
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel 2,5-furandicarboxylic acid-based bis(cyclic carbonate) for the synthesis of biobased non-isocyanate polyurethanes†
Lu Zhang a,
Xiaolan Luoa,
Yusheng Qin*ab and
Yebo Li*a
a,
Xiaolan Luoa,
Yusheng Qin*ab and
Yebo Li*a
aDepartment of Food, Agricultural and Biological Engineering, Ohio Agricultural Research and Development Center, The Ohio State University, 1680 Madison Ave, Wooster, OH 44691-4096, USA. E-mail: li.851@osu.edu; Tel: +1-330-263-3855
bKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: ysqin@ciac.ac.cn; Tel: +86-0431-85262567
First published on 22nd December 2016
Abstract
This paper describes the synthesis of a new biobased bis(cyclic carbonate) derived from 2,5-furandicarboxylic acid (FDCA) with the incorporation of CO2. The bis(cyclic carbonate) was then used to synthesize non-isocyanate polyurethanes (NIPUs) via polyaddition reactions with a series of diamines. The chemical structures of the bis(cyclic carbonate) and the NIPUs were characterized by Fourier transform infrared spectroscopy (FT-IR) and proton nuclear magnetic resonance spectroscopy (1H NMR). The number-average molecular weights (Mn) of the NIPUs were between 3900 g mol−1 and 7000 g mol−1 as determined by gel permeation chromatography (GPC). The thermal properties of the NIPUs were investigated by differential scanning calorimetry (DSC) and thermogravimetric analyses (TGA). The results showed that the NIPUs synthesized in this study had a degradation temperature for 5% weight loss (T5%) in the range of 240 °C and 279 °C, indicating good thermal stability. The NIPUs were also found to be fully amorphous with a broad range of glass transition temperatures (Tg) from 63 °C to 113 °C, depending on the chemical structures of the diamines used. The rigid chemical moiety of cycloaliphatic diamine led to a higher Tg of the NIPUs than the flexible carbon chains of linear aliphatic diamines. This study demonstrated a new method for the synthesis of biobased NIPUs, with satisfactory properties, from FDCA, which is an important platform chemical derived from cellulosic biomass.
1. Introduction
Globally, polyurethane (PU) is the sixth most consumed polymer with an annual production of over 12 million tons.1,2 Conventional PUs are synthesized from polyols and isocyanates. By changing formulations with different polyols and isocyanates, a variety of PUs with specific properties can be developed for a broad range of industrial applications, including coatings, foams, paints, thermoplastics, adhesives, and sealants.3–6 However, isocyanates are considered to be hazardous to the environment and human health.4,7,8 Moreover, isocyanates are typically produced from the reactions of amines with phosgene, which can be extremely dangerous to humans.6,9,10 Environmental and health hazard concerns over the use of isocyanates have led to an increasing demand for the development of environmentally-friendly processes for PU production without using toxic isocyanates.8The development of novel non-isocyanate polyurethanes (NIPUs) has received increased attention in recent years. Generally, linear NIPUs can be synthesized from three synthetic routes: the copolymerization of an aziridine with CO2, the transurethanization between a bis(carbamate) and a diol, and the ring-opening polyaddition between a bis(cyclic carbonate) and a diamine.6 The third route is currently the most studied one because of its large potential for industrial production, and numerous bis(cyclic carbonate)s are being tested in laboratories for the use as precursors for NIPU production.8 Because bis(cyclic carbonate)s are generally synthesized from the reaction of diepoxides with CO2, the production of NIPU via this route is considered to be an important strategy for chemical fixation of CO2.11 It is predicted that the production of cyclic carbonates and their derivatives would be able to reduce CO2 emissions by 120 million tons per year if the production process could be improved.12 The NIPU prepared via this route is also called poly(hydroxyurethane) (PHU) owing to the formation of hydroxyl groups that result from the opening of cyclic carbonate rings.10 The hydroxyl groups can form hydrogen bonds to the carbonyl oxygens within the urethane linkages, accounting for a number of characteristics of PHU, such as improved hydrophilicity, reduced crystallinity, and increased resistance to organic solvents.13–15
Due to the increasing environmental concerns associated with the use of fossil fuel-based PUs, a number of synthetic strategies have been investigated for the synthesis of PHUs from renewable resources. Most of the currently available biobased PHUs are derived from vegetable oils, especially soybean oil and linseed oil.11,16 Vegetable oils are fatty acid triglycerides with various degrees of unsaturation which can be epoxidized by a peroxide or a peracid. Epoxidized vegetable oils can further react with CO2 to yield vegetable oil-derived poly(cyclic carbonate)s. Eventually, biobased PHUs can be synthesized by the reaction between these poly(cyclic carbonate)s and diamines or triamines. However, the uncontrolled functionality and varied structures of vegetable oils sometimes result in ill-defined networks of the resulting cross-linked PHUs.17,18 For this reason, several attempts have been made to synthesize linear PHUs from vegetable oils. For example, unsaturated fatty acid diesters can be obtained by the transesterification of vegetable oils with diols. These diesters can be epoxidized, and then carbonated and, subsequently, reacted with diamines to generate linear PHUs.18 Although substantial progress has been made on the synthesis of PHUs from vegetable oils, the problems related to their potential competition with food production remain a concern.19 In this regard, the focus of the research on biobased PHUs is shifting toward the synthesis of PHUs from non-food renewable resources. Untill now, only a few synthetic routes have been put forward for the synthesis of biobased PHUs from non-food feedstocks, such as lignin-derived bisphenols and limonene.11,20
The compound 2,5-furandicarboxylic acid (FDCA) is a promising non-food renewable building block that can be obtained from cellulosic biomass.21 It has been considered to be a potential replacement for petroleum-based terephthalic acid for the production of polyesters and resins.19,22 Considering its dicarboxylic acid structure and biobased property, FDCA is anticipated to be a potential building block for synthesis of biobased PHUs. As far as we know, there has not been any report on the use of FDCA for the synthesis of PHUs. As shown in Scheme 1, we are proposing an innovative synthetic route to produce the biobased PHUs from an FDCA-derived bis(cyclic carbonate) and diamines. The influence of the chemical structures of these PHUs on their properties is also discussed in this paper.
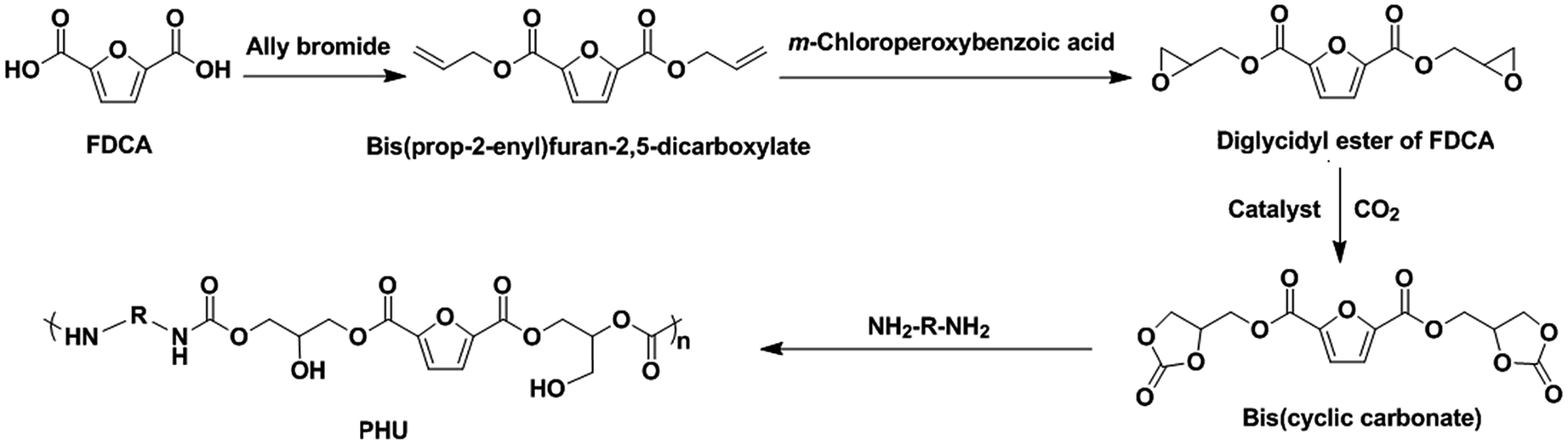 | ||
| Scheme 1 Synthetic strategy of PHU from FDCA with the integration of CO2. | ||
2. Experimental
2.1 Materials
FDCA was provided by the Dagaote Technology Co. (Mianyang, Sichuan, China). N,N-Dimethylformamide (DMF), dichloromethane, m-chloroperoxybenzoic acid (70–75%), tetraethylammonium bromide (TEAB), 1,6-hexanediamine, 1,8-diaminooctane, isophorondiamine, allyl bromide, dimethyl sulfoxide-d6 (DMSO-d6), and chloroform-d (CDCl3) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Fisher Scientific (Hampton, NH, USA).2.2 Synthesis of diglycidyl ester of FDCA (DGF)
DGF was synthesized from FDCA in two steps. Firstly, bis(prop-2-enyl)furan-2,5-dicarboxylate (FDCE) was synthesized via the dehydrobromination reaction between FDCA (50 g) and allyl bromide. Subsequently, the obtained FDCE (38 g) was epoxidized with m-chloroperoxybenzoic acid to produce DGF (24.5 g). The detailed synthetic procedures of FDCE and DGF were described in previous literature.23 The yields of FDCE and DGF in this study were 50% and 57%, respectively. The chemical structures of FDCE and DGF were confirmed by Fourier transform infrared spectroscopy (FT-IR) and nuclear magnetic resonance spectroscopy (1H NMR).![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H), 3013 (allyl group C–H), 2938 (–CH2–), 1719 (CO), 1652 (allyl group –CC–), 1572 (furan ring –CC–), 1271 (ester –C–O–), 1229 (furan ring –C–O–). 1H NMR (400 MHz, DMSO-d6, ppm, Fig. 2): 7.48 (2H, s, Ha), 6.07–5.98 (2H, m, Hc), 5.44–5.39 (2H, m, Hd), 5.32–5.29 (2H, m, Hd), 4.82 (4H, d, Hb).C–H), 3006 (oxirane ring C–H), 2943 (–CH2–), 1737 (CO), 1573 (furan ring –CC–), 1278 (ester –C–O–), 1232 (furan ring –C–O–), 868 (oxirane ring –C–O–). 1H NMR (400 MHz, CDCl3, ppm, Fig. 2): 7.29 (2H, s, Ha), 4.68 (H, d, Hb), 4.65 (H, d, Hb), 4.24–4.22 (2H, m, Hb), 3.37–3.35 (2H, m, Hc), 2.93 (2H, t, Hd), 2.75 (2H, q, Hd).
C–H), 3013 (allyl group C–H), 2938 (–CH2–), 1719 (CO), 1652 (allyl group –CC–), 1572 (furan ring –CC–), 1271 (ester –C–O–), 1229 (furan ring –C–O–). 1H NMR (400 MHz, DMSO-d6, ppm, Fig. 2): 7.48 (2H, s, Ha), 6.07–5.98 (2H, m, Hc), 5.44–5.39 (2H, m, Hd), 5.32–5.29 (2H, m, Hd), 4.82 (4H, d, Hb).C–H), 3006 (oxirane ring C–H), 2943 (–CH2–), 1737 (CO), 1573 (furan ring –CC–), 1278 (ester –C–O–), 1232 (furan ring –C–O–), 868 (oxirane ring –C–O–). 1H NMR (400 MHz, CDCl3, ppm, Fig. 2): 7.29 (2H, s, Ha), 4.68 (H, d, Hb), 4.65 (H, d, Hb), 4.24–4.22 (2H, m, Hb), 3.37–3.35 (2H, m, Hc), 2.93 (2H, t, Hd), 2.75 (2H, q, Hd).2.3 Synthesis of bis(cyclic carbonate)
An FDCA-derived five-membered bis(cyclic carbonate) was synthesized for the first time via the insertion of CO2 into DGF. To prepare this bis(cyclic carbonate), DGF (10.00 g) and TEAB (1 mol% with respect to DGF) were dissolved in dichloromethane (30 mL) that was dried over molecular sieves beforehand. The resulting solution was then loaded into a stainless steel reactor (50 mL), which was equipped with a magnetic stirrer and a pressure gauge under a nitrogen atmosphere. CO2 was added to the reactor until the pressure in the reactor reached 4.0 MPa. The reactants were stirred at 120 °C for 24 h. The reactor was then cooled down to room temperature and depressurized to atmospheric pressure by releasing the remaining CO2. The crude bis(cyclic carbonate) product was washed with dichloromethane to remove TEAB and unreacted DGF, and then pure bis(cyclic carbonate) was recovered as a light brown solid after drying in a vacuum oven at 30 °C for 24 h. The yield of the bis(cyclic carbonate) was 86%. The CO2 fixation for the bis(cyclic carbonate) was 24.7 wt% based on the 1H-NMR analysis. The chemical structure of the bis(cyclic carbonate) was confirmed by FT-IR and 1H NMR analyses. FT-IR (KBr, cm−1): 3155 and 3120 (furan ringC–H), 2925 (–CH2–), 1778 (cyclic carbonate ring CO), 1737 (ester CO), 1575 (furan ring –CC–), 1273 (ester –C–O–), 1231 (furan ring –C–O–), 1100 (cyclic carbonate ring –C–O–). 1H NMR (400 MHz, DMSO-d6, Fig. 2): 7.44 (2H, s, Ha), 5.19–5.14 (2H, m, Hc), 4.65–4.60 (4H, m, Hb), 4.53 (2H, q, Hd), 4.40 (2H, q, Hd).
2.4 Synthesis of PHUs
With the successful synthesis of the FDCA-derived bis(cyclic carbonate), a series of PHUs were synthesized via a polyaddition reaction with different diamines (1,6-hexanediamine, 1,8-diaminooctane, and isophorondiamine) at different temperatures (140 °C, 160 °C, and 180 °C). Typically, the bis(cyclic carbonate) (2.00 g, 5.62 mmol) and a diamine (5.62 mmol) were introduced into a glass reactor and then were heated to a fixed reaction temperature using an oil bath under a nitrogen atmosphere. DMF (1 mL) was used as a solvent for the reactions that were conducted at 140 °C and 160 °C to reduce the high viscosity of the reaction mixture, while no solvent was used for the reactions conducted at 180 °C. The reactions were terminated when the characteristic absorption peak of the cyclic carbonate groups disappeared in the FT-IR spectra. After the reactions, the PHUs were obtained as brown brittle solids. The PHUs were then crushed into small particles and dried in a vacuum oven at 50 °C for 24 h to remove DMF and the moisture that the PHUs might have absorbed from the atmosphere. The yields of the PHUs were between 81% and 98%. The chemical structures and thermal properties of the PHU products were characterized without further purification.O), 1576 (furan ring –CC–), 1529 (urethane –NH–), 1266 (ester –C–O–), 1222 (furan ring –C–O–). 1H NMR (400 MHz, DMSO-d6, Fig. 4): 8.68–8.38 (2H, m, Hf), 7.51–7.31 (2H, m, Ha), 5.10–4.95 (H, m, He1), 4.76–4.68 (H, m, Hc1), 4.58–4.07 (7H, m, Hb1, Hb2, Hc2, and Hd2), 4.05–3.86 (2H, m, Hd1), 3.82–3.73 (H, m, He2), 3.16–2.89 (4H, m, Hg), 1.60–1.31 (8H, m, Hh).O), 1577 (furan ring –CC–), 1528 (urethane –NH–), 1268 (ester –C–O–), 1222 (furan ring –C–O–). 1H NMR (400 MHz, DMSO-d6): 8.60–8.43 (2H, m), 7.46–7.38 (2H, m), 5.11–4.95 (H, m), 4.76–4.68 (H, m), 4.60–4.07 (7H, m), 4.05–3.92 (2H, m), 3.82–3.74 (H, m), 3.14–2.90 (4H, m), 1.59–1.33 (12H, m).O), 1575 (furan ring –CC–), 1534 (urethane –NH–), 1278 (ester –C–O–), 1224 (furan ring –C–O–). 1H NMR (400 MHz, DMSO-d6): 8.60–8.02 (2H, m), 7.46–7.35 (2H, m), 5.11–4.95 (H, m), 4.77–4.67 (H, m), 4.59–4.05 (7H, m), 4.04–3.88 (2H, m), 3.82–3.73 (H, m), 3.14–2.65 (2H, m), 1.58–1.33 (16H, m).2.5 Characterization methods
3. Results and discussion
3.1 Synthesis and characterization of FDCE and DGF
In this study, a two-step epoxidation process was used to prepare DGF from FDCA as described in the literature.24 Alternatively, a one-step procedure could also be used to prepare DGF via the esterification between FDCA and epihalohydrins, such as epichlorohydrin and epibromohydrin. However, the direct esterification of FDCA with epihalohydrins would inevitably result in the generation of oligomers with different degrees of polymerization, making it difficult for product separation.23,25The FT-IR spectra of FDCA, FDCE, and DGF are displayed in Fig. 1. Compared to the spectrum of FDCA, the broad absorption band ascribed to the hydroxyl stretch of the carboxyl groups between around 2500 cm−1 and 3000 cm−1 was absent in the spectrum of FDCE. The structure of FDCE was also confirmed by the characteristic absorption peaks of the –C–H stretch of methylene groups at 2938 cm−1, the C–H stretch of the allyl groups at 3013 cm−1, and the –CC– stretch of the allyl groups at 1652 cm−1. In addition, the absorption peak of the carbonyl stretch was shifted from 1665 cm−1 to 1719 cm−1 in the process, due to the carboxyl acid groups of FDCA being transformed to carboxylate ester groups. After FDCE was oxidized to DGF, the characteristic –CC– absorption peak of the allyl groups disappeared, and a new peak appeared at 868 cm−1, which was ascribed to the characteristic absorption peak of the epoxy rings.
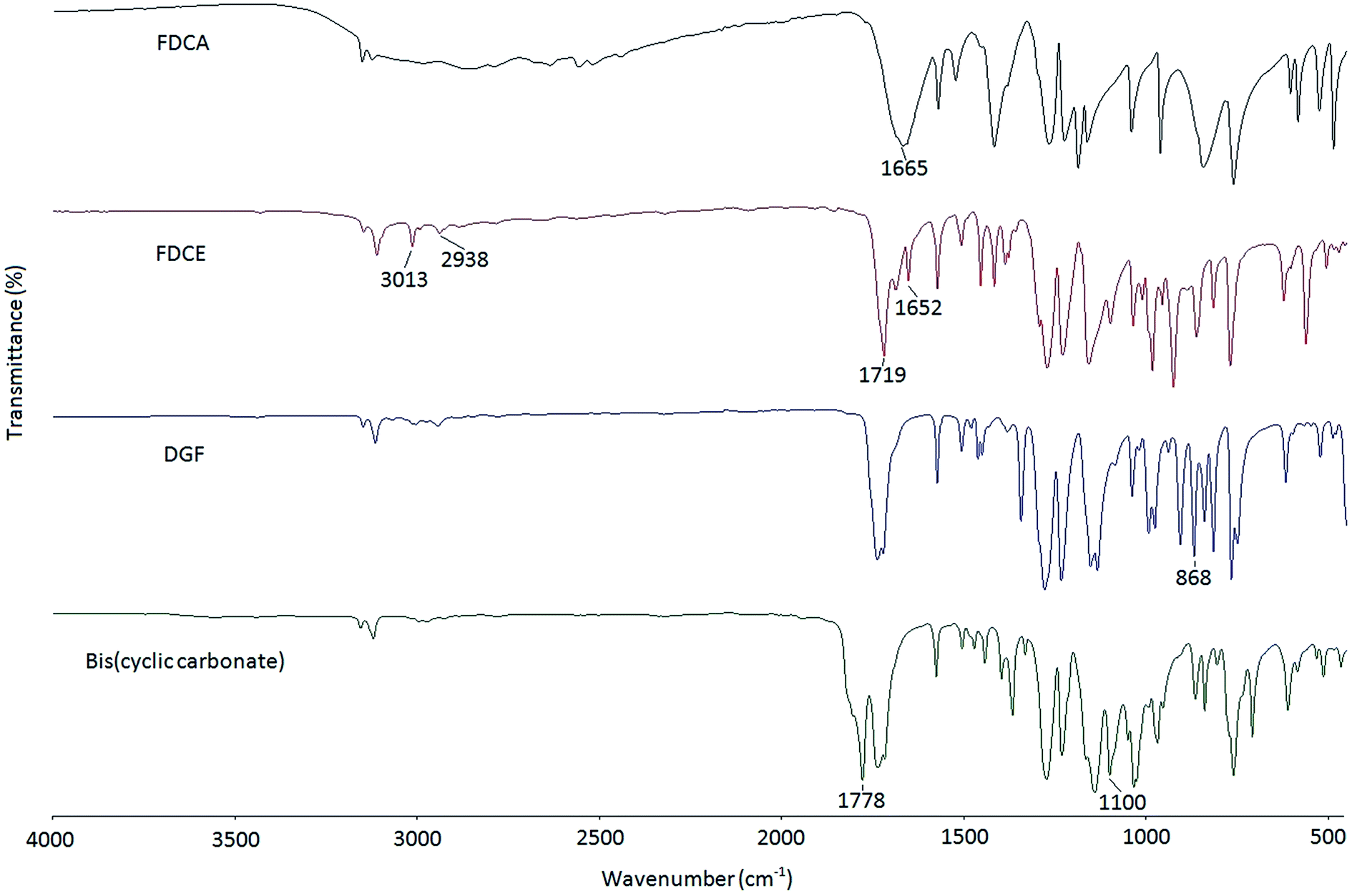 | ||
| Fig. 1 FT-IR spectra of FDCA, FDCE, DGF and the FDCA-derived bis(cyclic carbonate). | ||
The 1H NMR spectra of FDCE and DGF are shown in Fig. 2. In the 1H NMR spectrum of FDCE, a resonance signal shown at 7.48 ppm was ascribed to the protons on the furan ring. The signals at 5.29–5.44, and 5.98–6.07 ppm were ascribed to the olefinic protons of the allyl groups. The methylene protons between the double bonds and the ester groups gave doublets at 4.82 ppm due to their coupling interactions with the adjacent olefinic protons. After epoxidation, the signals corresponding to the olefinic protons of the allyl groups totally disappeared in the 1H NMR spectrum of DGF, indicating the total conversion of FDCE. New signals observed at 2.75 and 2.93 ppm were ascribed to the methylene protons on oxirane rings, and the signal at 3.35–3.37 ppm was ascribed to the methine protons on epoxide rings. The doublets of methylene protons adjacent to the ester groups were shifted from 4.82 ppm to 4.22–4.24 and 4.65–4.68 ppm upon the substitution of double bonds to epoxide groups. The signal corresponding to the protons on the furan ring did not shift substantially due to the similar chemical environment.
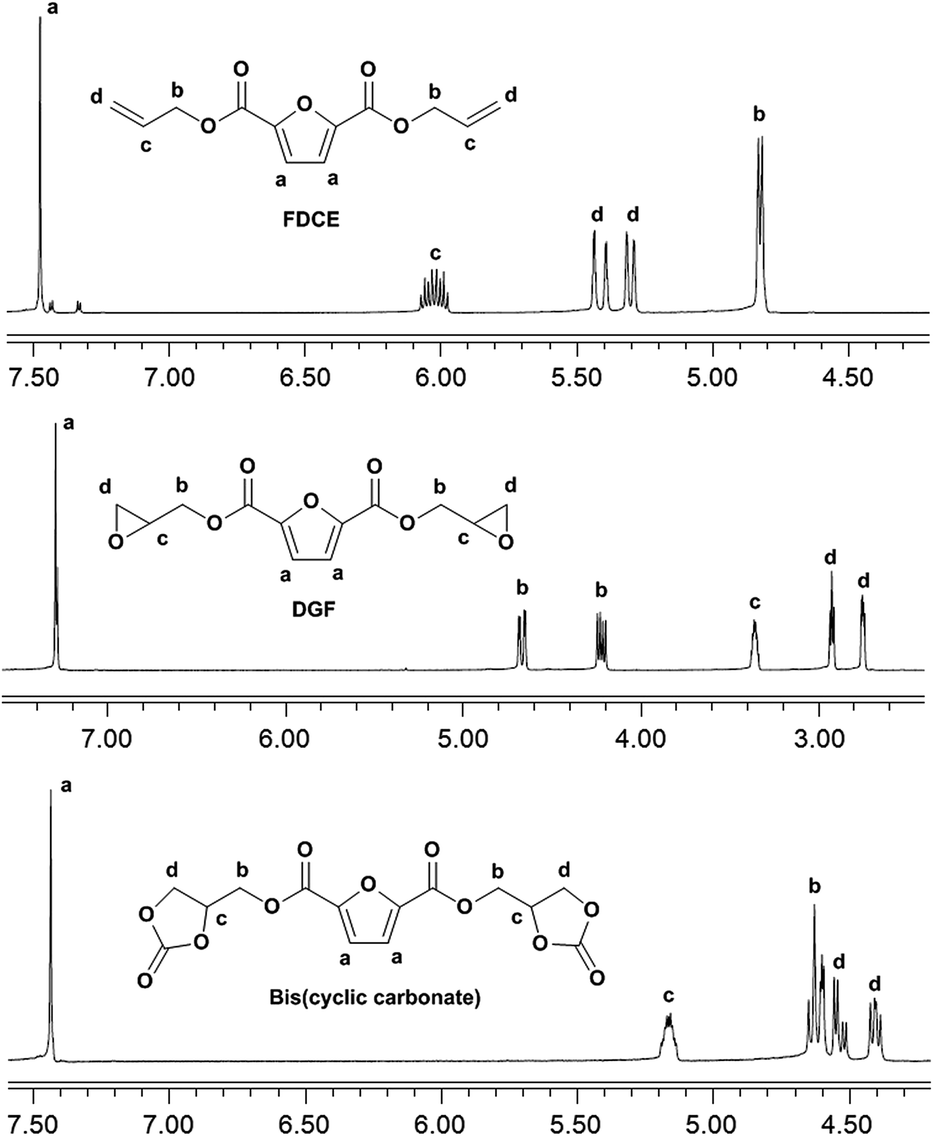 | ||
| Fig. 2 1H NMR spectra of FDCE, DGF and the FDCA-derived bis(cyclic carbonate). | ||
3.2 Synthesis of and characterization FDCA-derived bis(cyclic carbonate)
A five-membered bis(cyclic carbonate) was obtained via the cycloaddition reaction of DGF with CO2 catalyzed by TEAB (Scheme 2). TEAB is considered to be an efficient catalyst for the cycloaddition of epoxides with CO2. The highly reactive bromide anions of TEAB attack the epoxide leading to the opening of epoxy rings, followed by the generation of a bromo-alkoxide. The bromo-alkoxide then attacks the carbon atom of CO2 to produce five-membered cyclic carbonate rings.26,27 In this reaction, the reactant and the catalyst were soluble in dichloromethane, while the bis(cyclic carbonate) was not. Therefore, a simple washing and drying procedure could be used for product separation and catalyst recycling. Finally, pure bis(cyclic carbonate) was obtained.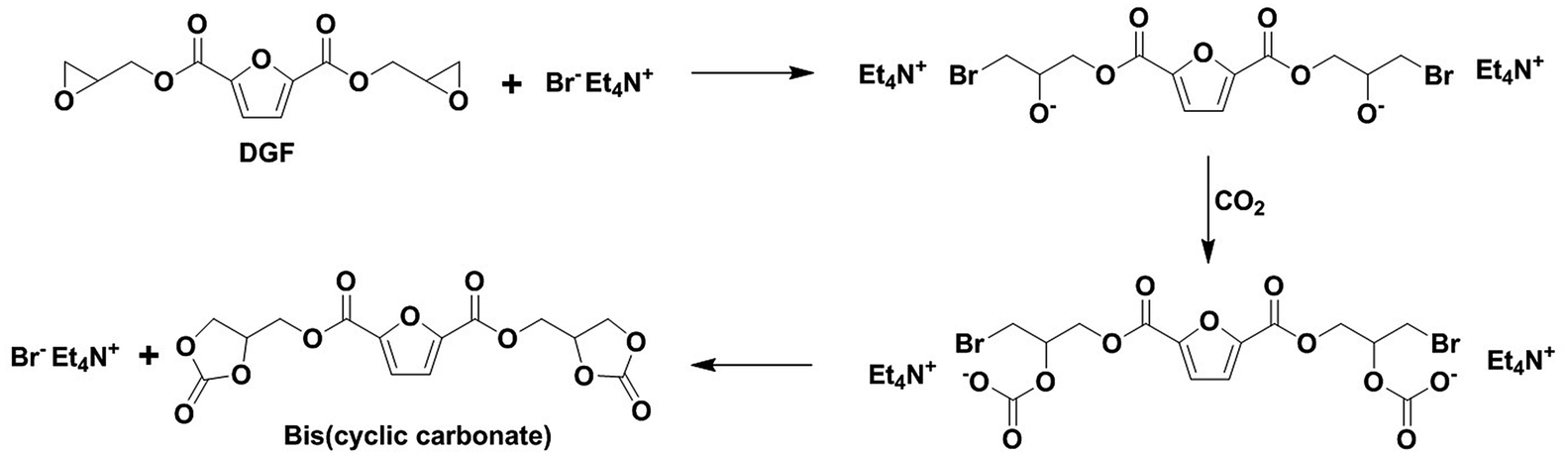 | ||
| Scheme 2 Mechanism of the carbonation of DGF catalyzed by TEAB. | ||
The chemical structure of the bis(cyclic carbonate) was confirmed by FT-IR and 1H NMR analyses. As shown in the FT-IR spectrum of the bis(cyclic carbonate), the characteristic absorption peak of the epoxide groups disappeared, and two new characteristic absorption peaks, which were ascribed to the CO and C–O bonds on the five-membered cyclic carbonate rings, appeared at 1778 and 1100 cm−1, respectively (Fig. 1). This indicated the successful conversion of the oxirane groups into cyclic carbonate groups. In the 1H NMR spectrum of the FDCA-derived bis(cyclic carbonate) shown in Fig. 2, after the insertion of CO2 into the epoxide rings, the signal of the methine protons originally located on the epoxide rings was shifted from 3.35–3.37 ppm to 5.14–5.19 ppm, which was the characteristic signal region of the methine protons of the cyclic carbonate rings. Similarly, the signals of the methylene protons of the epoxide rings were shifted from 2.75 and 2.93 ppm to 4.40 and 4.53 ppm, respectively, upon the carbonation of the epoxide groups. The signal at 4.60–4.65 ppm was assigned to the methylene protons adjacent to the cyclic carbonate groups and the signal at 7.44 ppm was assigned to the protons on the furan ring. The 1H NMR results further validated the cycloaddition of oxirane groups with CO2 for the formation of cyclic carbonate groups. No diepoxy or mono-epoxy compound was detected, indicating that the bis(cyclic carbonate) product obtained was of high purity.
3.3 Synthesis and characterization of FDCA-derived PHUs
A series of PHUs were synthesized from the polyaddition reaction of the bis(cyclic carbonate) and three different diamines. The synthesis of PHU is usually carried out at elevated temperatures to allow for faster reaction rates. However, side reactions such as crosslinking of the furan rings and the degradation of carbamate to urea or amide, may occur at high temperatures. In addition, high reaction temperatures may also cause the evaporation of diamines. For these reasons, it is advised that PHU synthesis should not be carried out at temperatures above 180 °C. In this study, three widely studied diamines, 1,6-hexanediamine, 1,8-diaminooctane, and isophorondiamine, were used to synthesize PHUs at three different temperatures to study the effects of diamine structure, as well as reaction temperature, on PHU properties, including molecular weight and thermal properties. The synthesis of PHUs can be carried out either in solvent or without solvent. When the reaction was conducted at 140 °C or 160 °C, a small amount of solvent was added to reduce the viscosity of the reactant mixture, which is necessary for further polymerization. At 180 °C, the reactions can be carried out without solvent owing to the decreased viscosity of the reaction mixture at this higher temperature. Since most PHUs are soluble in polar aprotic solvents due to the hydrophilicity of the hydroxyl groups, DMF was used as the solvent in this study.Fig. 3 shows the FT-IR spectra of the reaction mixture of the FDCA-derived bis(cyclic carbonate) and a diamine at different reaction times. At the beginning of the reaction, a broad characteristic band appeared at 3319 cm−1, indicating the formation of the hydroxyl groups resulting from the opening of the cyclic carbonate rings. Meanwhile, due to the unreacted bis(cyclic carbonate) presented at the early stage of the reaction, two characteristic peaks of the cyclic carbonate groups were observed at 1792 and 1050 cm−1 in the FT-IR spectrum after 6 h of reaction. As the reaction time increased, the intensity of these two peaks gradually decreased. After four days of reaction, these peaks almost disappeared, indicating the consumption of the bis(cyclic carbonate).
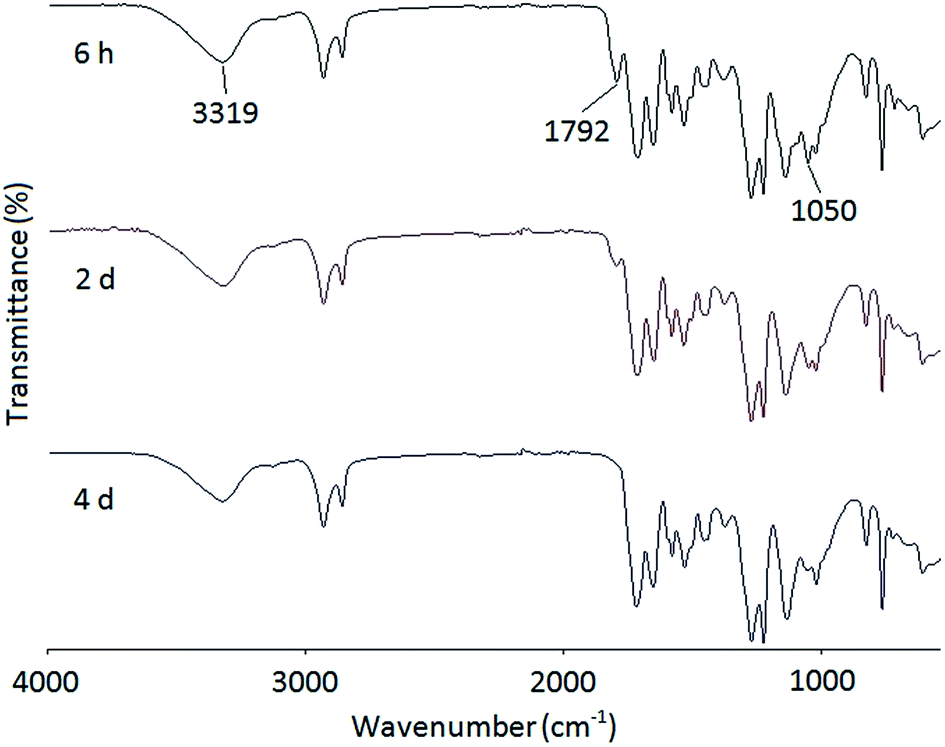 | ||
| Fig. 3 FT-IR spectra of the reaction mixture of the FDCA-derived bis(cyclic carbonate) and 1,8-diaminooctane at 140 °C and different reaction times. | ||
The chemical structures of the PHUs were further confirmed by 1H NMR analysis. Fig. 4 is an example of the 1H NMR spectrum of a typical PHU synthesized from 1,6-hexanediamine. The signals at 8.38–8.68 (f) ppm were assigned to the protons on the urethane groups, confirming the urethane structure of the final product. The signals at 1.31–1.60 (h), and 2.89–3.16 (g) ppm were assigned to the methylene protons on the alkyl groups. It was known that both primary and secondary hydroxyl groups could be formed along the PHU backbone due to the two different ring-opening pathways of the cyclic carbonates. In Fig. 4, two signals were observed at 4.95–5.10 (e1) and 3.73–3.82 (e2) ppm, corresponding to the formation of the primary and secondary hydroxyl groups, respectively. A small signal corresponding to the methine protons on the cyclic carbonate rings was also observed at 5.14–5.19 ppm, indicating the incomplete conversion of the bis(cyclic carbonate).
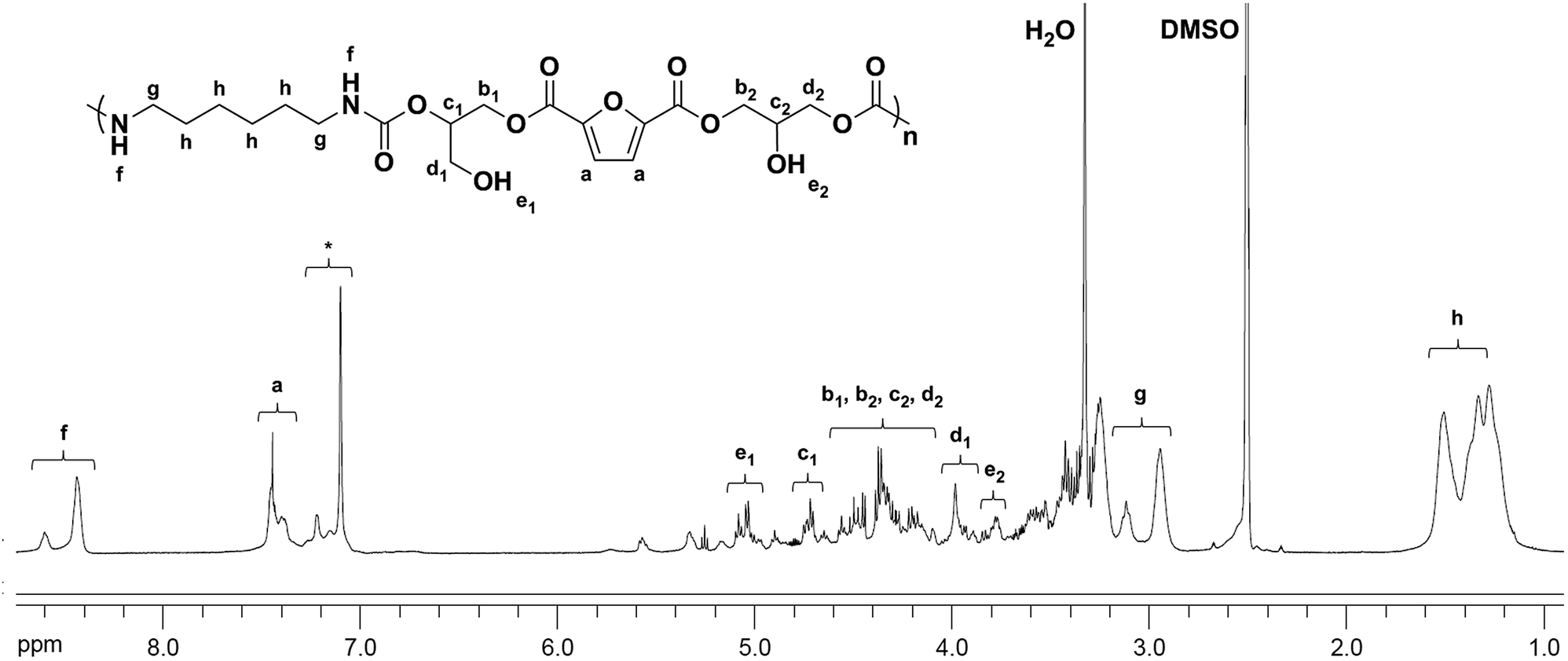 | ||
| Fig. 4 1H NMR spectra of PHU synthesized from the bis(cyclic carbonate) and 1,6-hexanediamine at 180 °C for 8 h (*: impurity). | ||
The product yield was determined by comparing the integral areas of the methine protons of cyclic carbonate groups and the protons of urethane groups in the 1H NMR spectra. Table 1 summarizes the yields of the PHUs synthesized from the FDCA-derived bis(cyclic carbonate) and different diamines at different reaction times and temperatures.
| Diamine | Temperature (°C) | Time | Yield (%) | Mn (g mol−1) | PDI | Tg (°C) |
|---|---|---|---|---|---|---|
| 1,6-Hexanediamine | 140 | 5.5 d | 87 | 6500 | 4.0 | 66 |
| 1,6-Hexanediamine | 160 | 2 d | 87 | 7000 | 4.3 | 65 |
| 1,6-Hexanediamine | 180 | 8 h | 88 | 7000 | 3.5 | 66 |
| 1,8-Diaminooctane | 140 | 4 d | 85 | 5200 | 2.3 | 65 |
| 1,8-Diaminooctane | 160 | 2 d | 89 | 6800 | 4.0 | 65 |
| 1,8-Diaminooctane | 180 | 8 h | 88 | 7000 | 2.6 | 63 |
| Isophorondiamine | 140 | 8 d | 86 | 4700 | 3.8 | 107 |
| Isophorondiamine | 160 | 3 d | 88 | 3900 | 3.3 | 113 |
| Isophorondiamine | 180 | 15 h | 81 | 5500 | 4.3 | 106 |
As expected, temperature had an effect on the reactivity of the polyaddition between the FDCA-derived bis(cyclic carbonate) and diamines. When the reactions were conducted at low temperatures, longer times were required for these reactions to reach similar conversions of the bis(cyclic carbonate) than that at high temperatures (Table 1). In this study, when the experiment was carried out at 80 °C, the polyaddition reaction barely occurred. However, according to the literature, it was reported that high yields of PHUs could be obtained from the polyaddition between a terephthalic acid-derived bis(cyclic carbonate) and 1,6-hexanediamine at room temperature.28 This result indicated that the FDCA-derived bis(cyclic carbonate) had much lower reactivity with diamines compared to the terephthalic acid-derived bis(cyclic carbonate) even though they had similar chemical structures. The low reactivity of the FDCA-derived bis(cyclic carbonate) might be attributed to the electron-donating effect of the oxygen atom on the furan ring. The electron-donating substitute on the bis(cyclic carbonate) decreased the electrophilicity of the carbonyl carbon on the cyclic carbonate rings, making it unfavorable for the nucleophilic polyaddition of the bis(cyclic carbonate) with diamines.
At 140 °C, the reaction of the bis(cyclic carbonate) with 1,8-diaminooctane was slightly faster than that with 1,6-hexanediamine, probably due to the unhindered character of the longer carbon chain of 1,8-diaminooctane.29 When the reactions were conducted at 160 °C and 180 °C, the reactivity of the bis(cyclic carbonate) with these two aliphatic diamines seemed to be similar. In contrast to these two types of aliphatic diamines, isophorondiamine showed much lower reactivity to the bis(cyclic carbonate) due to its higher steric hindrance.
The PHUs synthesized in this study were found to be resistant to a variety of organic solvents, including dichloromethane, acetone, and tetrahydrofuran, regardless of the diamine used and reaction temperature, and only partially soluble in DMF, DMSO, and acetonitrile. When dissolved in DMF, partial precipitations of the PHUs were observed in large quantities especially when the PHUs were synthesized from aliphatic diamines at high reaction temperatures. On the other hand, when using isophorondiamine, the PHUs obtained were almost completely soluble in DMF with only small amounts of precipitates observed. The insoluble parts of the PHUs were probably byproducts formed by the crosslinking of furan rings, and the results indicated that high reaction temperatures and flexible structures of diamine seemed to make the crosslinks more significant.
The molecular weights and distributions of the PHUs were determined by GPC analysis using DMF as a solvent (ESI, Fig. S1–S3†). The results showed that the number-average molecular weights (Mn) of the PHUs were between 3900 g mol−1 and 7000 g mol−1, and the polydispersity indexes (PDI) were in the range from 2.3 to 4.3 (Table 1). In comparison to the PHUs (a Mn of 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 g mol−1 and a PDI of 1.8) produced from terephthaloyl bis(cyclic carbonate) and 1,6-hexanediamine,28 the PHUs synthesized from the FDCA-derived bis(cyclic carbonate) and 1,6-hexanediamine in this study had relatively lower Mn, which resulted from the low reactivity of the FDCA-derived bis(cyclic carbonate) due to the electron-donating effect of its furan ring. However, when synthesized from isophorondiamine, the PHUs produced in this study had slightly larger Mn than those, according to the literature, produced from terephthaloyl bis(cyclic carbonate).30 As shown in Table 1, the Mn of the PHUs strongly depended on the structures of the diamines that were used. The PHUs formed with the linear aliphatic diamines had larger Mn than those formed with isophorondiamine. The relatively lower Mn of the PHUs formed with isophorondiamine could be explained by the low reactivity of the isophorondiamine due to its steric hindrance. Reaction temperature was another important factor that influenced the Mn of the PHUs. The Mn of the PHUs produced from the linear aliphatic diamines were lower when prepared at 140 °C, but no significant difference was observed between the PHUs prepared at 160 °C and 180 °C, and the PDI were higher when prepared at 160 °C than at the other two temperatures. For the PHUs produced from isophorondiamine, different patterns of the temperature effects on the Mn and PDI were observed. The Mn of the PHUs synthesized from isophorondiamine appeared to be relatively larger when produced at 180 °C, but high reaction temperature also led to high PDI.
200 g mol−1 and a PDI of 1.8) produced from terephthaloyl bis(cyclic carbonate) and 1,6-hexanediamine,28 the PHUs synthesized from the FDCA-derived bis(cyclic carbonate) and 1,6-hexanediamine in this study had relatively lower Mn, which resulted from the low reactivity of the FDCA-derived bis(cyclic carbonate) due to the electron-donating effect of its furan ring. However, when synthesized from isophorondiamine, the PHUs produced in this study had slightly larger Mn than those, according to the literature, produced from terephthaloyl bis(cyclic carbonate).30 As shown in Table 1, the Mn of the PHUs strongly depended on the structures of the diamines that were used. The PHUs formed with the linear aliphatic diamines had larger Mn than those formed with isophorondiamine. The relatively lower Mn of the PHUs formed with isophorondiamine could be explained by the low reactivity of the isophorondiamine due to its steric hindrance. Reaction temperature was another important factor that influenced the Mn of the PHUs. The Mn of the PHUs produced from the linear aliphatic diamines were lower when prepared at 140 °C, but no significant difference was observed between the PHUs prepared at 160 °C and 180 °C, and the PDI were higher when prepared at 160 °C than at the other two temperatures. For the PHUs produced from isophorondiamine, different patterns of the temperature effects on the Mn and PDI were observed. The Mn of the PHUs synthesized from isophorondiamine appeared to be relatively larger when produced at 180 °C, but high reaction temperature also led to high PDI.
Thermal properties of the PHUs synthesized from the FDCA-derived bis(cyclic carbonate) and diamines were investigated by DSC and TGA analyses. Typical DSC thermograms of the FDCA-derived PHUs are presented in Fig. 5. No endothermic or exothermic phenomenon was observed from the thermograms, indicating that these PHUs were fully amorphous. According to literatures, most PHUs were able to form amorphous phases, which could be attributed to the presence of hydroxyl groups along the polymer backbones.3,31 Since both primary and secondary hydroxyl groups could be formed, depending on which direction the five-membered cyclic carbonate rings were opened, the random distribution of the primary and secondary hydroxyl groups could hinder the ordering between the polymer chains, therefore preventing the crystallization of PHUs.17,32,33
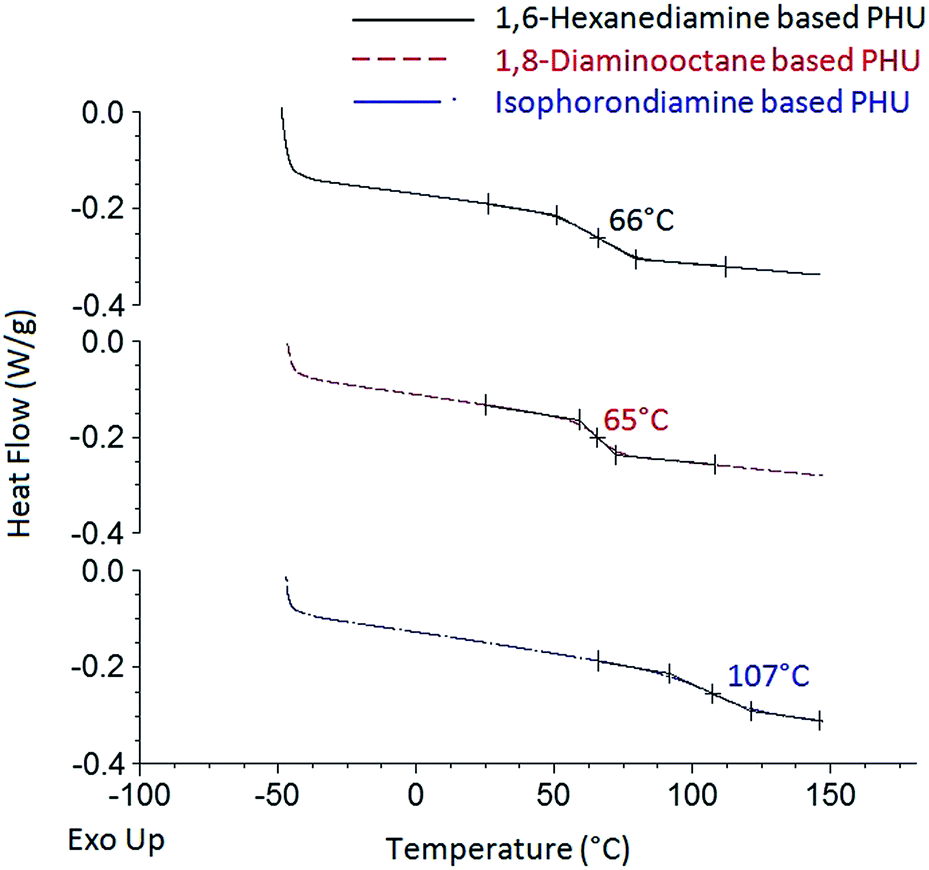 | ||
| Fig. 5 DSC thermograms of the FDCA-derived PHUs synthesized from three different diamines at 140 °C. | ||
The glass transition phenomenon was observed in the DSC thermograms. The midpoints of the transition temperatures in the heating cycles were interpreted as the glass transition temperatures (Tg). As shown in Table 1, the Tg of the PHUs ranged from 63 °C to 113 °C, depending on the chemical structures of the diamines used. Most studies agreed that the high molecular flexibility between the hydroxyurethane groups led to lower Tg of PHUs.1,28,30,34,35 The octamethylene moiety of the 1,8-diaminooctane-based PHUs should have higher flexibility of the polymer chains than the hexamethylene moiety of the 1,6-hexanediamine-based ones. However, the PHUs synthesized from 1,8-diaminooctane showed similar Tg compared to those synthesized from 1,6-hexanediamine. In contrast, the PHUs synthesized from isophoronediamine gave the highest Tg. The rigid cycloaliphatic units of the isophoronediamine-based PHUs gave rather low flexibility to the polymer chains and thus resulted in the highest Tg. Although the effects of reaction temperature and molecular weight of the PHUs on their Tg were observed in some studies,1 these effects were not clearly observed in this study.
Fig. 6 presents typical TGA curves of the FDCA-derived PHUs from three different diamines. All of these PHUs were stable up to 200 °C. The slight weight losses from 150 °C to 200 °C could be attributed to the evaporation of residual diamines or oligomers. Table 2 lists the decomposition temperatures at 5% weight losses (T5%) of these PHUs. The T5% were in the range of 240 °C and 279 °C, which is similar to the PHUs synthesized from terephthaloyl bis(cyclic carbonate).28 As shown in the TGA curves, the weights of the PHUs dropped quickly at two temperature ranges, indicating that the PHUs degraded in a two-step process (Fig. 6). In the first step, the maximum degradation rates occurred at a temperature between 307 °C and 327 °C with weight losses between 20% and 34%. In the second step, the maximum degradation rates occurred between 401 °C and 455 °C, which were associated with weight losses of 65–75%. The first step of degradation was due to the degradation of the urethane bonds, which are known to decompose starting at approximately 200 °C.10 In this step, the decomposition of urethanes may occur via three possible mechanisms: the degradation to isocyanates and alcohols; the formation of primary amines, CO2, and olefins; and the formation of secondary amines and CO2.36 The second step of degradation most likely occurred due to the scission of the furan rings.
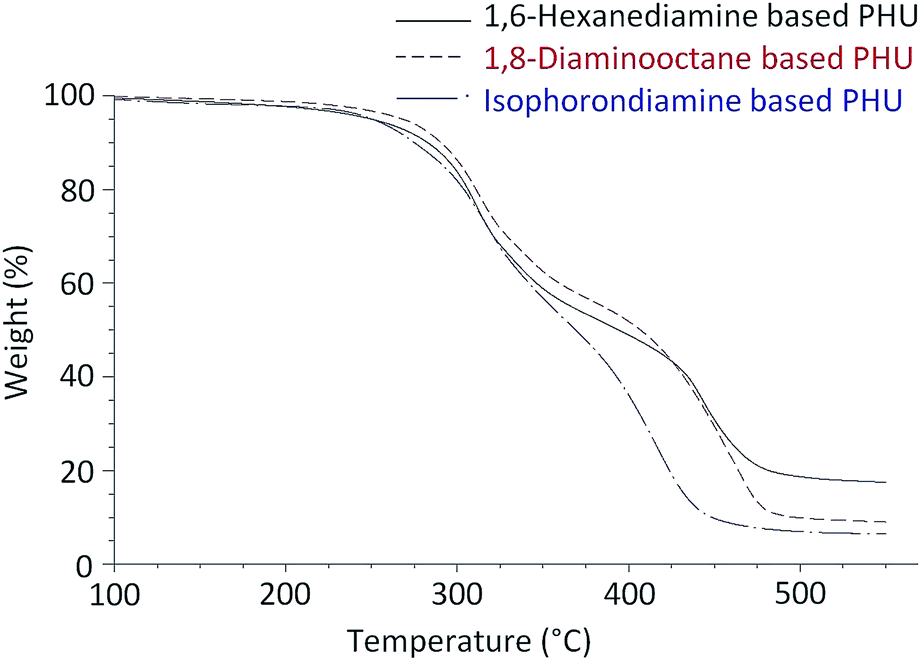 | ||
| Fig. 6 TGA curves of the FDCA-derived PHUs synthesized from three different diamines at 180 °C. | ||
| Sample | T5% (°C) | First step of degradation | Second step of degradation | |||
|---|---|---|---|---|---|---|
| Diamine | Temperature (°C) | Tmax (°C) | Weight loss (%) | Tmax (°C) | Weight loss (%) | |
| 1,6-Hexanediamine | 140 | 240 | 311 | 24 | 437 | 68 |
| 1,6-Hexanediamine | 160 | 254 | 307 | 25 | 438 | 72 |
| 1,6-Hexanediamine | 180 | 251 | 311 | 23 | 443 | 65 |
| 1,8-Diaminooctane | 140 | 279 | 312 | 20 | 451 | 73 |
| 1,8-Diaminooctane | 160 | 262 | 320 | 25 | 443 | 72 |
| 1,8-Diaminooctane | 180 | 269 | 313 | 21 | 455 | 74 |
| Isophorondiamine | 140 | 254 | 327 | 30 | 418 | 75 |
| Isophorondiamine | 160 | 256 | 326 | 34 | 401 | 69 |
| Isophorondiamine | 180 | 251 | 320 | 29 | 416 | 75 |
The thermal stability of PHU is known to depend on its chemical structure.37 It was reported that the thermal stability of PHUs synthesized from phenoxycarbonyloxymethyl ethylene carbonate and linear aliphatic diamines increased along with the increase of the number of methylene groups in the diamines.34 Similarly, the FDCA-derived PHUs synthesized from 1,8-diaminooctane exhibited higher T5% than those of the analogs synthesized from 1,6-hexanediamine, indicating the higher thermal stability of the 1,8-diaminooctane-based PHUs. The T5% of the PHUs produced from isophoronediamine were found to be similar to those produced from 1,6-hexanediamine. Tmax are defined as the temperatures at which the maximum rates of degradation occurred in each degradation step. As shown in Table 2, the FDCA-derived PHUs produced from 1,8-diaminooctane had Tmax similar to those produced from 1,6-hexanediamine in the first step of degradation. However, in the second step of degradation, the Tmax of the PHUs produced from 1,8-diaminooctane were slightly higher, and the weight losses corresponding to the Tmax were also slightly larger. In contrast to the PHUs produced from the linear aliphatic diamines, the PHUs produced from isophoronediamine showed slightly higher Tmax in the first step of degradation, and much lower Tmax in the second step of degradation.
4. Conclusions
FDCA has been recognized as a promising renewable platform chemical for developing biobased polymeric materials. In this study, a new biobased bis(cyclic carbonate) was, for the first time, successfully synthesized from FDCA through its epoxidation followed by the integration of CO2. The feasibility of using this bis(cyclic carbonate) as a monomer to react with various diamines for the development of biobased PHUs via a non-isocyanate pathway was also explored in this study. The chemical structures and thermal properties of these FDCA-derived PHUs were characterized using FT-IR, 1H NMR, GPC, DSC, and TGA analyses. According to the TGA results, the FDCA-derived PHUs exhibited good thermal stability. The PHUs were also found to be fully amorphous, and the Tg were highly dependent on the chemical structures of the diamines used. Due to the rigid cycloaliphatic structure, the PHUs synthesized from cycloaliphatic diamine showed higher Tg compared to those synthesized from linear aliphatic diamines.In sum, this study provided a novel route for the synthesis of biobased PHUs from non-food renewable resources coupled with an efficient method for CO2 fixation. A major drawback related to the PHU synthesis would be the necessity of a high reaction temperature in order to achieve a reasonable reaction rate due to the low reactivity of the FDCA-derived bis(cyclic carbonate). It would be of great interest to investigate the effect of catalysts on the polyaddition reaction between this bis(cyclic carbonate) and diamines in future studies.
Acknowledgements
The authors would like to thank Mrs Mary Wicks (Department of Food, Agricultural and Biological Engineering, The Ohio State Uinversity) for reading through the manuscript and providing useful suggestions.Notes and references
- S. Benyahya, B. Boutevin, S. Caillol, V. Lapinte and J.-P. Habas, Polym. Int., 2012, 61, 918–925 CrossRef CAS.
- E. Jiménez, B. Cabañas and G. Lefebvre, Environment, energy and climate change I: Environmental chemistry of pollutants and wastes, Springer, 2015 Search PubMed.
- J. L. J. Van Velthoven, L. Gootjes, D. S. Van Es, B. A. J. Noordover and J. Meuldijk, Eur. Polym. J., 2015, 70, 125–135 CrossRef CAS.
- L. Annunziata, A. K. Diallo, S. Fouquay, G. Michaud, F. Simon, J.-M. Brusson, J.-F. Carpentier and S. M. Guilllaume, Green Chem., 2014, 16, 1947–1956 RSC.
- M. Helou, J.-F. Carpentier and S. M. Guillaume, Green Chem., 2011, 13, 266–271 RSC.
- O. Figovsky, L. Shapovalov, A. Leykin, O. Birukova and R. Potashnikova, Sci. Isr.–Technol. Advantages, 2012, 14, 95–106 CAS.
- V. M. Lombardo, E. A. Dhulst, E. K. Leitsch, N. Wilmot, W. H. Heath, A. P. Gies, M. D. Miller, J. M. Torkelson and K. A. Scheidt, Eur. J. Org. Chem., 2015, 2015, 2791–2795 CrossRef CAS.
- G. Rokicki, P. G. Parzuchowski and M. Mazurek, Polym. Adv. Technol., 2015, 26, 707–761 CrossRef CAS.
- V. Besse, R. Auvergne, S. Carlotti, G. Boutevin, B. Otazaghine, S. Caillol, J.-P. Pascault and B. Boutevin, React. Funct. Polym., 2013, 73, 588–594 CrossRef CAS.
- C. Carr, L. Bonnet and L. Averous, RSC Adv., 2014, 4, 54018–54025 RSC.
- M. Bähr, A. Bitto and R. Mülhaupt, Green Chem., 2012, 14, 1447–1454 RSC.
- M. Aresta and A. Dibenedetto, Catal. Today, 2004, 98, 455–462 CrossRef CAS.
- O. Figovsky and L. Shapovalov, Macromol. Symp., 2002, 187, 325–332 CrossRef CAS.
- I. Javni, D. P. Hong and Z. S. Petrovic, J. Appl. Polym. Sci., 2008, 108, 3867–3875 CrossRef CAS.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2000, 39, 860–867 CrossRef.
- D. Miloslavskiy, E. Gotlib, O. Figovsky and D. Pashin, Int. Lett. Chem., Phys. Astron., 2014, 8, 20–29 CrossRef.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef CAS PubMed.
- A. Boyer, E. Cloutet, T. Tassaing, B. Gadenne, C. Alfos and H. Cramail, Green Chem., 2010, 12, 2205–2213 RSC.
- J. Zhang, J. Li, Y. Tang, L. Lin and M. Long, Carbohydr. Polym., 2015, 130, 420–428 CrossRef CAS PubMed.
- Q. Chen, K. Gao, C. Peng, H. Xie, Z. K. Zhao and M. Bao, Green Chem., 2015, 17, 4546–4551 RSC.
- M. Rose, D. Weber, B. V. Lotsch, R. K. Kremer, R. Goddard and R. Palkovits, Microporous Mesoporous Mater., 2013, 181, 217–221 CrossRef CAS.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- J. Deng, X. Liu, C. Li, Y. Jiang and J. Zhu, RSC Adv., 2015, 5, 15930–15939 RSC.
- S. Ma, X. Liu, Y. Jiang, L. Fan, J. Feng and J. Zhu, Sci. China: Chem., 2014, 57, 379–388 CrossRef CAS.
- S. Ma, X. Liu, Y. Jiang, Z. Tang, C. Zhang and J. Zhu, Green Chem., 2013, 15, 245–254 RSC.
- M. Bähr and R. Mülhaupt, Green Chem., 2012, 14, 483–489 RSC.
- J. Rintjema, L. P. Carrodeguas, V. Laserna, S. Sopeña and A. W. Kleij, Carbon dioxide and organometallics, Springer, 2016, vol. 53 Search PubMed.
- A. Steblyanko, W. Choi, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2375–2380 CrossRef CAS.
- L. Maisonneuve, A. S. More, C. Alfos, Y. Landais, T. Tassaing, E. Grau and H. Cramail, RSC Adv., 2014, 4, 25795–25803 RSC.
- S. Benyahya, J.-P. Habas, R. Auvergne, V. Lapinte and S. Caillol, Polym. Int., 2012, 61, 1666–1674 CrossRef CAS.
- V. Besse, F. Camara, F. Méchin, E. Fleury, S. Caillol, J. Pascault and B. Boutevin, Eur. Polym. J., 2015, 71, 1–11 CrossRef CAS.
- R. H. Lambeth and T. J. Henderson, Polymer, 2013, 54, 5568–5573 CrossRef CAS.
- J. L. J. Van Velthoven, Bio-based polyamide and poly(hydroxy urethane) coating resins: synthesis, characterization, and properties, Eindhoven University of Technology, 2015 Search PubMed.
- L. Ubaghs, N. Fricke, H. Keul and H. Hocker, Macromol. Rapid Commun., 2004, 25, 517–521 CrossRef CAS.
- X. Sheng, G. Ren, Y. Qin, X. Chen, X. Wang and F. Wang, Green Chem., 2014, 17, 373–379 RSC.
- I. Javni, Z. S. Petrović, A. Guo and R. Fuller, J. Appl. Polym. Sci., 2000, 77, 1723–1734 CrossRef CAS.
- M. Kim, H. Kim, C. Ha, D. Park and J. Lee, J. Appl. Polym. Sci., 2001, 81, 2735–2743 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25045a |
| This journal is © The Royal Society of Chemistry 2017 |