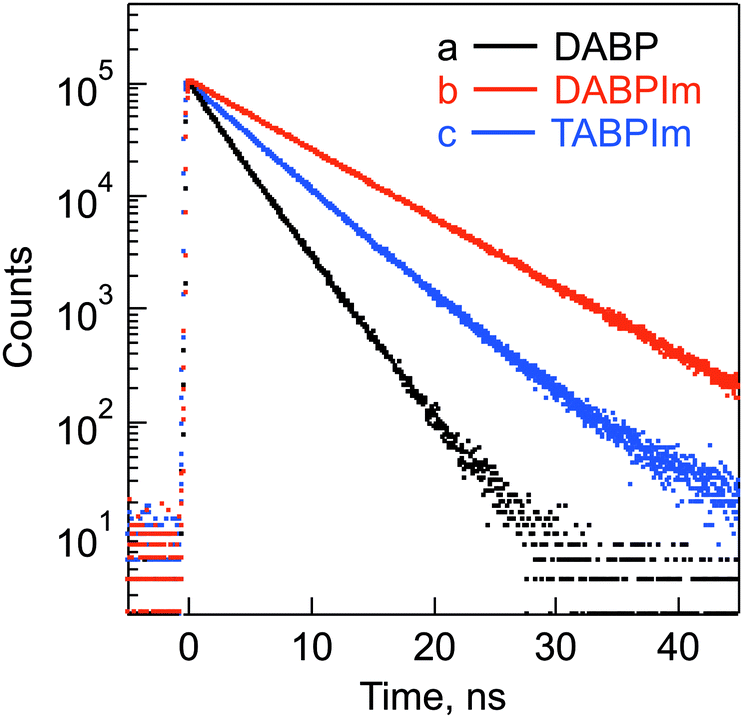 | ||
| Fig. 5 Fluorescence decay profiles of (a) DABP in toluene, (b) DABPIm in toluene and (c) TABPIm in CH2Cl2 (λex = 404 nm). | ||
The absolute fluorescence quantum yields (ΦFL) of these compounds were evaluated and summarized in Table 3. The ΦFL of DABP (0.083) is much smaller than that of BP (0.25), whereas an increase in ΦFL of DABPIm (0.33) was observed by the substitution of the imide unit. This value is also comparable to BPIm (ΦFL = 0.36). Additionally, ΦFL of TABPIm (0.11) was much smaller than that of DABPIm. The ΦFL values decreased by introducing nitrogen atoms, whereas an increased trend of ΦFL values was observed by the substitution of the imide unit (vide infra).
Then, we also evaluated the quantum yields of the intersystem crossing pathway (ΦISC) by 1O2 phosphorescence measurements using energy transfer from the triplet excited states of these derivatives to O2, assuming a negligible quenching process from the singlet excited states.66 Singlet oxygen was generated for these molecules employed as a sensitizer in oxygen-saturated toluene solution and detected by 1O2 phosphorescence at 1270 nm. We employed C60 as the standard considering the reported ΦISC value of fullerene C60 (0.96).67 Zinc tetraphenylporphyrin (ZnTPP) was also used as the standard compound of TABPIm (ΦISC = 0.88).68 It should be noted that the efficient energy transfer from the triplet excited states of the BP derivatives to O2 occurs without additional photochemical processes because the quenching fraction in O2-saturated toluene solution is close to unity (see: ESI,† Fig. S14). In addition, the sum of the above three quantum yields including the fluorescence (FL), intersystem crossing (ISC) and internal conversion (IC) pathways is close to unity within the experimental error. The quantum yields of the internal conversion (IC) pathways (ΦIC) can be therefore calculated by subtracting ΦFL and ΦISC from 1. The summarized ΦISC and ΦIC are shown in Table 3. For example, the ΦISC of DABP (0.88) is larger than that of BP (0.53), which is probably ascribed to the spin–orbit coupling caused by the introduction of nitrogen atoms onto the BP core. In contrast, by introducing the imide unit onto the DABP core, ΦISC of DABPIm decreases to 0.51 as compared to DABP (ΦISC = 0.88). The much enhanced ΦIC of TABPIm relative to ΦFL and ΦISC may be due to the vibrational relaxation of nitrogen atoms at the 1 and 2 positions. Nanosecond transient absorption measurements also demonstrated triplet–triplet (T–T) absorption signals of DABP (ESI,† Fig. S15) and DABPIm (ESI,† Fig. S14) because of high ΦISC.
To quantitatively discuss the excited-state dynamics, we calculated the net rate constants of these three processes including fluorescence emission kFL, intersystem crossing kISC and internal conversion kIC as shown in Table 3. According to the well-known theory of photochemistry, kFL is highly relevant to the molar extinction coefficients.69 As a result, the molar absorption coefficients (ε0–0) were estimated from the 0–0 bands in the absorption spectra (Table 3). For example, the ε0–0 value of DABP (950 cm−1 M−1) is somewhat larger than that of BP (600 cm−1 M−1), which corresponds to the kFL values because the kFL of DABP (2.9 × 107 s−1) is larger than that of BP (1.3 × 106 s−1).
However, as discussed above, the ΦFL of DABP (0.08) is smaller than the ΦFL of BP (0.25) regardless of the increase in kFL. This is probably attributable to the much more enhanced kISC of DABP (vide infra). It's well-known that kISC is largely dependent on the energy gaps (ΔEST) between the lowest singlet (S1) and triplet excited states (T1) through which crossing takes place. To quantitatively evaluate the values of these derivatives, the values of ΔEST were calculated by steady-state spectroscopic data and summarized in Table 3. However, similar ΔEST values do not correspond to the increased kISC values. The possible reason may be spin–orbit coupling due to the introduction of nitrogen atoms.
In addition, in the case of DABPIm, kFL (4.6 × 107 s−1) is similar to that of DABP (kFL = 2.9 × 107 s−1), whereas the kISC of DABPIm (7.1 × 107 s−1) is much smaller than that of DABP (3.0 × 108 s−1). Consequently, the enhanced ΦFL of DABPIm relative to DABP is due to the decreased kISC. Considering the above-mentioned ΔEST values of DABPIm (0.77 eV) and DABP (1.20 eV), this is not coincident with the trend of kISC values. Therefore, to further investigate the ISC pathways, we also examined the electronic configurations, which are responsible for the steady-state fluorescence and phosphorescence spectra. In both the fluorescence and phosphorescence peaks of DABPIm and DABP, the emission peaks became red-shifted and broadened with increasing solvent polarities (ESI,† Fig. S11 and S16). These are assigned to be the usual solvent effects on 1π,π* and 3π,π* states. According to the selection rules of ISC (El-Sayed's rule), ISC from 1π,π* to 3π,π* is forbidden.69
However, the out-of-plane vibrational mode provokes mixing between the singlet and triplet states.69 As stated above, the DABP spectrum (spectrum b in Fig. 1B) has characteristic sharp and strong peaks and this separation (ca. 500–700 cm−1) may correspond to the aromatic or C–H out-of-plane wagging mode.69 In contrast, DABPIm shows different vibrational modes with separations (∼1200–1400 cm−1), which may be attributable to the aromatic or C–H in-plane mode.69 This results in the deceleration of ISC. Separately, the pictures of DABP and DABPIm optimized by DFT calculations successfully support the vibrational trends (ESI,† Fig. S17).50 Thus, we can conclude that the introduction of imide units onto the DABP core (i.e., DABPIm) decelerates ISC, which leads to the enhanced ΦFL of DABPIm (Table 3).
Conclusions
Benzo[ghi]perylene (BP) derivatives substituted by nitrogen atoms and the imide unit were newly synthesized to examine on the electrochemical property and excited-state dynamics. The introduction of the electron-withdrawing units stabilized the energy levels of the HOMO and LUMO in DABP and DABPIm, whereas the nitrogen lone-pair of electrons in the 1 and 2 positions of TABPIm induced the increased HOMO level. These results are successfully explained by DFT calculations and are in good agreement with the spectroscopic and electrochemical behaviours. With regard to the excited-state dynamics of these derivatives, the ΦFL values decreased by introducing nitrogen atoms onto the BP core. In contrast, the increased trend of ΦFL values was observed by the substitution of the imide unit. The much enhanced ΦIC of TABPIm was also observed. This may be due to the vibrational relaxation of nitrogen atoms at the 1 and 2 positions. These phenomena were successfully explained by the analysed kinetic discussion. Such characteristic electrochemical and photophysical properties of N-substituted PAH derivatives provide a new perspective for the construction of efficient molecular electronics and energy conversion devices.Experimental section
General methods
Benzo[ghi]perylene was purchased from Wako Pure Chemical Industries. It was used for spectroscopic and electrochemical measurements with recrystallization from chloroform and hexane. All solvents and reagents of the best grade available were purchased from commercial suppliers and were used without further purification. Column flash chromatography was performed on silica gel (Kanto Chemical Silica gel 60 N, 40–50 μm or 100–210 μm). Preparative recycling gel permeation chromatography was performed using a high pressure liquid chromatography apparatus Japan Analytical Industry LC-9204 using chloroform as an eluent at room temperature. We used an LC-9204 apparatus equipped with a pump (JAI PI-60, flow rate 2.5 mL min−1), a UV detector (JAI UV-3740) and two columns (JAIGEL 2H and 1H, 40 × 600 mm for each). All experiments except for the single crystal X-ray diffraction measurements were performed at room temperature. 1H NMR and 13C NMR spectra were recorded on a 400 MHz JEOL JNM-A400, JNM-Al400, or JNM-ECX 400 spectrometer, using the solvent peak as the reference standard, with chemical shifts given in parts per million. CDCl3 was used as the NMR solvent. MALDI-TOF mass spectra were recorded on a Bruker Ultraflex. The ab initio calculations were performed with the Gaussian 09 program. The molecular geometries were optimized at a B3LYP/6-31G(d) level of theory. The molecular orbitals and vibrations were calculated at the same level.Spectroscopic measurements
UV/vis absorption spectra were recorded on a Perkin Elmer (Lamda 750) UV-VIS-NIR spectrophotometer. Fluorescence and phosphorescence emission spectra were recorded on Perkin Elmer (LS-55) spectrofluorophotometer. Fluorescence lifetimes were measured on a HORIBA Scientific time-correlated single-photon counting system (FluoroCube) with a laser light (DeltaDiode, laser diode head, 404 nm) as an excitation source. The absolute fluorescence quantum yields were measured by a Hamamatsu Photonics C9920-02 system equipped with an integrating sphere and a red-sensitive multichannel photodetector (PMA-12): excitation wavelength = 300 nm. The absolute fluorescence quantum yields were corrected for the spectral response of the photodetector. The wavelength range of the emission measurements is 300–950 nm. Nanosecond transient absorption measurements were carried out using a Unisoku TSP-2000 flash spectrometer. A Surelite-I Nd-YAG (Continuum, 4–6 ns fwhm) laser with the second harmonic at 532 nm was employed for the flash photo-irradiation. The photodynamics was monitored by continuous exposure to a xenon lamp (150 W) as a probe light and a photomultiplier tube (Hamamatsu R-2949) as a detector. Each sample solution was purged with Ar for at least 20 min prior to the measurements. All experiments were performed at room temperature.Electrochemical measurements
Cyclic voltammograms were recorded on an Iviumstat 20 V/2.5 A potentiostat by using a three electrode system. A platinum electrode was used as the working electrode. A platinum wire served as the counter electrode, and a saturated calomel electrode was used as the reference electrode. The ferrocene/ferrocenium redox couple was used as an internal standard. All solutions were purged prior to electrochemical and spectral measurements by using nitrogen gas.1O2 phosphorescence measurements
The 1O2* emission spectrum was measured by the following setup. The sample solution in toluene was bubbled by O2 for 20 minutes before measurement. A 355 nm laser pulse was irradiated (EKSPLA 2210A, 1 kHz operation, 6 mW) on the solution. Emission in the NIR region from the sample solution was chopped at 84 Hz frequency, then introduced to the monochromator (Shimadzu, SPG-120IR) and detected by a photodiode (New focus, model 2153). The diode signal was accumulated by the digital lock-in amplifier (NF Electronic instruments LI5640), which was operated in the chopper frequency, and then transferred to the PC. The PC controlled the monochromator to obtain the emission spectrum. The ΦISC value in solution was measured following a general method with C60 (0.96)67 or zinc tetraphenylporphyrin (ZnTPP) (0.88)68 as the standard.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP16K14067, JP17H05162, JP17H05270 to T. H., JP17K14476 to H. S., JP26102012 to T. T. This work was performed under the Cooperative Research Program of “Network Joint Research Centre for Materials and Devices”.References
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed.
- K. Müllen and J. P. Rabe, Acc. Chem. Res., 2008, 41, 511–520 CrossRef PubMed.
- M. D. Watson, A. Fechtenkotter and K. Müllen, Chem. Rev., 2001, 101, 1267–1300 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- G. Li, H. Phan, T. S. Herng, T. Y. Gopalakrishna, C. Liu, W. Zeng, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 5012–5016 CrossRef CAS PubMed.
- D. Jänsch, I. Ivanov, Y. Zagranyarski, I. Duznovic, M. Baumgarten, D. Turchinovich, C. Li, M. Bonn and K. Müllen, Chem. – Eur. J., 2017, 23, 4870–4875 CrossRef PubMed.
- H. Hayashi, N. Aratani and H. Yamada, Chem. – Eur. J., 2017, 23, 7000–7008 CrossRef CAS PubMed.
- R. Dong, M. Pfeffermann, D. Skidin, F. Wang, Y. Fu, A. Narita, M. Tommasini, F. Moresco, G. Cuniberti, R. Berger, K. Müllen and X. Feng, J. Am. Chem. Soc., 2017, 139, 2168–2171 CrossRef CAS PubMed.
- T. Dumslaff, B. Yang, A. Maghsoumi, G. Velpula, K. S. Mali, C. Castiglioni, S. De Feyter, M. Tommasini, A. Narita, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 4726–4729 CrossRef CAS PubMed.
- F. D'Souza and O. Ito, Chem. Soc. Rev., 2012, 41, 86–96 RSC.
- L. Chen, K. S. Mali, S. R. Puniredd, M. Baumgarten, K. Parvez, W. Pisula, S. De Feyter and K. Müllen, J. Am. Chem. Soc., 2013, 135, 13531–13537 CrossRef CAS PubMed.
- W. Zhang, W. Jin, T. Fukushima, N. Ishii and T. Aida, Angew. Chem., Int. Ed., 2009, 48, 4747–4750 CrossRef CAS PubMed.
- D. Khim, K.-J. Baeg, J. Kim, M. Kang, S.-H. Lee, Z. Chen, A. Facchetti, D.-Y. Kim and Y.-Y. Noh, ACS Appl. Mater. Interfaces, 2013, 5, 10745–10752 CAS.
- S. J. Kang, S. Ahn, J. B. Kim, C. Schenck, A. M. Hiszpanski, S. Oh, T. Schiros, Y.-L. Loo and C. Nuckolls, J. Am. Chem. Soc., 2013, 135, 2207–2212 CrossRef CAS PubMed.
- P. E. Hartnett, A. Timalsina, H. S. S. R. Matte, N. Zhou, X. Guo, W. Zhao, A. Facchetti, R. P. H. Chang, M. C. Hersam, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2014, 136, 16345–16356 CrossRef CAS PubMed.
- D. Kato, H. Sakai, N. V. Tkachenko and T. Hasobe, Angew. Chem., Int. Ed., 2016, 55, 5230–5234 CrossRef CAS PubMed.
- T. Sakuma, H. Sakai, Y. Araki, T. Mori, T. Wada, N. V. Tkachenko and T. Hasobe, J. Phys. Chem. A, 2016, 120, 1867–1875 CrossRef CAS PubMed.
- Y. Yamamoto, H. Sakai, J. Yuasa, Y. Araki, T. Wada, T. Sakanoue, T. Takenobu, T. Kawai and T. Hasobe, Chem. – Eur. J., 2016, 22, 4263–4273 CrossRef CAS PubMed.
- H. Sakai, T. Kubota, J. Yuasa, Y. Araki, T. Sakanoue, T. Takenobu, T. Wada, T. Kawai and T. Hasobe, Org. Biomol. Chem., 2016, 14, 6738–6743 CAS.
- H. Sakai, T. Kubota, J. Yuasa, Y. Araki, T. Sakanoue, T. Takenobu, T. Wada, T. Kawai and T. Hasobe, J. Phys. Chem. C, 2016, 120, 7860–7869 CAS.
- H. Sakai, S. Shinto, J. Kumar, Y. Araki, T. Sakanoue, T. Takenobu, T. Wada, T. Kawai and T. Hasobe, J. Phys. Chem. C, 2015, 119, 13937–13947 CAS.
- H. Osaki, C.-M. Chou, M. Taki, K. Welke, D. Yokogawa, S. Irle, Y. Sato, T. Higashiyama, S. Saito, A. Fukazawa and S. Yamaguchi, Angew. Chem., Int. Ed., 2016, 55, 7131–7135 CrossRef CAS PubMed.
- T. Seki, S. Yagai, T. Karatsu and A. Kitamura, J. Org. Chem., 2008, 73, 3328–3335 CrossRef CAS PubMed.
- L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2008, 41, 1674–1684 CrossRef CAS PubMed.
- Y. S. Zhao, H. Fu, A. Peng, Y. Ma, Q. Liao and J. Yao, Acc. Chem. Res., 2009, 43, 409–418 CrossRef PubMed.
- S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC.
- T. Kato, T. Yasuda, Y. Kamikawa and M. Yoshio, Chem. Commun., 2009, 729–739 RSC.
- T. Hasobe, J. Phys. Chem. Lett., 2013, 4, 1771–1780 CrossRef CAS PubMed.
- T. Hasobe, Phys. Chem. Chem. Phys., 2012, 14, 15975–15987 RSC.
- T. Hasobe, Phys. Chem. Chem. Phys., 2010, 12, 44–57 RSC.
- B. A. Jones, M. J. Ahrens, M.-H. Yoon, A. Facchetti, T. J. Marks and M. R. Wasielewski, Angew. Chem., Int. Ed., 2004, 43, 6363–6366 CrossRef CAS PubMed.
- J. Li, J.-J. Chang, H. S. Tan, H. Jiang, X. Chen, Z. Chen, J. Zhang and J. Wu, Chem. Sci., 2012, 3, 846–850 RSC.
- B. Yoo, T. Jung, D. Basu, A. Dodabalapur, B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, Appl. Phys. Lett., 2006, 88, 082104 CrossRef.
- T. Aoki, H. Sakai, K. Ohkubo, T. Sakanoue, T. Takenobu, S. Fukuzumi and T. Hasobe, Chem. Sci., 2015, 6, 1498–1509 RSC.
- T. Hasobe, K. Ida, H. Sakai, K. Ohkubo and S. Fukuzumi, Chem. – Eur. J., 2015, 21, 11196–11205 CrossRef CAS PubMed.
- H. Sakai, K. Ohkubo, S. Fukuzumi and T. Hasobe, Chem. – Asian J., 2016, 11, 613–624 CrossRef CAS PubMed.
- T. J. Sisto, Y. Zhong, B. Zhang, M. T. Trinh, K. Miyata, X. Zhong, X. Y. Zhu, M. L. Steigerwald, F. Ng and C. Nuckolls, J. Am. Chem. Soc., 2017, 139, 5648–5651 CrossRef PubMed.
- P. E. Hartnett, S. M. Dyar, E. A. Margulies, L. E. Shoer, A. W. Cook, S. W. Eaton, T. J. Marks and M. R. Wasielewski, Chem. Sci., 2015, 6, 402–411 RSC.
- S. M. M. Conron, L. E. Shoer, A. L. Smeigh, A. B. Ricks and M. R. Wasielewski, J. Phys. Chem. B, 2013, 117, 2195–2204 CrossRef PubMed.
- T. M. Wilson, M. J. Tauber and M. R. Wasielewski, J. Am. Chem. Soc., 2009, 131, 8952–8957 CrossRef CAS PubMed.
- V. M. Blas-Ferrando, J. Ortiz, K. Ohkubo, S. Fukuzumi, F. Fernandez-Lazaro and Á. Sastre-Santos, Chem. Sci., 2014, 5, 4785–4793 RSC.
- V. M. Blas-Ferrando, J. Ortiz, L. Bouissane, K. Ohkubo, S. Fukuzumi, F. Fernandez-Lazaro and Á. Sastre-Santos, Chem. Commun., 2012, 48, 6241–6243 RSC.
- S. Fukuzumi, K. Ohkubo, J. Ortiz, A. M. Gutierrez, F. Fernandez-Lazaro and Á. Sastre-Santos, Chem. Commun., 2005, 3814–3816 RSC.
- A. J. Jimenez, R. M. K. Calderon, M. S. Rodriguez-Morgade, D. M. Guldi and T. Torres, Chem. Sci., 2013, 4, 1064–1074 RSC.
- F. J. Céspedes-Guirao, K. Ohkubo, S. Fukuzumi, Á. Sastre-Santos and F. Fernández-Lázaro, J. Org. Chem., 2009, 74, 5871–5880 CrossRef PubMed.
- H. Horinouchi, H. Sakai, Y. Araki, T. Sakanoue, T. Takenobu, T. Wada, N. V. Tkachenko and T. Hasobe, Chem. – Eur. J., 2016, 22, 9631–9641 CrossRef CAS PubMed.
- M. Supur, Y. M. Sung, D. Kim and S. Fukuzumi, J. Phys. Chem. C, 2013, 117, 12438–12445 CAS.
- R. K. Dubey, M. Niemi, K. Kaunisto, K. Stranius, A. Efimov, N. V. Tkachenko and H. Lemmetyinen, Inorg. Chem., 2013, 52, 9761–9773 CrossRef CAS PubMed.
- K. Ida, H. Sakai, K. Ohkubo, Y. Araki, T. Wada, T. Sakanoue, T. Takenobu, S. Fukuzumi and T. Hasobe, J. Phys. Chem. C, 2014, 118, 7710–7720 CAS.
- S. Hirayama, H. Sakai, Y. Araki, M. Tanaka, M. Imakawa, T. Wada, T. Takenobu and T. Hasobe, Chem. – Eur. J., 2014, 20, 9081–9093 CrossRef CAS PubMed.
- M. Takase, T. Narita, W. Fujita, M. S. Asano, T. Nishinaga, H. Benten, K. Yoza and K. Müllen, J. Am. Chem. Soc., 2013, 135, 8031–8040 CrossRef CAS PubMed.
- J. Wei, B. Han, Q. Guo, X. Shi, W. Wang and N. Wei, Angew. Chem., Int. Ed., 2010, 49, 8209–8213 CrossRef CAS PubMed.
- M. A. Biasutti, S. De Feyter, S. De Backer, G. B. Dutt, F. C. De Schryver, M. Ameloot, P. Schlichting and K. Müllen, Chem. Phys. Lett., 1996, 248, 13–19 CrossRef CAS.
- B. He, A. B. Pun, L. M. Klivansky, A. M. McGough, Y. Ye, J. Zhu, J. Guo, S. J. Teat and Y. Liu, Chem. Mater., 2014, 26, 3920–3927 CrossRef CAS.
- W. Jiang, Y. Li and Z. Wang, Chem. Soc. Rev., 2013, 42, 6113–6127 RSC.
- D. Wu, W. Pisula, M. C. Haberecht, X. Feng and K. Müllen, Org. Lett., 2009, 11, 5686–5689 CrossRef CAS PubMed.
- W. Jiang, H. Qian, Y. Li and Z. Wang, J. Org. Chem., 2008, 73, 7369–7372 CrossRef CAS PubMed.
- G.-J. Zhao and K.-L. Han, J. Phys. Chem. A, 2009, 113, 4788–4794 CrossRef CAS PubMed.
- F. Masetti, U. Mazzucato and J. B. Birks, Chem. Phys., 1975, 9, 301–306 CrossRef CAS.
- B. N. Bandyopadhyay and A. Harriman, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 663–674 RSC.
- C. A. Parker and T. A. Joyce, Trans. Faraday Soc., 1966, 62, 2785–2792 RSC.
- T. Sander, H. G. Löhmannsröben and H. Langhals, J. Photochem. Photobiol., A, 1995, 86, 103–108 CrossRef CAS.
- S. Alibert-Fouet, I. Seguy, J.-F. Bobo, P. Destruel and H. Bock, Chem. – Eur. J., 2007, 13, 1746–1753 CrossRef CAS PubMed.
- T. Caronna, F. Fontana, G. Longhi, A. Mele, I. N. Sora and L. Vigano, ARKIVOC, 2009, 8, 145–155 Search PubMed.
- A. Chouai, S. E. Wicke, C. Turro, J. Bacsa, K. R. Dunbar, D. Wang and R. P. Thummel, Inorg. Chem., 2005, 44, 5996–6003 CrossRef CAS PubMed.
- M. Pineiro, A. L. Carvalho, M. M. Pereira, A. M. d. A. R. Gonsalves, L. G. Arnaut and S. J. Formosinho, Chem. – Eur. J., 1998, 4, 2299–2307 CrossRef CAS.
- J. W. Arbogast, A. P. Darmanyan, C. S. Foote, F. N. Diederich, R. L. Whetten, Y. Rubin, M. M. Alvarez and S. J. Anz, J. Phys. Chem., 1991, 95, 11–12 CrossRef CAS.
- H. Goerner, J. Phys. Chem., 1982, 86, 2028–2035 CrossRef CAS.
- N. J. Turro, Modern Molecular Photochemistry, University Science Books, Sausalito, 1991 Search PubMed.
- W. R. Dawson and J. L. Kropp, J. Phys. Chem., 1969, 73, 1752–1758 CrossRef CAS.
- W. R. Dawson and M. W. Windsor, J. Phys. Chem., 1968, 72, 3251–3260 CrossRef CAS.
- M. Fujitsuka, A. Watanabe, O. Ito, K. Yamamoto and H. Funasaka, J. Phys. Chem. A, 1997, 101, 4840–4844 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, 1H NMR spectra, 13C NMR spectra, MALDI-TOF mass spectra, cyclic voltammograms, and absorption and fluorescence spectra. CCDC 1565206. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00301c |
| This journal is © the Partner Organisations 2017 |