
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectroscopic studies on the comparative refolding of guanidinium hydrochloride denatured hen egg-white lysozyme and Rhizopus niveus lipase assisted by cationic single-chain/gemini surfactants via artificial chaperone protocol
Nuzhat Gull*a,
Mohd Ishtikharbd,
Md. Sayem Alam c,
Syedah Noorul Sabah Andrabia and
Rizwan Hasan Khan*d
c,
Syedah Noorul Sabah Andrabia and
Rizwan Hasan Khan*d
aDepartment of Chemistry, Govt. Degree College for Women, M. A. Road, Srinagar-190001, India. E-mail: nuzhatwahid@gmail.com; Tel: +91-9697508005
bDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai, India
cCentral Leather Research Institute, Chennai, India
dInterdisciplinary Biotechnology Unit, Aligarh Muslim University, Aligarh-202002, India. E-mail: rizwanhkhan1@gmail.com; Fax: +91-571-2721776; Tel: +91-571-2727388
First published on 6th June 2017
Abstract
Referred to as second generation surfactants, the gemini surfactants have shown promise in various potential areas of surfactant application. Here we report on the comparative refolding of hen egg white lysozyme (HEWL)/Rhizopus niveus lipase (RNL) by cationic gemini (G5, G6)/single-chain surfactant (CTAB) in the artificial chaperone assisted two step method. The studies were carried out in an aqueous medium at a physiological pH of 7.4 using dynamic light scattering (DLS), circular dichroism (CD) and fluorescence spectroscopy. The results indicate that very small concentrations of gemini surfactants, at which the single-chain homologue was found to be ineffective, refolded the GdCl denatured enzymes. A perusal of DLS data indicates that against the hydrodynamic radius (Rh) of 2.0 ± 0.06 nm/3.5 ± 0.12 nm for the native lysozyme/RNL, the Rh of the enzymes when the refolding was attempted by simple dilution was found to be 5.1 ± 0.16/38.2 ± 0.98 nm. Hydrodynamic radii very near to the native enzyme, i.e., 2.4 ± 0.08 nm/2.2 ± 0.06 nm for lysozyme and 5.2 ± 0.20 nm/4.5 ± 0.22 nm for RNL, were recorded by using 0.005 mM G5/0.005 mM G6 in combination with methyl-β-cyclodextrin. Like dilution the CTAB assisted refolding was also found not to be very impressive and the Rh observed was far beyond the native value. The CD and fluorescence studies faithfully corroborate with the DLS data. The results obtained from the multi-technique approach are associated with the stronger forces in gemini surfactants owing to the presence of two charged head groups and two hydrocarbon tails. Keeping in view the results, it is strongly suggested that the gemini surfactants assisted artificial chaperone protocol may be effectively used in the refolding of proteins produced in the genetically engineered cells and may also be used in circumventing diseases resulting from protein aggregation/misfolding.
Introduction
Recombinant DNA technology has made available several simple techniques that have made accessible unlimited and inexpensive sources of otherwise rare proteins. Practical interest in “protein refolding problem” stems from the fact that proteins overproduced by genetically engineered cells are often obtained in non-native forms (e.g. inclusion bodies).1,2 Although the inclusion bodies can be solubilised in concentrated chaotropes but it results in a protein devoid of its native conformation. As the proteins must fold into its characteristic and functional three dimensional structures in order to function,3,4 the denaturant concentration has to be lowered to allow the formation of the native protein conformation. When proper conditions for refolding are identified the correct refolding process competes, often in disadvantage, with misfolding and aggregation.5,6 Protein misfolding and aggregation pose a serious problem in the industrial process of producing the recombinant proteins.7–9 In order to develop efficient folding processes the competition between folding and aggregation in ‘in vitro’ proteins has to be understood. Aggregation may be due to the association of hydrophobic surfaces that are exposed during the refolding process.10 It is now known that within the cell of a living organism there are a number of auxiliary factors that assist in protein folding, including folding catalysts and molecular chaperones.11 Chaperones appear to act sequentially in protein folding by binding to folding intermediates that are in various stages of folding and then passing them on to next chaperone or chaperone complex in the cascade eventually releasing a competent native protein.12–16 In addition, they may maintain newly synthesised proteins in an unfolded conformation suitable for translocation across membranes and bind to non native proteins during cellular stress, among other functions.17 Thus the chaperones catalyse correct protein folding by interacting with the non native protein intermediates and preventing non-specific aggregation.An approach for controlling the competition between renaturation and aggregation has been described by Rozema and Gellman.18,19 This method employs small molecules, a surfactant and cyclodextrin, to guide the folding process. These low molecular weight assistants were referred to as “artificial chaperones” because the development of this technique was inspired by the mechanism of GroEL/GroES chaperone system. In the first step of this approach the detergent forms a complex with the denatured protein preventing aggregation while in the second step cyclodextrin selectively binds the detergent system stripping it from the protein that is then able to refold. Though the cationic single chain surfactants have been established as refolding agents,18–20 the role of gemini surfactants is an area that demands thorough investigation.21,22
Novel class of surfactants, called gemini surfactants, consist of two identical moieties covalently linked by a spacer group at or near the ionic head group, possess properties such as low critical micellar concentration, strong hydrophobic micro-domain, low kraft temperature etc. that makes them superior to conventional monomeric surfactants.23,24 Referred to as second generation surfactants, the geminis have shown promise in various potential areas of surfactant application showing strong surface activity and better solubility, wetting foaming and lime soap dispersion capability. They can be of orders of magnitude more surface active then the conventional single-chain counterparts.25 The comparative interaction of single-chain and gemini surfactants have made it amply clear that the gemini surfactants have higher affinity for proteins then their conventional single-chain surfactant counterparts26–30 and can be efficiently utilised in the refolding process via the artificial chaperone protocol.31,32
Based on the above facts the present study is directed towards the comparative refolding of hen egg white lysozyme (HEWL)/Rhizopus niveus lipase (RNL) by CTAB (C16H33(CH3)2Br−) and its gemini counterparts (bis-cetyl-di-methylammonium) pentane dibromide/(biscetyldimethylammonium) hexane dibromide (C16H33(CH3)2N+–(CH2)5–N+(CH3)2C16H33)·2Br−/(C16H33(CH3)2N+–(CH2)6–N+(CH3)2C16H33)·2Br−, designated as G5/G6, Fig. 1, using dynamic light scattering (DLS), circular dichroism (CD) and fluorescence spectroscopy as the probing techniques. HEWL (Fig. 2A) comprises of two structural domains; one α-domain consisting of four α-helices and a C-terminal 310 helix, and a β-domain consisting of a triple-stranded antiparallel β-sheet, a 310 helix, and a long loop. A short double-stranded antiparallel β-sheet links the two domains, as does one of the four-disulphide bridges. The active site lies between the two domains. The protein is known to be structurally robust in solution.33 Lysozyme is a component of tears and mucous and prevents bacterial infection. It disrupts bacteria by hydrolyzing the cell walls.
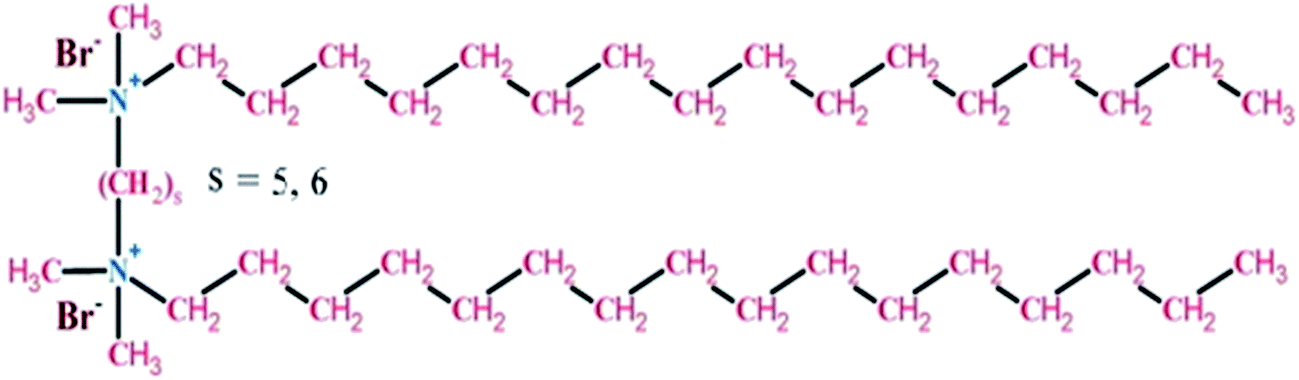 | ||
| Fig. 1 The structure of gemini surfactant (bis-cetyl-di-methylammonium) pentane dibromide, where S = 5 denoted by G5 chemically represented as (C16H33(CH3)2N+–(CH2)5–N+(CH3)2C16H33)·2Br− Similarly if S = 6 it is represented by (C16H33(CH3)2N+–(CH2)6–N+(CH3)2C16H33)·2Br and is denoted by G6. | ||
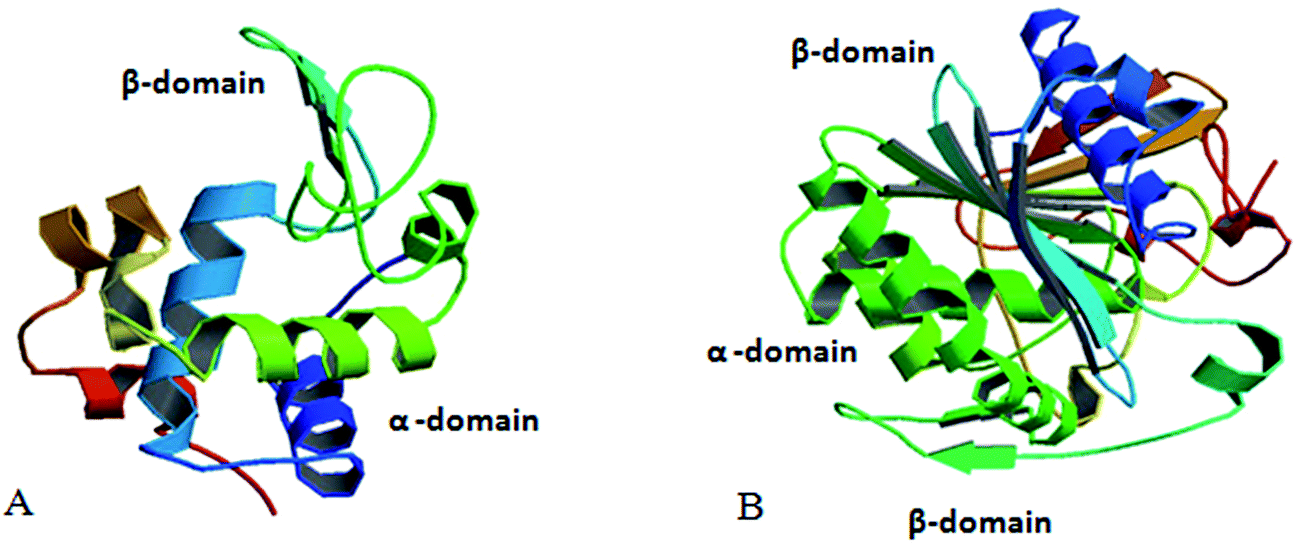 | ||
| Fig. 2 (A) Structure of hen egg white lysozyme (adopted from RCSB PDB – 5AMY submitted by Zander, U. with the DOI: 10.2210/pdb5amy/pdb). (B) Structure of Rhizopus niveus lipase (adopted from Kohno et al. (1996) J. Biochem. (Tokyo) 120, 505–510, PubMed: 8902613). | ||
Lipases (Fig. 2B) occur in plants, mammals and microorganisms, where their physiological role is believed to be digestive. Besides hydrolysis they also catalyse the synthesis of long-chain acrylic-glycerol. Lipase from Rhizopus niveus is superior in its 1–3 position specificity and has been used to produce cacao butter substitute. Lipase shows an α/β-hydrolase fold, a common 3-D fold in several other hydrolases. An α/β-hydrolase fold consists of a core of eight parallel β-sheets surrounded on both sides by α-helices. In recent times lipase so receive much attention due to their extensive use in biotechnology.34
Materials and methods
Materials
HEWL (L6876), RNL (62310), guanidinium hydrochloride (GdCl), methyl-β-cyclodextrin and CTAB (Sigma) were used as received. Gemini surfactant with spacer chain –(CH2)5– and –(CH2)6–, designated as G5 and G6, respectively were synthesized and characterized as described elsewhere.35 All other reagents and buffer components used were of analytical grade. Double distilled water was used throughout the study. Stock solutions of lysozyme, CTAB, G5 and G6 were prepared in 60 mM sodium phosphate buffer (pH 7.4) and utilized to prepare the samples of desired concentrations. The critical micellar concentration (cmc) of CTAB, G5 and G6 was determined to be 0.010 mM, 0.0066 mM, 0.0079 mM determined by surface tension measurements, respectively as described elsewhere.36 Denaturation of HEWL/RNL was carried out with guanidine hydrochloride (GdCl). For CD and fluorescence studies the initial concentration of HEWL/RNL and GdCl were 40 mg ml−1 and 6 M, respectively in the denatured stock solution. The concentration of HEWL/RNL was confirmed by determining its absorbance at 280 nm on a Perkin Elmer (Lambda 25) double beam spectrophotometer using the respective extinction coefficient of 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 940/34755 cm−1 M−1 or alternatively by the method of Gill and Vonhippel.37 After leaving the stock solution overnight, two sets of solutions were then prepared. In one set, the denatured stock solution was diluted to 60 mM GdCl and 0.4 mg ml−1 HEWL/RNL. In a parallel set, the dilution of denatured stock solution to the same final GdCl and HEWL/RNL concentration was carried out in presence of G5/G6 and methyl-β-cyclodextrin to get the final cyclodextrin concentration of 500 μM at 5 μM/100 μM CTAB and 5 μM G5/G6. The gemini surfactant and methyl-β-cyclodextrin were added sequentially in quick succession. For DLS 200 mg ml−1 HEWL/RNL in 6 M GdCl stock solution is diluted to 2 mg ml−1 HEWL/RNL in 60 mM GdCl. Solutions were thoroughly mixed and left for 20 h and assayed for conformational changes using DLS, CD and fluorescence studies.
940/34755 cm−1 M−1 or alternatively by the method of Gill and Vonhippel.37 After leaving the stock solution overnight, two sets of solutions were then prepared. In one set, the denatured stock solution was diluted to 60 mM GdCl and 0.4 mg ml−1 HEWL/RNL. In a parallel set, the dilution of denatured stock solution to the same final GdCl and HEWL/RNL concentration was carried out in presence of G5/G6 and methyl-β-cyclodextrin to get the final cyclodextrin concentration of 500 μM at 5 μM/100 μM CTAB and 5 μM G5/G6. The gemini surfactant and methyl-β-cyclodextrin were added sequentially in quick succession. For DLS 200 mg ml−1 HEWL/RNL in 6 M GdCl stock solution is diluted to 2 mg ml−1 HEWL/RNL in 60 mM GdCl. Solutions were thoroughly mixed and left for 20 h and assayed for conformational changes using DLS, CD and fluorescence studies.
DLS measurement
DLS measurements were carried out at 830 nm by using DynaPro-TC-04 dynamic light scattering equipment (Protein Solutions, Wyatt Technology, Santa Barbara, CA) equipped with a temperature-controlled microsampler. The samples were spun at 10000 rpm for 10 min and were filtered through 0.10 μm Whattman syringe directly into a 12 μL quartz cuvette. Mean hydrodynamic radius (Rh) and polydispersity were analyzed using Dynamics 6.10.0.10 software at optimized resolution. The Rh values were estimated on the basis of an autocorrelation analysis of scattered light intensity data based on translation diffusion coefficient by Stoke's–Einstein relationship.38| Rh = kT/6πηD | (i) |
Circular dichroism (CD) measurements
CD spectra were recorded on a Jasco J-815 model spectropolarimeter, equipped with a microcomputer. The instrument was calibrated with (+)-10-camphorsulfonic acid. All the CD measurements were carried out at 25 °C with a thermostatically controlled cell holder attached to a Neslab RTE-110 water bath with an accuracy of ±0.1 °C. Changes in the secondary structure of the protein were monitored in the far-UV region (200–250 nm) using 0.1 cm path length of cell. The signal from the reference sample containing buffer and the detergent was subtracted from the CD signal for all measurements. The high-tension voltage for the spectra obtained was found to be less than 600 V. Spectra were collected with a scan speed of 20 nm min−1 and response time of 1 s. Each spectrum was the average of four scans. The results are expressed in terms of mean residue ellipticity (MRE) expressed in units of deg cm2 dmol−1, defined as| MRE = θobs/10 × n × Cp × l | (ii) |
Fluorescence measurements
The fluorescence spectra were collected at 25 °C with a 1 cm path length cell using a Shimadzu (5301PC) spectrophotometer equipped with a constant temperature holder attached to a Neslab RTE-110 water bath, with an accuracy of ±0.1 °C. The excitation and emission slits were set at 5 nm. The reference sample consisting of the buffer and the detergent did not give any fluorescence signal. Intrinsic fluorescence was measured by exciting the protein solution at 280 nm and 295 nm, and emission spectra were recorded in the range of 300–400 nm.Results and discussion
Dynamic light scattering (DLS) analysis
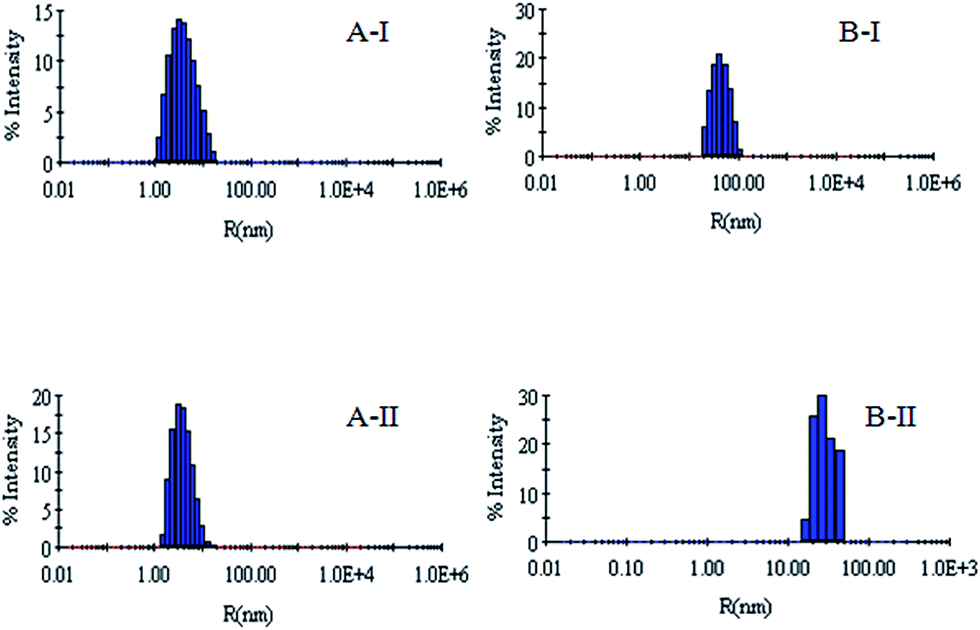
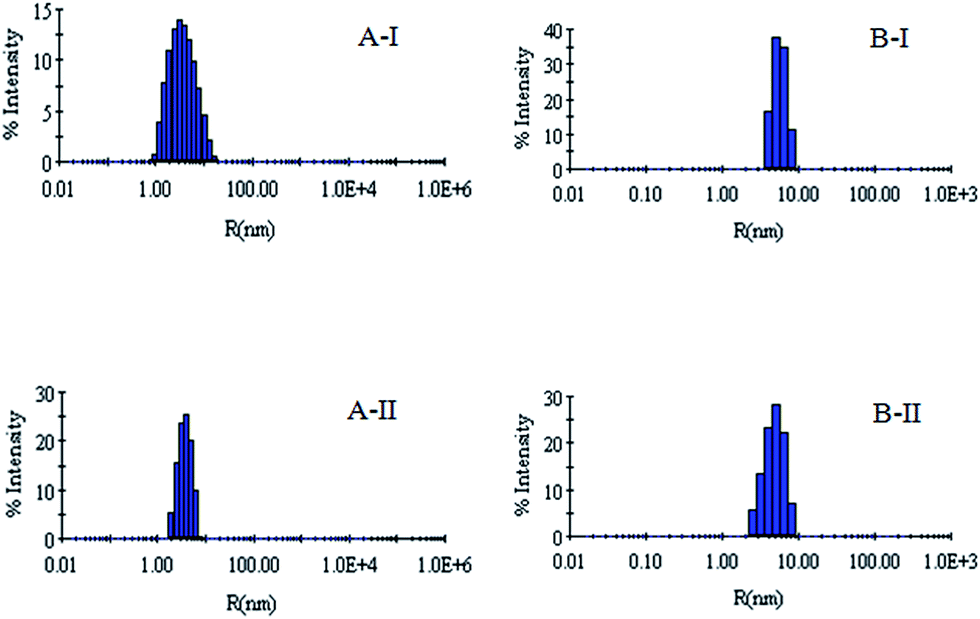
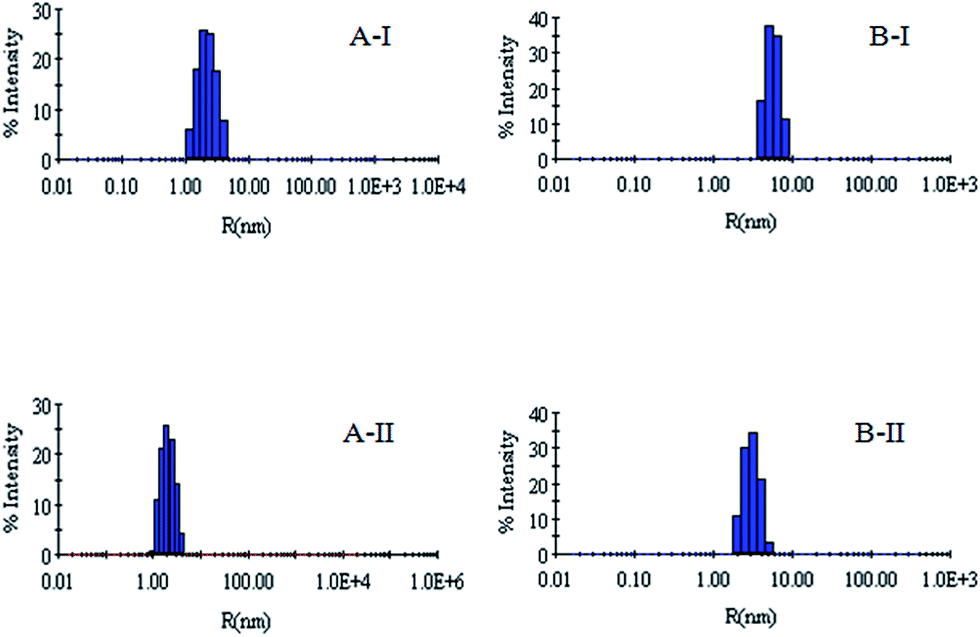
DLS has been used to monitor the changes in the dimensions of the proteins during denaturation and renaturation.39 This has been extended here to study the variations in the hydrodynamic radius of serum albumins during its artificial chaperone assisted refolding.The variations in the hydrodynamic radii (Rh) of the enzymes are depicted in Fig. 3–8 and the data have been tabulated (Table 1). The values of hydrodynamic radii of native HEWL and RNL, as indicated by the Fig. 3AI and BI were recorded at 2.1 ± 0.06 nm and 3.5 ± 0.07 nm respectively.40 Hydrodynamic radii equal to (7.6 ± 0.22) nm/85.0 ± 2.08 nm are observed upon denaturation of the native samples of HEWL/RNL by 6 M GdCl as given in Fig. 3AII and BII. Dilution of GdCl denatured HEWL/RNL to 60 mM GdCl and 2 mg ml−1 enzyme concentration leads to the hydrodynamic radii of 5.1 ± 0.16 nm/38.2 ± 1.10 nm as evident from Fig. 4AI and BI and Table 1. Large Rh values of the diluted enzymes compared to the native samples is attributed to the formation of aggregates as dilution is an aggregation prone pathway. Aggregation occurs due to the association of the hydrophobic surfaces that are exposed during the refolding process when attempted by simple dilution. No significant change in hydrodynamic radius is observed when refolding is attempted by the addition of methyl-β-cyclodextrin as evident from Fig. 4AII and BII and Table 1. Fig. 5AI and AII and 6AI and AII, indicate that the addition of 5/100 mM CTAB and 5 μM G5/G6 reduces the hydrodynamic radius of HEWL to 4.7 ± 0.12 nm/4.4 ± 0.12 nm and 4.2 ± 0.10 nm/4.1 ± 0.12 nm respectively, compared to the hydrodynamic radius of 5.1 ± 0.16 nm for the diluted sample (Table 1). In case of RNL compared to the Rh of 38.2 ± 1.10 nm for the diluted sample the Rh observed upon the addition of 5/100 mM CTAB and 5 μM G5/G6 is 19.0 ± 0.84 nm/11.3 ± 0.32 nm and 9.5 ± 0.19 nm/8.1 ± 0.12 nm, respectively as indicated by Fig. 5BI and BII and 6BI and BII. This reduction is suggested to be due to the reason that the addition of the surfactants checks the aggregate formation as hydrophobic tail of the surfactant interacts with the exposed hydrophobic portions of enzyme and leads to the formation of enzyme–surfactant complex. The results obtained with the single-chain surfactant CTAB and methyl-β-cyclodextrin are not very significant and the hydrodynamic radii equal to 4.4 ± 0.12 nm and 4.0 ± 0.10 nm, respectively are recorded when 5/100 mM CTAB and methyl-β-cyclodextrin are sequentially added to the denatured sample as depicted by Fig. 7AI and AII. RNL follows the similar trend and the respective hydrodynamic radii recorded upon addition of 5/100 mM CTAB and methyl-β-cyclodextrin are 7.8 ± 0.14 nm and 6.7 ± 0.12 nm as determined from Fig. 7BI and BII. As illustrated by Fig. 8AI and AII hydrodynamic radii very near to the native HEWL, i.e., 2.4 ± 0.08 nm/2.2 ± 0.06 nm, is observed when in addition to G5/G6, methyl-β-cyclodextrin is added to the denatured protein and RNL in this case shows a respective hydrodynamic radius of 5.8 ± 0.10 nm/4.5 ± 0.08 nm as shown in Fig. 8BI and BII. This later decrease in the hydrodynamic radii is attributed to the fact that methyl-β-cyclodextrin strips off the surfactant, breaking the protein–surfactant complex and thus completes refolding.
| Sample name | Dynamic light scattering results | Circular dichroism results | ||||
|---|---|---|---|---|---|---|
| HEWL | RNL | HEWL | RNL | |||
| Rh (nm) | Pd (%) | Rh (nm) | Pd (%) | α-Helix (%) | α-Helix (%) | |
| Native protein/enzyme | 2.0 ± 0.06 | 17.7 | 3.5 ± 0.07 | 11.8 | 45.58 | 22.50 |
| GdCl denatured protein/enzyme | 7.6 ± 0.22 | 39.6 | 85.0 ± 2.08 | 30.6 | 01.17 | 00.48 |
| Diluted protein/enzyme | 5.1 ± 0.16 | 24.7 | 38.2 ± 1.10 | 23.9 | 32.82 | 13.84 |
| Diluted protein/enzyme-CD | 4.8 ± 0.14 | 22.1 | 30.1 ± 1.44 | 17.8 | 33.06 | 15.27 |
| Diluted protein/enzyme-CTAB 5 mM | 4.7 ± 0.12 | 21.3 | 19.0 ± 0.84 | 17.6 | 33.90 | 16.19 |
| Diluted protein/enzyme-CTAB 5 mM-CD | 4.4 ± 0.12 | 20.8 | 7.8 ± 0.14 | 16.4 | 36.24 | 17.36 |
| Diluted protein/enzyme-CTAB 100 mM | 4.4 ± 0.14 | 20.7 | 11.3 ± 0.32 | 19.8 | 34.71 | 16.76 |
| Diluted protein/enzyme-CTAB 100 mM-CD | 4.0 ± 0.10 | 21.2 | 6.7 ± 0.12 | 20.1 | 38.05 | 18.09 |
| Diluted protein/enzyme-G5 | 4.2 ± 0.10 | 19.5 | 9.5 ± 0.19 | 15.1 | 34.10 | 15.84 |
| Diluted protein/enzyme-G5-CD | 2.4 ± 0.08 | 18.0 | 5.8 ± 0.10 | 16.4 | 41.18 | 18.88 |
| Diluted protein/enzyme-G6 | 4.1 ± 0.12 | 19.9 | 8.1 ± 0.12 | 18.7 | 34.11 | 16.91 |
| Diluted protein/enzyme-G6-CD | 2.2 ± 0.06 | 19.2 | 4.5 ± 0.08 | 17.4 | 42.13 | 19.87 |
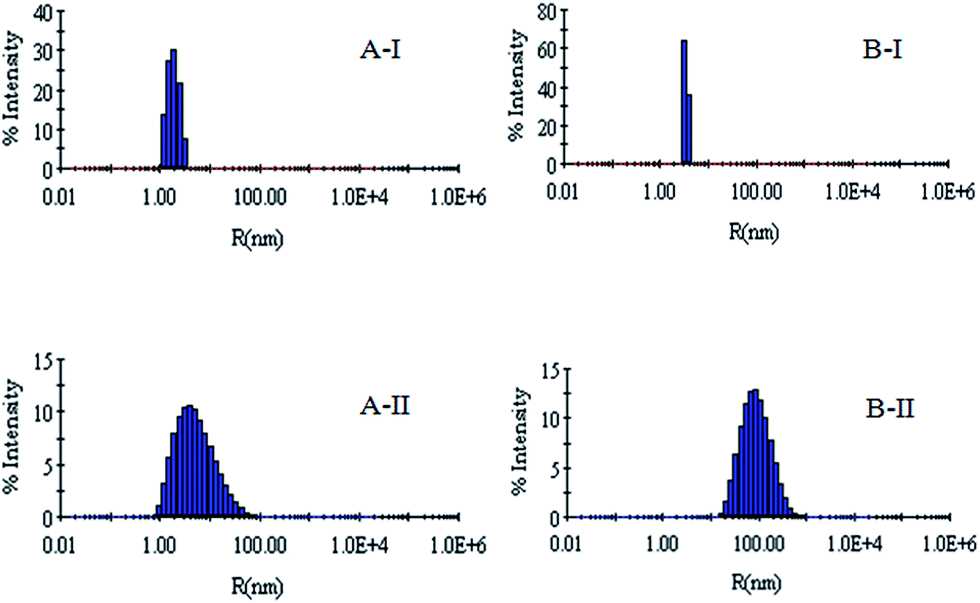 | ||
| Fig. 3 (AI) Hydrodynamic radius (Rh) of lysozyme in native state obtained at pH 7.4 in 60 mM sodium phosphate buffer. (AII) Hydrodynamic radius (Rh) of lysozyme denatured by 6 M GdCl. (BI) Hydrodynamic radius (Rh) of RNL in native state obtained at pH 7.4 in 60 mM sodium phosphate buffer. (BII) Hydrodynamic radius (Rh) of RNL denatured by 6 M GdCl. | ||
 | ||
| Fig. 4 (AI) Hydrodynamic radius (Rh) of the denatured lysozyme sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer (AII) hydrodynamic radius (Rh) of the denatured lysozyme sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 500 μM methyl-β-cyclodextrin. (BI) Hydrodynamic radius (Rh) of the denatured RNL sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer (BII) hydrodynamic radius (Rh) of the denatured RNL sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 500 μM methyl-β-cyclodextrin. | ||
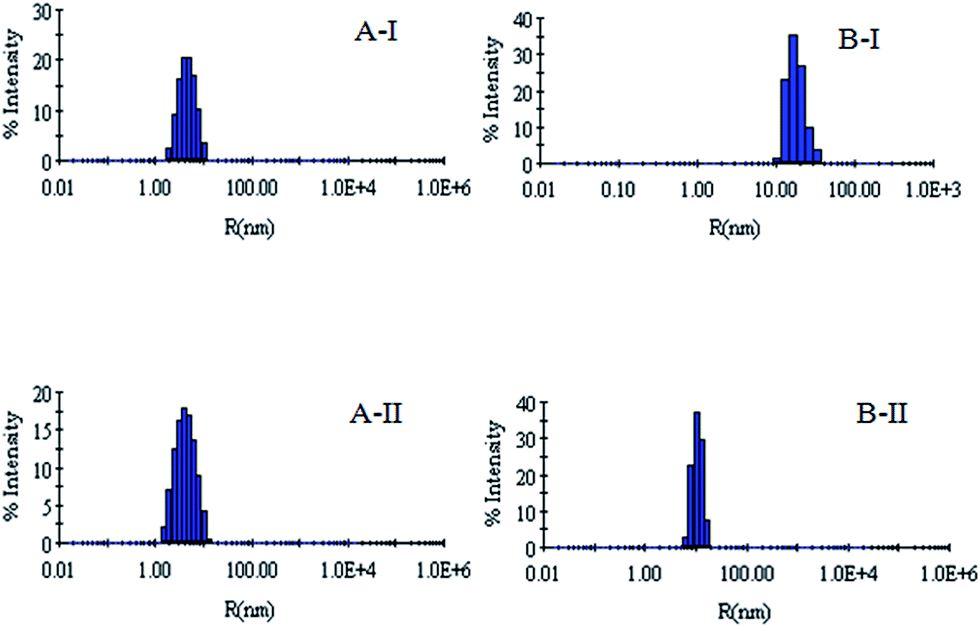 | ||
| Fig. 5 (AI) Hydrodynamic radius (Rh) of the denatured lysozyme sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 5 mM CTAB. (AII) Hydrodynamic radius (Rh) of the denatured lysozyme sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 100 mM CTAB. (BI) Hydrodynamic radius (Rh) of the denatured RNL sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 5 mM CTAB. (BII) Hydrodynamic radius (Rh) of the denatured RNL sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 100 mM CTAB. | ||
 | ||
| Fig. 6 (AI) Hydrodynamic radius (Rh) of the denatured lysozyme sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 0.005 mM G5 (AII) hydrodynamic radius (Rh) of the denatured lysozyme sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 0.005 mM G6. (BI) Hydrodynamic radius (Rh) of the denatured RNL sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 0.005 mM G5 (BII) hydrodynamic radius (Rh) of the denatured RNL sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 0.005 mM G6. | ||
 | ||
| Fig. 7 (A) Hydrodynamic radius (Rh) of the denatured lysozyme sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 5 mM CTAB + 500 μM methyl-β-cyclodextrin. (AII) Hydrodynamic radius (Rh) of the denatured lysozyme sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 100 mM CTAB + 500 μM methyl-β-cyclodextrin. (BI) Hydrodynamic radius (Rh) of the denatured RNL sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 5 mM CTAB + 500 μM methyl-β-cyclodextrin. (BII) Hydrodynamic radius (Rh) of the denatured RNL sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 100 mM CTAB + 500 μM methyl-β-cyclodextrin. | ||
 | ||
| Fig. 8 (AI) Hydrodynamic radius (Rh) of the denatured lysozyme sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 0.005 mM G5 and 500 μM methyl-β-cyclodextrin. (AII) Hydrodynamic radius (Rh) of the denatured lysozyme sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 0.005 mM G6 and 500 μM methyl-β-cyclodextrin. (BI) Hydrodynamic radius (Rh) of the denatured RNL sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 0.005 mM G5 and 500 μM methyl-β-cyclodextrin. (BII) Hydrodynamic radius (Rh) of the denatured RNL sample diluted to 60 mM GdCl with 60 mM sodium phosphate buffer in the presence of 0.005 mM G6 and 500 μM methyl-β-cyclodextrin. | ||
That the conformational changes are significant and are observed at considerably lower concentrations in gemini surfactants compared to the single-chain surfactant are attributed to the chemistry of gemini surfactants. It was established that the micellization in the case of gemini surfactants occurs at much lower concentration and the micelles formed have a stronger hydrophobic core compared to CTAB.33 The hydrophobic interactions as a result of the strong hydrophobic micro-domain of the gemini surfactants are enhanced to such an extent that they compress or refold the protein at very low concentrations as indicated by the decreased hydrodynamic radius.
Far-UV circular dichroism analysis
Circular dichroism (CD) spectroscopy is an important technique in the structural characterization of proteins, and especially for secondary structure determination. Secondary structural elements, such as α-helices, β-turns, and random coil structures, all make bands of individual shapes and magnitudes in the far-ultra-violet region.41 CD measurements were, therefore, performed to monitor the changes of secondary structure generated by interactions of serum albumins with G5/G6. Alterations of ellipticity at 222 nm (θ222 nm/mdeg) are used to probe varying α-helical contents.42Comparative variations in the ellipticity at 222 nm (θ222 nm/mdeg)/α-helical content with varying single-chain/gemini surfactants are depicted in Fig. 9A–D. Fig. 9A and C shows the typical far-UV CD spectra of HEWL/RNL in the presence of 5/100 μM CTAB and 5 μM G5 with and without 500 μM methyl-β-cyclodextrin. The comparative variations with CTAB and G6 are illustrated in Fig. 9B and D.
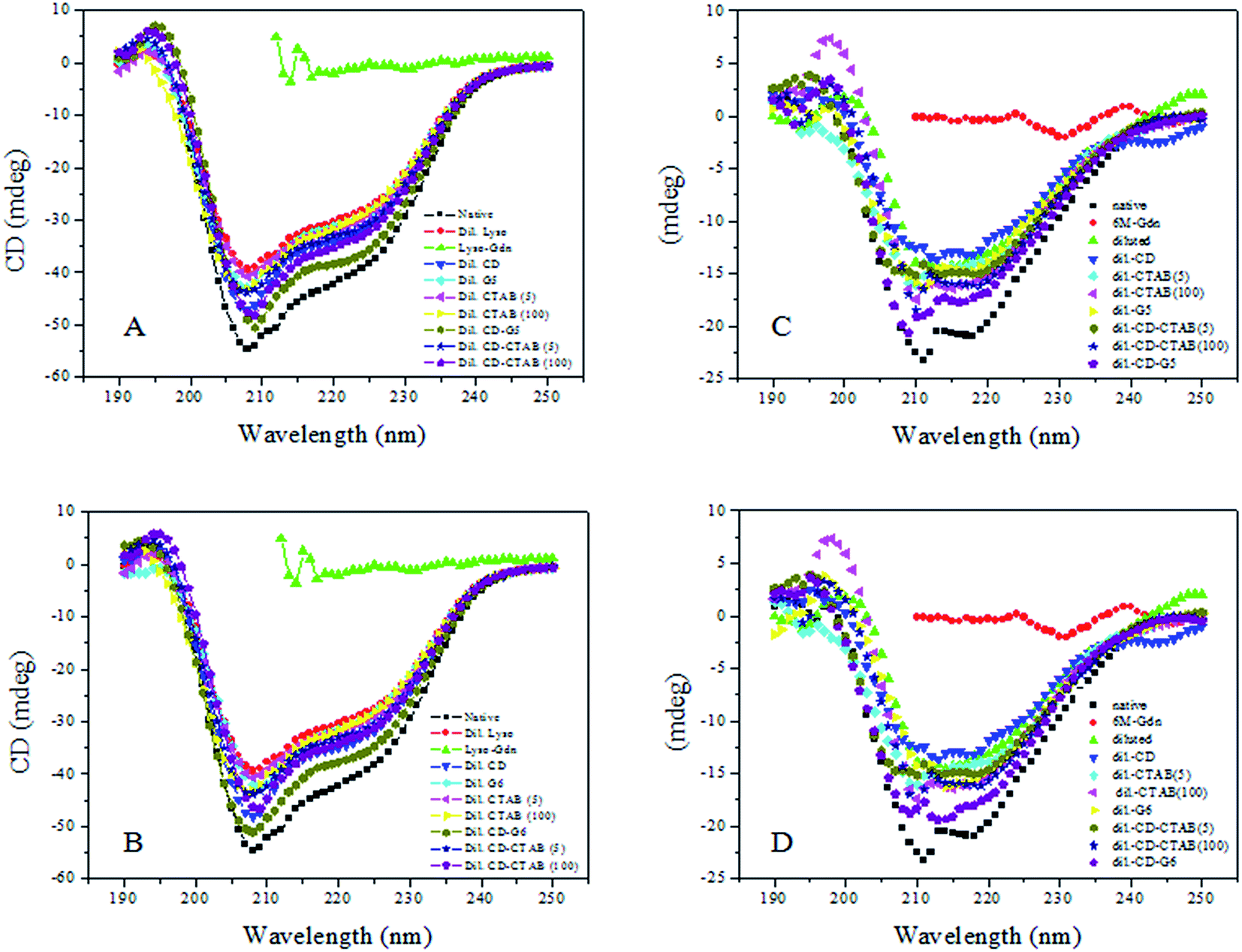 | ||
| Fig. 9 Far-UV CD spectra of HEWL and RNL at pH 7.4 in the native state, denatured state obtained by adding 6 M GdCl, denatured sample diluted to 60 mM GdCl with 60 mM phosphate buffer of pH 7.4 in the absence of the surfactant, and in the presence of methyl-β-cyclodextrin, 5 mM CTAB, 5 mM CTAB + 500 μM methyl-β-cyclodextrin, 100 mM CTAB, 100 mM CTAB + 500 μM methyl-β-cyclodextrin and similarly with 0.005 M gemini surfactants, 0.005 mM gemini surfactant + 500 μM methyl-β-cyclodextrin. Figures represents (A) gemini surfactant G5 with HEWL, (B) represents gemini surfactant G6 with HEWL, (C) gemini surfactant G5 with RNL, (D) represents gemini surfactant G6 with RNL. A cell of path length of 1 mm was used to obtain an average of four spectra. All the spectra have been corrected for their respective blanks. | ||
Surfactant does not contribute to the CD signal in the range of 200–250 nm, thus the observed CD is solely due to the peptide bonds of enzyme. HEWL/RNL (40 mg ml−1) denatured with 6 M GdCl loses the secondary structure almost completely as evident from the Fig. 7. Although the refolding of the enzymes by dilution of the denatured sample to 60 mM GdCl and 0.4 mg ml−1 enzyme concentration increases the α-helical content, but being an aggregation prone pathway it is far off from the native protein. It is clear from the figures that methyl-β-cyclodextrin added to the diluted enzyme, in the absence of the gemini surfactant, does not improve the helical content much. The maximum increase in the helical content is registered when the denatured enzyme is treated with 5 μM G5/G6 + methyl-β-cyclodextrin. Concentrations of the single-chain surfactant as low as the gemini surfactant (5 μM) were found to be ineffective while no significant increase in the helical content, compared to the dilution pathway is detected and even upon increasing the [CTAB] concentration 1000 times (5 μM)/20000 times (100 μM), the results were still not very impressive.
CD data indicates that against the respective native α-helical content of 45%/22.5% in HEWL/RNL the helical content in the denatured enzymes is lost almost completely. The dilution pathway increases this content to 32%/13% while by employing the gemini surfactant assisted artificial chaperone protocol i.e. carrying the dilution in the presence of 5 μM G5/G6 + methyl-β-cyclodextrin the helical content rises to 41%/42% in HEWL and 19%/20% in case of RNL, respectively. In case of CTAB the α-helical content rises to 36%/38% in presence of 5 μM/100 μM CTAB + methyl-β-cyclodextrin, respectively. In case of RNL 17%/18% α-helical content was observed by employing 5 μM/100 μM CTAB + methyl-β-cyclodextrin. In the absence of methyl-β-cyclodextrin the α-helical content observed in HEWL and RNL with 5 μM/100 μM CTAB is 33.9%/34.7% and 16%/17%, respectively.
The present investigation indicates that micromolar concentration of the gemini surfactant, at which the single-chain surfactant is ineffective, may be effectively used in recovering the α-helical content from the GdCl denatured proteins. The hydrophobic surfaces of the protein, upon dilution to non-denaturing concentrations of GdCl, are exposed resulting in an increase in the intermolecular forces, thereby leading to aggregation. The helicity of the protein, thus, does not increase appreciably when refolding is attempted by simple dilution. The surfactant upon interacting with the proteins forms the protein–surfactant complex and diminishes the intermolecular interactions that lead to aggregation, relative to intra-molecular interactions that drive folding. Upon the addition of methyl-β-cyclodextrin the transient association of the protein with the surfactant is broken and proper refolding is initiated. The significant increase in the α-helical content observed at much smaller concentrations of the gemini surfactants G5/G6 compared to the single-chain surfactant CTAB is attributed to the stronger hydrophobic interactions between the protein and the surfactant molecules owing to the stronger hydrophobic core of G5/G6 compared to CTAB.43 This results in a significant rise in the helicity when gemini surfactant + methyl-β-cyclodextrin is introduced in the solution used to dilute GdCl denatured proteins. These results corroborate the CD data and support our view point that the gemini surfactants may be effectively used in protein refolding via the artificial chaperone protocol.
Intrinsic fluorescence analysis
Fluorescence spectroscopy is widely employed to study proteins and peptides. Aromatic amino acids tryptophan, tyrosine, and phenylalanine offer intrinsic fluorescence probes of protein conformation, dynamics, and intermolecular interactions.44 Both HEWL and RNL are multi-tryptophan enzymes. The molecule of HEWL contains six tryptophan residues while three tryptophan residues are present in RNL. Upon interaction with substrates or inhibitors in solution, the tryptophan residues are perturbed as reflected by changes in UV, CD and fluorescence.45,46The emission spectra of HEWL/RNL in the absence and presence of 5/100 μM CTAB, 5 μM G5 and 500 μM methyl-β-cyclodextrin are presented in Fig. 10. As evident from figures, fluorescence gets significantly quenched as the native enzyme is denatured with 6 M GdCl. On diluting the denatured sample, the enzyme exhibits an increase in the α-helical content, as evident from CD spectra, resulting in increase in the fluorescence intensity (Fig. 10). This is due to the fact that, due to the increase in secondary structure more number of hydrophobic sites are made available on dilution compared to denatured sample where the secondary structure is completely lost. As dilution is an aggregation prone pathway the intensity is much less than that of the native curve. There is no significant rise in fluorescence intensity when the diluted sample is treated with methyl-β-cyclodextrin implying that no further rise in secondary structure is observed. Fluorescence intensity very near to the native curve is observed when the denatured sample is treated with a combination of 5 μM G5/G6 and 500 μM methyl-β-cyclodextrin.
 | ||
| Fig. 10 Fluorescence emission spectra of lysozyme at pH 7.4 in the native state, denatured state obtained by adding 6 M GdCl, denatured sample diluted to 60 mM GdCl with 60 mM phosphate buffer of pH 7.4 in the absence of the surfactant, and in the presence of methyl-β-cyclodextrin, 5 mM CTAB, 5 mM CTAB + methyl-β-cyclodextrin, 100 mM CTAB, 100 mM CTAB + methyl-β-cyclodextrin, and similarly with 0.005 mM gemini surfactants, 0.005 mM gemini surfactants + 500 μM methyl-β-cyclodextrin. Figures represents (A) gemini surfactant G5 with HEWL, (B) represents gemini surfactant G6 with HEWL, (C) gemini surfactant G5 with RNL, (D) represents gemini surfactant G6 with RNL. A cell of path length of 1 cm was used to obtain an average of four spectra. All the spectra have been corrected for their respective blanks. | ||
The results with 5 μM/100 μM CTAB are not as impressive as that of the gemini surfactants despite the fact that concentration of the single-chain surfactant employed is 1000/20000 times greater than that of gemini surfactant. Fluorescence data thus faithfully corroborates with the data obtained from CD and DLS studies and confirms the fact that maximum recovery of the helical content is observed by employing the combination of gemini surfactant and methyl-β-cyclodextrin via the artificial chaperone protocol.
Conclusions
The role of gemini surfactant in the refolding of denatured samples of HEWL/RNL has been investigated using well recognized spectroscopic techniques, i.e., CD, DLS and intrinsic fluorescence, in the present study. The results presented clearly indicate that very small concentrations of gemini surfactant efficiently refold the enzymes. The results have been attributed to the fact that gemini surfactants act as efficient capturing agents owing to their architecture, and thus form enzyme–surfactant complexes at very low concentrations thereby circumventing aggregation. This leads to a comparable hydrodynamic radius to the native enzyme after subjecting the GdCl denatured enzyme to artificial chaperone assisted refolding using gemini surfactants as capturing agent and greater recovery of the α-helical content compared to simple dilution/use of single-chain surfactants. Thus, it is suggested that gemini surfactants may effectively be used in the protein refolding and may address one of the most pressing demand of biotechnology industry for the development of efficient and inexpensive folding aides and may also prove fruitful for drug delivery.Author contributions
Conceived and designed the experiments: NG, MI, RHK. Performed the experiments: MI, MSA. Analyzed the data: NG, MI, NSA, RHK. Contributed reagents/materials/analysis tools: RHK, MI, MSA. Wrote the paper: NG, MI, RHK.Acknowledgements
Financial assistance from the Indian Council of Medical Research (ICMR), Government of India to M. I. in the form of a research fellowship (BIC/11/12/2013) is acknowledged. Encouragement from the Principal and faculty Dept. of chemistry, Govt. Degree College for Women, M. A. Road, Srinagar to N. G. and S. N. S. is gratefully acknowledged. We are thankful to Interdisciplinary Biotechnology Unit, Aligarh Muslim University, Aligarh for the use of their facilities.References
- E. D. B. Clark, Curr. Opin. Biotechnol., 1998, 9(2), 157–163 CrossRef PubMed.
- F. A. Marston, Biochem. J., 1986, 240(1), 1–12 CrossRef CAS PubMed.
- M. L. Anson, Adv. Protein Chem., 1945, 2, 361–386 CrossRef CAS.
- C. B. Anfinson, Science, 1973, 181, 223–230 Search PubMed.
- R. Jaenicke and R. Rudolph, in Proceedings of the 28th Conference of German Biochemical Society, ed. R. Jaenicke, Elsevier/North-Holland Biomedical Press, Amesterdam, 1980, p. 525 Search PubMed.
- P. Horowitz and N. L. Criscimagna, J. Biol. Chem., 1986, 261, 15652–15658 CAS.
- J. L. Cleland, in Protein Folding, In Vivo and In Vitro; ACS Symposium Series, American Chemical Society, Washington DC, 1993, p. 526 Search PubMed.
- E. D. B. Clark, Curr. Opin. Biotechnol., 2001, 12, 202–207 CrossRef CAS PubMed.
- In Protein Refolding, ACS Symposium Series 470, ed. G. Geourgiou and E. D. B. Clark, American Chemical Society, Washington D.C., 1991, p. 169 Search PubMed.
- A. L. Fink, Folding Des., 1998, 3(1), 9–23 CrossRef.
- M. J. Getting and P. Sambrook, Nature, 1992, 355, 33–45 CrossRef PubMed.
- A. L. Horwich, G. W. Farr and W. A. Fenton, Chem. Rev., 2006, 106, 1917–1930 CrossRef CAS PubMed.
- S. Walter and J. Buchner, Angew. Chem., Int. Ed., 2002, 41, 1098–1113 CrossRef CAS PubMed.
- K. Richter and J. Buchner, J. Cell. Physiol., 2001, 188, 281–290 CrossRef CAS PubMed.
- H. Grallert and J. Buchner, J. Struct. Biol., 2001, 135(2), 95–103 CrossRef CAS PubMed.
- D. Thirumalai and G. H. Lorimer, Annu. Rev. Biophys. Biomol. Struct., 2001, 30, 245–269 CrossRef CAS PubMed.
- P. F. Anthony, Physiol. Rev., 1999, 79, 425–449 Search PubMed.
- D. Rozema and S. H. Gellman, J. Am. Chem. Soc., 1995, 117, 2373–2374 CrossRef CAS.
- D. Rozema and S. H. Gellman, J. Biol. Chem., 1996, 271, 3478–3487 CrossRef CAS PubMed.
- D. Rozema and S. H. Gellman, Biochemistry, 1996, 35(49), 15760–15771 CrossRef CAS PubMed.
- D. Nath and M. Rao, Eur. J. Biochem., 2001, 268, 5471–5478 CrossRef CAS PubMed.
- Y. Moriyama and K. Takeda, Langmuir, 2005, 21(12), 5524–5528 CrossRef CAS PubMed.
- R. Zana, Adv. Colloid Interface Sci., 2002, 97, 205–253 CrossRef CAS PubMed.
- M. J. Rozen, CHEMTECH, 1993, 23, 30–33 Search PubMed.
- M. J. Rosen and D. J. Tracy, J. Surfactants Deterg., 1998, 1, 547–554 CrossRef CAS.
- M. S. Ali, N. Gull, J. M. Khan, V. K. Aswal, R. H. Khan and Kabir-ud-Din, J. Colloid Interface Sci., 2010, 352(2), 436–443 CrossRef CAS PubMed.
- N. Gull, P. Sen, R. H. Khan and Kabir-ud-Din, Langmuir, 2009, 25, 11686–11691 CrossRef CAS PubMed.
- N. Gull, P. Sen, R. H. Khan and Kabir-ud-Din, J. Biochem., 2009, 145, 67–77 CrossRef CAS PubMed.
- Y. Pi, Y. Shang, C. Peng, H. Liu, Y. Hu and J. Jiang, Biopolymers, 2006, 83, 243–249 CrossRef CAS PubMed.
- Y. Li and X. Wang, J. Phys. Chem. B, 2006, 110, 8499–8505 CrossRef CAS PubMed.
- N. Gull, M. A. Mir, J. M. Khan, R. H. Khan, G. M. Rather and A. A. Dar, J. Colloid Interface Sci., 2011, 364, 157–162 CrossRef CAS PubMed.
- N. Gull, J. M. Khan, M. Ishtikhar, A. Qadeer, R. A. Khan, M. Gull and R. H. Khan, J. Colloid Interface Sci., 2015, 439, 170–176 CrossRef CAS PubMed.
- T. Aria and W. Norde, Colloids Surf., B, 1990, 51, 1–15 CrossRef.
- K.-E. Jeager, B. W. Dijkstre and M. T. Reetz, Annu. Rev. Microbiol., 1999, 53, 315–351 CrossRef PubMed.
- Kabir-Ud-Din, W. Fatma and Z. A. Khan, J. Phys. Chem. B, 2007, 111, 8860–8867 CrossRef CAS PubMed.
- M. A. Mir, N. Gull, J. M. Khan, R. H. Khan, A. A. Dar and G. M. Rather, J. Phys. Chem. B, 2010, 114, 3197–3204 CrossRef CAS PubMed.
- S. C. Gill and P. H. S. von Hippel, Anal. Biochem., 1989, 182, 319–326 CrossRef CAS PubMed.
- K. Rezwan, L. P. Meier, M. J. Vörös, M. Textor and L. J. Gauckler, Langmuir, 2004, 20, 10055–10061 CrossRef CAS PubMed.
- D. F. Nicoli and G. B. Benedek, Biopolymers, 1976, 15, 2421–2437 CrossRef CAS PubMed.
- B. Saha, J. Saikia and G. Das, RSC Adv., 2013, 3, 7867–7879 RSC.
- S. Brahms and J. Brahms, J. Mol. Biol., 1980, 13, 149–178 CrossRef.
- K. M. Sharon, Biochim. Biophys. Acta, 2005, 1751(2), 119–139 Search PubMed.
- C. Sun, J. Yang, X. Wu, X. Huang, F. Wang and S. Liu, Biophys. J., 2005, 88, 3518–3524 CrossRef CAS PubMed.
- Y. Chen and M. D. Barkley, Biochemistry, 1998, 37, 9976–9982 CrossRef CAS PubMed.
- V. I. Teichberg and N. Sharon, FEBS Lett., 1970, 70(2), 171–174 CrossRef.
- R. W. Woody, Methods Enzymol., 1950, 246, 34–71 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |