DOI:
10.1039/C6RA22297H
(Paper)
RSC Adv., 2017,
7, 6467-6473
Three-dimensional nitrogen and boron codoped graphene for carbon dioxide and oils adsorption
Received
6th September 2016
, Accepted 7th January 2017
First published on 10th January 2017
Abstract
Three-dimensional heteroatom-doped graphene macroporous structures possess superior features, such as the large pore volume, numerous surface active sites and the high specific surface area. These properties are useful for the preparation of high-performance adsorbents. We introduce a facile synthesis route for the preparation of three-dimensional nitrogen and boron codoped graphene aerogels (BN-GAs) via a solvothermal method at 180 °C for 6 h. The BN-GAs possess mesostructures with 2.5–11 nm pores, a high surface area (169.9 m2 g−1), and contain nitrogen and boron. Based on the unique feature of the large specific surface area and the high nitrogen and boron contents, the BN-GAs show a high CO2 capture of 2.1–2.9 mmol g−1 (273 K, 1.0 bar) and excellent absorption capacities of up to 23 g g−1 for different oils and solvents. These results suggest that the BN-GAs may serve as a potential adsorbent for CO2 and oil leakage capture.
1. Introduction
Water pollution and global warming are the main environmental problems that challenge humanity. Water pollution is caused mainly by frequent oil and chemical solvent leakages. Three major measures have been used to address oil spillage and chemical solution leakage accidents: chemical methods (in situ burning), physical methods (various adsorbents)1,2 and biological methods (biodegradation).3,4 However, burning may cause secondary pollution, and biodegradation suffers from intrinsic limitations, including a high cost and a long operational cycle. Solid adsorbents are of low cost and are easy to maintain, and thus, exhibit important potential in practical applications. Global warming is caused by greenhouse gas (carbon dioxide (CO2), methane (CH4), nitrous oxide (N2O), hydrofluorocarbons (HCFs)) emissions. CO2 that is derived from fossil fuel combustion is the most important greenhouse gas. Stabilizing CO2 levels in the atmosphere and establishing an efficient CO2 capture and sequestration technology is of vital significance. CO2 capture measures include mainly solid adsorbent adsorption and alkali solution adsorption.5–8 CO2 adsorption by using alkali solutions is the most mature technology of the actual applications, but it exhibits intrinsic limitations, including energy consumption costs, operational safety, and equipment service life.9 Therefore, physical adsorption is also the best option to solve this problem.
Traditional solid absorbents consist of activated carbon,10 metal–organic frameworks,11,12 mesoporous silica/carbon,13,14 and zeolites,15–17 and have been studied for CO2 capture and the removal of oil after spillage. However, the high cost and inconvenience of recovery restricts the industrial, large-scale applications of zeolite materials. Activated carbon can overcome these weaknesses but it has a low adsorption capacity and a poor selectivity for the uptake of CO2 and spilled oils. Metal–organic framework materials exhibit a much higher capture capacity and selectivity than the two former adsorbents. The synthetic polymer process is complicated and their ecological impact remains unclear. Hence, it is critical to develop environmentally friendly CO2 and oil sorbents with a high sorption capacity, selectivity, and good recyclability.
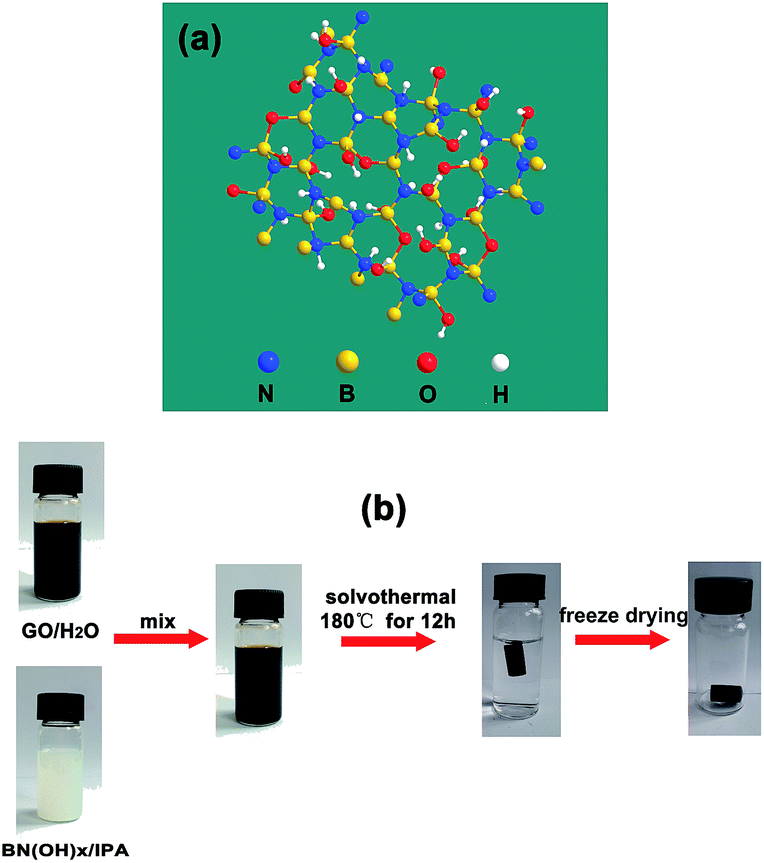
Graphene, which consists of a single layer of carbon atoms that are packed tightly into a two-dimensional sheet of sp2-hybridized carbon, has been developed extensively in recent decades because of its extraordinary electron mobility, good mechanical properties, and thermal conductivity.18 Self-assembled graphene oxide (GO) with different functional materials to synthesize multifunctional graphene-based aerogels with synergistic performance, such as inorganic nanoparticles (Fe,19 Co,20 Ni,21) doped graphene aerogels, graphene/carbon nanotube composite aerogels, graphene/polymers (polyaniline (PANI),22 polyvinylidene fluoride (PVDF),23 polystyrene (PS),24) composite aerogels, and heteroatom (N,25 B,26,27 S,28) doped graphene aerogels have attracted significant attention because of their applications in environmental remediation, supercapacitors, hydrogen storage, flexible electronics, and sensors. As a sorbent, heteroatom-doped graphene aerogels display excellent performance over traditional materials because of two advantages: (1) the inherent characteristics of graphene aerogels, such as a larger specific surface area, porosity, and low apparent density;27 (2) their ability to create adsorption active sites by heteroatoms or carbon atoms around heteroatoms to promote the adsorption of acidic gas (CO2, sulfur dioxide (SO2)) and their intrinsic hydrophobicity and oleophylic property.29,30 Hydroxylated boron nitride (BN(OH)x) materials are structural analogues of graphene two-dimensional materials in which C atoms are replaced by alternating boron (B) and nitrogen (N) atoms and with surface functionalization with hydroxyl groups (Fig. 1a).31 The BN(OH)x has been used to load anticancer drugs because of their high degree of hydroxylation. However, the use of BN(OH)x as functional materials mixed with graphene to prepare graphene-based aerogel has never been reported.
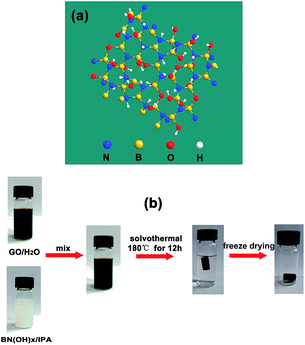 |
| | Fig. 1 (a) Atomic structure of BN(OH)x. (b) Schematic diagram illustrating the procedure to fabricate BN-GAs. | |
Herein, we demonstrate an efficient method for the large-scale synthesis of a novel heterostructure that is comprised of BN(OH)x nanosheets that are interconnected with N and B codoped graphene aerogels (BN-GAs). The advantages brought by the introduction of BN(OH)x can be summarized as follows: (a) BN(OH)x acts as the swelling agent to effectively prevent GO from restacking during the solvothermal process, thus producing a large pore volume. (b) The doping of nitrogen and boron atoms destroys the electrical neutrality of carbon materials and the uneven distribution of the charge, which enables the surface of the composites produce the adsorption active site. These advantages impart the BN-GAs with excellent adsorption capacity towards CO2 and oils. Fig. 1b shows that the BN-GAs are prepared by using a one-step solvothermal method, during which the reduction of GO (rGO) and the doping of nitrogen and boron is achieved simultaneously.
2. Experimental section
2.1 Materials
Graphite powder (99.9 wt%, average particle size: 2 μm, Adamas), cyanoguanidine (99 wt%, Adamas), boric acid (99 wt%, Greagent), concentrated sulfuric acid, concentrated nitric acid, potassium permanganate, hydrogen peroxide, hydrochloric acid and other chemicals were obtained from Sinopharm Chemical Reagent Company. All chemicals were purchased and used directly without any further purification.
2.2 Synthesis of GO
GO was prepared from natural graphite flakes using a modified Hummers' method.29
2.3 Synthesis of BN(OH)x
BN(OH)x was prepared via thermal substitution process.31 In a typical synthesis, cyanoguanidine was pyrolysis in a tubular furnace at 500 °C for 4 h under N2 flow of 50 mL min−1 with a ramp rate of 2.2 °C min−1 and nature cool to room temperature, and a yellow solid (g-C3N4) was obtained. Then 3 g g-C3N4 powders and 0.9 g boric acid were dissolved in 100 mL deionized water, ultrasonic dispersion for 1 hour. It was then vaporized at 100 °C in air for several hours, and a mixture of well mixed g-C3N4/boric acid was obtained. The mixture was pyrolysis in a tubular furnace at 800 °C for 1 h under N2 flow of 50 mL min−1 with a ramp rate of 3.3 °C min−1 and cool naturally to room temperature to obtain BN(OH)x.
2.4 Synthesis of BN-GAs
BN-GAs were prepared by a combined solvothermal assembly and freeze-drying process. In a typical procedure, a 10 mL GO (4 mg mL−1) aqueous dispersion and a 10 mL BN(OH)x (2 mg mL−1) isopropanol dispersion were treated by sonication for 30 min, followed by intimate mixing with mild ultrasound for 10 min. The resulting stable suspension was sealed in a Teflon-lined autoclave, and was treated solvothermally at 180 °C for 6 h. A black cylinder-like organogel was produced and washed with deionized water several times to remove the isopropanol. Finally, the corresponding hydrogel was freeze overnight, followed by vacuum drying at −60 °C for 72 h, then the aerogel BN-GAX–Y was obtained, where X–Y is the mass ratio of GO to BN(OH)x. For comparison, GA (undoped) was also prepared using the same procedure.
2.5 Materials characterization
Photographs of the BN-GA materials were recorded using a COOLPIX S260 digital camera (Nikon, Japan). The morphology and microstructure were examined using S-4800 field-emission scanning electron microscopy (FE-SEM, Hitachi, Japan). Transmission electron microscopy (TEM) measurements were performed on an EOL-2100 (JEOL, Japan) electron microscope at an operating voltage of 200 kV. Atomic force microscopy (AFM) views were obtained using a Dimension 3100 (Vecco, USA) microscope. Powder X-ray diffraction (XRD) measurements were carried out on a D/max-2200/PC (Rigaku, Japan) with Cu Kα λ = 1.54 Å, a voltage of 40 kV and a current of 40 mA. Raman spectra were obtained using a SENTERRA instrument (Bruker, Germany) with 532 nm line of Ar-ion laser and approximately 5 mW power. The chemical nature of the composites was characterized via X-ray photoelectron spectroscopy (XPS) data that were acquired using an AXIS UltraDLD apparatus (Shimadzu-Kratos, Japan). Monochromatic Al Kα X-ray (hν = 1486.6 eV) was used for analysis, with a photoelectron takeoff angle of 90° to the surface plane. The textural characteristics were quantified by measuring the N2 adsorption/desorption isotherms at −196 °C in a 3H-2000PM2 specific surface & pore size analysis instrument (Beishide, China). All samples were outgassed at 150 °C under vacuum for 6 h prior to the N2 adsorption measurements. The specific surface area was determined by using the Brunauer–Emmett–Teller (BET) model for the N2 adsorption data in the relative pressure (p/p0) range of 0.05–0.20. The total pore volume (Vtotal) was estimated from the amount of N2 adsorbed at p/p0 ∼0.99, whereas the mesopore volume (Vmeso) and pore size distribution were calculated using the Barrett–Joyner–Halenda model.
2.6 Gas adsorption measurements
The CO2 adsorption equilibrium and kinetics of the as-prepared materials were measured volumetrically in a 3H-2000PM2 specific surface and pore-size analysis instrument. The adsorption isotherms were recorded at 0 and 25 °C, and up to 1 bar. The adsorption temperature was controlled by using a Dewar bottle with a circulating jacket that was connected to a thermostatic bath using water as the coolant. Approximately 100 mg of adsorbent sample was used in the adsorption studies. Before each adsorption experiment, the samples were degassed at 150 °C under vacuum for 6 h to remove any moisture and organics. N2 adsorption isotherms at 25 °C were measured using an identical procedure. Ultrahigh-purity (99.99%) gas sources were used. The expected experimental uncertainties in the amounts adsorbed, as estimated using error propagation in all the measured variables, was 0.02 mmol g−1.
2.7 Oil absorption experiments
The absorption capacity of graphene aerogel for various oils and organic solvents was determined by mass measurement. Firstly, the initial sample mass, Wi, was determined. The sample was placed in different oils or organic solvents. After samples and surface oils or solvents were removed with filter paper, the sample was weighed and its mass, Wf, was recorded. The oil absorption capacity (Q) was calculated from:
3. Results and discussion
3.1 Morphology and characterization
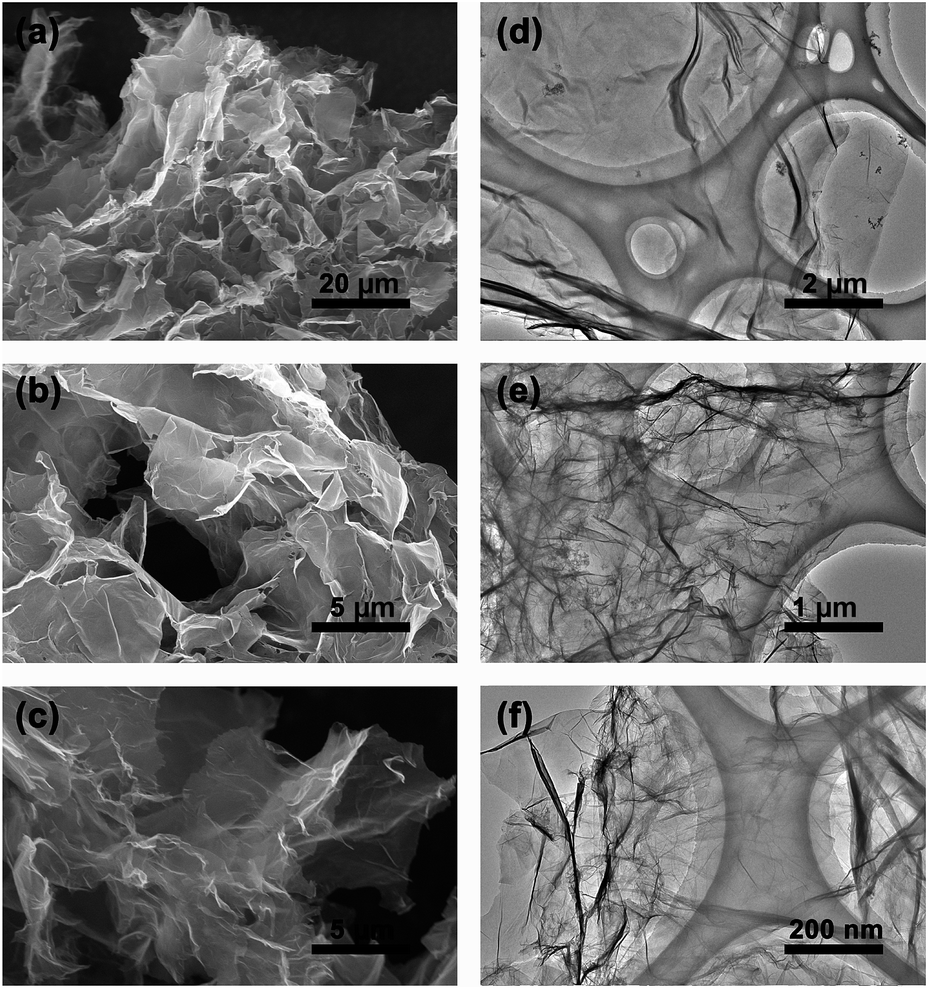
The three-dimensional (3D) morphology of GA and BN-GA2–1 samples was confirmed by SEM and TEM. The GA sample had a very smooth surface with relatively small pore structure (Fig. 2a–c). In contrast, the BN-GA2–1 sample showed an interconnected framework of graphene nanosheets with a porous structure (Fig. 3a). Furthermore, Fig. 3b exhibited that the BN-GA2–1 sample had a well stacked layer structure, and had a considerable number of pore structures in the wall layers. Fig. 3c showed the morphology of BN-GA2–1 with a rough surface, which was beneficial to improvement the adsorption capacity of the material. The TEM images indicated BN-GA2–1 sample had more disordered packing structure with respect to GA (Fig. 2d–f and 3d, e), which indicated that the introduction of BN(OH)x made the graphene sheets stacking more disordered and generated more pore structure. Fig. 3f showed the BN(OH)x had a nanoscale lamellar structure.
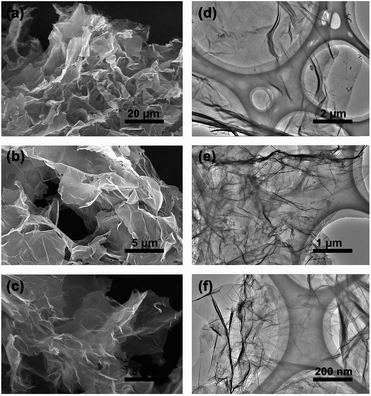 |
| | Fig. 2 (a, b, c) SEM and (d, e, f) TEM images of the GA. | |
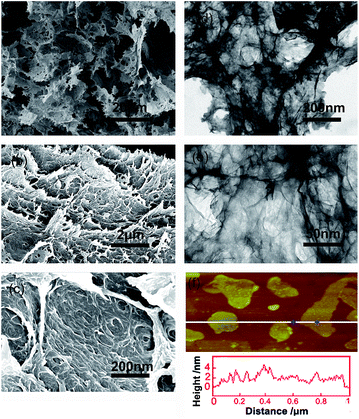 |
| | Fig. 3 (a, b, c) SEM and (d, e) TEM images of the BN-GA2–1 architecture. (f) AFM height image of the BN(OH)x aqueous suspension (0.05 mg mL−1). | |
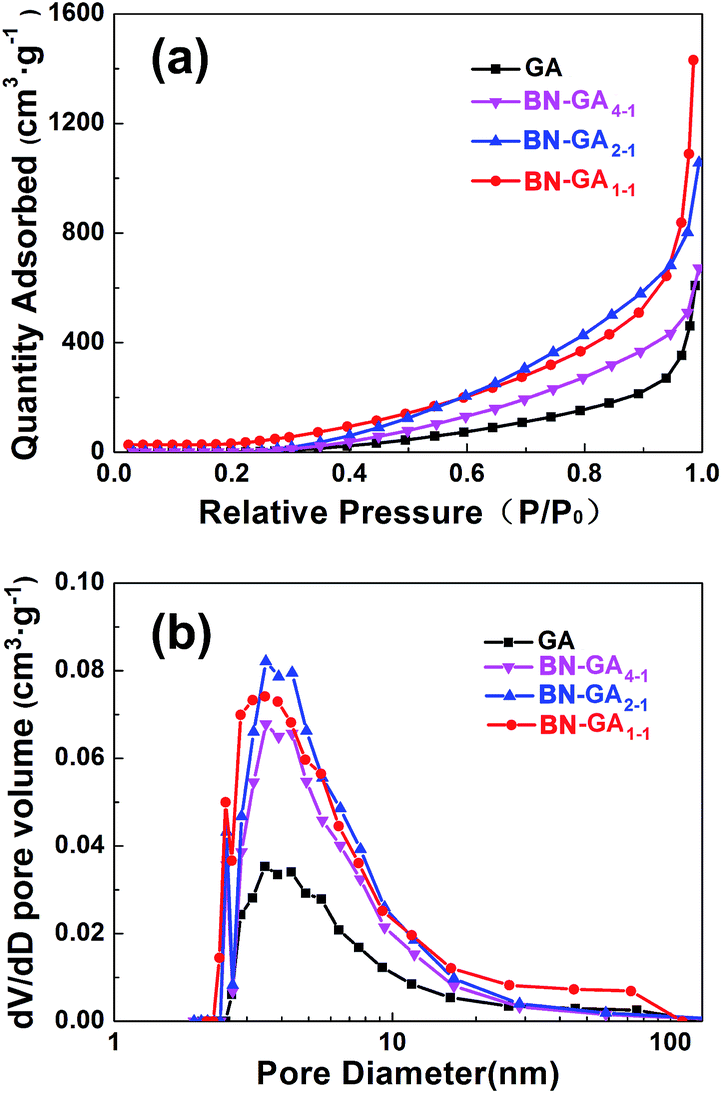
The N2 adsorption isotherm was a type-IV isotherm based on a significant hysteresis loop, which indicated that BN-GAs possessed a typical mesoporous structure (Fig. 4a). From the adsorption branch of the isotherm, the GA, BN-GA4–1, BN-GA2–1, and BN-GA1–1 specific surface areas were 52.3, 65.7, 112.4, and 169.9 m2 g−1 as calculated by a multi-point BET method, and the most probable pore width was 2.5–11 nm as calculated using the Barrett–Joyner–Halenda model. The pore-width distribution was concentrated in the range of a typical mesoporous structure (Fig. 4b). It was speculated that a mesoporous structure with a pronounced distribution of 2.5–11 nm appeared on the surface of the neighboring stacked graphene nanosheets on the BN-GAs. The total pore volumes of GA, BN-GA4–1, BN-GA2–1, and BN-GA1–1, for pore widths ranging from 2 to 167 nm, were 0.94, 1.04, 1.61, and 2.21 cm3 g−1, respectively (Table 1). Our experimental data indicated that the specific surface area of the BN-GA1–1 was 169.9 m2 g−1, which was substantially higher than that of the raw GAs and could be attributed to the presence of BN(OH)x sheets in the composite. The large pore volumes and specific surface areas of BN-GAs should guarantee the adsorption performance.
Table 1 Textural properties of GA and BN-GAs
| Sample |
Textual properties |
| SBET (m2 g−1) |
Vtotal (cm3 g−1) |
| GA |
52.3 |
0.94 |
| BN-GA4–1 |
65.7 |
1.04 |
| BN-GA2–1 |
112.4 |
1.61 |
| BN-GA1–1 |
169.9 |
2.21 |
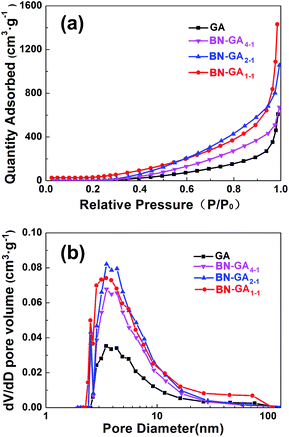 |
| | Fig. 4 (a) N2 sorption isotherms (b) BJH pore distribution of GA and BN-GAs. | |
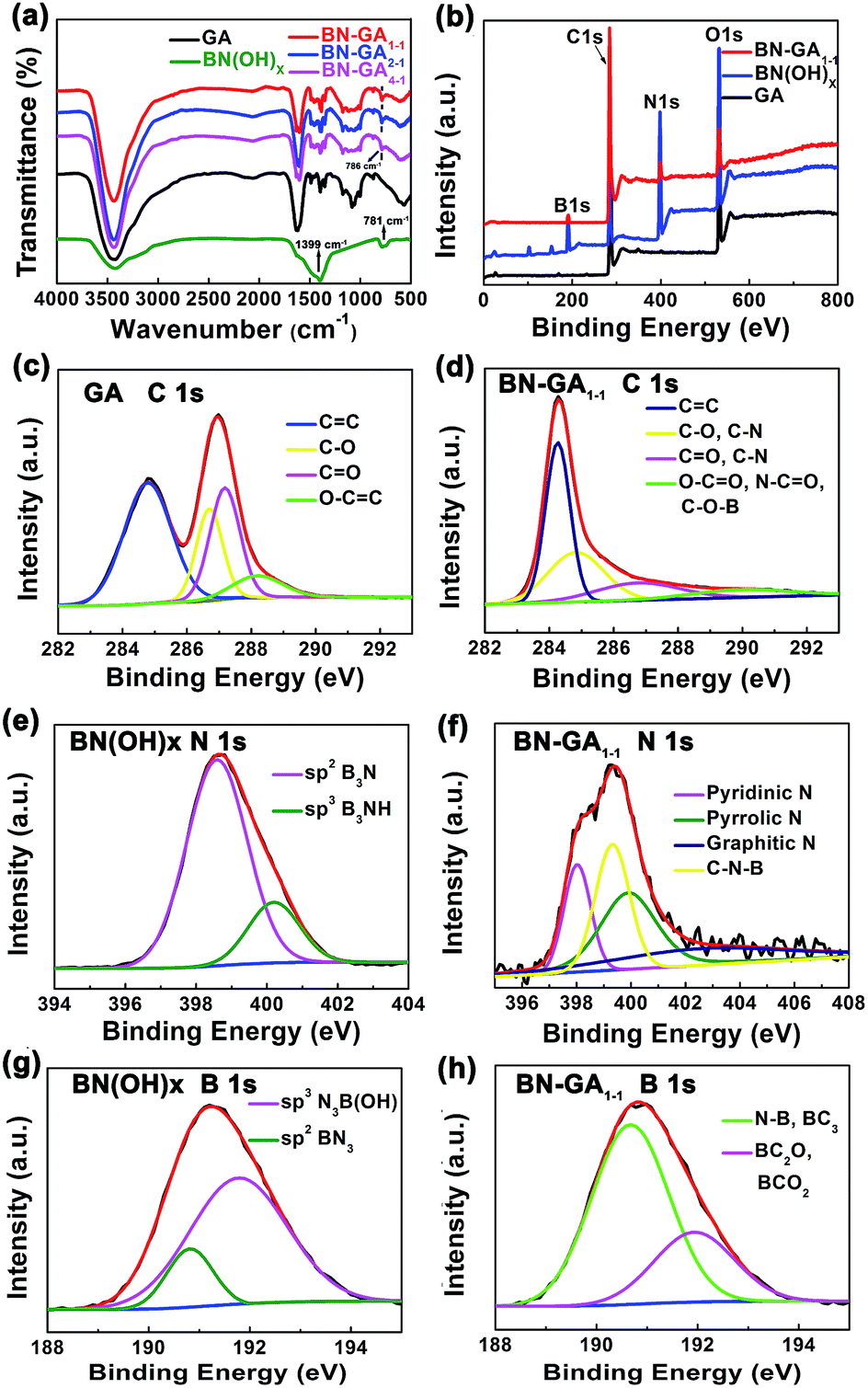
To confirm that the BN(OH)x was doped in the BN-GAs during the solvothermal process, we obtained FTIR, XPS, XRD and Raman spectra. The FTIR data was shown in Fig. 5a. For BN-800-1, the broad band at 3425 cm−1 was attributed to O–H stretching vibrations of hydroxyl groups. The peaks at 1399 and 781 cm−1 belonged to stretching and bending modes of B–N vibrations, respectively.32 For the GO, the IR absorption peaks at 1622 and 1080 cm−1 corresponded to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–O stretching vibrations, respectively.32,33 The IR adsorption peak at 1389 cm−1 due to the O–H deformation vibration.34 Obviously, the B–N bending vibration at 786 cm−1 appeared in all BN-GAX samples. The B–N stretching vibration at 1399 cm−1 might overlap with the peak position of the O–H deformation vibration in the reduction of graphene oxide. Furthermore, the other characteristic absorption peaks of the reduction of graphene oxide were also found in all BN-GA samples, suggesting the successful doping of nitrogen and boron.
C and C–O stretching vibrations, respectively.32,33 The IR adsorption peak at 1389 cm−1 due to the O–H deformation vibration.34 Obviously, the B–N bending vibration at 786 cm−1 appeared in all BN-GAX samples. The B–N stretching vibration at 1399 cm−1 might overlap with the peak position of the O–H deformation vibration in the reduction of graphene oxide. Furthermore, the other characteristic absorption peaks of the reduction of graphene oxide were also found in all BN-GA samples, suggesting the successful doping of nitrogen and boron.
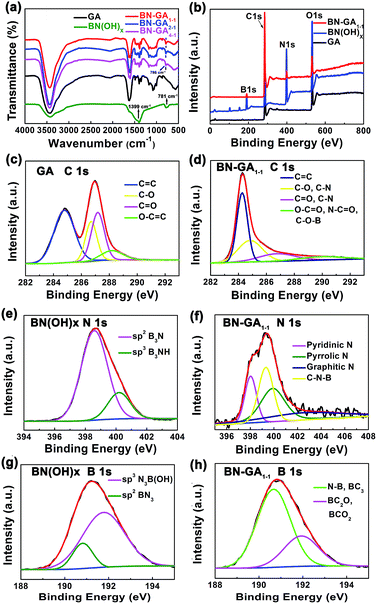 |
| | Fig. 5 (a) FTIR spectra of BN(OH)x, GA, BN-GAs. (b–h) XPS data of GA, BN(OH)x and BN-GA1–1. | |
XPS measurements of GA, BN(OH)x, and BN-GA1–1 were shown in Fig. 5b–h. We observed two additional peaks with binding energies of 191.1 eV (B 1s) and 399.3 eV (N 1s),25–27,30,31 which confirmed the presence of boron and nitrogen, as shown in the XPS survey spectra of the BN-GA1–1 sample (Fig. 5b). Fig. 5c showed the C 1s spectrum of GA which exhibited four peaks of CC (284.8 eV), C–O (286.7 eV), CO (287.2 eV) and O–CO (288.2 eV).19,30 Fig. 5e showed the N 1s spectrum of BN(OH)x which exhibited two peaks of B3N (398.6 eV) and B3NH (400.2 eV). The B 1s spectrum of BN(OH)x exhibited two peaks of BN3 (190.8 eV) and N3B(OH) (191.8 eV), as shown in Fig. 5g.31 Four peaks (284.3, 284.9, 286.8 and 289.9 eV) in the C 1s spectrum of BN-GA1–1 correspond to the sp2 C and the chemical environment of carbon atoms bonded to carbon, nitrogen, and boron in graphene (Fig. 5b).30,35 The asymmetric N 1s XPS peak of BN-GA1–1 (Fig. 5f) could be fitted to four peaks of pyridinic N (398.1 eV), pyrrolic N (399.9 eV) graphitic N (402.6 eV), and C–N–B (399.3 eV).25,30,31 Two peaks at 190.8 eV (B–N, BC3) and 192.1 eV (BC2O, BCO2) could be identified by fitting the B 1s XPS spectra of BN-GA1–1 (Fig. 5h). Which suggested that a certain amount of boron was bonded to nitrogen as B–N, with carbon in the form of B–C, and with oxygen in the form of B–O, respectively.30,31 The above spectral observation indicated the successful doping of BN(OH)x.
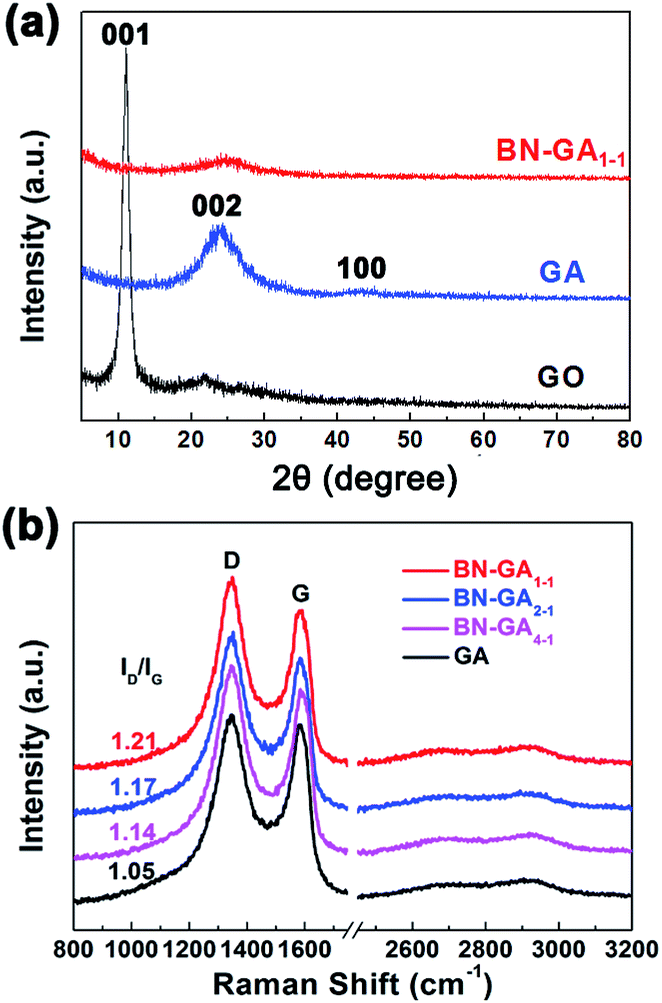
The XRD patterns indicated that the BN-GA1–1 contained rGO and BN(OH)x (Fig. 6a). The weak peaks at 24.2 (002) and 43.1 (100) could be attributed to rGO, the disappearance of the peak at 11.0 (001) that corresponded to GO indicated that the GO reduction was complete.36–38 Very weak graphene sheet peaks of BN-GA1–1 were detected, which suggested that graphene sheets had a highly disordered stacking with a low degree of graphitization because the BN(OH)x sheets in the graphene sheets prevented the graphene sheets from restacking.36,39 The Raman spectra of GA and BN-GAs exhibited two typical peaks (D band and G band) and a small broad peak of a two-dimensional band (Fig. 6b). The D band at around 1346 cm−1 corresponded to the defects of the graphene, and the G-band at around 1580 cm−1 provided evidence for the existence of sp2-hybridized carbon atoms and ordered graphitic structure.2 The ratios of the D band to G band integrated intensities (ID/IG) were 1.05 (GA), 1.14 (BN-GA4–1), 1.17 (BN-GA2–1) and 1.21 (BN-GA1–1), which indicated the disordered degree of graphene increased with the increasing of BN(OH)x doping amount.27,37,40
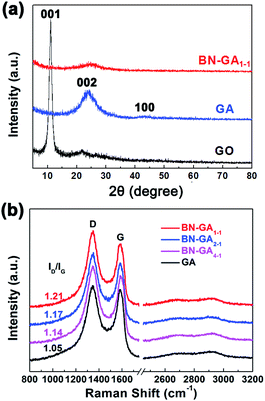 |
| | Fig. 6 (a) XRD patterns of the GO, GA and BN-GA1–1. (b) Raman spectra of the GA and BN-GAs. | |
3.2 Oil and organic solvents capture capacity
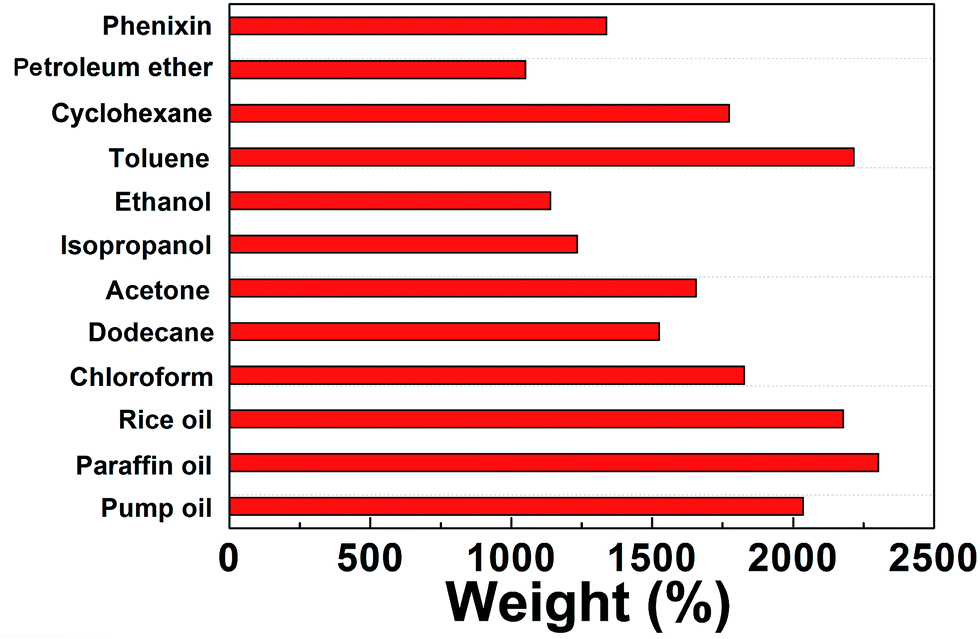
BN-GAs are hydrophobic and have a high porosity, so they should have excellent absorption capacities for oil spills and organic contaminants.41–44 The adsorption properties of the monoliths were evaluated for a range of organic solvents and oils. The BN-GA1–1 exhibited a high and rapid absorption for dodecane (Fig. 7). Once it contacted an oil film (dodecane) dispersed on the water surface, the BN-GA1–1 absorbed the surrounding oil layer immediately to remove the oil film and gave rise to the emergence of a transparent water surface after a few seconds. The absorption capacities of the monoliths for different oils and solvents varied from 10 to 23 times their own weight, depending on the density and viscosity of the oils and organic solvents (Fig. 8). The excellent absorption capability could be attributed to the porous structure and large hydrophobic surface area of the reduced graphene and to the abundant amount of adsorption active sites, which had a considerably larger nitrogen, boron, and hydroxyl group content. The adsorbed oils in the BN-GAs could be extracted by using an acetone solvent followed by drying, whereas absorbed organic solvents were removed readily by heating. Thus, the BN-GAs could be used repeatedly in oil–water separation, water purity, and environmental protection.
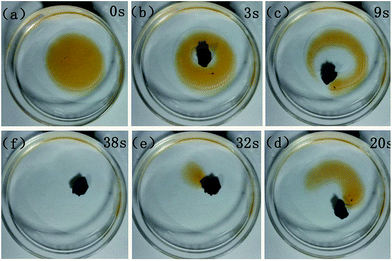 |
| | Fig. 7 Absorption of dodecane from water with BN-GA1–1. The dodecane was dyed with Sudan red for clear observation. | |
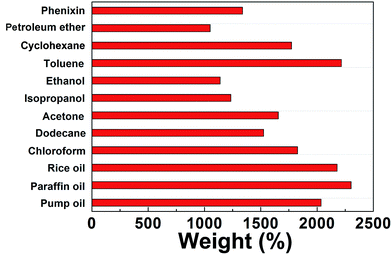 |
| | Fig. 8 Adsorption capacities of BN-GA1–1 for various oils and organic solvents. | |
3.3 CO2 capture capacity
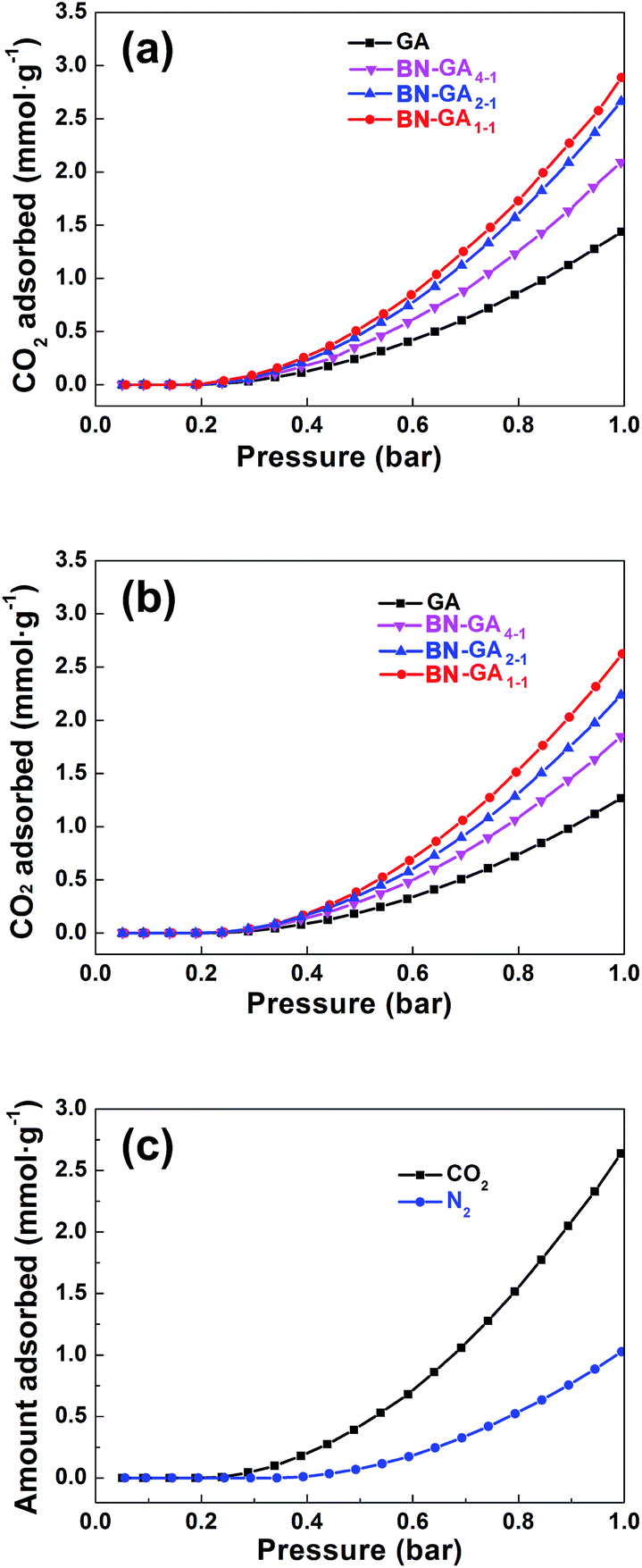
The BN-GAs also have excellent absorption capacity for CO2. The CO2 adsorption isotherms of the as-prepared GA and the BN-GA samples at 0 and 25 °C were shown in Fig. 9a and b, respectively. As expected, CO2 adsorption by the GA materials was dependent on their specific surface area, pore size, and adsorption active site (Table 2). GA, due to its low specific surface area (52.3 m2 g−1) and rare adsorption active sites, showed the lowest CO2 adsorption capacity of 1.4 and 1.3 mmol g−1 at 0 and 25 °C, respectively, whereas the BN-GA samples CO2 adsorption capacity at 0 °C and 25 °C was in the 2.1–2.9 mmol g−1 (61.6–127.6 mg g−1) and 1.8–2.6 mmol g−1 (57.2–114.4 mg g−1) range, respectively. The CO2/N2 selectivity of BN-GA1–1 was calculated from the single component CO2 and N2 adsorption isotherms (Fig. 9c). BN-GA1–1 showed a CO2/N2 saturation pressure selectivity of 2.6. BN-GAs exhibit, relative to the GA sample, better CO2 uptake and excellent CO2/N2 selectivity. This is due to the N, B codoped graphene aerogels with hydroxylated boron nitrides exhibit two specific characteristics that have a positive effect on CO2 adsorption. Indeed, the BN-GAs samples have a larger specific surface area, higher porosity, and more adsorption active sites, which contain a considerably larger nitrogen, boron, and hydroxyl group content.
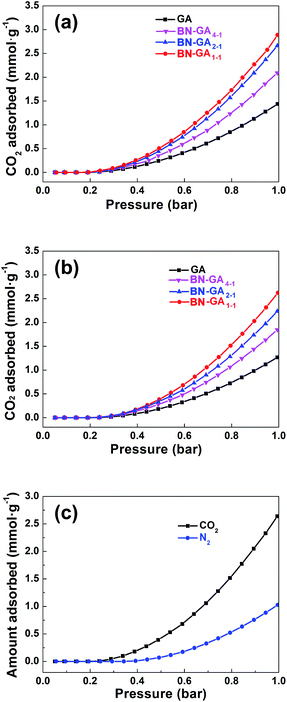 |
| | Fig. 9 CO2 adsorption isotherms at (a) 0 °C and (b) 25 °C for GA and BN-GAs, respectively. (c) CO2 and N2 adsorption isotherms of BN-GA1–1 at 25 °C. | |
Table 2 CO2 capture capacities of the GA and BN-GAs at different adsorption temperatures and 1 bar
| Sample |
CO2 uptake, mmol g−1 (mg g−1) |
| 0 °C |
25 °C |
| GA |
1.4 (61.6) |
1.3 (57.2) |
| BN-GA4–1 |
2.1 (92.4) |
1.8 (79.2) |
| BN-GA2–1 |
2.7 (118.8) |
2.2 (96.8) |
| BN-GA1–1 |
2.9 (127.6) |
2.6 (114.4) |
As a stable and nontoxic carbon material, the aerogels will not have any adverse effect on human and environment, GA and its composites as the adsorption material has been a lot of reported.45 The initial synthesis of BN(OH)x is used as a kind of safe, nontoxic, stable, biocompatible anticancer drug carrier.31 Thereby, BN-GAs which prepared by solvothermal method with GO and BN(OH)x are safe, stable and nontoxic. In these three-dimensional materials, the stacked rGO layers provided skeleton structure and the introduction of BN(OH)x sheets increases the porosity, the surface roughness and the adsorption active sites. As for the process of CO2 and oils adsorption, it will not cause reactions to produce toxic and harmful substances. Meanwhile, BN-GAs have the advantages of operation safety, low energy consumption, easy maintenance and regeneration as a solid adsorbent, indicating excellent industrial application potentials.
4. Conclusion
In summary, we have synthesized self-assembled BN-GAs hybrids using a facile solvothermal reduction and freeze-drying of GO and BN(OH)x dispersions in a mixture of isopropanol and water. Because of the BN(OH)x hybrid 3D macroporous structure and high surface area, the resulting BN-GAs show excellent adsorption performance for CO2, oil, and organic solvents and outstanding CO2/N2 selectivity, which may provide BN-GAs as a 3D porous structure for other efficient energy-storage and environmental protection devices.
Acknowledgements
This work was supported by the National Sciences & Technology Major Project of China (No. 2011ZX05011-004) and the National Nature Sciences Foundation of China (No. 41502122).
Notes and references
- K. C. Kemp, H. Seema, M. Saleh, N. H. Le, K. Mahesh, V. Chandra and K. S. Kim, Nanoscale, 2013, 5, 3149 RSC.
- C. Chen, R. Li, L. Xu and D. Yan, RSC Adv., 2014, 4, 17393 RSC.
- R. M. Atlas and T. C. Hazen, Environ. Sci. Technol., 2011, 45, 6709 CrossRef CAS PubMed.
- K. Rahman, J. Thahira-Rahman, P. Lakshmanaperumalsamy and I. Banat, Bioresour. Technol., 2002, 85, 257 CrossRef CAS PubMed.
- A. B. Rao and E. S. Rubin, Environ. Sci. Technol., 2002, 36, 4467 CrossRef CAS PubMed.
- D. Camper, J. E. Bara, D. L. Gin and R. D. Noble, Ind. Eng. Chem. Res., 2008, 47, 8496 CrossRef CAS.
- J. E. Bara, D. E. Camper, D. L. Gin and R. D. Noble, Acc. Chem. Res., 2009, 43, 152 CrossRef PubMed.
- C.-H. Yu, C.-H. Huang and C.-S. Tan, Aerosol Air Qual. Res., 2012, 12, 745 CAS.
- C. Zhang, W. Song, Q. Ma, L.-J. Xie and X. Zhang, Energy Fuels, 2016, 30, 4181 CrossRef CAS.
- H. Yang, M. Gong and Y. Chen, J. Nat. Gas Chem., 2011, 20, 460 CrossRef CAS.
- Q. Yang, S. Vaesen, F. Ragon, A. D. Wiersum, D. Wu, A. Lago, T. Devic, C. Martineau, F. Taulelle and P. L. Llewellyn, Angew. Chem., Int. Ed., 2013, 52, 10316 CrossRef CAS PubMed.
- C. Yang, U. Kaipa, Q. Z. Mather, X. Wang, V. Nesterov, A. F. Venero and M. A. Omary, J. Am. Chem. Soc., 2011, 133, 18094 CrossRef CAS PubMed.
- G.-P. Hao, W.-C. Li, D. Qian, G.-H. Wang, W.-P. Zhang, T. Zhang, A.-Q. Wang, F. Schüth, H.-J. Bongard and A.-H. Lu, J. Am. Chem. Soc., 2011, 133, 11378 CrossRef CAS PubMed.
- I. Tan, A. Ahmad and B. Hameed, J. Hazard. Mater., 2009, 164, 473 CrossRef CAS PubMed.
- J.-S. Lee, J.-H. Kim, J.-T. Kim, J.-K. Suh, J.-M. Lee and C.-H. Lee, J. Chem. Eng. Data, 2002, 47, 1237 CrossRef CAS.
- F. Su, C. Lu, S.-C. Kuo and W. Zeng, Energy Fuels, 2010, 24, 1441 CrossRef CAS.
- G. J. Suppes, M. A. Dasari, E. J. Doskocil, P. J. Mankidy and M. J. Goff, Appl. Catal., A, 2004, 257, 213 CrossRef CAS.
- X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey and H. Zhang, Small, 2011, 7, 1876 CrossRef CAS PubMed.
- S. Yang, X. Song, P. Zhang, J. Sun and L. Gao, Small, 2014, 10, 2270 CrossRef CAS PubMed.
- R. Wang, C. Xu, J. Sun, L. Gao and H. Yao, ACS Appl. Mater. Interfaces, 2014, 6, 3427 CAS.
- L. Ren, K. S. Hui and K. N. Hui, J. Mater. Chem. A, 2013, 1, 5689 CAS.
- Y.-C. Yong, X.-C. Dong, M. B. Chan-Park, H. Song and P. Chen, ACS Nano, 2012, 6, 2394 CrossRef CAS PubMed.
- R. Li, C. Chen, J. Li, L. Xu, G. Xiao and D. Yan, J. Mater. Chem. A, 2014, 2, 3057 CAS.
- Y. Lin, G. J. Ehlert, C. Bukowsky and H. A. Sodano, ACS Appl. Mater. Interfaces, 2011, 3, 2200 CAS.
- Y. Zhao, C. Hu, Y. Hu, H. Cheng, G. Shi and L. Qu, Angew. Chem., Int. Ed., 2012, 124, 11533 CrossRef.
- Y. Su, Y. Zhang, X. Zhuang, S. Li, D. Wu, F. Zhang and X. Feng, Carbon, 2013, 62, 296 CrossRef CAS.
- Z. S. Wu, A. Winter, L. Chen, Y. Sun, A. Turchanin, X. Feng and K. Müllen, Adv. Mater., 2012, 24, 5130 CrossRef CAS PubMed.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496 CrossRef CAS PubMed.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- M. Sevilla, P. Valle-Vigón and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781 CrossRef CAS.
- Q. Weng, B. Wang, X. Wang, N. Hanagata, X. Li, D. Liu, X. Wang, X. Jiang, Y. Bando and D. Golberg, ACS Nano, 2014, 8, 6123 CrossRef CAS PubMed.
- H. Yang, H. Zhu, M. M. Hendrix, N. J. Lousberg, W. G. De, A. C. Esteves and J. H. Xin, Adv. Mater., 2013, 25, 1150 CrossRef CAS PubMed.
- S. Tao, Y. Wang and Y. An, J. Mater. Chem., 2011, 21, 11901 RSC.
- H. Hu, Z. Zhao, W. Wan, Y. Gogotsi and J. Qiu, Adv. Mater., 2013, 25, 2219 CrossRef CAS PubMed.
- J. Zhou, Z. Zhang, W. Xing, J. Yu, G. Han, W. Si and S. Zhuo, Electrochim. Acta, 2015, 153, 68 CrossRef CAS.
- S. Chowdhury and R. Balasubramanian, Ind. Eng. Chem. Res., 2016, 55, 7906 CrossRef CAS.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324 CrossRef CAS PubMed.
- M. Z. Iqbal and A. A. Abdala, RSC Adv., 2013, 3, 24455 RSC.
- S. Yang, X. Song, P. Zhang, J. Sun and L. Gao, Small, 2014, 10, 2270 CrossRef CAS PubMed.
- L. Xu, G. Xiao, C. Chen, R. Li, Y. Mai, G. Sun and D. Yan, J. Mater. Chem. A, 2015, 3, 7498 CAS.
- S. Kabiri, D. N. Tran, T. Altalhi and D. Losic, Carbon, 2014, 80, 523 CrossRef CAS.
- Y. He, Y. Liu, T. Wu, J. Ma, X. Wang, Q. Gong, W. Kong, F. Xing, Y. Liu and J. Gao, J. Hazard. Mater., 2013, 260, 796 CrossRef CAS PubMed.
- X. Song, L. Lin, M. Rong, Y. Wang, Z. Xie and X. Chen, Carbon, 2014, 80, 174 CrossRef CAS.
- X. Gui, Z. Zeng, Z. Lin, Q. Gan, R. Xiang, Y. Zhu, A. Cao and Z. Tang, ACS Appl. Mater. Interfaces, 2013, 5, 5845 CAS.
- M. Yusuf, F. M. Elfghi, S. A. Zaidi, E. C. Abdullah and M. A. Khan, RSC Adv., 2015, 5, 50392 RSC.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence