DOI:
10.1039/C6RA24949C
(Paper)
RSC Adv., 2017,
7, 1045-1055
In situ repair of graphene defects and enhancement of its reinforcement effect in polyvinyl alcohol hydrogels
Received
9th October 2016
, Accepted 13th November 2016
First published on 4th January 2017
Abstract
Defects cause unexpected and unsatisfactory effects in graphene when it is used as a reinforcement in composites. Herein, γ-ray irradiation was introduced as a facile and efficient method to simultaneously repair the defects of reduced graphene oxide (rGO) and enhance the reinforcement effect of graphene in a polyvinyl alcohol (PVA) matrix. Under the action of γ-ray irradiation, the surfaces of rGO were activated and free radicals were produced on PVA chains. At higher irradiation doses, random chain scissions of the PVA main-chain were linked with the reactive groups around the edge of defects on the rGO, leading to the repair of the crystalline graphene, which was evidenced by the decrease of ID/IG and stronger 2D bands of the irradiated PVA/rGO hydrogel composites. Thus, the reinforcement effect of graphene is enhanced by γ-ray irradiation. The tensile and compressive strength of the PVA/rGO hydrogel composites achieved their maximum values at irradiation doses of 100 kGy and 150 kGy, respectively, which were 44% and 171% higher than the nonirradiated composites, and 113% and 336% higher than those of pure PVA hydrogel, respectively.
1. Introduction
Poly(vinyl alcohol) (PVA) can form intriguing hydrogels with high water content, which are considered very promising biomaterials for biomedical and tissue engineering for repairing diseased or damaged cartilage, meniscuses, nucleus pulposus, and human lenses because of their excellent biocompatibility, nontoxicity, good water-absorption ability and lubrication.1–5 Unfortunately, the pure PVA hydrogel suffers from poor mechanical strength, which has hindered its development and applications. Integrating the hydrogels with nanofillers to form nanocomposites is regarded as an effective method to enhance the mechanical strength and toughness of hydrogels.6–9
Graphene, a unique allotrope of carbon, is a one-atom planar sheet composed of sp2-hybridized carbon atoms arranged in a honeycomb lattice.10,11 Among the remarkable physiochemical properties11–15 observed in graphene, the extremely high strength reported in monolayer graphene, of 1000 ± 100 GPa for the modulus of elasticity and 130 GPa for the intrinsic strength,12 shows great potential for reinforcing its nanocomposites for various applications, such as energy devices and biomedicine.16,17 However, graphene-based nanocomposites cannot usually achieve the expected reinforcing level. For example, Yang et al.18 synthesized PVA/graphene hydrogels by freeze–thaw method, where only a 16% and a 32% increase in modulus and tensile stress, respectively, were demonstrated for the composites containing 3.5 wt% graphene. Thayummanavan prepared PVA/sodium alginate-m-graphene nanocomposites19 and PVA/sodium dodecyl sulfate/sodium alginate-m-graphene nanocomposites,20 and just a 40% and a 112% increase in tensile strength, respectively, were obtained with 0.5 wt% graphene content. Similar to other low-dimensional carbon nanostructures like fullerenes and carbon nanotubes,21,22 disorder and defects in graphene, resulting from the synthesis process or the transfer process,23 strongly affect the mechanical properties, so controlling defects is recognized as a promising method to tailor or modify the intrinsic properties of graphene.24 If these lattice defects can be repaired within its nanocomposites, the resulting graphene could exhibit a better reinforcing effect.
Recently, γ-ray irradiation has been successfully utilized for the surface functionalization of graphene. Moreover, it has been reported that irradiation can further limit the aggregations of graphene sheets owing to its mild irradiation reduction rate.25 Gupta et al.26 found that the functionalization of reduced graphene oxide (rGO) nanosheets with poly(ethylene glycol) 200 (PEG200) through γ-radiolysis led to an increase in the d spacing of the graphene sheets and a decrease in the defect density of the carbon network in the rGO, and the PEG200-functionalized rGO was used as an effective lubricant to reduce both the friction coefficient and the wear of sliding steel surfaces. Dumee et al.27 demonstrated that the graphene obtained from the reduction of graphene oxide (GO) through γ-ray irradiation showed less change in morphology than that obtained from chemically reduced GO, and the decrease in the D/G ratio from the Raman spectra after γ-ray irradiation indicated the recovery of the pristine graphene. Shi et al.28 also found that the D/G ratio decreased from 1.30 for the nonirradiated PVA/GO hydrogels to 0.93 for the composites treated with an irradiation dose of 200 kGy, suggesting the repair of defects by the recovery of the aromatic structure. Kim et al.23 reported that the surface potential of graphene could be engineered by introducing atomic-scale defects via irradiation with high-energy particles.
γ-Ray irradiation has also been studied as a clean method for the modification of hydrogel materials. When used in polymer composites, irradiation results in either cross-linking or degradation of polymers. When the PVA hydrogels were irradiated in aqueous solution, the energy of irradiation was partly absorbed by water, and the resulting H· and ·OH were abstracted hydrogen atoms from the PVA chain, leading to the coupling of two polymer radicals and hence the formation of cross-linking.29,30 Moreover, cross-linking was also formed by the reaction of PVA· produced from PVA by the direct action of irradiation.30 Degradation of the polymer occurred with an excessive dose of irradiation as a result of the scission of the C–C bonds of the main chain.31 Zhang et al.32 demonstrated that PVA radiolysis was initiated by ·OH and H·, leading to chain scission and formation of ketones/enols. Shi et al.33 demonstrated that the mechanical strength of PVA/poly(vinyl pyrrolidone) (PVP) hydrogels increased first and then decreased with increasing irradiation dose, and hydrogels treated with an irradiation dose of 100 kGy showed the best mechanical performance and the lowest friction coefficient.
It is expected that during γ-ray irradiation, the C–C scission chain from PVA induced by irradiation could bridge the defects in graphene to repair its crystalline structure by linking with the radicals around the edge of the defects in the graphene, which would increase the strength of graphene and its nanocomposites. Furthermore, the interfacial interaction between the graphene and the PVA matrix is thought to be enhanced owing to the increased interfacial activities of graphene upon γ-ray irradiation.34 Thus, in the present study, PVA/rGO hydrogel composites were synthesized by the freeze–thaw method followed by γ-ray irradiation with various total doses. Changes in the microstructure of the hydrogel composites were characterized after irradiation. The thermal stability and degree of crystallinity of the PVA/rGO hydrogel composites were assessed as a function of irradiation dose. Additionally, the mechanical performances of the PVA/rGO hydrogel composites after γ-ray irradiation were investigated.
2. Experimental
2.1. Materials
The reduced graphene oxide was purchased from Nanjing FAME Bearing Co. Ltd., Nanjing, China. It was prepared by improved Hummers' method and reduced by hydrazine. PVA, saponified with a polymerization degree of 1700, was obtained from Kuraray Co. Ltd., Tokyo, Japan.
2.2. Preparation of PVA/rGO hydrogels
To prepare the PVA/rGO composite hydrogels, PVA powder was dissolved in deionized water at 95 °C for 10 h to form an aqueous solution. A certain amount of rGO power was dispersed in deionized water by ultrasonic agitation for 20 min. Then, the rGO aqueous suspension was dripped into the PVA solution, and the mixed solution was stirred at 95 °C for 1 h. After being held at a higher temperature to remove air bubbles, the mixed solution was poured into molds and subjected to five cycles of freezing at −20 °C for 18 h and thawing at room temperature for 6 h. The amount of rGO powder was 0.1 wt% of the PVA powder, and the polymer concentration was kept at 15% (w/w). After that, the as-prepared hydrogels immersed in oxygen-free deionized water were then exposed to 60Co γ-ray irradiation at a dose rate of 0.89 kGy h−1 at room temperature across a series of total doses comprising 50, 100, 150, and 200 kGy. Nonirradiated PVA/rGO hydrogels were also prepared as controls to evaluate the impact of the irradiation dose on the material properties in terms of microstructure, and mechanical and friction properties.
2.3. Characterization techniques
Scanning electron microscopy (SEM; FEI Quanta 250FEG, Hillsboro, OR) was used to evaluate the morphology of the rGO powder and the fracture morphology of the PVA/rGO hydrogels after tensile tests. The hydrogel samples were placed in a freeze dryer (FD-1A-50, Beijing Boyikang Laboratory Instrument Co., Ltd., Beijing, China) for at least 2 days to remove all water, and then the samples were sputter-coated with a layer of gold for SEM observations. Field-emission transmission electron microscopy (FETEM) images of rGO were recorded using a Tecnai G2 20 LaB6 instrument (FEI, Hillsboro, OR). An FM-Nanoview 1000 atomic force microscope (AFM; Suzhou Flyingman Precision Instrument Co., Ltd., Suzhou, China) was used to determine the thickness of the rGO sheets. X-ray diffraction (XRD) patterns were obtained with a Bruker-AXS D8 Advance X-ray diffractometer (Bruker, Karlsruhe, Germany) with Cu Kα radiation (λ = 0.1541 nm) from 5° to 60° (in steps of 0.02°). Fourier transform infrared (FT-IR) spectroscopy (Nicolet MAGNA-IR 750, Madison, WI) was performed in the range of 4000–550 cm−1 with a resolution of 4 cm−1. Raman spectra were recorded on a LabRAM ARAMIS Raman microscope system (Horiba JobinYvon, Villeneuve d'Ascq, France) using green (532 nm) laser excitation. X-ray photoelectron spectroscopy (XPS) analysis was performed with a PHI QUANTERA II photoelectron spectrometer (ULVAC PHI, Kanagawa, Japan). Survey spectra were obtained at 100 eV pass energy, whereas high-resolution peak scans were performed at a 20 eV pass energy.
2.4. Water content
For all of the hydrogel samples, the water content, W, was determined according to the equation:| |
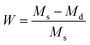 | (1) |
where Ms and Md represent the masses of the fully swollen and completely dried samples, respectively. Five independent samples were tested for each set of hydrogels (n = 5).
2.5. Thermal properties and crystallinity
Thermogravimetric analysis (TGA) was carried out using a TGA/SDTA851E thermal analyzer from 50 to 600 °C at a heating rate of 10 °C min−1, under a N2 atmosphere. Differential scanning calorimetry (DSC; Mettler Toledo, Greifensee, Switzerland, model DSC823e) was carried out from 50 to 300 °C at a heating rate of 10 °C min−1, under a N2 atmosphere. The speed of the nitrogen gas flow was 30 mL min−1. The crystallinity degree was calculated using the heat of fusion of a perfect crystal of PVA (ΔHf* = 138.6 J g−1):35| |
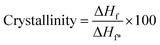 | (2) |
2.6. Mechanical property tests
The mechanical property tests were carried out on a universal testing machine (Instron 3367, USA). The tensile properties were determined at a strain rate of 10 mm min−1. Unconfined compression tests were also carried out at a rate of 4 mm min−1 until the compression strain reached 65% because the PVA/rGO hydrogels are a soft material. Three independent samples were tested for each set of hydrogels (n = 3).
3. Results and discussion
3.1. Characterization of rGO
Fig. 1a and b show that the surface morphology of the rGO appears like cotton wool, and some crumpling is observed because of the very small thickness. Based on the TEM image shown in Fig. 1b, the rGO sheets show a restacked few-layer structure of about 2–3 layers. Fig. 1c shows a typical atomic force microscopy (AFM) image of rGO deposited onto a cleaved mica substrate from an aqueous dispersion and the corresponding height profile of the graphene nanosheets. The thickness of the rGO nanosheets is about 0.83 nm (Fig. 1c), which contain at least two atom layers.36
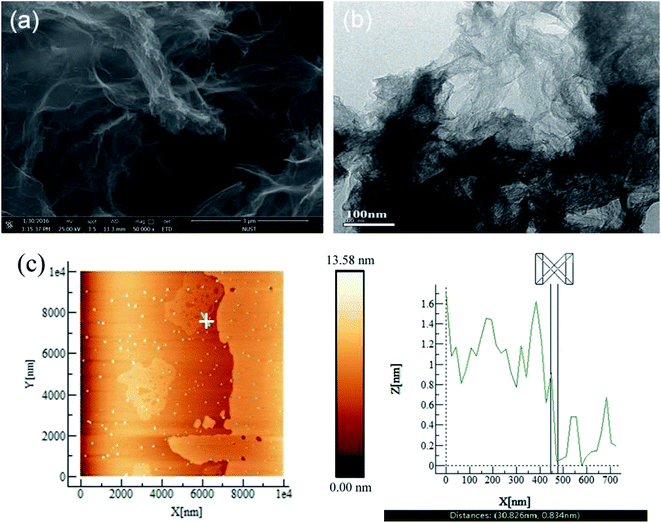 |
| | Fig. 1 (a) SEM, (b) TEM and (c) AFM images of rGO. | |
3.2. Changes in the microstructure of PVA/rGO composites after irradiation
Fig. 2 shows the XRD patterns of pure PVA and PVA/rGO hydrogels treated with different irradiation doses. The typical characteristic diffraction peak of pure PVA is observed at about 2θ = 19.3°. It is clear that after the incorporation of rGO into the PVA matrix, the XRD patterns of the PVA/rGO composites show only the characteristic diffraction peak from PVA, whereas the characteristic peaks of rGO do not appear. It may be because the additive amount of rGO is very small, and its diffraction is very weak compared to PVA. Moreover, an intensified peak at 2θ = 22.9° for the PVA/rGO composites is observed, which demonstrates the intercalated nature of the dispersed rGO sheets.19,20 After the γ-ray irradiation is applied to the composites, the positions of the diffraction peaks shift little (Table 1), but their intensities decrease significantly compared to the nonirradiated composite sample. The results imply that the γ-ray irradiation leads to a great decrease in the crystallinity degree of the PVA/rGO hydrogel composites. With the increase of irradiation dose, the peak intensity decreases gradually, but the change is not very significant.
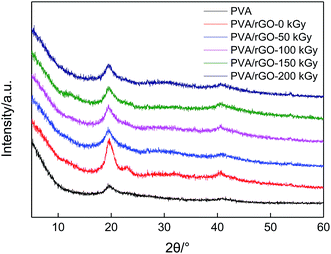 |
| | Fig. 2 XRD patterns of pure PVA, and PVA/rGO hydrogels treated with different irradiation doses. | |
Table 1 Summary of the results from XRD, DSC and TGA for various hydrogel samples
| Samples |
XRD |
DSC |
TGA |
| 2θ/° |
Tm/°C |
Crystallinity/% |
Tp-second/°C |
Tp-third/°C |
| PVA/rGO-0 kGy |
19.6 |
226.4 |
37.9% |
360.0 |
436.7 |
| PVA/rGO-50 kGy |
19.5 |
219.8 |
26.0% |
367.3 |
439.2 |
| PVA/rGO-100 kGy |
19.4 |
219.7 |
24.2% |
370.3 |
440.5 |
| PVA/rGO-150 kGy |
19.5 |
210.6 |
3.6% |
365.3 |
437.8 |
| PVA/rGO-200 kGy |
19.4 |
207.1 |
1.8% |
365.0 |
437.7 |
FT-IR spectroscopy was performed to investigate the interaction between the rGO sheets and the polymer matrix. Fig. 3 gives the FT-IR spectra of rGO, pure PVA, and the nonirradiated and irradiated PVA/rGO hydrogels. In the rGO spectrum, all the characteristic peaks are very weak. The characteristic peaks at about 1567 cm−1 and 1117 cm−1 correspond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O stretching vibrations, respectively, indicating that there are a small number of residual oxygen-containing functional groups in the rGO. In the spectrum of PVA, the absorption peak at 3000–3700 cm−1 is due to the symmetrical stretching vibration of hydroxyl groups, which demonstrates the strong intermolecular and intramolecular hydrogen bonding in PVA.37 It is well known that both the –OH and C–O stretching bands are sensitive to the hydrogen bonding.18 When the rGO is incorporated into PVA, the –OH stretching peak of the composite is shifted to a lower wavenumber. This is because the rGO cuts off the hydrogen bonding between the polymer molecules, and new hydrogen bonds are formed between the rGO sheets and the PVA matrix. Significant decreases in the peak intensities of –OH stretching and C–O stretching (at around 1091 cm−1) are found with the increase of irradiation dose. It can be explained by the increasing interfacial interactions between rGO and PVA and the removal of oxygen-containing functional groups from the rGO by γ-ray irradiation, suggesting the successful grafting of PVA on the rGO.
O and C–O stretching vibrations, respectively, indicating that there are a small number of residual oxygen-containing functional groups in the rGO. In the spectrum of PVA, the absorption peak at 3000–3700 cm−1 is due to the symmetrical stretching vibration of hydroxyl groups, which demonstrates the strong intermolecular and intramolecular hydrogen bonding in PVA.37 It is well known that both the –OH and C–O stretching bands are sensitive to the hydrogen bonding.18 When the rGO is incorporated into PVA, the –OH stretching peak of the composite is shifted to a lower wavenumber. This is because the rGO cuts off the hydrogen bonding between the polymer molecules, and new hydrogen bonds are formed between the rGO sheets and the PVA matrix. Significant decreases in the peak intensities of –OH stretching and C–O stretching (at around 1091 cm−1) are found with the increase of irradiation dose. It can be explained by the increasing interfacial interactions between rGO and PVA and the removal of oxygen-containing functional groups from the rGO by γ-ray irradiation, suggesting the successful grafting of PVA on the rGO.
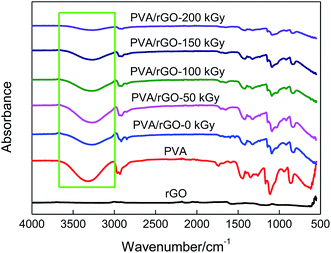 |
| | Fig. 3 FTIR spectra of rGO, pure PVA, and the nonirradiated and irradiated PVA/rGO hydrogels. | |
Raman spectroscopy was used to further characterize the structural changes in the PVA/rGO composites. Fig. 4 shows the Raman spectra of the rGO, pure PVA and PVA/rGO hydrogel composite. Two remarkable peaks located at 1358 and 1594 cm−1 correspond to the D and G bands for the rGO, respectively. The D band is an indicator of disorder resulting from structural defects, whereas the G band is attributed to the ordered sp2 carbon domains.38 For the pure PVA, the peak at 1430 cm−1 is ascribed to the stretching vibration of –CH in the PVA molecules, and the most intense band centered at 2912 cm−1 is assigned to the stretching vibrations of –CH2.39 In the Raman spectra of the PVA/rGO composite, both characteristic peaks of PVA and rGO appear. The ratio of D to G-band intensity, ID/IG, is used to qualitatively characterize the defect concentration in graphitic material.40 The ID/IG was 0.9 and 1.5 for the rGO and PVA/rGO composite, respectively. The increase of the D/G ratio suggests the generation of a large number of sp2 carbon domains with a smaller average size in PVA/rGO.41 Fig. 5 presents the effect of irradiation dose on the Raman spectra of PVA/rGO hydrogel composites. It is obvious that the D bands become weaker after γ-ray irradiation, whereas the G bands downshift when compared to the nonirradiated PVA/rGO hydrogels. As shown in Fig. 6, the ID/IG decreases significantly after γ-ray irradiation, and it gradually decreases from 0.7 to 0.3 with the increase of the irradiation dose from 50 to 200 kGy. The significant reduction of ID/IG indicates the recovery of the rGO crystalline microstructure, which is ascribed to the reduced pre-dominance of the boundaries of the rGO crystallites on the rGO inner crystal plane.27 This also implies that γ-ray irradiation removes the oxygen-containing functional groups present in rGO, as the increased interfacial activities of the rGO sheets induced by irradiation facilitate interactions with the free radicals generated in the PVA, which leads to an increase in the dimensions of the in-plane sp2 domains, followed by the decreasing disorder.26 The 2D band originates from the splitting in the π-electron dispersion and is ascribed to the interactions between the basal planes of graphite. Generally, the position and shape of the 2D band are very sensitive to the number of graphene layers and the stacking order of the graphene sheets along the c axis.42 As shown in Fig. 5, γ-ray irradiation produces a small displacement in the 2D band of the PVA/rGO hydrogel composites owing to the resulting structural changes in rGO sheets in the process of γ-ray irradiation. The 2D band becomes sharp for the irradiated PVA/rGO hydrogel composites, and an increase in the 2D band intensity is observed with the increase of irradiation dose, indicating the gradually recovery of the graphene layers. The results imply the successful grafting of PVA chains on rGO and the restoration of sp2-hybridized carbon bonds in graphitic structures.43
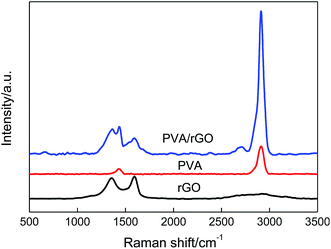 |
| | Fig. 4 Raman spectra of rGO, pure PVA, and PVA/rGO hydrogel. | |
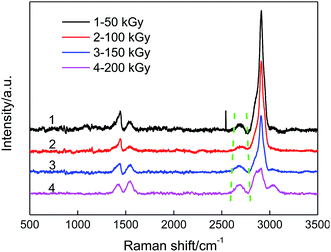 |
| | Fig. 5 Raman spectra of PVA/rGO hydrogel composites treated with different irradiation doses. | |
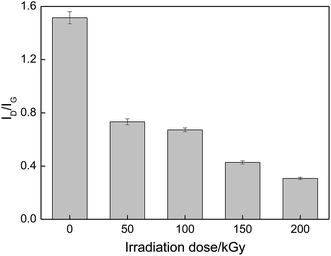 |
| | Fig. 6 Effect of irradiation dose on ID/IG of PVA/rGO hydrogel composites. | |
Highlights on the C 1s and O 1s peaks are shown in the low resolution XPS spectra of rGO, PVA/rGO-0 kGy, and PVA/rGO-200 kGy (Fig. 7), and the fitting curves of the C 1s and O 1s for the different bands are shown in Fig. 8. The C/O ratio of the rGO is 9.36. When rGO is incorporated into the PVA hydrogel, the intensity of the O 1s peak increases significantly. After irradiation, the intensity of the O 1s peak decreases remarkably, and the C/O ratio of PVA/rGO-200 kGy (4.15) is 61% higher than that of the nonirradiated PVA/rGO hydrogel composite (2.58). The C 1s XPS spectrum of rGO in Fig. 8a is split into three components attributable to various types of carbon atoms: the aromatic C–C (sp2-hybridized carbon), C–O (hydroxyl and epoxy), and CO (carbonyl and carboxyl) groups, centered at 284.7, 285.7, and 288.2 eV, respectively. The peak intensity corresponding to C–O and CO is much weaker than that of C–C, indicating the presence of a small number of residual oxygen-containing functional groups in the rGO, which is consistent with the FT-IR results. In the O 1s spectra of rGO (Fig. 8d), the two deconvoluted components are observed at 531.9 and 533.4 eV, which are assigned to C–O and CO, respectively. The C 1s XPS spectrum of PVA/rGO hydrogel composite also shows these three peaks, corresponding to C–C (283.3 eV), C–O (284.7 eV), and CO (286.4 eV) (Fig. 8b). The intensities of the oxygen-containing functional groups of the composite are increased significantly compared to those of rGO, especially for the C–O (Fig. 8e), which is attributed to the presence of abundant hydroxyl groups on PVA molecular chains. After irradiation, the strong C–C band of the composites indicates the stability of the symmetry of the sp2 framework, whereas the intensities of the oxygen-containing functional groups decrease significantly (Fig. 8c). In the O 1s spectra of the irradiated PVA/rGO composite (Fig. 8f), an approximately one-half decrease in the peak intensities of both C–O and CO was clearly observed compared to those of the nonirradiated PVA/rGO hydrogel composites (Fig. 8f). The results demonstrate that the oxygen-containing functional groups are eliminated by γ-ray irradiation.
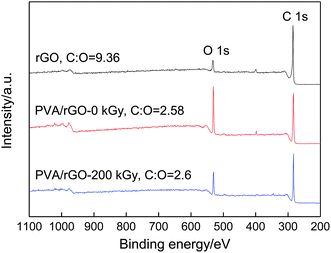 |
| | Fig. 7 XPS survey scans of rGO, PVA/rGO-0 kGy and PVA/rGO-200 kGy hydrogels. | |
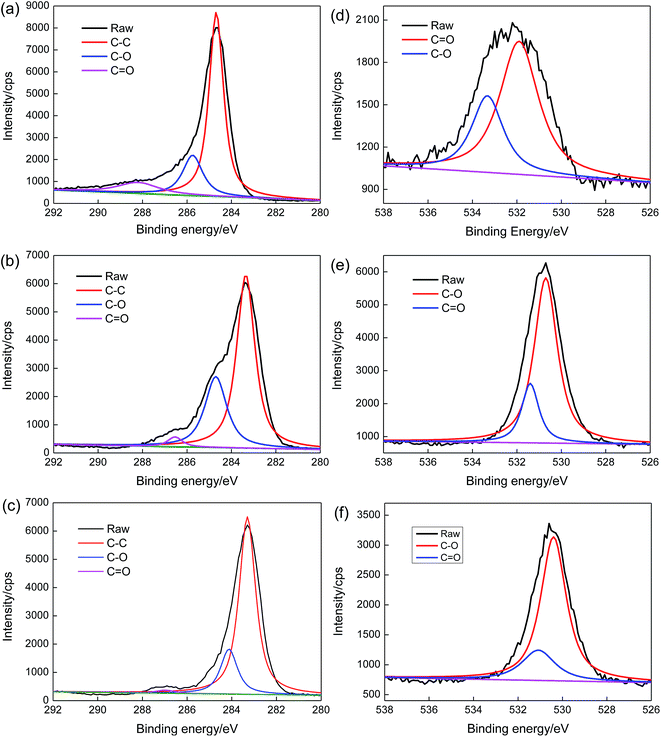 |
| | Fig. 8 (a–c) C 1s region and (d–f) O 1s regions of (a and d) rGO, (b and e) PVA/rGO-0 kGy, and (c and f) PVA/rGO-200 kGy. | |
Fig. 9 presents the effect of irradiation dose on the water contents of PVA/rGO hydrogel composites. The water content of the nonirradiated PVA/rGO hydrogel is higher than that of pure PVA hydrogel (∼81.3%),44 which is because the residual oxygen-containing functional groups on the rGO are able to absorb water molecules. It is clearly shown in the figure that the water contents of the PVA/rGO hydrogel composites decrease significantly with the increase of the irradiation dose. The loss of the oxygen-containing functional groups makes the rGO more hydrophobic; on the other hand, the γ-ray irradiation promotes great cross-linking of the polymer chains, which would lead to a denser network structure of the hydrogels and leave less space for the water molecules to inhabit.33
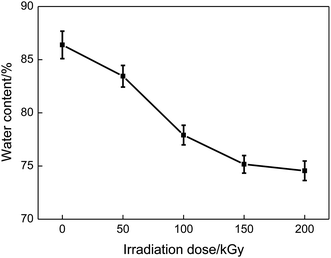 |
| | Fig. 9 Effect of irradiation dose on the water content of PVA/rGO hydrogels. | |
3.3. Effects of irradiation dose on the thermal stability and degree of crystallinity of PVA/rGO hydrogel composites
TGA and corresponding DTG were performed to investigate the effect of irradiation dose on the thermal stability of the PVA/rGO hydrogel composites. As shown in Fig. 10a, both nonirradiated and irradiated PVA/rGO composites show a three-step weight loss process. The first weight-loss process below 250 °C is associated with the evaporation of water entrapped in the molecules. The second weight-loss process at about 360 °C is ascribed to –OH collapse and partial main chain decomposition. The third weight-loss process at approximately 440 °C is due to residual main chain decomposition. It is clear that all the TGA curves of the hydrogels shift up to a higher temperature after irradiation compared to the nonirradiated PVA/rGO hydrogel. The peak temperature (Tp) of the DTG curve represents the temperature corresponding to the maximum weight-loss rate. As shown in Fig. 10b and Table 1, the Tp values of the PVA/rGO composite irradiated with a dose of 100 kGy appear at about 370.3 and 440.5 °C, which are increased by 7.3 and 3.8 °C, respectively, compared to those of the nonirradiated samples. These values are also higher than those reported for graphene or graphene oxide-based polymer nanocomposites.18,45–48 The improvement in thermal stability of the irradiated PVA/rGO composites is mainly because of the strong interfacial interactions between the rGO and the PVA matrix and the physical barrier effect of graphene.47 The repair of defects on the rGO sheets by irradiation would make the pyrolysis products diffuse more slowly.
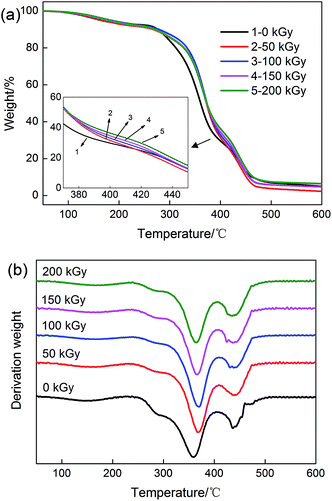 |
| | Fig. 10 (a) TGA and (b) DTG curves of PVA/rGO hydrogels treated with different irradiation doses. | |
Since PVA is a kind of semicrystalline polymer, its mechanical performance is strongly dependent on the degree of crystallinity and load transfer from PVA to rGO sheets. DSC curves were used to investigate the thermal behavior and the degree of crystallinity of PVA/rGO hydrogel composites in the present study (Fig. 11). The crystallinity of the PVA/rGO hydrogel composite (37.9%) is slightly higher than that of pure PVA hydrogel (35.1%) reported in our previously study,44 because the interactions between the rGO and the PVA matrix constrain the PVA molecules to undergo an ordered arrangement, which is necessary for the formation of crystallite in the PVA matrix.49 As summarized in Table 1, both melting temperature and the calculated crystallinity of the PVA/rGO hydrogel composites were found to decrease with the increase of the irradiation dose. When the applied irradiation dose is 200 kGy, the melting temperature and degree of crystallinity of the composite decrease by 19.3 °C and 36.1%, respectively, compared to those of the nonirradiated PVA/rGO composite. On the contrary, irradiation leads to an increase in the degree of crystallinity of GO/ultrahigh-molecular-weight polyethylene (UHMWPE) composites owing to the formation of perfect crystals during γ-ray irradiation.50 In the case of the present study, the presence of the rGO within the PVA matrix impedes the formation of hydrogen bonds among the PVA chains. γ-Ray irradiation promotes great cross-linking between the rGO and the PVA matrix, which breaks the symmetry and regularity of the polymer chains and hinders the crystal growth. As a result, the melting temperature and degree of crystallinity of the PVA/rGO hydrogel composites decrease after irradiation.
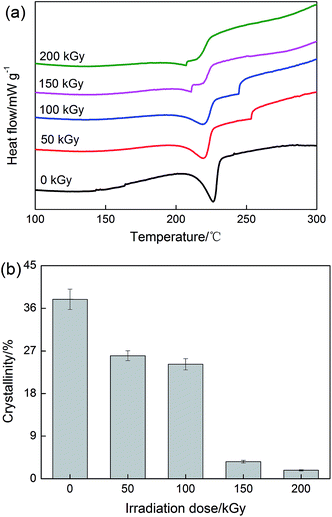 |
| | Fig. 11 (a) DSC curves for various PVA/rGO hydrogel composites, (b) effect of irradiation dose on crystallinity of the composites. | |
3.4. Mechanical enhancement mechanism of PVA/rGO hydrogel composites by γ-ray irradiation
Fig. 12a shows the tensile stress–strain relationship for the PVA/rGO hydrogel composites treated with various irradiation doses, and the relevant properties are plotted in Fig. 12b. The tensile strength of the PVA/rGO hydrogel composites increases significantly and then decreases as the irradiation dose increases from 50 to 200 kGy, which is similar to the results obtained for the pure PVA hydrogel51 and PVA/PVP blend hydrogels.33 The elongation at break decreases with the increase of irradiation doses owing to cross-linking. The PVA/rGO hydrogel composites achieve the maximum increase of 44% in tensile strength (∼1.41 MPa) at an irradiation dose of 100 kGy, compared to the nonirradiated samples (∼0.98 MPa), and this value is about 113% higher than that of the nonirradiated pure PVA (∼0.66 MPa) reported in our previous study.52
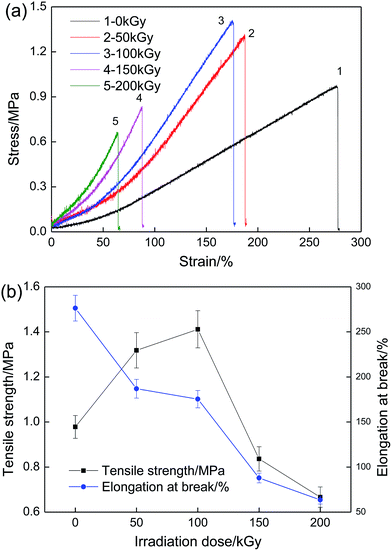 |
| | Fig. 12 (a) Tensile stress–strain curves of PVA/rGO hydrogel composites treated with various irradiation doses and (b) effect of irradiation dose on tensile strength and elongation at break. | |
Fig. 13 gives the compressive strength of the PVA/rGO hydrogel composites as a function of irradiation dose. It is shown that the compressive strength also increases first and then decreases with the increase of irradiation dose, and the maximum compressive strength of about 1.57 MPa is obtained when the applied irradiation dose is 150 kGy, which is a 171% enhancement compared to that of the nonirradiated PVA/rGO composites (∼0.58 MPa), and is an approximately 336% improvement compared to the nonirradiated pure PVA hydrogel (∼0.36 MPa).52
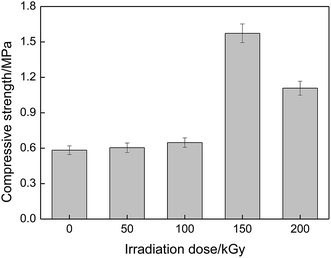 |
| | Fig. 13 Compressive strength of PVA/rGO hydrogel composites as a function of irradiation dose. | |
The fracture surfaces of the PVA/rGO hydrogel composites treated with various irradiation doses were investigated by SEM after tensile testing. As clearly shown in Fig. 14, all PVA/rGO hydrogel composites show a similar layered-structure with uniformly dispersed rGO sheets in the PVA matrix. The formation of this layered-structure is attributed to the intercalation of rGO in the PVA matrix, and the orientations of the rGO sheets tend to be parallel to the tensile direction, indicating the existence of strong interfacial interactions between rGO and PVA so that the rGO are coated with the PVA matrix. With the increase of irradiation dose, the layered structure becomes clearer, whereas the matrix becomes denser, which is ascribed to the great cross-linking induced by γ-ray irradiation. Such a layered structure is different from that of the films of the polymer/rGO nanocomposites. They tend to align parallel to the film surface with the addition of rGO, and could exhibit preferential orientation within the film, leading to the formation of the ultrastrong paper-like structure. However, these materials are brittle and break at small elongation.53,54
 |
| | Fig. 14 SEM images of the fracture surface of PVA/rGO hydrogel composites treated with irradiation doses of (a) 0, (b) 50, (c) 100, (d) 150, and (e) 200 kGy. | |
There are defects present on the rGO sheets (Fig. 15a), as demonstrated by the AFM and Raman results. At a low γ-ray irradiation dose, the reactivity of the rGO increases, especially at the sites of functional groups around the edge of defects (Fig. 15b). Then the reactive groups on the irradiated rGO react with PVA free radicals produced by the action of irradiation, which could generate chemical covalent bands between PVA and rGO, which is confirmed by FT-IR and Raman spectroscopy. At a higher irradiation dose, the random scission of C–C bonds of the PVA main-chain occurs, and the resulting shortened chains with higher mobility are likely to link the reactive groups around the edge of the defects on the rGO by forming C–C bands between PVA and rGO to repair the crystalline graphene plate (Fig. 15c). For the polymer, H· and ·OH produced from water during γ-ray irradiation couple two polymer radicals by hydrogen abstraction from the PVA chain, and then the PVA· can also react with each other under irradiation, leading to the cross-linking in the polymer.29,30 As a result, the mechanical performances of PVA/rGO are improved significantly by γ-ray irradiation, but are then decreased because of the degradation of the polymer by the excessive dose of irradiation. Interestingly, in comparison, the critical irradiation dose for the compressive strength transition for the PVA/rGO hydrogel is higher, and the percentage increase in compressive strength compared to the nonirradiated samples is larger than the pure PVA51 or PVA/PVP hydrogels.33 That is because the repair of defects on the rGO sheet by PVA chains occurs at a higher dose of irradiation, and the rGO with a lower defect concentration could exhibit a better reinforcing effect on its nanocomposites. The rGO with repaired defects exhibits better physical barrier effects in the hydrogel composites, which efficiently improve the thermal stability. The enhanced interfacial interactions between rGO and PVA molecular chains owing to the high total dose of irradiation severely break the symmetry and regularity of the polymer chains, which accounts for the remarkable decrease in crystallinity.
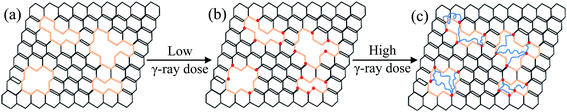 |
| | Fig. 15 The proposed repair mechanism for the crystalline graphene plate under γ-ray irradiation. (a) Defects present on graphene sheets. (b) The reactive groups on the edge of the defects are activated at low γ-ray irradiation dose. (c) The PVA scission chains link the reactive groups around the edge of defects on the graphene. | |
4. Conclusions
γ-Ray irradiation was used to repair defects on graphene and improve the reinforcement effect of graphene in PVA hydrogels. The increasing irradiation doses led to greater interfacial interactions between the rGO and the PVA matrix, which increased the thermal stability of PVA/rGO hydrogel composites and decreased the crystallinity degree. PVA/rGO hydrogel composites achieved their maximum values of tensile strength and compressive strength at an irradiation dose of 100 kGy and 150 kGy, respectively, which were increased by 40% and 167% compared to those for the nonirradiated composites. The improvement in mechanical performances is attributed to the repair of the crystalline graphene plate by the chemical covalent bonding between the shortened carbon chains from PVA and the reactive groups around the edge of the irradiated rGO.
Acknowledgements
This project was supported by the National Natural Science Foundation of China (Grants 51575278 and 11172142), the Fundamental Research Funds for the Central Universities (No. 30910612203 and 30915014103), the Learning-Research-Production prospective study project of Jiangsu (No. BY2016004-08), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
References
- M. Wang, Y. Li, J. Wu, F. Xu, Y. Zuo and J. A. Jansen, J. Biomed. Mater. Res., Part A, 2008, 85, 418–426 CrossRef PubMed.
- M. I. Baker, S. P. Walsh, Z. Schwartz and B. D. Boyan, J. Biomed. Mater. Res., Part B, 2012, 100, 1451–1457 CrossRef PubMed.
- A. Joshi, G. Fussell, J. Thomas, A. Hsuan, A. Lowman, A. Karduna, E. Vresilovic and M. Marcolongo, Biomaterials, 2006, 27, 176–184 CrossRef CAS PubMed.
- Y. Shi and D. S. Xiong, Wear, 2013, 305, 280–285 CrossRef CAS.
- G. Leone, M. Consumi, G. Greco, C. Bonechi, S. Lamponi, C. Rossi and A. Magnani, J. Biomed. Mater. Res., Part B, 2011, 97, 278–288 CrossRef PubMed.
- J. N. Coleman, U. Khan and Y. K Gunko, Adv. Mater., 2006, 18, 689–706 CrossRef CAS.
- Y. Q. Xiang, Z. Q. Peng and D. J. Chen, Eur. Polym. J., 2006, 42, 2125–2132 CrossRef CAS.
- W. C. Lin, W. Fan, A. Marcellan, D. Hourdet and C. Creton, Macromolecules, 2010, 43, 2554–2563 CrossRef CAS.
- K. Haraguchi, Curr. Opin. Solid State Mater. Sci., 2007, 11, 47–54 CrossRef CAS.
- K. S. Novoselov, Z. Jiang, Y. Zhang, S. V. Morozov, H. L. Stormer, U. Zeitler, J. C. Maan, G. S. Boebinger, P. Kim and A. K. Geim, Science, 2007, 315, 1379 CrossRef CAS PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- C. G. Lee, X. D. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone and P. Kim, Solid State Commun., 2008, 146, 351–355 CrossRef CAS.
- A. A. Balandin, S. Ghosh, W. Z. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- D. G. Papageorgiou, I. A. Kinloch and R. J. Young, Carbon, 2015, 95, 460–484 CrossRef CAS.
- S. Goenka, V. Sant and S. Sant, J. Controlled Release, 2014, 173, 75–88 CrossRef CAS PubMed.
- X. M. Yang, L. Li, S. M. Shang and X. M. Tao, Polymer, 2010, 51, 3431–3435 CrossRef CAS.
- N. Thayummanavan, P. B. Tambe, G. Joshi and M. Shukla, Compos. Interfaces, 2014, 21, 487–506 CrossRef.
- N. Thayummanavan, P. B. Tambe and G. Joshi, Cellul. Chem. Technol., 2015, 49, 69–80 Search PubMed.
- R. Saito, G. Dresselhaus and M. S. Dresselhaus, Chem. Phys. Lett., 1992, 195, 537–542 CrossRef CAS.
- C. Gomez-Navarro, P. J. De Pablo, J. Gomez-Herrero, B. Biel, F. J. Garcia-Vidal, A. Rubio and F. Flores, Nat. Mater., 2005, 4, 534–539 CrossRef CAS PubMed.
- J. H. Kim, J. H. Hwang, J. Suh, S. Tongay, S. Kwon, C. C. Hwang, J. Wu and J. Y. Park, Appl. Phys. Lett., 2015, 106, 171604 Search PubMed.
- Z. Q. Luo, J. Z. Shang, S. H. Lim, D. H. Li, Q. H. Xiong, Z. X. Shen, J. Y. Lin and T. Yu, Appl. Phys. Lett., 2010, 97, 233111 CrossRef.
- M. Q. Sun, G. C. Wang, X. W. Li and C. Z. Li, J. Power Sources, 2014, 245, 436–444 CrossRef CAS.
- B. Gupta, N. Kumar, K. Panda, A. A. Melvin, S. Joshi, S. Dash and A. K. Tyagi, J. Phys. Chem. C, 2016, 120, 2139–2148 CAS.
- L. F. Dumee, C. F. Feng, L. He, Z. F. Yi, F. S. She, Z. Peng, W. M. Gao, C. Banos, J. B. Davies, C. Huynh, S. Hawkins, M. C. Duke, S. Gray, P. D. Hodgson and L. X. Kong, Carbon, 2014, 70, 313–318 CrossRef CAS.
- Y. Shi, D. S. Xiong, J. L. Li and N. Wang, J. Phys. Chem. C, 2016, 120, 19442–19453 CAS.
- C. Von Sonntag, E. Bothe, P. Ulanski and D. J. Deeble, Radiat. Phys. Chem., 1995, 46, 527–532 CrossRef CAS.
- I. Sakurada and Y. Ikada, Bull. Inst. Chem. Res., Kyoto Univ., 1964, 42, 22–31 CAS.
- I. Sakurada and Y. Ikada, Bull. Inst. Chem. Res., Kyoto Univ., 1963, 41, 103–113 CAS.
- S. J. Zhang and H. Q. Yu, Water Res., 2004, 38, 309–316 CrossRef CAS PubMed.
- Y. Shi, D. S. Xiong and J. F. Zhang, Tribol. Int., 2014, 78, 60–67 CrossRef CAS.
- M. J. Martínez–Morlanesa, P. Castellb, P. J. Alonsoc, M. T. Martinezb and J. A. Puértolas, Carbon, 2012, 50, 2442–2452 CrossRef.
- Y. Shi, D. S. Xiong, Y. T. Liu, N. Wang and X. D. Zhao, Mater. Sci. Eng., C, 2016, 65, 172–180 CrossRef CAS PubMed.
- T. Kuilla, S. Bhadra, D. Yao, N. H. Kim, S. Bose and J. H. Lee, Prog. Polym. Sci., 2010, 35, 1350–1375 CrossRef CAS.
- X. D. Qi, X. L. Yao, S. Deng, T. N. Zhou and Q. Fu, J. Mater. Chem. A, 2014, 2, 2240–2249 CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401–187405 CrossRef CAS PubMed.
- N. A. Zubair, N. A. Rahman, H. N. Lim, R. M. Zawawi and Y. Sulaiman, RSC Adv., 2016, 6, 17720–17727 RSC.
- J. J. Chen, J. L. Li, D. S. Xiong, Y. He, Y. J. Ji and Y. K. Qin, Appl. Surf. Sci., 2016, 361, 49–56 CrossRef CAS.
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, Nat. Nanotechnol., 2009, 4, 25–29 CrossRef CAS PubMed.
- G. Srinivas, Y. W. Zhu, R. Piner, N. Skipper, M. Ellerby and R. Ruoff, Carbon, 2010, 48, 630–635 CrossRef CAS.
- C. Vallés, J. D. Núñez, A. M. Benito and W. K. Maser, Carbon, 2010, 50, 835–844 CrossRef.
- Y. Shi, D. S. Xiong, Y. Peng and N. Wang, eXPRESS Polym. Lett., 2015, 10, 125–138 CrossRef.
- X. X. Sheng, D. L. Xie, W. X. Cai, X. Y. Zhang, L. Zhong and H. P. Zhang, Ind. Eng. Chem. Res., 2015, 54, 649–658 CrossRef CAS.
- R. Surudzic, A. Jankovic, N. Bibic, M. Vukasinovic-Sekulic, A. Peric-Grujic, V. Miskovic-Stankovic, S. J. Park and K. Y. Rhee, Composites, Part B, 2016, 85, 102–112 CrossRef CAS.
- C. Bao, Y. Q. Guo, L. Song and Y. Hu, J. Mater. Chem., 2011, 21, 13942–13950 RSC.
- J. Liang, Y. Huang, L. Zhang, Y. Wang, Y. Ma and T. Guo, Adv. Funct. Mater., 2009, 19, 2297–2302 CrossRef CAS.
- S. K. Sharma, J. Prakash and P. K. Pujari, Phys. Chem. Chem. Phys., 2015, 17, 29201–29209 RSC.
- G. D. Huang, Z. F. Ni, G. M. Chen, W. C. Pang and Y. W. Zhao, Int. J. Polym. Anal. Charact., 2016, 21, 417–427 CrossRef CAS.
- R. Y. Ma, D. S. Xiong, Y. Peng, J. B. Jin, L. McCann, Z. M. Jin and J. Fisher, J. Med. Biomech., 2009, 24, 347–351 CAS.
- Y. Shi, D. S. Xiong, J. L. Li and N. Wang, RSC Adv., 2016, 6, 82467–82477 RSC.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
- C. M. Chen, Q. H. Yang, Y. G. Yang, W. Lv, Y. F. Wen, P. X. Hou, M. Z. Wang and H. M. Cheng, Adv. Mater., 2009, 21, 3007–3011 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence















