
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAerobic oxidation and EATA-based highly enantioselective synthesis of lamenallenic acid†
Xingguo
Jiang
a,
Yufeng
Xue
a and
Shengming
Ma
*ab
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, P. R. China. E-mail: masm@sioc.ac.cn
bDepartment of Chemistry, Fudan University, 220 Handan Lu, Shanghai 200433, P.R. China
First published on 8th February 2017
Abstract
By utilizing the recently developed EATA (enantioselective allenylation of terminal alkynes) reaction and the aerobic oxidation of alcohols, lamenallenic acid, a naturally occurring axially chiral allene, has been synthesized with a high ee value and E/Z selectivity from methyl 5-hexynoate and (E)-10-dodecenal, which could be prepared easily from 1-decyne and 1-pentyne, respectively, with (S)-α,α-dimethylprolinol as the chiral source.
Introduction
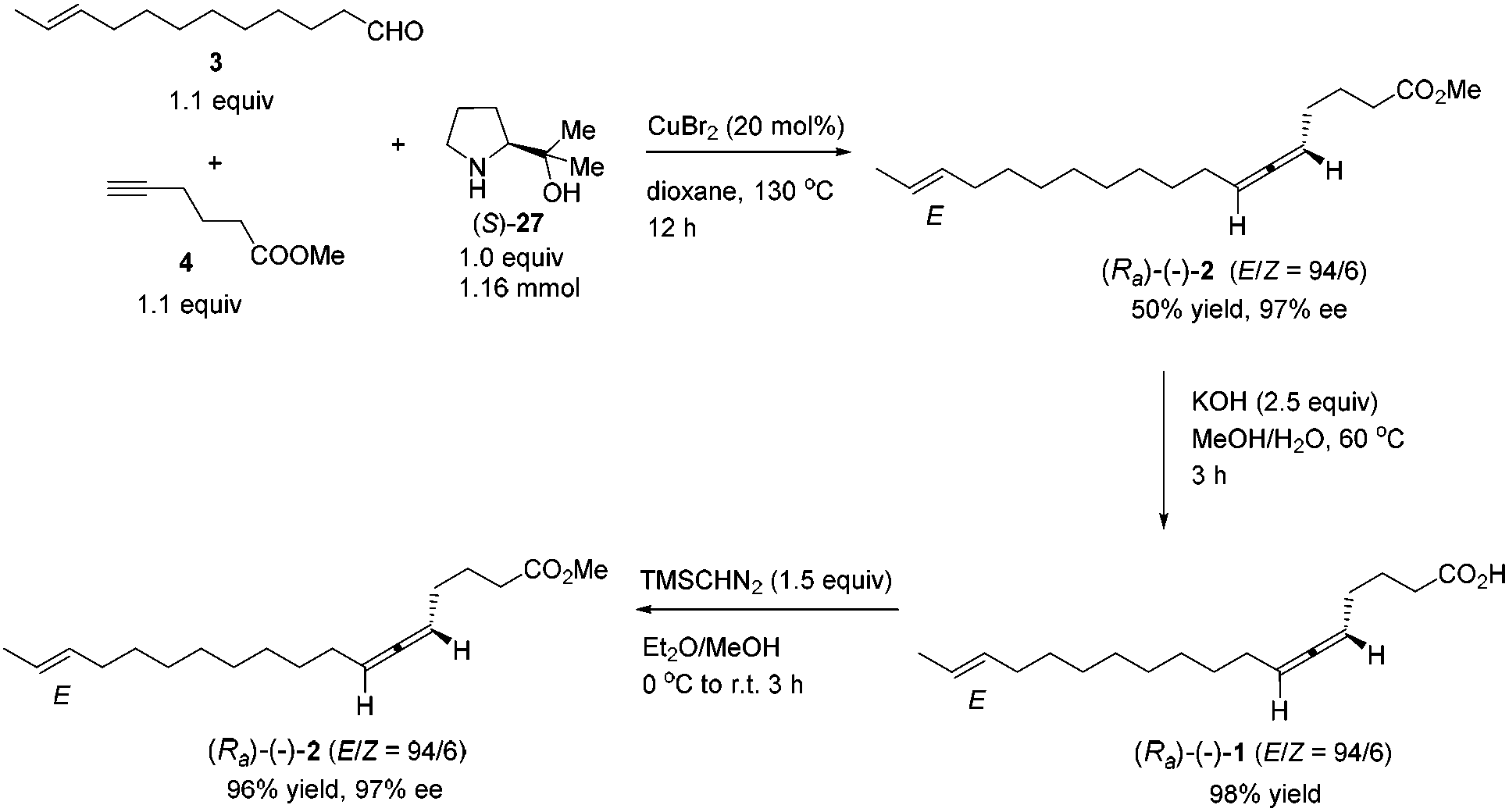
Currently, about 150 natural products containing an allenic or cumulenic structure have been isolated and characterized.1 With a typical structure of 1,3-disubstitued allene, lamenallenic acid was isolated from Lamium Purpureum L. (Labiatae) seed oil in 1967.2 This molecule contains a trans double bond, an axially chiral allene unit, and a free carboxylic acid group (Scheme 1). According to the literature,2 the natural product was laevorotatory and the configuration was expected to be Ra by comparing the specific rotation with (−)-laballenic acid of similar structure, which was previously determined to be Ra by the Landor group.3 Although the structure does not look too complicated, due to the lack of efficient methods for the highly stereoselective construction of the allene unit, in 1972, the Landor group finished the first total synthesis of racemic lamenallenic acid from 10-undecenoic acid and the THP-ether of 4-pentyn-2-enol in 12 steps.4 The allenic unit was synthesized by the reaction of 2,14-hexadecadien-4-yn-1-ol and LiAlH4. By adding cyclohexylidene-α-D-glucofuranose as the chiral source in this step, (−)-lamenallenic acid was obtained with a very low ee as judged by comparing the specific optical rotation of its (−)-methyl ester ([α]20D = −5.8 (c = 5.6, EtOH)) to the reported data for methyl ester of the natural lamenallenoic acid ([α]25D = −50 (c = 0.28, EtOH) at 589 nm).2 Thus, the known synthesis lacks efficiency and high stereoselectivity. Obviously, the challenges for synthesizing the naturally occurring lamenallenic acid are the highly stereoselective construction of the trans C–C double bond5 and the Ra-axially chiral allene unit.6 According to the EATA reaction recently developed by our group,7 we reasoned that methyl lamenallenate (Ra)-2 may be synthesized from (E)-10-dodecenal 3 and methyl 5-hexynoate 4 (Scheme 1).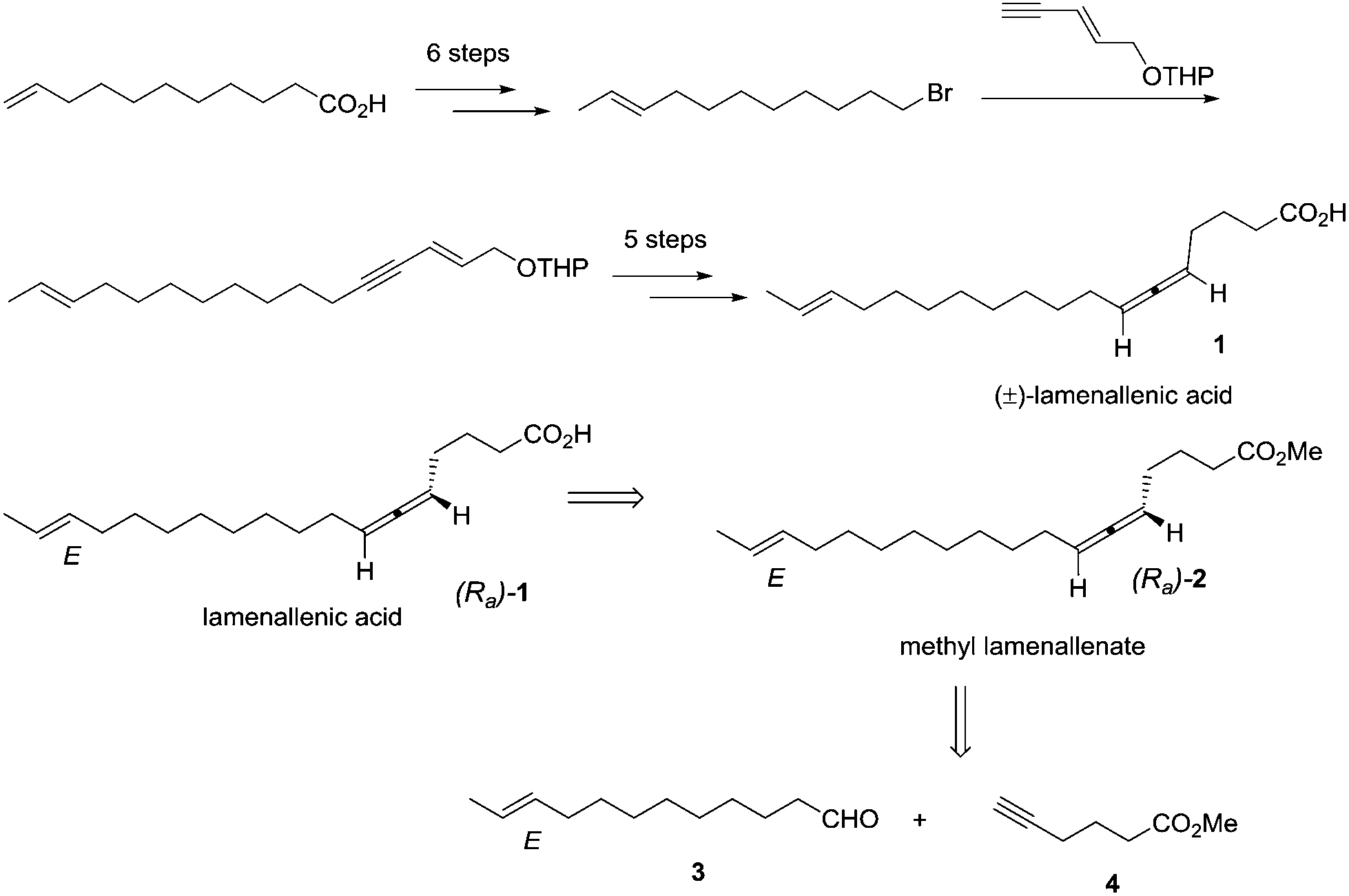 | ||
| Scheme 1 Reported synthesis and our retrosynthetic analysis of lamenallenic acid. | ||
Results and discussion
After some unsuccessful initial attempts to construct the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond with a high E/Z ratio involving compounds 6–16 (for details, see the ESI†), we observed that the reduction of 10-dodecynol THP ether 20, which was synthesized from 1-decyne 17 in 4 steps, with Li/NH3 (l)8 gave (E)-10-dodecenol THP ether 21 successfully in 98% yield with an E/Z ratio of 96/4 (Scheme 2). After deprotection, the trans dodec-10-enol 16 underwent an aerobic oxidation reaction using Fe(NO3)3·9H2O/TEMPO/NaCl9,10 as catalysts to give trans 10-dodecynal 3.
C double bond with a high E/Z ratio involving compounds 6–16 (for details, see the ESI†), we observed that the reduction of 10-dodecynol THP ether 20, which was synthesized from 1-decyne 17 in 4 steps, with Li/NH3 (l)8 gave (E)-10-dodecenol THP ether 21 successfully in 98% yield with an E/Z ratio of 96/4 (Scheme 2). After deprotection, the trans dodec-10-enol 16 underwent an aerobic oxidation reaction using Fe(NO3)3·9H2O/TEMPO/NaCl9,10 as catalysts to give trans 10-dodecynal 3.
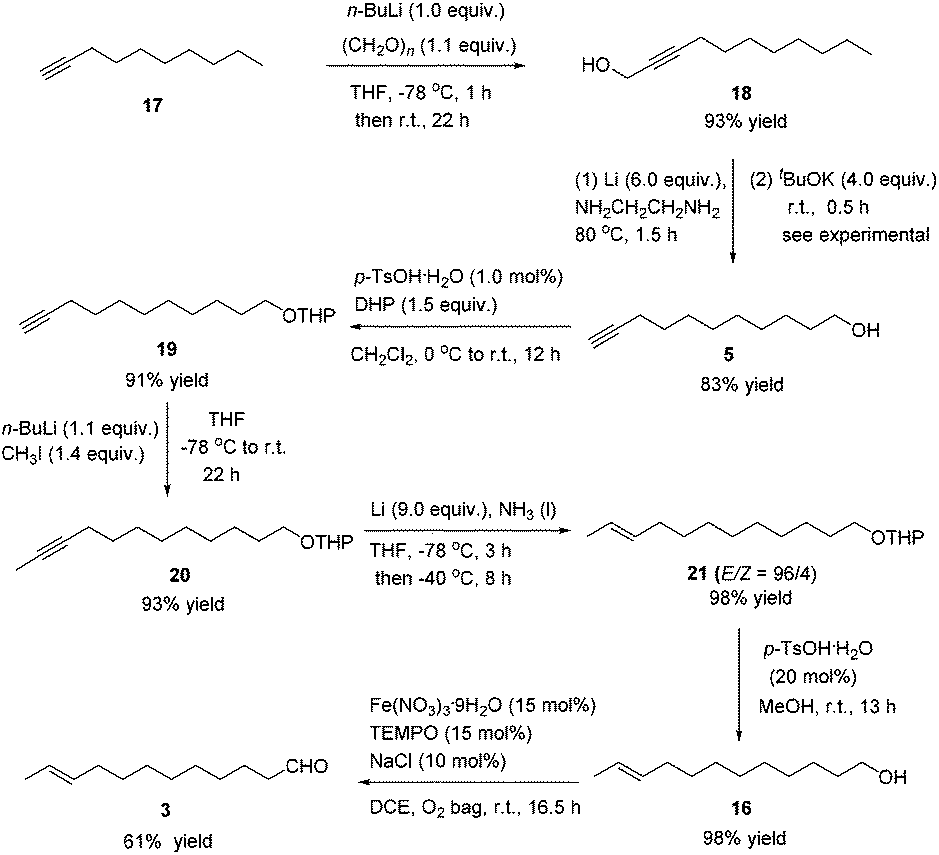 | ||
| Scheme 2 Synthetic route to (E)-10-dodecenal 3. | ||
Methyl 5-hexynoate 4 was then synthesized from 1-pentyne 22 in 4 steps (Scheme 3): 5-hexyn-1-ol 24 was prepared from 1-pentyne 22 through a similar process for the preparation of alkynol 5 from 17, which was further oxidized to 5-hexynoic acid 25 by applying the Fe(NO3)3·9H2O/TEMPO/KCl-catalyzed aerobic oxidation reaction11 resulting in 86% yield with an O2 bag and in 85% yield with an air bag. The esterification reaction with DCC, DMAP and MeOH in CH2Cl2 afforded methyl 5-hexynoate 4 in 77% yield.
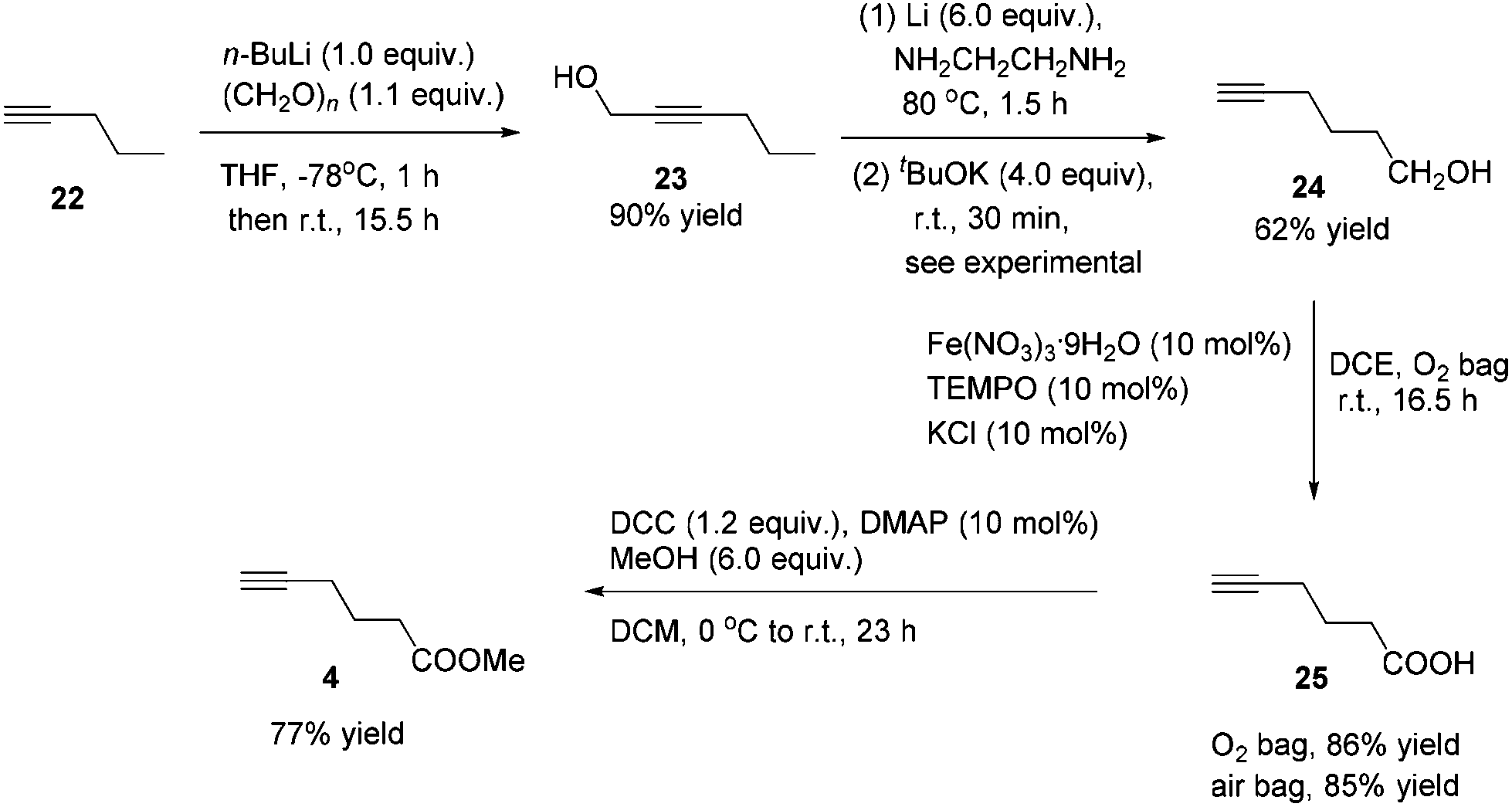 | ||
| Scheme 3 The synthetic route to methyl 5-hexynoate 4. | ||
We then tested the EATA reaction of the terminal alkyne 4 with E-enal 3: the reaction under the reported conditions7 with 20 mol% of CuBr2, 1.5 equiv. of alkyne 4, 1.5 equiv. of aldehyde 3 and 1.0 equiv. of (S)-diphenyl prolinol 26 in dioxane at 130 °C successfully afforded 47% yield of methyl lamenallenate (Ra)-2 with 93% ee. The yield and ee were almost not affected when reducing the amount of aldehyde from 1.5 to 1.1 (entries 1–3, Table 1). With 1.0 equiv. of aldehyde, the yield dropped to 45% (entry 4, Table 1). Increasing the amount of chiral amine (S)-26 to 1.5 equiv. or alkyne to 2.0 equiv. did not significantly improve the yield or ee (entries 5 and 6, Table 1). However, changing the chiral amino alcohol from (S)-diphenyl prolinol (S)-26 to (S)-dimethyl prolinol (S)-27 increased the yield to 59% and ee to 97% (entry 7, Table 1). The ee remained high when reducing both terminal alkyne 4 and aldehyde 3 to 1.1 equiv. (entries 8–10, Table 1).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | X | y | z | R | Yieldb of (Ra)-2 (%) | ee (%) |
| a The reaction was conducted on the 0.1 mmol scale with 0.02 mmol of CuBr2, x equiv. of 4, y equiv. of 3 and z equiv. of chiral amine in 0.8 mL of dioxane at 130 °C in a sealed tube for 12 h. b NMR yield. Isolated yield is given in parentheses. | ||||||
| 1 | 1.5 | 1.5 | 1.0 | Ph | 52 (47) | 93 |
| 2 | 1.5 | 1.2 | 1.0 | Ph | 50 (48) | 92 |
| 3 | 1.5 | 1.1 | 1.0 | Ph | 50 (48) | 92 |
| 4 | 1.5 | 1.0 | 1.0 | Ph | 48 (45) | 92 |
| 5 | 1.5 | 1.0 | 1.5 | Ph | 48 (48) | 91 |
| 6 | 2.0 | 1.1 | 1.0 | Ph | 51 (48) | 93 |
| 7 | 1.5 | 1.5 | 1.0 | Me | 59 (59) | 97 |
| 8 | 1.5 | 1.1 | 1.0 | Me | 57 (56) | 97 |
| 9 | 1.2 | 1.1 | 1.0 | Me | 55 (54) | 97 |
| 10 | 1.1 | 1.1 | 1.0 | Me | 55 (55) | 96 |
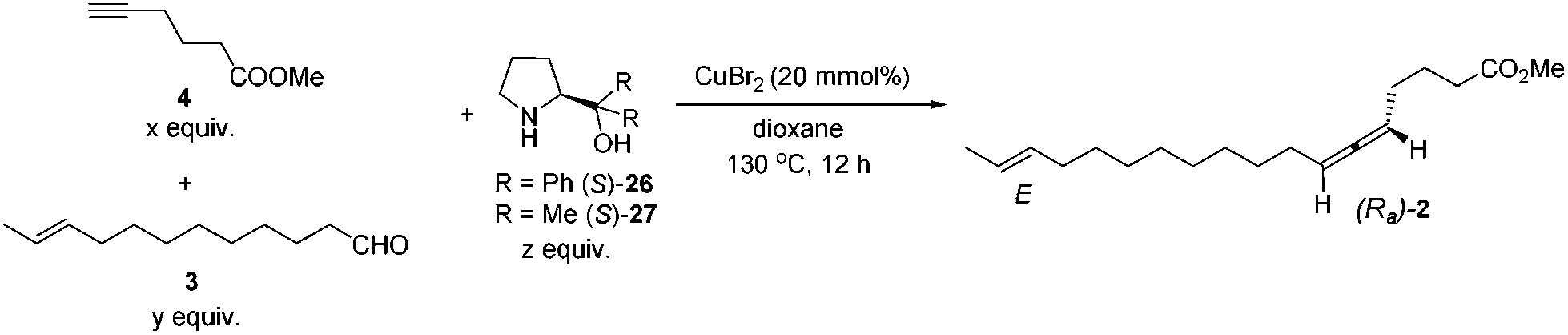
By applying 20 mol% of CuBr2, 1.1 equiv. of alkyne 4, 1.1 equiv. of aldehyde 3 and 1.0 equiv. of (S)-dimethyl prolinol (S)-27 in dioxane at 130 °C as the standard conditions, the reaction on the 1.16 mmol scale afforded methyl lamenallenate (Ra)-(−)-2 in 50% yield with 97% ee ([α]29.3D = −52.8 (c = 0.305, EtOH)) (lit:2 [α]25D−50 (c = 0.28, EtOH)) (Scheme 4). KOH was used in MeOH at 60 °C to execute the hydrolysis to produce (−)-lamenallenic acid (Ra)-(−)-1 in 98% yield ([α]29.4D = −55.2 (c = 0.995, EtOH), E/Z = 94/6). For determination of ee of the synthesized (−)-lamenallenic acid, the prepared acid was subjected to a methylation reaction to reproduce (−)-methyl lamenallenate (Ra)-(−)-2 with 97% ee. Thus, the synthesis of (−)-lamenallenic acid with 97% ee was accomplished.
 | ||
| Scheme 4 Synthesis of (−)-lamenallenic acid. | ||
Conclusions
In conclusion, we finished the highly enantioselective total synthesis of (−)-lamenallenic acid with high optical purity in 9 steps from 1-decyne and 1-pentyne. By applying (S)-dimethyl prolinol as the chiral source, the EATA reaction of (E)-10-dodecenal and methyl 5-hexynoate, which were prepared from two simple terminal alkynes by routine chemical transformations and iron-catalyzed aerobic oxidation reactions developed by our group, was accomplished with high ee. This has nicely demonstrated the direct tolerance of functional groups such as the CC bond and carboxylate. Based on our previous report,7 the reaction of a terminal alkyne with an aldehyde in the presence of (S)-prolinol leads to (Ra)-allene; thus, we confirmed the absolute configuration of the allene moiety in natural lamenallenic acid. Compared to the known synthesis, the current synthesis enjoys a high efficiency and high enantioselectivity. Further studies are being pursued in this laboratory.
Experimental
General information
1H and 13C nuclear magnetic resonance spectra were recorded with an instrument operated at 400 MHz for 1H NMR and 100 MHz for 13C NMR spectra. THF and dioxane were dried over sodium wire and distilled freshly before use. DCE was used directly without further treatment unless mentioned otherwise. (S)-Dimethyl prolinol (S)-2712 was synthesized by following the reported procedure. Other reagents were used as received without further treatment.CAUTION: Oxygen in use with combination with organic solvents; remove all ignition sources including sources of sparks, static, or flames since oxygen increases the intensity of any fire. Inhalation of pure oxygen should be avoided as well.
Experimental details and analytical data
To a 250 mL flame-dried three-necked flask were added 17 (11.6421 g, 95% purity, 80 mmol) and anhydrous THF (80 mL). A solution of n-BuLi (2.5 M in hexane, 32.0 mL, 80 mmol) was added dropwise at −78 °C under an Ar atmosphere in 25 min. After 1 h, paraformaldehyde (2.6426 g, 88 mmol) was added into the flask and the resulting mixture was stirred at −78 °C for 1 h. The reaction was then allowed to warm to room temperature naturally and stirred for 22 h. A saturated NH4Cl aqueous solution (40 mL) was added to quench the reaction. Et2O (200 mL) and H2O (100 mL) were added. The organic layer was separated and the aqueous phase was extracted by ethyl ether (60 × 3 mL). The combined organic layer was washed with brine and dried over anhydrous Na2SO4. After filtration and evaporation, the residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 20/1 to 5/1) to afford 1813 (12.5673 g, 93%) as an oil: 1H NMR (400 MHz, CDCl3) δ 4.28–4.22 (m, 2 H, OCH2), 2.21 (tt, J1 = 7.0 Hz, J2 = 2.1 Hz, 2 H, C

![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2), 1.87 (brs, 1 H, OH), 1.51 (quint, J = 7.2 Hz, 2 H, CH2), 1.42–1.20 (m, 10 H, 5 × CH2), 0.88 (t, J = 6.8 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 86.6, 76.7, 51.3, 31.8, 29.14, 29.06, 28.8, 28.6, 22.6, 18.7, 14.0; IR (neat) ν (cm−1) 3336, 2925, 2855, 2289, 2226, 1462, 1376, 1228, 1137, 1012; MS (ESI) m/z (%): 191 ((M + Na)+).
CCH2), 1.87 (brs, 1 H, OH), 1.51 (quint, J = 7.2 Hz, 2 H, CH2), 1.42–1.20 (m, 10 H, 5 × CH2), 0.88 (t, J = 6.8 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 86.6, 76.7, 51.3, 31.8, 29.14, 29.06, 28.8, 28.6, 22.6, 18.7, 14.0; IR (neat) ν (cm−1) 3336, 2925, 2855, 2289, 2226, 1462, 1376, 1228, 1137, 1012; MS (ESI) m/z (%): 191 ((M + Na)+).
To a 250 mL three-necked flask were added a Li rod (2.0820 g, 300.0 mmol) and NH2CH2CH2NH2 (200 mL) under an Ar atmosphere. The resulting mixture was stirred for 1.5 h at 80 °C with a preheated oil bath. After being cooled to room temperature naturally, tBuOK (22.4435 g, 200.0 mmol) was added and the mixture was stirred for 30 min at room temperature. 18 (8.4130 g, 50.0 mmol) was added dropwise within 5 min. After being stirred at room temperature for 2 h, the reaction mixture was poured into a 1 L beaker containing a mixture of ice and water (200 mL). Et2O (300 mL) was added to separate the organic layer and the aqueous phase was extracted with ethyl ether (100 × 3 mL). The combined organic layer was washed sequentially with 1 M aqueous HCl (80 mL), a saturated NaHCO3 aqueous solution (80 mL), and brine. After being dried over anhydrous Na2SO4, the mixture was filtered and evaporated. The residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 9/1 to 3/1) to afford 515 (7.0109 g, 83%) as an oil: 1H NMR (400 MHz, CDCl3) δ 3.63 (t, J = 6.6 Hz, 2 H, OCH2), 2.18 (td, J1 = 7.1 Hz, J2 = 2.8 Hz, 2 H, C
 CCH2), 1.94 (t, J = 2.6 Hz, 1 H, CCH), 1.70–1.48 (m, 5 H, OH + 2 × CH2), 1.44–1.22 (m, 10 H, 5 × CH2); 13C NMR (100 MHz, CDCl3) δ 84.7, 68.0, 62.9, 32.7, 29.4, 29.3, 29.0, 28.6, 28.4, 25.6, 18.3; IR (neat) ν (cm−1) 3308, 2928, 2856, 2116, 1462, 1055; MS (ESI) m/z (%): 169 (M + H)+.
CCH2), 1.94 (t, J = 2.6 Hz, 1 H, CCH), 1.70–1.48 (m, 5 H, OH + 2 × CH2), 1.44–1.22 (m, 10 H, 5 × CH2); 13C NMR (100 MHz, CDCl3) δ 84.7, 68.0, 62.9, 32.7, 29.4, 29.3, 29.0, 28.6, 28.4, 25.6, 18.3; IR (neat) ν (cm−1) 3308, 2928, 2856, 2116, 1462, 1055; MS (ESI) m/z (%): 169 (M + H)+.
To a 100 mL Schlenk flask were added 5 (1.6815 g, 10.0 mmol) and CH2Cl2 (20 mL). p-TsOH·H2O (19.0 mg, 0.1 mmol) and DHP (1.37 mL, d = 0.922 g mL−1, 1.2618 g, 15.0 mmol) were added at 0 °C. Then, the resulting mixture was allowed to warm up to room temperature and stirred for 12 h. A saturated NaHCO3 aqueous solution (5 mL) was added to quench the reaction. Et2O (50 mL) and H2O (50 mL) were added. The organic phase was separated and the aqueous phase was extracted with ethyl ether (25 × 3 mL). The combined organic layer was washed with brine and dried over anhydrous Na2SO4. After filtration and evaporation, the residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl ether = 100/1 to 50/1) to afford 1916 (2.2915 g, 91%) as an oil: 1H NMR (400 MHz, CDCl3) δ 4.60–4.55 (m, 1 H, CH of the THP group), 3.91–3.83 (m, 1 H, one proton of the THP group), 3.77–3.69 (dt, J1 = 9.6 Hz, J2 = 7.0 Hz, 1 H, one proton of OCH2), 3.54–3.46 (m, 1 H, one proton of the THP group), 3.42–3.34 (dt, J1 = 9.6 Hz, J2 = 6.8 Hz, 1 H, one proton of OCH2), 2.18 (td, J1 = 7.1 Hz, J2 = 2.8 Hz, 2 H, C
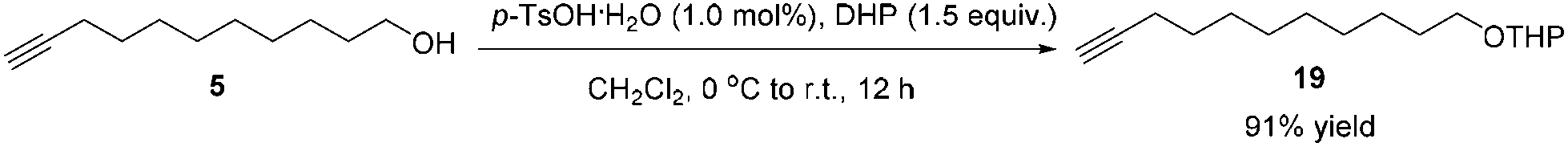 CCH2), 1.94 (t, J = 2.6 Hz, 1 H, CCH), 1.89–1.78 (m, 1 H, one proton of the THP group), 1.76–1.67 (m, 1 H, one proton of the THP group), 1.64–1.47 (m, 8 H, four protons of the THP group + 2 × CH2), 1.44–1.24 (m, 10 H, 5 × CH2); 13C NMR (100 MHz, CDCl3) δ 98.8, 84.7, 68.0, 67.6, 62.3, 30.7, 29.7, 29.4, 29.0, 28.7, 28.4, 26.1, 25.4, 19.6, 18.3; IR (neat) ν (cm−1) 3310, 2930, 2856, 2117, 1460, 1350, 1260, 1202, 1121, 1076, 1029; MS (EI) m/z (%): 252 (M+, 0.27), 85 (100).
CCH2), 1.94 (t, J = 2.6 Hz, 1 H, CCH), 1.89–1.78 (m, 1 H, one proton of the THP group), 1.76–1.67 (m, 1 H, one proton of the THP group), 1.64–1.47 (m, 8 H, four protons of the THP group + 2 × CH2), 1.44–1.24 (m, 10 H, 5 × CH2); 13C NMR (100 MHz, CDCl3) δ 98.8, 84.7, 68.0, 67.6, 62.3, 30.7, 29.7, 29.4, 29.0, 28.7, 28.4, 26.1, 25.4, 19.6, 18.3; IR (neat) ν (cm−1) 3310, 2930, 2856, 2117, 1460, 1350, 1260, 1202, 1121, 1076, 1029; MS (EI) m/z (%): 252 (M+, 0.27), 85 (100).
To a 250 mL flame-dried three-neck flask were added 19 (2.0210 g, 8.0 mmol) and anhydrous THF (25 mL) under an Ar atmosphere. A solution of n-BuLi (2.5 M in hexanes, 3.52 mL, 8.8 mmol) was added dropwise at −78 °C in 5 min. After 30 min, a solution of CH3I (0.69 mL, d = 2.3 g mL−1, 1.5897 g, 11.2 mmol) in anhydrous THF (20 mL) was added dropwise within 10 min at −78 °C. The resulting mixture was allowed to warm up to room temperature naturally and stirred for 22 h. A saturated NH4Cl aqueous solution (30 mL) was added to quench the reaction. Et2O (200 mL) and H2O (100 mL) were added. The organic layer was separated and the aqueous phase was extracted by ethyl ether (50 × 3 mL). The combined organic layer was washed with brine and dried over anhydrous Na2SO4. After filtration and evaporation, the residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl ether = 30/1) to afford 208 (1.9848 g, 93%) as an oil: 1H NMR (400 MHz, CDCl3) δ 4.61–4.54 (m, 1 H, CH of the THP group), 3.92–3.82 (m, 1 H, one proton of the THP group), 3.73 (dt, J1 = 9.6 Hz, J2 = 6.9 Hz, 1 H, one proton of OCH2), 3.55–3.45 (m, 1 H, one proton of the THP group), 3.38 (dt, J1 = 9.6 Hz, J2 = 6.7 Hz, 1 H, one proton of OCH2), 2.14–2.07 (m, 2 H, CH2), 1.88–1.80 (m, 1 H, one proton of the THP group), 1.78 (t, J = 2.4 Hz, 3 H, CH3), 1.76–1.67 (m, 1 H, one proton of the THP group), 1.64–1.42 (m, 8 H, four protons of the THP group + 2 × CH2), 1.41–1.22 (m, 10 H, 5 × CH2); 13C NMR (100 MHz, CDCl3) δ 98.8, 79.3, 75.2, 67.6, 62.3, 30.7, 29.7, 29.41, 29.38, 29.1, 29.0, 28.8, 26.2, 25.5, 19.6, 18.7, 3.4; IR (neat) ν (cm−1) 2928, 2856, 1460, 1350, 1260, 1202, 1122, 1076, 1031; MS (EI) m/z (%): 266 (M+, 0.94), 85 (100).
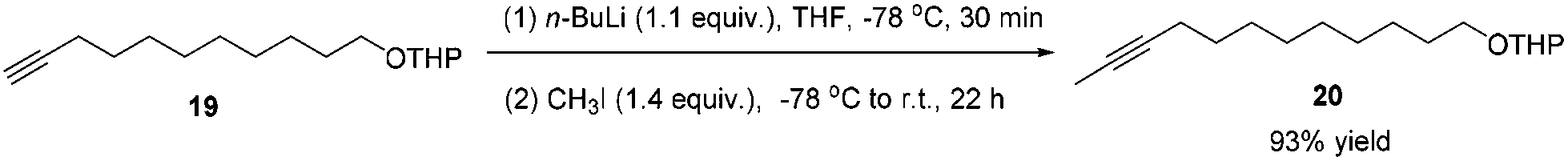
To a 500 mL three-necked flask was introduced NH3 gas at −78 °C to collect ∼150 mL of liquid NH3. 20 (4.5080 g, 17 mmol), THF (85 mL) and a Li rod (1.0178 g, 153 mmol) were added sequentially at −78 °C. After stirring at −78 °C for 3 h, the resulting mixture was then warmed to −40 °C and stirred for 8 h until the completion of the reaction as monitored by TLC. A saturated NH4Cl aqueous solution (40 mL) was added to quench the reaction. The flask was opened to air overnight to allow the NH3 (l) to evaporate. Et2O (40 mL) and H2O (20 mL) were added and the organic layer was separated. The aqueous phase was extracted by ethyl ether (30 × 3 mL). The combined organic layer was washed with brine and dried over anhydrous MgSO4. After filtration and evaporation, the residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl ether = 50/1) to afford 2117 (E/Z = 96/4) (4.4510 g, 98%) as an oil: 1H NMR (400 MHz, CDCl3) δ 5.47–5.35 (m, 2 H, CH
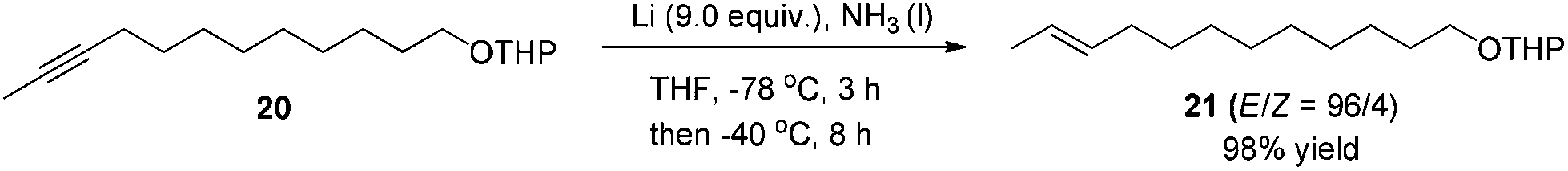 CH), 4.59–4.56 (m, 1 H, CH of the THP group), 3.91–3.83 (m, 1 H, one proton of the THP group), 3.73 (dt, J1 = 10.0 Hz, J2 = 7.0 Hz, 1 H, one proton of OCH2), 3.53–3.46 (m, 1 H, one proton of the THP group), 3.38 (dt, J1 = 9.6 Hz, J2 = 6.7 Hz, 1 H, one proton of the THP group), 2.00–1.91 (m, 2 H, CH2), 1.88–1.78 (m, 1 H, one proton of the THP group), 1.76–1.68 (m, 1 H, one proton of the THP group), 1.66–1.62 (m, 3 H, CH3), 1.62–1.46 (m, 6 H, four protons of the THP group + CH2), 1.40–1.21 (m, 12 H, 6 × CH2); 13C NMR (100 MHz, CDCl3) δ 131.6, 124.5, 98.8, 67.6, 62.3, 32.6, 30.8, 29.7, 29.6, 29.5, 29.44, 29.43, 29.1, 26.2, 25.5, 19.7, 17.9; IR (neat) ν (cm−1) 2925, 2854, 1458, 1321, 1260, 1202, 1122, 1077, 1030; MS (EI) m/z (%): 268 (M+, 0.33), 85 (100). The following signals are discernible for 21 (Z): 1H NMR (400 MHz, CDCl3) δ 2.07–2.00 (m, 2 H, CH2).
CH), 4.59–4.56 (m, 1 H, CH of the THP group), 3.91–3.83 (m, 1 H, one proton of the THP group), 3.73 (dt, J1 = 10.0 Hz, J2 = 7.0 Hz, 1 H, one proton of OCH2), 3.53–3.46 (m, 1 H, one proton of the THP group), 3.38 (dt, J1 = 9.6 Hz, J2 = 6.7 Hz, 1 H, one proton of the THP group), 2.00–1.91 (m, 2 H, CH2), 1.88–1.78 (m, 1 H, one proton of the THP group), 1.76–1.68 (m, 1 H, one proton of the THP group), 1.66–1.62 (m, 3 H, CH3), 1.62–1.46 (m, 6 H, four protons of the THP group + CH2), 1.40–1.21 (m, 12 H, 6 × CH2); 13C NMR (100 MHz, CDCl3) δ 131.6, 124.5, 98.8, 67.6, 62.3, 32.6, 30.8, 29.7, 29.6, 29.5, 29.44, 29.43, 29.1, 26.2, 25.5, 19.7, 17.9; IR (neat) ν (cm−1) 2925, 2854, 1458, 1321, 1260, 1202, 1122, 1077, 1030; MS (EI) m/z (%): 268 (M+, 0.33), 85 (100). The following signals are discernible for 21 (Z): 1H NMR (400 MHz, CDCl3) δ 2.07–2.00 (m, 2 H, CH2).
To a 100 mL round bottom flask were added 21 (4.2570 g, 16.0 mmol), p-TsOH·H2O (603.7 mg, 3.2 mmol), and MeOH (32 mL). The reaction was stirred for 13 h until the completion of the reaction as monitored by TLC. A saturated NaHCO3 aqueous solution (40 mL) was added to quench the reaction. After filtration, MeOH was removed by evaporation. Et2O (40 mL) and H2O (20 mL) were added and the organic layer was separated. The aqueous phase was extracted by ethyl ether (30 × 3 mL). The combined organic layer was washed with brine (30 mL) and dried over anhydrous MgSO4. After filtration and evaporation, the residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 5/1 to 2/1) to afford 168 (2.8620 g, 98%) as an oil: 1H NMR (400 MHz, CDCl3) δ 5.47–5.35 (m, 2 H, CH
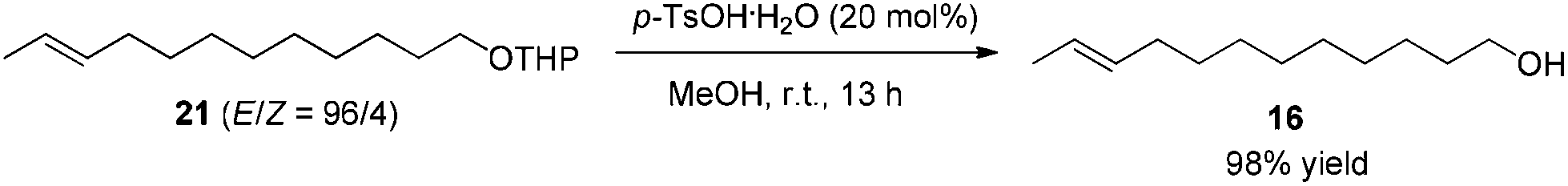 CH), 3.62 (t, J = 6.6 Hz, 2 H, OCH2), 1.99–1.92 (m, 2 H, CH2), 1.80 (brs, 1 H, OH), 1.64 (dd, J1 = 3.6 Hz, J2 = 1.2 Hz, 3 H, CH3), 1.56 (quint, J = 7.0 Hz, 2 H, CH2), 1.39–1.20 (m, 12 H, 6 × CH2); 13C NMR (100 MHz, CDCl3) δ 131.6, 124.5, 62.9, 32.7, 32.5, 29.55, 29.53, 29.41, 29.38, 29.1, 25.7, 17.9; IR (neat) ν (cm−1) 3364, 2924, 2854, 1459, 1377, 1056; MS (EI) m/z (%): 184 (M+, 0.54), 55 (100).
CH), 3.62 (t, J = 6.6 Hz, 2 H, OCH2), 1.99–1.92 (m, 2 H, CH2), 1.80 (brs, 1 H, OH), 1.64 (dd, J1 = 3.6 Hz, J2 = 1.2 Hz, 3 H, CH3), 1.56 (quint, J = 7.0 Hz, 2 H, CH2), 1.39–1.20 (m, 12 H, 6 × CH2); 13C NMR (100 MHz, CDCl3) δ 131.6, 124.5, 62.9, 32.7, 32.5, 29.55, 29.53, 29.41, 29.38, 29.1, 25.7, 17.9; IR (neat) ν (cm−1) 3364, 2924, 2854, 1459, 1377, 1056; MS (EI) m/z (%): 184 (M+, 0.54), 55 (100).
To a 50 mL Schlenk flask were added Fe(NO3)3·9H2O (145.6 mg, 0.36 mmol), TEMPO (56.5 mg, 0.36 mmol), NaCl (14.1 mg, 0.24 mmol), 16 (442.5 mg, 2.4 mmol), and DCE (9.6 mL) sequentially under the atmosphere of oxygen from a gas bag. The reaction was then carried out at room temperature for 16.5 h. The crude reaction mixture was filtered through a short column of silica gel eluted with ethyl ether (3 × 40 mL). After evaporation, the residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 30/1) to afford 318 (267.2 mg, 61%) as an oil: 1H NMR (400 MHz, CDCl3) δ 9.76 (t, J = 1.8 Hz, 1 H, CHO), 5.47–5.35 (m, 2 H, CH
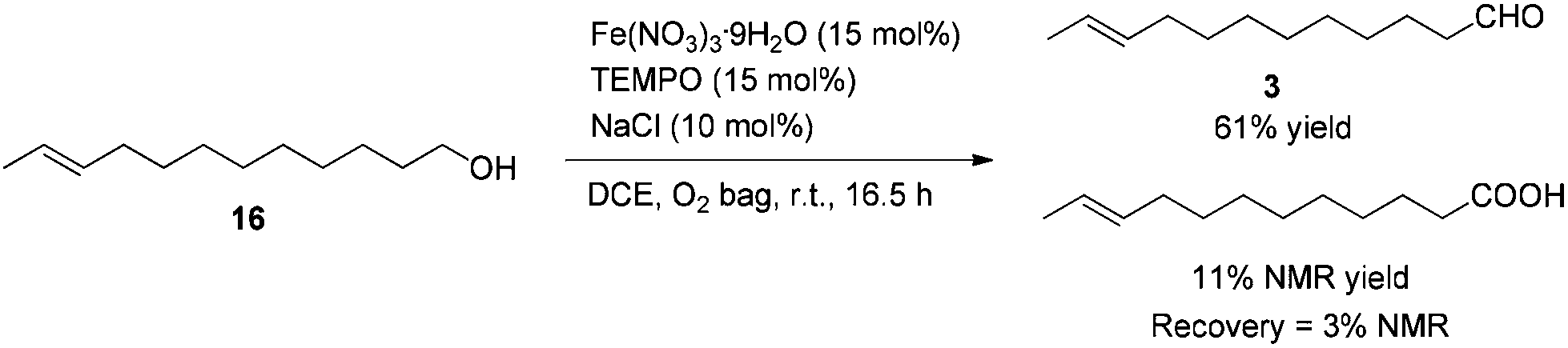 CH), 2.42 (td, J1 = 7.4 Hz, J2 = 2.0 Hz, 2 H, CH2), 1.99–1.90 (m, 2 H, CHCHCH2), 1.68–1.58 (m, 5 H, CH3 + CH2), 1.38–1.21 (m, 10 H, 5 × CH2); 13C NMR (100 MHz, CDCl3) δ 203.0, 131.5, 124.6, 43.9, 32.5, 29.5, 29.3, 29.2, 29.1, 29.0, 22.0, 17.9; IR (neat) ν (cm−1) 2925, 2854, 2715, 1727, 1455, 1411, 1013; MS (EI) m/z (%): 182 (M+, 0.67), 55 (100).
CH), 2.42 (td, J1 = 7.4 Hz, J2 = 2.0 Hz, 2 H, CH2), 1.99–1.90 (m, 2 H, CHCHCH2), 1.68–1.58 (m, 5 H, CH3 + CH2), 1.38–1.21 (m, 10 H, 5 × CH2); 13C NMR (100 MHz, CDCl3) δ 203.0, 131.5, 124.6, 43.9, 32.5, 29.5, 29.3, 29.2, 29.1, 29.0, 22.0, 17.9; IR (neat) ν (cm−1) 2925, 2854, 2715, 1727, 1455, 1411, 1013; MS (EI) m/z (%): 182 (M+, 0.67), 55 (100).
Compound 23 was prepared following the same procedure as preparation of compound 18. The reaction of 22 (3.9134 g, 98% purity, 56.3 mmol), n-BuLi (2.5 M in hexanes, 22.5 mL, 56.3 mmol), and paraformaldehyde (1.9583 g, 95% purity, 61.9 mmol) in anhydrous THF (56 mL) afforded 2319 (4.9558 g, 90%) as an oil: 1H NMR (400 MHz, CDCl3) δ 4.29–4.23 (m, 2 H, OCH2), 2.20 (tt, J1 = 7.2 Hz, J2 = 2.2 Hz, 2 H, C
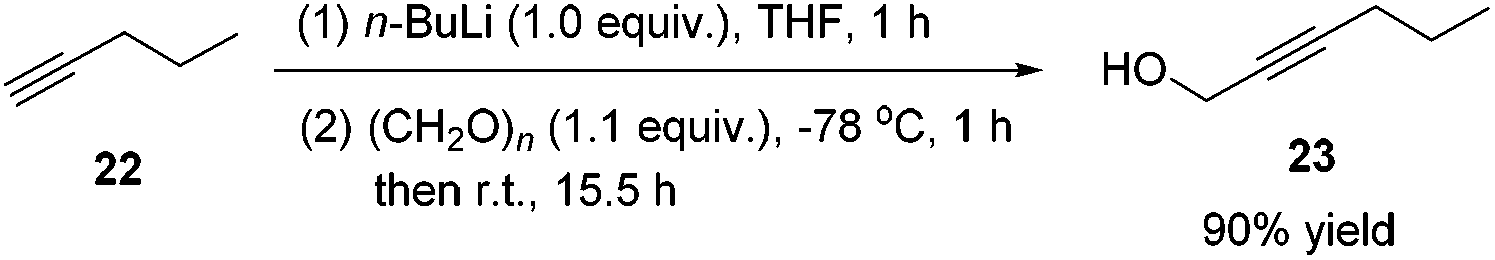 CCH2), 1.56–1.49 (m, 3 H, CH2 + OH), 0.98 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (100 MHz, CDCl3) δ 86.2, 78.4, 51.1, 21.9, 20.6, 13.4; IR (neat) ν (cm−1) 3329, 2963, 2935, 2872, 2289, 2227, 1457, 1431, 1380, 1338, 1136, 1032, 1003; MS (ESI) m/z (%): 98 (M+, 15.61), 83 (100).
CCH2), 1.56–1.49 (m, 3 H, CH2 + OH), 0.98 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (100 MHz, CDCl3) δ 86.2, 78.4, 51.1, 21.9, 20.6, 13.4; IR (neat) ν (cm−1) 3329, 2963, 2935, 2872, 2289, 2227, 1457, 1431, 1380, 1338, 1136, 1032, 1003; MS (ESI) m/z (%): 98 (M+, 15.61), 83 (100).
Compound 24 was prepared following the same procedure as preparation of compound 5. The reaction of the Li rod (417.3 mg, 60.0 mmol), tBuOK (4.4892 g, 40.0 mmol), and 23 (981.7 mg, 10.0 mmol) in NH2CH2CH2NH2 (40 mL) afforded 2420 (608.8 mg, 62%) as an oil: 1H NMR (400 MHz, CDCl3) δ 3.68 (t, J = 6.4 Hz, 2 H, OCH2), 2.24 (td, J1 = 6.8 Hz, J2 = 2.8 Hz, 2 H,
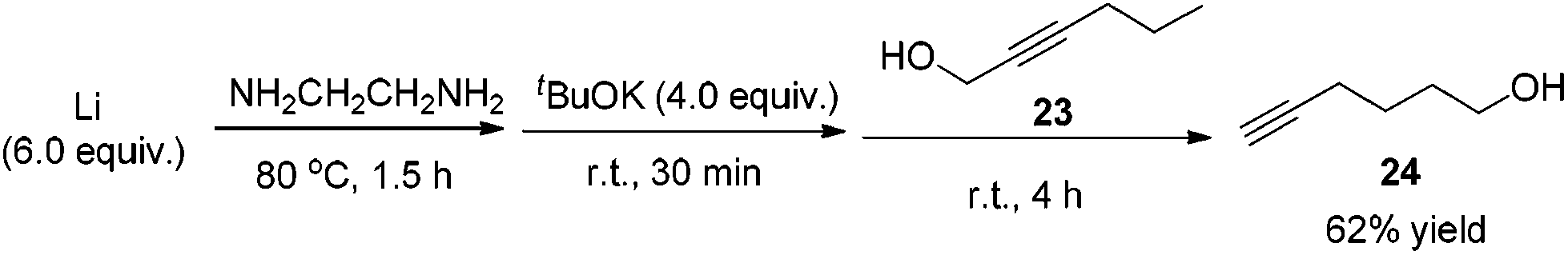 CCH2), 1.97 (t, J = 2.6 Hz, 1 H, CH), 1.74–1.51 (m, 5 H, 2 × CH2 + OH); 13C NMR (100 MHz, CDCl3) δ 84.2, 68.5, 62.0, 31.5, 24.6, 18.1; IR (neat) ν (cm−1) 3295, 2941, 2868, 2116, 1455, 1434, 1329, 1260, 1161, 1060, 1039; MS (ESI) m/z (%): 98 (M+, 4.42), 79 (100).
CCH2), 1.97 (t, J = 2.6 Hz, 1 H, CH), 1.74–1.51 (m, 5 H, 2 × CH2 + OH); 13C NMR (100 MHz, CDCl3) δ 84.2, 68.5, 62.0, 31.5, 24.6, 18.1; IR (neat) ν (cm−1) 3295, 2941, 2868, 2116, 1455, 1434, 1329, 1260, 1161, 1060, 1039; MS (ESI) m/z (%): 98 (M+, 4.42), 79 (100).
To a Schlenk tube were added Fe(NO3)3·9H2O (40.6 mg, 0.1 mmol), TEMPO (15.7 mg, 0.1 mmol), KCl (7.7 mg, 0.1 mmol), 24 (97.8 mg, 1.0 mmol), and DCE (4.0 mL) sequentially under the atmosphere of oxygen from a gas bag. The reaction was then carried out at room temperature for 12 h until the completion of the reaction as monitored by TLC (petroleum ether/ethyl acetate = 5/1). The crude reaction mixture was filtered through a short column of silica gel eluted with ethyl ether (3 × 25 mL). After evaporation, the residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 6/1 to 2/1) to afford 2521 (95.9 mg, 86%) as an oil: 1H NMR (400 MHz, CDCl3) δ 10.34 (brs, COOH), 2.52 (t, J = 7.4 Hz, 2 H, CH2), 2.29 (td, J1 = 7.0 Hz, J2 = 2.8 Hz, 2 H,
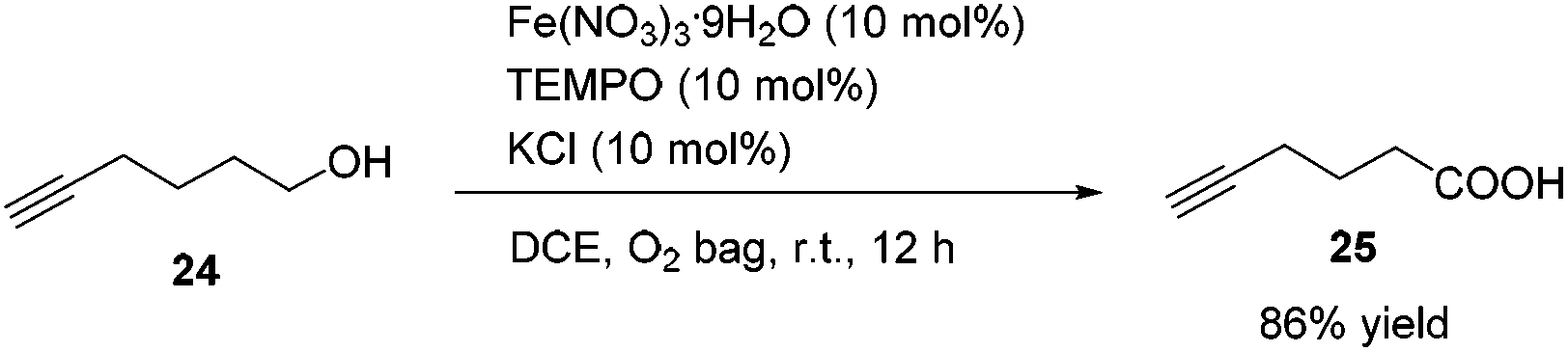 CCH2), 2.00 (t, J = 2.8 Hz, 1 H, CCH), 1.86 (quint, J = 7.2 Hz, 2 H, CH2); 13C NMR (100 MHz, CDCl3) δ 179.7, 83.0, 69.3, 32.5, 23.2, 17.7; IR (neat) ν (cm−1) 3400–2800, 3297, 2118, 1703, 1413, 1242, 1205, 1156, 1052; MS (ESI) m/z (%): 112 (M+, 1.68), 70 (100).
CCH2), 2.00 (t, J = 2.8 Hz, 1 H, CCH), 1.86 (quint, J = 7.2 Hz, 2 H, CH2); 13C NMR (100 MHz, CDCl3) δ 179.7, 83.0, 69.3, 32.5, 23.2, 17.7; IR (neat) ν (cm−1) 3400–2800, 3297, 2118, 1703, 1413, 1242, 1205, 1156, 1052; MS (ESI) m/z (%): 112 (M+, 1.68), 70 (100).
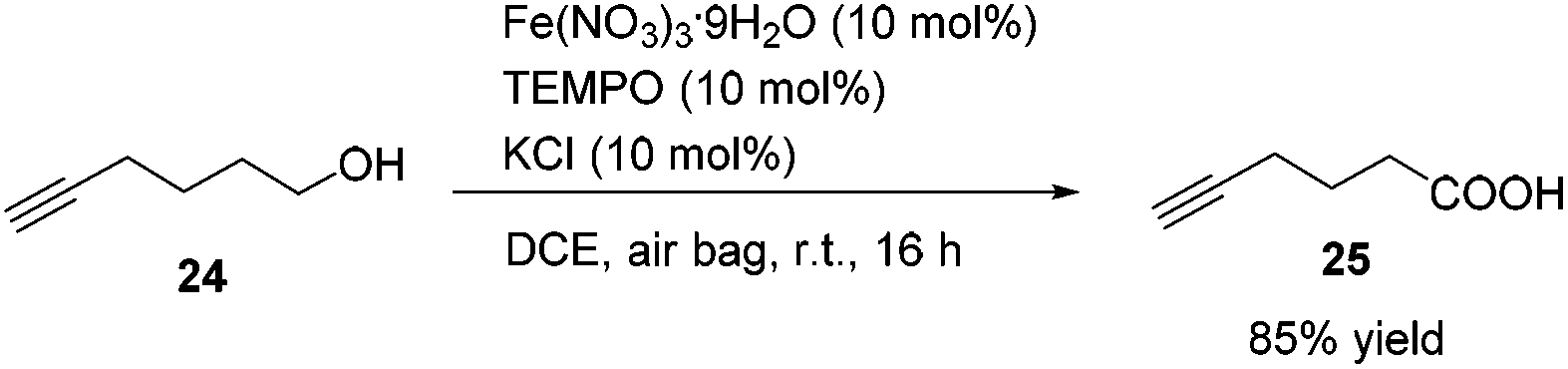 CH), 1.86 (quint, J = 7.2 Hz, 2 H, CH2); 13C NMR (100 MHz, CDCl3) δ 179.7, 83.0, 69.3, 32.6, 23.2, 17.7.
CH), 1.86 (quint, J = 7.2 Hz, 2 H, CH2); 13C NMR (100 MHz, CDCl3) δ 179.7, 83.0, 69.3, 32.6, 23.2, 17.7.
To a round bottom flask were added 25 (1.1163 g, 10.0 mmol)/DCM (5 mL) and DMAP (122.2 mg) sequentially. MeOH (2.43 mL, 0.791 g mL−1, 1.9224 g, 60.0 mmol)/DCM (5 mL), DCC (2.4800 g, 12.0 mmol), and DCM (10 mL) were added sequentially at 0 °C. The resulting mixture was then allowed to warm up to room temperature and stirred for 23 h. The crude reaction mixture was filtered through a short column of silica gel eluted with DCM (3 × 10 mL) and evaporated. H2O (20 mL) and Et2O (20 mL) were added and the organic layer was separated. The aqueous phase was extracted by ethyl ether (20 × 3 mL). The combined organic layer was washed with brine and dried over anhydrous MgSO4. After filtration and evaporation, the residue was purified via chromatography on silica gel (eluent: petroleum ether/ethyl ether = 25/1) to afford 422 (967.0 mg, 77%) as an oil: 1H NMR (400 MHz, CDCl3) δ 3.69 (s, 3H, OCH3), 2.47 (t, J = 7.4 Hz, 2 H, CH2), 2.27 (td, J1 = 6.9 Hz, J2 = 2.7 Hz, 2 H, CH2), 1.98 (t, J = 2.8 Hz, 1 H,
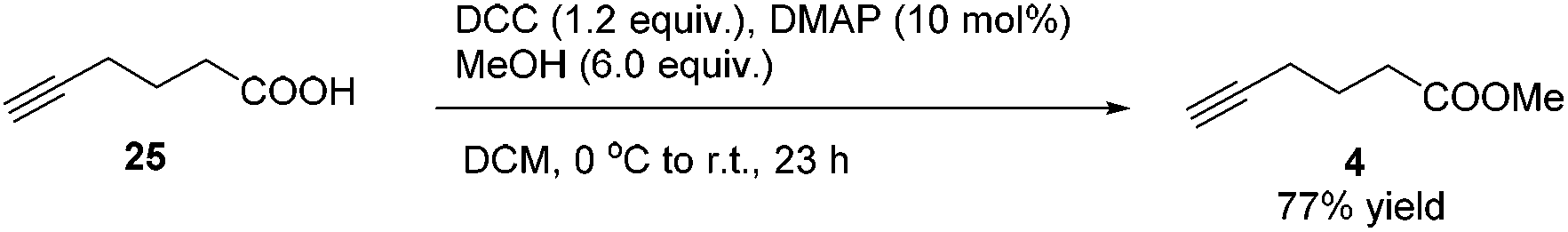 CH), 1.86 (quint, J = 7.2 Hz, 2 H, CH2); 1H NMR (400 MHz, CDCl3) δ 173.5, 83.2, 69.1, 51.6, 32.6, 23.5, 17.8; IR (neat) ν (cm−1) 3295, 2954, 2118, 1736, 1438, 1370, 1318, 1223, 1161, 1058; MS (ESI) m/z (%): 126 (M+, 25.52), 82 (100).
CH), 1.86 (quint, J = 7.2 Hz, 2 H, CH2); 1H NMR (400 MHz, CDCl3) δ 173.5, 83.2, 69.1, 51.6, 32.6, 23.5, 17.8; IR (neat) ν (cm−1) 3295, 2954, 2118, 1736, 1438, 1370, 1318, 1223, 1161, 1058; MS (ESI) m/z (%): 126 (M+, 25.52), 82 (100).
To a flame dried Schlenk tube were added CuBr2 (51.6 mg, 0.23 mmol), (S)-2712 (150.1 mg, 1.16 mmol), 4 (161.6 mg, 1.28 mmol)/dioxane (4.6 mL), and 3 (233.3 mg, 1.28 mmol)/dioxane (4.6 mL) sequentially. The Schlenk tube was then sealed with a screw cap of polytetrafluoroethylene. The resulting mixture was then stirred at 130 °C with a preheated oil bath. After 12 h, the reaction was complete as monitored by TLC (petroleum ether/ethyl acetate = 5/1) and the resulting mixture was diluted with 30 mL of Et2O and washed with an aqueous solution of hydrochloric acid (3 M, 20 mL). The organic layer was separated and the aqueous phase was extracted with ethyl ether (20 × 2 mL). The combined organic layer was washed with brine and dried over anhydrous Na2SO4. After filtration and evaporation, the residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl ether = 200/1) to afford (Ra)-(−)-22,4 (E/Z = 94/6) (168.3 mg, 50%) as an oil: 97% ee (HPLC conditions: CHIRALPAK AD-RH (0.46 × 15 cm, 5 μm), MeCN/H2O = 62/38, 0.7 mL min−1, λ = 214 nm, tR(major) = 48.9 min, tR(minor) = 60.2 min); [α]29.3D = −52.8 (c = 0.305, EtOH) (lit:2 [α]25D = −50 (c = 0.28, EtOH)); 1H NMR (400 MHz, CDCl3) δ 5.47–5.35 (m, 2 H, CH
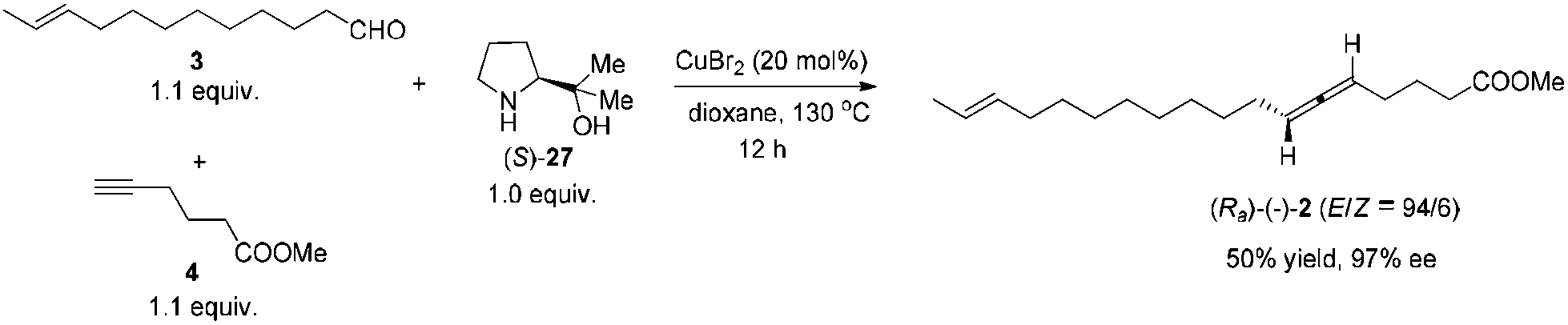 CH), 5.13–5.00 (m, 2 H, CHCCH), 3.67 (s, 3 H, CH3), 2.36 (t, J = 7.4 Hz, 2 H, CH2), 2.06–1.92 (m, 6 H, 3 × CH2), 1.74 (quint, J = 7.4 Hz, 2 H, CH2), 1.66–1.62 (m, 3 H, CH3), 1.43–1.22 (m, 12 H, 6 × CH2); 13C NMR (100 MHz, CDCl3) δ 204.0, 174.1, 131.6, 124.5, 91.5, 89.7, 51.5, 33.3, 32.6, 29.6, 29.5, 29.4, 29.18, 29.15, 29.08, 28.9, 28.3, 24.3, 17.9; IR (neat) ν (cm−1) 2924, 2853, 1962, 1741, 1436, 1365, 1313, 1243, 1209, 1154, 1059, 1019, 966, 876; MS (EI) m/z (%): 292 (M+, 4.79), 154 (100). The following signals are discernible for (Ra)-(−)-2 (Z): 1.61–1.59 (m, 3 H, CH3).
CH), 5.13–5.00 (m, 2 H, CHCCH), 3.67 (s, 3 H, CH3), 2.36 (t, J = 7.4 Hz, 2 H, CH2), 2.06–1.92 (m, 6 H, 3 × CH2), 1.74 (quint, J = 7.4 Hz, 2 H, CH2), 1.66–1.62 (m, 3 H, CH3), 1.43–1.22 (m, 12 H, 6 × CH2); 13C NMR (100 MHz, CDCl3) δ 204.0, 174.1, 131.6, 124.5, 91.5, 89.7, 51.5, 33.3, 32.6, 29.6, 29.5, 29.4, 29.18, 29.15, 29.08, 28.9, 28.3, 24.3, 17.9; IR (neat) ν (cm−1) 2924, 2853, 1962, 1741, 1436, 1365, 1313, 1243, 1209, 1154, 1059, 1019, 966, 876; MS (EI) m/z (%): 292 (M+, 4.79), 154 (100). The following signals are discernible for (Ra)-(−)-2 (Z): 1.61–1.59 (m, 3 H, CH3).
To a flame dried Schlenk tube were added CuBr2 (18.0 mg, 0.08 mmol), (S)-26 (50.8 mg, 0.2 mmol), (R)-26 (50.9 mg, 0.2 mmol), 4 (75.7 mg, 0.6 mmol)/dioxane (1.6 mL), and 3 (80.2 mg, 0.44 mmol)/dioxane (1.6 mL) sequentially. The Schlenk tube was then sealed with a screw cap of polytetrafluoroethylene. The resulting mixture was then heated at 130 °C with a preheated oil bath. The reaction was complete after 12 h as monitored by TLC (petroleum ether/ethyl acetate = 5/1). After being cooled to room temperature, the resulting mixture was diluted with 15 mL of Et2O and washed with an aqueous solution of hydrochloric acid (3 M, 10 mL). The organic layer was separated, and the aqueous phase was extracted by ethyl ether (10 mL). The combined organic layer was washed with brine and dried over anhydrous Na2SO4. After filtration and evaporation, the residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl ether = 200/1) to afford (rac)-(±)-24 (60.7 mg, 52%) as an oil: 1H NMR (400 MHz, CDCl3) δ 5.47–5.35 (m, 2 H, CH
 CH), 5.14–4.99 (m, 2 H, CHCCH), 3.67 (s, 3 H, CH3), 2.36 (t, J = 7.4 Hz, 2 H), 2.06–1.92 (m, 6 H, 3 × CH2), 1.74 (quint, J = 7.4 Hz, 2 H, CH2), 1.66–1.61 (m, 3 H, CH3), 1.43–1.22 (m, 12 H, 6 × CH2); 13C NMR (100 MHz, CDCl3) δ 204.0, 174.1, 131.7, 124.5, 91.5, 89.8, 51.5, 33.3, 32.6, 29.6, 29.5, 29.4, 29.19, 29.17, 29.1, 28.9, 28.3, 24.3, 17.9; IR (neat) ν (cm−1) 2924, 2853, 1962, 1741, 1436, 1365, 1313, 1242, 1207, 1154, 1060, 1018, 966, 876; MS (EI) m/z (%): 292 (M+, 6.07), 80 (100).
CH), 5.14–4.99 (m, 2 H, CHCCH), 3.67 (s, 3 H, CH3), 2.36 (t, J = 7.4 Hz, 2 H), 2.06–1.92 (m, 6 H, 3 × CH2), 1.74 (quint, J = 7.4 Hz, 2 H, CH2), 1.66–1.61 (m, 3 H, CH3), 1.43–1.22 (m, 12 H, 6 × CH2); 13C NMR (100 MHz, CDCl3) δ 204.0, 174.1, 131.7, 124.5, 91.5, 89.8, 51.5, 33.3, 32.6, 29.6, 29.5, 29.4, 29.19, 29.17, 29.1, 28.9, 28.3, 24.3, 17.9; IR (neat) ν (cm−1) 2924, 2853, 1962, 1741, 1436, 1365, 1313, 1242, 1207, 1154, 1060, 1018, 966, 876; MS (EI) m/z (%): 292 (M+, 6.07), 80 (100).
To a round bottom flask were added sequentially KOH (56.1 mg, 1.0 mmol), a solvent mixture (2 mL, MeOH/H2O = 4/1 by volume), and (Ra)-(−)-2 (117.0 mg, 0.4 mmol)/solvent mixture (2 mL, MeOH/H2O = 4/1 by volume). The resulting mixture was then stirred at 60 °C. The reaction was complete after 3 h as monitored by TLC. HCl (3 M, 0.4 mL) was added dropwise after the mixture was cooled with an ice bath. MeOH was removed by using a rotary evaporator. CH2Cl2 (30 mL) and water (20 mL) were added. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (10 mL × 3). The combined organic phase was washed with brine and dried over anhydrous Na2SO4. After filtration and evaporation, the residue was purified by chromatography on silica gel to afford (Ra)-(−)-12,4 (E/Z = 94/6) (109.3 mg, 98%) (eluent: petroleum ether/ethyl acetate = 10/1 to 2/1) as an oil: [α]29.4D = −55.2 (c = 0.995, EtOH); 1H NMR (400 MHz, CDCl3) δ 10.93 (brs, 1 H, COOH), 5.47–5.34 (m, 2 H, CH
 CH), 5.14–5.00 (m, 2 H, CHCCH), 2.40 (t, J = 7.4 Hz, 2 H, CH2), 2.08–1.92 (m, 6 H, 3 × CH2), 1.75 (quint, J = 7.4 Hz, 2 H, CH2), 1.66–1.62 (m, 3 H, CH3), 1.43–1.21 (m, 12 H, 6 × CH2); 13C NMR (100 MHz, CDCl3) δ 204.0, 180.3, 131.7, 124.5, 91.6, 89.6, 33.3, 32.6, 29.6, 29.5, 29.4, 29.18, 29.16, 29.1, 28.9, 28.2, 23.9, 17.9; IR (neat) ν (cm−1) 3500–2500, 2923, 2853, 1962, 1707, 1438, 1413, 1287, 1240, 1204, 1158, 1056, 964, 875; MS (EI) m/z (%): 278 (M+, 1.17), 140 (100). The following signals are discernible for (Ra)-(−)-1 (Z): 1.61–1.58 (m, 3 H, CH3).
CH), 5.14–5.00 (m, 2 H, CHCCH), 2.40 (t, J = 7.4 Hz, 2 H, CH2), 2.08–1.92 (m, 6 H, 3 × CH2), 1.75 (quint, J = 7.4 Hz, 2 H, CH2), 1.66–1.62 (m, 3 H, CH3), 1.43–1.21 (m, 12 H, 6 × CH2); 13C NMR (100 MHz, CDCl3) δ 204.0, 180.3, 131.7, 124.5, 91.6, 89.6, 33.3, 32.6, 29.6, 29.5, 29.4, 29.18, 29.16, 29.1, 28.9, 28.2, 23.9, 17.9; IR (neat) ν (cm−1) 3500–2500, 2923, 2853, 1962, 1707, 1438, 1413, 1287, 1240, 1204, 1158, 1056, 964, 875; MS (EI) m/z (%): 278 (M+, 1.17), 140 (100). The following signals are discernible for (Ra)-(−)-1 (Z): 1.61–1.58 (m, 3 H, CH3).
To a round bottom flask were added (Ra)-(−)-1 (55.8 mg, 0.2 mmol) and a solvent mixture (5 mL, Et2O/MeOH = 4/1 by volume). TMSCHN2 (2 M in hexane, 0.15 mL, 0.3 mmol) was added dropwise at 0 °C. The reaction mixture was then warmed up to room temperature naturally. When the reaction was complete as monitored by TLC after 3 h, the solvent was evaporated under vacuum, and the residue was purified by chromatography on silica gel to afford (Ra)-(−)-22,4 (E/Z = 94/6) (56.2 mg, 96%) (eluent: petroleum ether/ethyl ether = 150/1) as an oil: 97% ee (HPLC conditions: CHIRALPAK AD-RH (0.46 × 15 cm, 5 μm), MeCN/H2O = 62/38, 0.7 mL min−1, λ = 214 nm, tR(major) = 47.1 min, tR(minor) = 60.0 min); [α]29.4D = −52.1 (c = 0.315, EtOH) (lit:2 [α]25D−50 (c = 0.28, EtOH)); 1H NMR (400 MHz, CDCl3) δ 5.47–5.34 (m, 2 H, CH
 CH), 5.13–5.00 (m, 2 H, CHCCH), 3.67 (s, 3 H, CH3), 2.36 (t, J = 7.6 Hz, 2 H, CH2), 2.06–1.91 (m, 6 H, 3 × CH2), 1.74 (quint, J = 7.2 Hz, 2 H, CH2), 1.67–1.61 (m, 3 H, CH3), 1.44–1.20 (m, 12 H, 6 × CH2); 13C NMR (100 MHz, CDCl3) δ 204.0, 174.0, 131.6, 124.5, 91.4, 89.7, 51.4, 33.3, 32.6, 29.6, 29.5, 29.4, 29.2, 29.14, 29.06, 28.9, 28.3, 24.3, 17.9. The following signals are discernible for (Ra)-(−)-1 (Z): 1.61–1.58 (m, 3 H, CH3).
CH), 5.13–5.00 (m, 2 H, CHCCH), 3.67 (s, 3 H, CH3), 2.36 (t, J = 7.6 Hz, 2 H, CH2), 2.06–1.91 (m, 6 H, 3 × CH2), 1.74 (quint, J = 7.2 Hz, 2 H, CH2), 1.67–1.61 (m, 3 H, CH3), 1.44–1.20 (m, 12 H, 6 × CH2); 13C NMR (100 MHz, CDCl3) δ 204.0, 174.0, 131.6, 124.5, 91.4, 89.7, 51.4, 33.3, 32.6, 29.6, 29.5, 29.4, 29.2, 29.14, 29.06, 28.9, 28.3, 24.3, 17.9. The following signals are discernible for (Ra)-(−)-1 (Z): 1.61–1.58 (m, 3 H, CH3).
Acknowledgements
Financial support from the National Natural Science Foundation of China (21232006) and the National Basic Research Program (2015CB856600) are greatly appreciated.Notes and references
- A. Hoffmann-Röder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196 CrossRef PubMed.
- K. L. Mikalaijczak, M. F. Rogers, C. R. Smith and I. A. Wolff, Biochem. J., 1967, 105, 1245 CrossRef.
- (a) S. R. Landor and N. Punja, Tetrahedron Lett., 1966, 7, 4905 CrossRef; (b) S. R. Landor, B. J. Miller, J. P. Regan and A. R. Tatchell, Chem. Commun., 1966, 585 RSC; (c) S. R. Landor, B. J. Miller and A. R. Tatchell, J. Chem. Soc. C, 1966, 1822 RSC; (d) R. J. D. Evans, S. R. Landor and J. P. Regan, Chem. Commun., 1965, 397 RSC.
- J. S. Cowie, P. D. Landor, S. R. Landor and N. Punja, J. Chem. Soc., Perkin Trans. 1, 1972, 2197 RSC.
- For some examples of metal catalyzed E-selective semi-reduction of alkynes: (a) B. M. Trost, Z. T. Ball and T. Jöge, J. Am. Chem. Soc., 2002, 124, 7922 CrossRef CAS PubMed; (b) D. Srimani, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2013, 52, 14131 CrossRef CAS PubMed; (c) M. K. Karunananda and N. P. Mankad, J. Am. Chem. Soc., 2015, 137, 14598 CrossRef CAS PubMed.
- For selected reviews on constructing axially chiral allenes: (a) N. Krause and A. Hoffmann-Röder, Tetrahedron, 2004, 60, 11671 CrossRef CAS; (b) M. Ogasawara, Tetrahedron: Asymmetry, 2009, 20, 259 CrossRef CAS; (c) R. K. Neff and D. E. Frantz, ACS Catal., 2014, 4, 519 CrossRef CAS; (d) J. Ye and S. Ma, Org. Chem. Front., 2014, 1, 1210 RSC.
- (a) X. Huang, T. Cao, Y. Han, X. Jiang, W. Lin, J. Zhang and S. Ma, Chem. Commun., 2015, 51, 6956 RSC; (b) X. Tang, X. Huang, T. Cao, Y. Han, X. Jiang, W. Lin, Y. Tang, J. Zhang and S. Ma, Org. Chem. Front., 2015, 2, 688 RSC; (c) X. Huang, C. Xue, C. Fu and S. Ma, Org. Chem. Front., 2015, 2, 1040 RSC.
- M. J. Cryle, P. Y. Hayes and J. J. De Voss, Chem. – Eur. J., 2012, 18, 15994 CrossRef CAS PubMed.
- S. Ma, J. Liu, S. Li, B. Chen, J. Cheng, J. Kuang, Y. Liu, B. Wan, Y. Wang, J. Ye, Q. Yu, W. Yuan and S. Yu, Adv. Synth. Catal., 2011, 353, 1005 CrossRef CAS; S. Ma, J. Liu, S. Li, B. Chen, J. Cheng, J. Kuang, Y. Liu, B. Wan, Y. Wang, J. Ye, Q. Yu, W. Yuan and S. Yu, CN Pat., 102336619, 2012 Search PubMed; S. Ma, J. Liu, S. Li, B. Chen, J. Cheng, J. Kuang, Y. Liu, B. Wan, Y. Wang, J. Ye, Q. Yu, W. Yuan and S. Yu, US Pat., 8748669, 2014 Search PubMed; S. Ma, J. Liu, S. Li, B. Chen, J. Cheng, J. Kuang, Y. Liu, B. Wan, Y. Wang, J. Ye, Q. Yu, W. Yuan and S. Yu, JP Pat., 5496366, 2014 Search PubMed; S. Ma, J. Liu, S. Li, B. Chen, J. Cheng, J. Kuang, Y. Liu, B. Wan, Y. Wang, J. Ye, Q. Yu, W. Yuan and S. Yu, EP Pat., 2599765, 2016 Search PubMed.
- For some reviews on metal-catalyzed aerobic oxidation of alcohols: (a) S. D. McCann and S. S. Stahl, Acc. Chem. Res., 2015, 48, 1756 CrossRef CAS PubMed; (b) B. L. Ryland and S. S. Stahl, Angew. Chem., Int. Ed., 2014, 53, 8824 CrossRef CAS PubMed; (c) J. Piera and J. E. Bäckvall, Angew. Chem., Int. Ed., 2008, 47, 3506 CrossRef CAS PubMed; (d) Q. Cao, L. M. Dornan, L. Rogan, N. L. Hughes and M. J. Muldoon, Chem. Commun., 2014, 50, 4524 RSC.
- (a) X. Jiang, J. Zhang and S. Ma, J. Am. Chem. Soc., 2016, 138, 8344 CrossRef CAS PubMed; (b) S. Ma and X. Jiang, Chinese patent pending, application No201610141434.2, 2016 Search PubMed.
- C. Zeng, D. Yuan, B. Zhao and Y. Yao, Org. Lett., 2015, 17, 2242 CrossRef CAS PubMed.
- J. A. Cabezas and A. C. Oehlschlager, Synthesis, 1999, 107 CrossRef CAS.
- (a) C. A. Brown and A. Yamashita, J. Am. Chem. Soc., 1975, 97, 891 CrossRef CAS; (b) J. Kuang, X. Xie and S. Ma, Synthesis, 2013, 592 CAS.
- Y. Xue and M. B. Zimmt, Chem. Commun., 2011, 47, 8832 RSC.
- T. Nakamura, K. Yonesu, Y. Mizuno, C. Suzuki, Y. Sakata, Y. Takuwa, F. Narab and S. Satoh, Bioorg. Med. Chem., 2007, 15, 3548 CrossRef CAS PubMed.
- N. Y. Plugar, G. G. Verba, A. A. Abduvakhabov and F. G. Kamaev, Chem. Nat. Compd., 1990, 26, 455 CrossRef.
- D.-H. Hu, J. He, Y.-W. Zhou, J.-T. Feng and X. Zhang, Molecules, 2012, 17, 12140 CrossRef CAS PubMed.
- R. J. Fox, G. Lalic and R. G. Bergman, J. Am. Chem. Soc., 2007, 129, 14144 CrossRef CAS PubMed.
- A.-C. Bédard and S. K. Collins, J. Am. Chem. Soc., 2011, 133, 19976 CrossRef PubMed.
- G. A. Krafft and J. A. Katzenellenbogen, J. Am. Chem. Soc., 1981, 103, 5459 CrossRef CAS.
- L. Qi, M. M. Meijler, S.-H. Lee, C. Sun and K. D. Janda, Org. Lett., 2004, 6, 1673 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR and HPLC spectra of all the products. See DOI: 10.1039/c6qo00785f |
| This journal is © the Partner Organisations 2017 |