
Synthesis of polycyclic sultams via a palladium-catalyzed reaction of 1-bromo-2-(cyclopropylidenemethyl)benzenes with 2-alkynylbenzenesulfonamides†
Xinxing
Gong
a,
Guangming
Li
*b and
Jie
Wu
*ac
aDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai 200433, China. E-mail: jie_wu@fudan.edu.cn
bXinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine, Shanghai 200092, China. E-mail: ligm68@126.com
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Linglin Road, Shanghai 200032, China
First published on 26th October 2016
Abstract
Polycyclic sultams are efficiently synthesized through a palladium-catalyzed tandem reaction of 1-bromo-2-(cyclopropylidenemethyl)benzenes with 2-alkynylbenzenesulfonamides. The transformation proceeds through double carbometallation with excellent chemoselectivity and regioselectivity, leading to a range of 7,7a-dihydro-6H-benzo[f]indeno[1,2-d][1,2]thiazepine 5,5-dioxides in moderate to good yields. During the reaction process, three new bonds and two rings including the seven-membered sultam ring are formed.
Introduction
Cyclic sulfonamides are widely present in both natural products and therapeutic agents, and their applications in organic synthesis and pharmaceutical research have been demonstrated.1 Tianeptine as a typical example is a drug, which is used for the treatment of major depressive episodes.2 Consistent with our continuing interest in the libraries’ construction and biological evaluation of natural product-like compounds,3 we are interested in the preparation of bioactive compounds with a core of cyclic sulfonamide.4 Recently, we identified that a cyclic sulfonamide has great potential in inducing apoptosis of hepatic stellate cells (HSCs) in vitro, and the activation of caspase-3 might be involved in this process.5 This promising result prompted us to look for more active hits. Therefore, the construction of diverse cyclic sulfonamides with complexity for further biological evaluations is in high demand.Recently, we have developed the strategy of double carbometallation for the generation of complex molecules.3b,e This approach is attractive, since excellent chemoselectivity and regioselectivity can be observed during the reaction process with the formation of multi-bonds and the competitive pathways are minimized.6 Usually, substrates with multi-active sites are involved in a tandem process.7 For example, the strategy was initially discovered for the reaction of 2-alkynylbenzenebromides with amines, which provided 5H-cyclopenta[c]quinolines in good yields with the formation of four new bonds via double carbometallation.6g Based on the alkynyl moiety and aryl bromide in 2-alkynylbenzenebromide, a range of polycycles was then produced when 2-alkynylbenzenebromides reacted with various coupling partners.6 Further studies revealed that the approach could be extended to 1-bromo-2-(cyclopropylidenemethyl)benzenes 1,8 which was used as a replacement of 2-alkynylbenzenebromide. The reactivity of alkylidenecyclopropanes has been demonstrated.9 We conceived that the cyclopropylidenemethyl group could be considered as an equivalence of an alkynyl moiety during the reaction process. For instance, we developed a facile route for the assembly of indeno[1,2-c]chromenes via a palladium-catalyzed tandem reaction of 1-bromo-2-(cyclopropylidenemethyl)benzenes 1 with 2-alkynylphenols (Scheme 1).8a This reaction worked well, and we did not observe the formation of benzofurans via direct cyclization of 2-alkynylphenol. As mentioned above, the excellent chemoselectivity and regioselectivity under the conditions produced the final outcome. Encouraged by this result, we envisioned that 2-alkynylbenzenesulfonamides 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 would be the choice for the reaction of 1 as well. As expected, cyclic sulfonamides 3A would be prepared if the reaction proceeded through a similar transformation.8a As described in Scheme 1, we postulated that an oxidative addition of compound 1 to palladium(0) would occur to produce Pd(II) A. Although the competitive reaction pathways including intramolecular 6-endo cyclization10 and direct C–N bond formation11 would happen, we believed that the presence of sulfonamide would direct the regioselective coordination and insertion of Pd(II) A to the triple bond of 2 to generate intermediate B. Subsequently, intermediate C would be formed via an intramolecular insertion of the double bond of alkylidenecyclopropane into the carbon–Pd(II) bond. Further C–N bond formation in the presence of a base would provide compound 3. After intramolecular rearrangement as similar as the reaction of 1-bromo-2-(cyclopropylidenemethyl)benzene with 2-alkynylphenol, compound 3A would be generated. To identify the practicability of this proposed route shown in Scheme 1, we therefore started to explore the palladium-catalyzed tandem reaction of 1 with 2.
10 would be the choice for the reaction of 1 as well. As expected, cyclic sulfonamides 3A would be prepared if the reaction proceeded through a similar transformation.8a As described in Scheme 1, we postulated that an oxidative addition of compound 1 to palladium(0) would occur to produce Pd(II) A. Although the competitive reaction pathways including intramolecular 6-endo cyclization10 and direct C–N bond formation11 would happen, we believed that the presence of sulfonamide would direct the regioselective coordination and insertion of Pd(II) A to the triple bond of 2 to generate intermediate B. Subsequently, intermediate C would be formed via an intramolecular insertion of the double bond of alkylidenecyclopropane into the carbon–Pd(II) bond. Further C–N bond formation in the presence of a base would provide compound 3. After intramolecular rearrangement as similar as the reaction of 1-bromo-2-(cyclopropylidenemethyl)benzene with 2-alkynylphenol, compound 3A would be generated. To identify the practicability of this proposed route shown in Scheme 1, we therefore started to explore the palladium-catalyzed tandem reaction of 1 with 2.
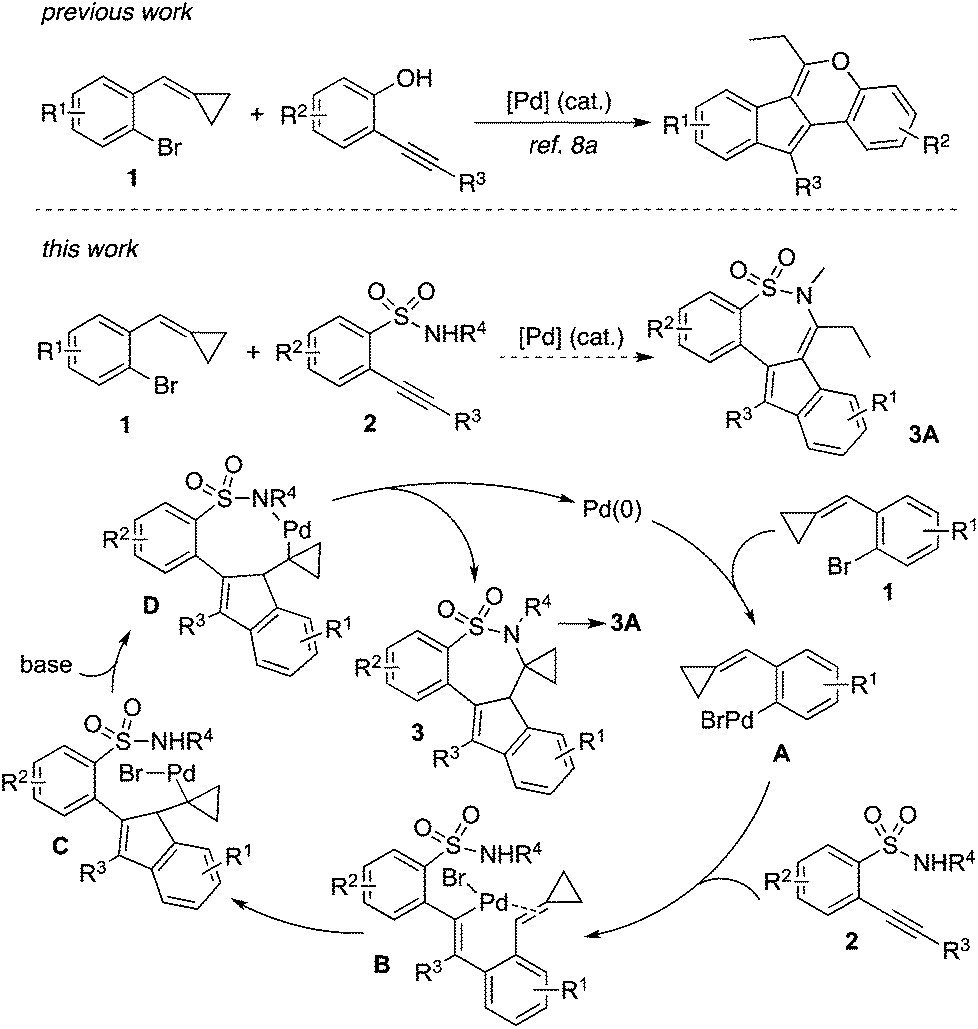 | ||
| Scheme 1 A proposed palladium-catalyzed reaction of 1-bromo-2-(cyclopropylidenemethyl)benzenes 1 with 2-alkynylbenzenesulfonamides 2. | ||
Results and discussion
Initially, the reaction of 1-bromo-2-(cyclopropylidenemethyl)benzene 1a and N-methyl-2-(phenylethynyl)benzenesulfonamide 2a was selected for reaction development (Table 1). At the beginning, the reaction was performed in the presence of palladium acetate (2.5 mol%), 1,1′-bis(diphenylphosphino)propane (DPPP, 5 mol%) and potassium carbonate in 1,4-dioxane under reflux (Table 1, entry 1). However, no reaction occurred under the conditions. Only a trace amount of the product was detected when the phosphine ligand was changed to XPhos (Table 1, entry 2). To our delight, a product was isolated in 16% yield when 1,1′-bis(diphenylphosphino)ferrocene (DPPF) was used as the ligand (Table 1, entry 3). Interestingly, the structure of this product was identified as compound 3a after X-ray crystallography analysis (see ESI†). We did not observe the formation of compound 3Aa, as illustrated above.8a In our previous report, the driving force for the ring-opening rearrangement of cyclopropane came from the electron pair of oxygen in 2-alkynylphenol.8a We reasoned that the weaker force from nitrogen in 2-alkynylbenzenesulfonamides provided less thrust to promote the ring-opening transformation. We further evaluated other phosphine ligands. Gratifyingly, the corresponding product 3a could be obtained in 40% yield when 1,1′-bis(dicyclohexylphosphino)ferrocene (L1) was employed as the ligand (Table 1, entry 6).a
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Pd] | Ligand | Base | Solvent | Yieldb (%) |
| a Reaction conditions: 1-bromo-2-(cyclopropylidenemethyl)benzene 1a (0.40 mmol), N-methyl-2-(phenylethynyl)benzenesulfonamide 2a (0.2 mmol), palladium catalyst (2.5 mol%), ligand (5 mol%), base (0.4 mmol), solvent (2.0 mL), reflux, N2, overnight. b Isolated yield based on N-methyl-2-(phenylethynyl)benzenesulfonamide 2a. c The reaction was performed at 80 °C. | |||||
| 1 | Pd(OAc)2 | DPPP | K2CO3 | Dioxane | NR |
| 2 | Pd(OAc)2 | XPhos | K2CO3 | Dioxane | Trace |
| 3 | Pd(OAc)2 | DPPF | K2CO3 | Dioxane | 16 |
| 4 | Pd(OAc)2 | PCy3 | K2CO3 | Dioxane | 23 |
| 5 | Pd(OAc)2 | SPhos | K2CO3 | Dioxane | 36 |
| 6 | Pd(OAc)2 | L1 | K2CO3 | Dioxane | 40 |
| 7 | Pd(OAc)2 | L1 | K3PO4 | Dioxane | NR |
| 8 | Pd(OAc)2 | L1 | DABCO | Dioxane | Trace |
| 9 | Pd(OAc)2 | L1 | KHCO3 | Dioxane | 10 |
| 10 | Pd(OAc)2 | L1 | Cs2CO3 | Dioxane | 24 |
| 11 | Pd(OAc)2 | L1 | KOAc | Dioxane | 25 |
| 12 | Pd2dba3 | L1 | K2CO3 | Dioxane | Trace |
| 13 | PdCl2 | L1 | K2CO3 | Dioxane | 33 |
| 14 | PdCl2(PPh3)2 | L1 | K2CO3 | Dioxane | 11 |
| 15 | PdCl2(dppf) | L1 | K2CO3 | Dioxane | 60 |
| 16 | Pd(TFA)2 | L1 | K2CO3 | Dioxane | 40 |
| 17 | PdCl2(dppf) | L1 | K2CO3 | DMF | NR |
| 18 | PdCl2(dppf) | L1 | K2CO3 | DMSO | NR |
| 19 | PdCl2(dppf) | L1 | K2CO3 | Toluene | 22 |
| 20 | PdCl2(dppf) | L1 | K2CO3 | AmylOH | Trace |
| 21 | PdCl2(dppf) | L1 | K2CO3 | Diglyme | 13 |
| 22c | PdCl2(dppf) | L1 | K2CO3 | Dioxane | 35 |
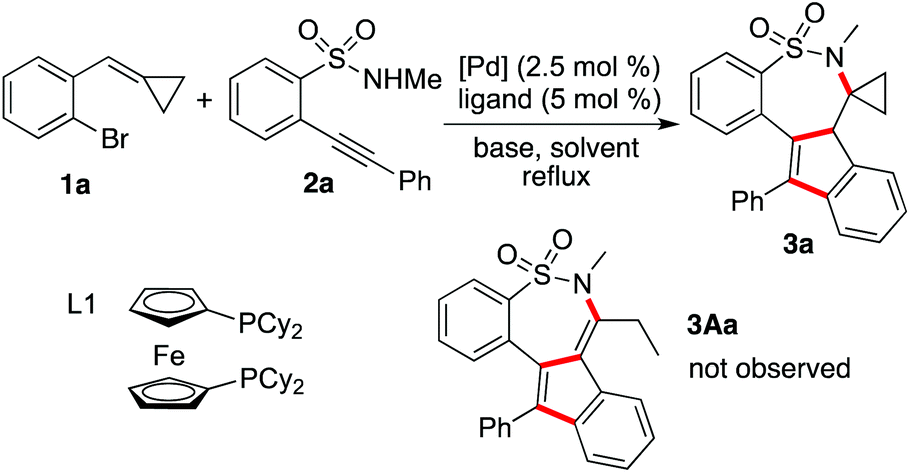
With this promising result in hand, we then explored the reaction in the presence of different bases (Table 1, entries 7–11) and no better yields were obtained. We also examined the reaction catalyzed by other palladium salts. It was found that the reaction worked efficiently when PdCl2(dppf) was employed (Table 1, entry 15), leading to the desired product 3a in 60% yield. Other solvents including DMF, DMSO, toluene, diglyme (diethylene glycol dimethyl ether), or AmylOH (2-methyl-2-butanol) were utilized in this transformation. However, the results were inferior. The yield of the corresponding product 3a was lower when the reaction was performed at 80 °C (Table 1, entry 22). The reaction was retarded when the amount of palladium catalyst was reduced (data not shown in Table 1).
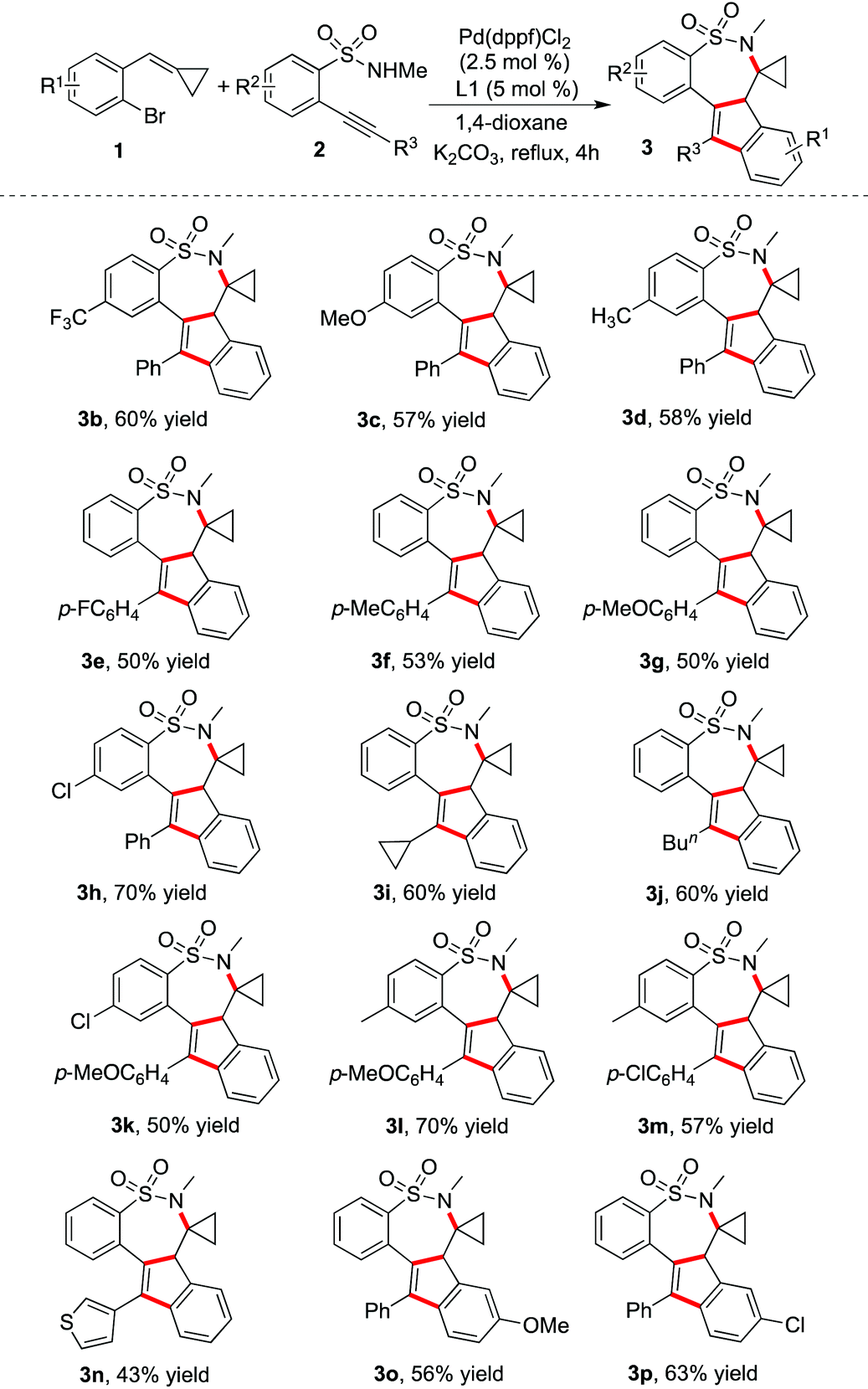
Under the above optimized conditions, the reaction scope was then investigated. Examples are presented in Table 2. Usually, the reactions went to completion in 4 hours. It was found that the palladium-catalyzed tandem reaction of 1-bromo-2-(cyclopropylidenemethyl)benzenes 1 with 2-alkynylbenzenesulfonamides 2 worked well, producing the corresponding 7,7a-dihydro-6H-benzo[f]indeno[1,2-d][1,2]thiazepine 5,5-dioxides in moderate to good yields. The polycyclic sultams could be constructed efficiently through double carbometallation with excellent chemoselectivity and regioselectivity. Different functional groups could be compatible under the standard conditions. For instance, the thiophenyl-substituted product 3n could be generated as expected. A range of compounds 2 with different substituents appended to the triple bond or the aromatic ring were good partners in the transformation as well. The electronic effects of R1 on the aromatic ring of compounds 1 were also explored, which demonstrated that both the electron-donating group and electron-withdrawing group were tolerated under the standard reaction conditions. Additionally, the substrates with the methyl or phenyl group changed from the cyclopropyl group were synthesized and examined under the reaction conditions. However, the reactions were complex and no desired products were generated. We reasoned that the cyclopropyl group might have an influence on controlling the coordination of Pd(II) in this transformation, which would promote the intramolecular insertion of a palladium intermediate into the double bond. In the meantime, we explored the reaction of N-methyl-1-(2-(phenylethynyl)phenyl)methanamine with 1-bromo-2-(cyclopropylidenemethyl)benzene 1a. No desired product through double carbometallation was observed.
| a Isolated yield based on 2-alkynylbenzenesulfonamide 2. |
|---|
|
Conclusions
In conclusion, we have developed an efficient route to polycyclic sultams through a palladium-catalyzed tandem reaction of 1-bromo-2-(cyclopropylidenemethyl)benzenes with 2-alkynylbenzenesulfonamides. The transformation proceeds through double carbometallation with excellent chemoselectivity and regioselectivity, leading to a range of 7,7a-dihydro-6H-benzo[f]indeno[1,2-d][1,2]thiazepine 5,5-dioxides in moderate to good yields. During the reaction process, three new bonds and two rings including the seven-membered sultam ring are formed.Acknowledgements
Financial support from the National Natural Science Foundation of China (no. 21372046 and 21532001) and the Opening Project of Gannan Medical University Collaborative Innovation Center for Gannan Oil-tea Camellia Industrial Development is gratefully acknowledged.Notes and references
- For selected examples: (a) A. K. Ganguly, S. S. Alluri, D. Caroccia, D. Biswas, C.-H. Wang, E. Kang, Y. Zhang, A. T. McPhail, S. S. Carroll, C. Burlein, V. Munshi, P. Orth and C. Strickland, J. Med. Chem., 2011, 54, 7176 CrossRef CAS PubMed; (b) L. Kiefer, T. Gorojankina, P. Dauban, H. Faure, M. Ruat and R. H. Dodd, Bioorg. Med. Chem. Lett., 2010, 20, 7483 CrossRef CAS PubMed; (c) P. Gilleron, N. Wlodarczyk, R. Houssin, A. Farce, G. Laconde, J.-F. Goossens, A. Lemoine, N. Pommery, J.-P. Henichart and R. Millet, Bioorg. Med. Chem. Lett., 2007, 17, 5465 CrossRef CAS PubMed; (d) G. Laconde, P. Depreux, P. Berthelot, N. Pommery and J.-P. Hénichart, Eur. J. Med. Chem., 2005, 40, 167 CrossRef CAS PubMed; (e) A. K. Ganguly, S. S. Alluri, D. Caroccia, D. Biswas, C.-H. Wang, E. Kang, Y. Zhang, A. T. McPhail, S. S. Carroll, C. Burlein, V. Munshi, P. Orth and C. Strickland, J. Med. Chem., 2011, 54, 7176 CrossRef CAS PubMed.
- S. Kasper and B. S. McEwen, CNS Drugs, 2008, 22, 15 CrossRef CAS PubMed.
- For reviews and accounts, see: (a) G. Qiu and J. Wu, Chem. Rec., 2016, 16, 19 CrossRef CAS PubMed; (b) Y. Luo, X. Pan, X. Yu and J. Wu, Chem. Soc. Rev., 2014, 43, 834 RSC; (c) G. Qiu, Y. Kuang and J. Wu, Adv. Synth. Catal., 2014, 356, 3483 CrossRef CAS; (d) L. He, H. Nie, G. Qiu, Y. Gao and J. Wu, Org. Biomol. Chem., 2014, 12, 9045 RSC; (e) G. Qiu and J. Wu, Synlett, 2014, 2703 CAS; (f) G. Qiu, Q. Ding and J. Wu, Chem. Soc. Rev., 2013, 42, 5257 RSC; (g) H. Wang, Y. Kuang and J. Wu, Asian J. Org. Chem., 2012, 1, 302 CrossRef CAS.
- (a) Q. Xiao, J. Sheng, Z. Chen and J. Wu, Chem. Commun., 2013, 49, 8647 RSC; (b) X. Gong, H. Xia and J. Wu, Org. Chem. Front., 2016, 3, 697 RSC; (c) Q. Xiao, Y. Luo, Y. Yan, Q. Ding and J. Wu, RSC Adv., 2013, 3, 5779 RSC; (d) H. Wang, Y. Luo, X. Hou and J. Wu, Dalton Trans., 2013, 42, 4410 RSC.
- J. Wang, G. Li, Y. Tong, Y. Deng, J. Wu and J. Fan, J. Practical Hepatology, 2015, 18, 344 Search PubMed.
- For selected examples, see: (a) X. Pan, Y. Luo, H. Xia and J. Wu, Chem. Commun., 2015, 51, 16483 RSC; (b) X. Pan, M. Chen, L. Yao and J. Wu, Chem. Commun., 2014, 50, 5891 RSC; (c) X. Gong, M. Chen, L. Yao and J. Wu, Chem. – Asian J., 2016, 11, 1613 CrossRef CAS PubMed; (d) X. Pan, Y. Luo, G. Liu, S. Pu and J. Wu, Adv. Synth. Catal., 2012, 354, 171 CrossRef CAS; (e) H. Wang, Y. Luo, B. Zhu and J. Wu, Chem. Commun., 2012, 48, 5581 RSC; (f) X. Pan, Y. Luo and J. Wu, Chem. Commun., 2011, 47, 8967 RSC; (g) Y. Luo, X. Pan and J. Wu, Org. Lett., 2011, 13, 1150 CrossRef CAS PubMed; (h) Y. Luo, L. Hong and J. Wu, Chem. Commun., 2011, 47, 5298 RSC; (i) Y. Luo and J. Wu, Chem. Commun., 2011, 47, 11137 RSC.
- For selected reviews, see: (a) L. F. Tietze, G. Brasche and K. Gericke, Domino Reactions in Organic Synthesis, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed; (b) J. H. Kim, Y. O. Ko, J. Bouffard and S.-g. Lee, Chem. Soc. Rev., 2015, 44, 2489 RSC; (c) L. F. Tietze, M. A. Düfert, T. Hungerland, K. Oum and T. Lenzer, Chem. – Eur. J., 2011, 17, 8452 CrossRef CAS PubMed; (d) J. Panteleev, L. Zhang and M. Lautens, Angew. Chem., Int. Ed., 2011, 50, 9089 CrossRef CAS PubMed.
- (a) X. Pan, Y. Luo, Y. Ding, X. Fan and J. Wu, Adv. Synth. Catal., 2014, 356, 1072 CrossRef CAS; (b) S. Li, Y. Luo, X. Wang, M. Guo and J. Wu, Chem. – Asian J., 2012, 7, 1691 CrossRef CAS PubMed and references cited therein.
- For selected reviews, see: (a) I. Nakamuran and Y. Yamamoto, Adv. Synth. Catal., 2002, 344, 111 CrossRef; (b) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2003, 103, 1213 CrossRef CAS PubMed; (c) E. Nakamura and S. Yamago, Acc. Chem. Res., 2002, 35, 867 CrossRef CAS PubMed; (d) L.-X. Shao and M. Shi, Curr. Org. Chem., 2007, 11, 1135 CrossRef CAS; (e) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed; (f) M. Shi, L.-X. Shao, J.-M. Lu, Y. Wei, K. Mizuno and H. Maeda, Chem. Rev., 2010, 110, 5883 CrossRef CAS PubMed.
- For selected examples, see: (a) T. M. Ha, B. Yao, Q. Wang and J. Zhu, Org. Lett., 2015, 17, 5256 CrossRef CAS PubMed; (b) M. Hellal and G. D. Cuny, Tetrahedron Lett., 2011, 52, 5508 CrossRef CAS; (c) D. K. Barange, V. R. Batchu, D. Gorja, V. R. Pattabiraman, L. K. Tatini, J. M. Babu and M. Pal, Tetrahedron, 2007, 63, 1775 CrossRef CAS.
- For selected examples, see: (a) P. Bhattacharya, K. Senapati, K. Chattopadhyay, S. M. Mandalb and A. Basak, RSC Adv., 2015, 5, 61562 RSC; (b) M. K. Ghorai, M. Sayyad, Y. Nanaji and S. Jana, Chem. – Asian J., 2015, 10, 1480 CrossRef CAS PubMed; (c) S. Shin, Y. Park, C. Kim, J. Son and P. Lee, J. Org. Chem., 2015, 80, 5859 CrossRef CAS PubMed; (d) J. Zhang, D. Cao, H. Wang, G. Zhao and Y. Shang, Tetrahedron, 2015, 71, 1785 CrossRef CAS; (e) S. Fang, X. Niu, Z. Zhang, Y. Sun, X. Si, C. Shan, L. Wei, A. Xu, L. Feng and C. Ma, Org. Biomol. Chem., 2014, 12, 6895 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and related data. CCDC 1491815. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00480f |
| This journal is © the Partner Organisations 2017 |