
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A comparative study of physicochemical and functional properties of silver carp myofibrillar protein glycated with glucose and maltodextrin
Shulai Liu,
Peicheng Zhao,
Jingjing Zhang,
Qiuhong Xu,
Yuting Ding and
Jianhua Liu*
Department of Food Science and Engineering, Ocean College, Zhejiang University of Technology, Hangzhou 310014, P. R. China. E-mail: jhliu@zjut.edu.cn; Fax: +86 571 88320237; Tel: +86 571 88320237
First published on 4th January 2017
Abstract
Myofibrillar protein (Mf) from silver carp (Hypophthalmichthys molitrix) was incubated with glucose and maltodextrin for 0–96 h at 50 °C and 75% relative humidity to obtain glycoconjugates in different periods of the Maillard reaction. Physicochemical properties, including molecular weight, sugars bound to Mf, available amino acid content and FT-IR properties all indicated that glycation occurred successfully. In addition, SDS-PAGE analysis and available amino acids of Maillard reaction productions (MRPs) suggested that glucose reacted more easily with Mf than maltodextrin. Functional properties of MRPs were superior to Mf and reached the highest value at 8–12 h, and then decreased significantly. Heat stability and solubility of Mf–maltodextrin in 0.5 M NaCl was better than for Mf–glucose, while the solubility in 0.1 M NaCl, and the emulsifying and foaming properties of Mf–glucose were in general better than for Mf–maltodextrin. The variety of saccharides and the process of the Maillard reaction had a great influence on the physicochemical and functional properties of MRPs.
1. Introduction
Fish protein has high nutritional value, which makes it an essential component of the diet. Myofibrillar protein (Mf) is a primary protein of fish, which can provide favorable functional properties, such as emulsifying properties, gel forming ability, and water-holding capacity. However, some chemical properties and the thermal stability of Mf are worse than for other vertebrates.1 When protein denaturation occurs during preparation, processing and storage, the properties of Mf, especially the solubility and emulsifying properties will decrease easily.2 Therefore, for the improvement of the additional economic value of fish muscle and the development of fishery, it is particularly important to inhibit or reduce fish protein denaturation, improve its functional properties and then manufacture high quality foodstuffs using fish muscle.Glycation is the covalent attachment of sugars to available amino groups of proteins to form neoglycoproteins through Amadori rearrangement steps in the Maillard reaction.3 Although various available chemical techniques have been developed to prepare synthetic glycoproteins, the production of glycoconjugates between proteins and saccharides using the Maillard reaction has received much attention in recent years. Owing to mild and safe conditions requiring no extraneous chemicals, this reaction is superior to other types of chemical modification for food proteins, and poses a promising application for protein modification in the food industry.4 Glycation is an effective method to improve the functional properties of fish proteins and even endows them with a novel functionality.5 The impact of glycation through the Maillard reaction on food proteins has been widely reported, such as improving the emulsifying properties of fish Mf glycated with alginate oligosaccharides,6 raising the solubility of carp Mf conjugated with ribose,7 increasing the heat stability of carp Mf glycated with dextran,8 enhancing the antioxidant effect of chicken Mf reacted with glucose and maltose,9 and improving the gel properties by glycation with konjac glucomannan.10 However, these research efforts paid less attention to the impact of the structure and molecular weight of saccharides, and the understanding of the relationships between the types of saccharides and functional properties of glycated Mf is seldom reported.
Glucose, a monosaccharide with small molecular weight, and maltodextrin (DE 20), a polysaccharide with a complex structure and large molecular weight, were chosen as sugar donors in the present study. Myofibrillar protein of silver carp (Hypophthalmichthys molitrix) was used to prepare glycoconjugates through the Maillard reaction, with glucose and maltodextrin, respectively. The aim of this research is to evaluate the changes in the physicochemical and functional properties of the MRPs. More importantly, due to the differences in structure and molecular weight of glucose and maltodextrin, physicochemical and functional properties of Mf–glucose (M–G) and Mf–maltodextrin (M–M) are compared and discussed in depth.
2. Experimental
2.1. Materials
Live silver carp was purchased at the local market in Hangzhou. Tris, sodium dodecyl sulfate (SDS), bovine serum albumin, glucose and maltodextrin (DE 20) were purchased from Sigma Company (St. Louis, MO, USA). The other chemicals were purchased from Hangzhou HuaLin Company (Hangzhou, China).2.2. Preparation of myofibrillar protein
The myofibrillar protein was prepared from the ordinary muscle of a cultured silver carp using the method of Saeki.11 Firstly, the fish was hit over head with a stick until it was dead. After that, its head and tail were cut. The viscera, skin and bone of the fish were removed to obtain fresh muscle, and then the red muscle and white muscle were separated. The white muscle was ground with a mincing machine and soaked in three volumes of 0.5% Triton X-100 (containing 0.05 M NaCl) for 10 min, and then the supernatant was decanted. The washed ground meat was then re-immersed in 8 volumes (based on initial muscle weight) of 0.05 M NaCl and 0.5% Triton X-100 and homogenized for 2 min at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm using a homogenizer (model F6/10, FLUKO Co. Ltd., Germany). After filtration through cotton gauze to remove impurities, the homogenate was centrifuged at 8000g for 10 min to collect the myofibrils. The precipitate was re-suspended in 0.05 M NaCl and centrifuged (8000g, 10 min) four times. The pure myofibrils obtained were filtered through nylon cloth. All operations were carried out at 4 °C.
000 rpm using a homogenizer (model F6/10, FLUKO Co. Ltd., Germany). After filtration through cotton gauze to remove impurities, the homogenate was centrifuged at 8000g for 10 min to collect the myofibrils. The precipitate was re-suspended in 0.05 M NaCl and centrifuged (8000g, 10 min) four times. The pure myofibrils obtained were filtered through nylon cloth. All operations were carried out at 4 °C.
2.3. Glycation of myofibrillar protein
The myofibril and saccharide (maltodextrin and glucose) powders with the weight ratio of 1:2 were distributed in 50 mM NaCl. The protein concentration of the mixture was adjusted to 6.0 mg mL−1. The mixture was frozen at −80 °C and then freeze-dried using a freeze-dryer (FD-1-50, Bo Yikang Co. Ltd., Beijing, China). The lyophilized samples were then immediately stored at −25 °C and used within 30 days.
The lyophilized mixture was then incubated at 50 °C for 0, 2, 5, 8, 12, 24, 48, 72 and 96 h and 75% relative humidity in an incubator cabinet (RXZ-288A, Jiangnan Co. Ltd., Zhejiang, China). After reaction, the protein powder was immediately dissolved in 0.05 M KCl-20 mM PBS (pH 7.4), and centrifuged (10000g, 20 min, 4 °C) four or five times to remove the unreacted saccharides. The precipitate was collected and freeze-dried for subsequent experiments and analysis.
2.4. Electrophoretic analysis
Sodium-dodecyl-sulphate polyacrylamide gel electrophoresis (SDS-PAGE) was used to measure the molecular weight changes of the Maillard reaction products (MRPs), using 7.5% separating gel and a 5% stacking gel. Gels were stained with Coomassie Brilliant Blue R250 and destained with a solution of methanol and acetic acid.2.5. Amount of saccharides bound to Mf
As the unreacted saccharides were removed after glycation, the MRPs were re-dissolved in 0.5 M NaCl containing 40 mM tris–HCl (pH 7.5). The amount of saccharides bound to Mf was determined by the phenol–sulfuric acid method.12 Each value of the binding saccharide to Mf was the mean of three replicates.2.6. Available amino acid groups
The amino acid groups were determined by the ortho-phthaldialdehyde (OPA) method with some modifications.13 The OPA reagent was prepared by mixing 50 mL of 0.1 M sodium borate buffer, 5 mL of 20% SDS, 80 mg of OPA (dissolved in 2 mL of methanol) and 20 μL of β-mercaptoethanol in water. The MRPs were dissolved in 40 mM tris–HCl (pH 7.5), obtaining 0.5 M NaCl and adjusted to 6 mg mL−1. Then, the proteins were precipitated with 7.5% trichloroacetic acid (at the final concentration) to remove the tris buffer, and re-dissolved in a 50 mM phosphate buffer (pH 7.5). The mixed solution (200 μL) was added to 4 mL OPA reagent, and the absorbency was read at 340 nm within 5 min.2.7. Fourier transform infrared spectroscopy (FT-IR)
The infrared absorption data of Mf and modified Mf were obtained in the range 4000–400 cm−1 in KBr pellets using a Nicolet spectrophotometer Protege 460 FT-IR at room temperature (20 °C). Twenty individual spectra of 64 scans were recorded for each sample at 4 cm−1 resolution.2.8. Solubility in media with different ionic strengths
The solubility and heat stability of MRPs were measured according to previous methods, with some modifications.14 The two types of MRPs were dispersed in 40 mM tris–HCl (pH 7.5), containing 0.5 M NaCl or 0.1 M NaCl. The protein concentration was adjusted to 1.0 mg mL−1. After being homogenized for 1 min at 12000 rpm, the solution was centrifuged at 5000g for 30 min. The protein solubility was expressed as the percentage of protein in the supernatant to the total protein content determined by Coomassie Brilliant Blue G250.
2.9. Heat stability
The evaluation of heat stability was based on the solubility of the protein. After centrifugation, the supernatant with 0.5 M NaCl was heated at 80 °C for 1 h, and then centrifuged at 5000g for 30 min. Heat stability was expressed as the percentage of protein in the supernatant, after being heated at 80 °C, to the protein in the supernatant before being heated.2.10. Emulsifying properties
The method of Pearce et al.15 was used, with some modifications. The MRPs were dissolved in 40 mM tris–HCl and 0.5 M NaCl (pH 7.5), respectively, and the protein concentration was 3 mg mL−1. The protein solution (6.0 mL) was mixed with 2.0 mL of pure corn oil in centrifuge tubes, and homogenized for 1 min at 13500 rpm in ice water. Then, 0.3 mL of the emulsion was immediately taken from the bottom of the tube, and diluted with 5.7 mL of 0.1% SDS at 25 °C. The turbidity of the emulsion was measured at a wavelength of 500 nm for evaluating the emulsifying activity, expressed as EA. After that, the emulsion was held for 10 min at constant temperature. The absorbance of the emulsion after standing was taken for dilution and turbidity measurement, as described above. The emulsifying stability was expressed as | ESI = A0 × T/(A0 − At). |
2.11. Foaming properties
The foaming properties of the MRPs were characterized through their foam height and stability measured in a tube, based on the method of Sánchez et al.,16 with some modifications. The foam formation and the foam stability were determined by optical measurements. The foams were produced with a homogenizer for 2 min at 17500 rpm, in 3 mL of solution (50 mM Tris–HCl – 0.5 M NaCl, pH 7.5), which contained 1.5% protein. The initial height of the solution and the foam height were recorded at intervals of 0, 2, 10, 20 and 30 min, using a caliper. The foaming capacity17 was expressed as the proportion of foam height at 0 min to solution height. The foaming stability (FS) was conveyed by the percentage of foam height at some time to 0 min. The measurement of the height was rapid and accurate to three digits after the decimal point.
2.12. Statistical analysis
All experiments were carried out with samples from at least three different lots of MRPs. Statistical analysis on a completely randomized design was performed by SPSS 13.0 for Windows software (SPSS Institute Inc., Cary, NC). Duncan's Multiple Range Test was applied to detect differences of means, and P <0.05 was considered to be statistically significant.3. Results and discussion
3.1. Analysis of SDS-PAGE
SDS-PAGE patterns of Mf and Mf glycated with glucose and maltodextrin are shown in Fig. 1. As reported, Mf was mainly composed of two myosin heavy chains, each with molecular weight of about 200 kDa, actin (about 43 kDa), tropomyosin (about 36 kDa) and four myosin light chains (about 25 kDa, respectively) (Fig. 1, lane 1).17 The SDS-PAGE patterns show the changes in the protein subunits during the glycation process. With the process of glycation, the electrophoretic mobility of the myosin heavy chain and actin decreased slightly. This agrees well with the report by Katayama et al.18 about myofibrillar proteins of shellfish glycation with glucose. The cross linking mainly between lysine residues of myofibrillar protein and sugars resulted in the increase of the molecular weight of the myosin heavy chain and actin. Since the mobility of bands in the SDS-PAGE depend on the molecular weight of protein subunits, which became heavier, the electrophoretic mobility of the corresponding bands became slower. In addition, in the early stage of glycation, it could be observed that the distance of the stripes mobility in Fig. 1b was more significant than in Fig. 1a, because of the larger molecular weight of maltodextrin than glucose. | ||
| Fig. 1 SDS-PAGE patterns of glycated myofibrillar proteins. (a) Mf–glucose glycoconjugates, (b) Mf–maltodextrin glycoconjugates. Lanes 1, 2, 3, 4, 5, 6, 7, 8 and 9: incubation for 0, 2, 5, 8, 12, 24, 48, 72 and 96 h, respectively. | ||
Furthermore, myosin, tropomyosin and actin in both Fig. 1a and b, were found to disappear gradually in the later stage of glycation (lane 7, 8 and 9), due to the possible degradation of the main components of myofibrillar proteins. Also, the loss of subunits in Fig. 1a was more serious than in Fig. 1b, probably because maltodextrin would protect Mf from degradation or denaturation to a certain extent.
3.2. Evaluation of the glycation progress
The Maillard reaction is the covalent attachment of sugars to available amino acid groups of protein (mainly lysine). With the process of glycation, the saccharides attached to Mf increased, while the available amino acid groups decreased.As shown in Fig. 2, the rapid loss of available amino acid groups occurred, and after 96 h of reaction, it decreased to 40% (Mf–glucose glycoconjugates) and 50% (Mf–maltodextrin glycoconjugates) of the initial values. These results indicate that the interaction between glucose/maltodextrin and Mf was severe. About half of the available amino acid groups were substituted in Mf by glucose and maltodextrin. This result was similar to Sato et al.,19 who reported that glycated myofibrillar proteins had a significant decrease in amino acid groups and increase in the saccharide content. Besides, the loss of available amino acid groups of Mf–glucose was greater than for maltodextrin; this is because glucose has the smaller molecular weight, leading to a faster reaction with Mf than maltodextrin.
 | ||
Fig. 2 Amount of saccharides bound to Mf ( ), and the decrease in available amino acid groups ( ), and the decrease in available amino acid groups ( ) in Mf during reactions with saccharides for different reaction times. (a) Mf–glucose glycoconjugates, (b) Mf–maltodextrin glycoconjugates. ) in Mf during reactions with saccharides for different reaction times. (a) Mf–glucose glycoconjugates, (b) Mf–maltodextrin glycoconjugates. | ||
The amount of saccharides bound to Mf increased simultaneously with the loss of the available amino acid groups. The binding glucose and maltodextrin increased markedly during the reaction and reached 22 μg mg−1 (Fig. 2a) and 105 μg mg−1 (Fig. 2b) after 96 h, respectively. Furthermore, it was observed that the amount of attached maltodextrin was more than glucose, because the molecular weight of maltodextrin is much larger than glucose.
3.3. FT-IR analysis
FT-IR spectroscopy is a particularly useful technique for the study of protein–carbohydrate systems,20 because the chemical fingerprints of proteins and carbohydrates do not overlap significantly in several identifiable regions of the mid-infrared spectrum.21The FT-IR spectra for Mf and Mf–glucose/maltodextrin mixture incubated in different periods are shown in Fig. 3a and b, respectively. The most distinctive spectral features for proteins are the strong amide I and II bands centered at approximately 1650 cm−1 and 1540 cm−1, respectively.22 For carbohydrates, a series of overlapping peaks located in the region of 1180–953 cm−1 results from vibration modes such as the stretching of C–C and C–O, and the bending mode of C–H bonds. These absorptions are weak in most proteins. In both Fig. 3a and b, it was found that the intensities of the peaks in the regions of 1650 cm−1 and 1540 cm−1, ascribed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N from amide I and II, increased with the progress of the reaction; this is because functional groups including NH2 may be lost, while the amount of those associated with Maillard products, such as the Amadori compound (CO), Schiff base (CN), and pyrazines (C–N), may be increased by the Maillard reaction, as reported by Farhat et al.20 In addition, in the spectra of proteins, the amide III band is at 1300–1200 cm−1; this band is very complex and results mainly from C–N stretching and N–H deformation.23
O and C–N from amide I and II, increased with the progress of the reaction; this is because functional groups including NH2 may be lost, while the amount of those associated with Maillard products, such as the Amadori compound (CO), Schiff base (CN), and pyrazines (C–N), may be increased by the Maillard reaction, as reported by Farhat et al.20 In addition, in the spectra of proteins, the amide III band is at 1300–1200 cm−1; this band is very complex and results mainly from C–N stretching and N–H deformation.23
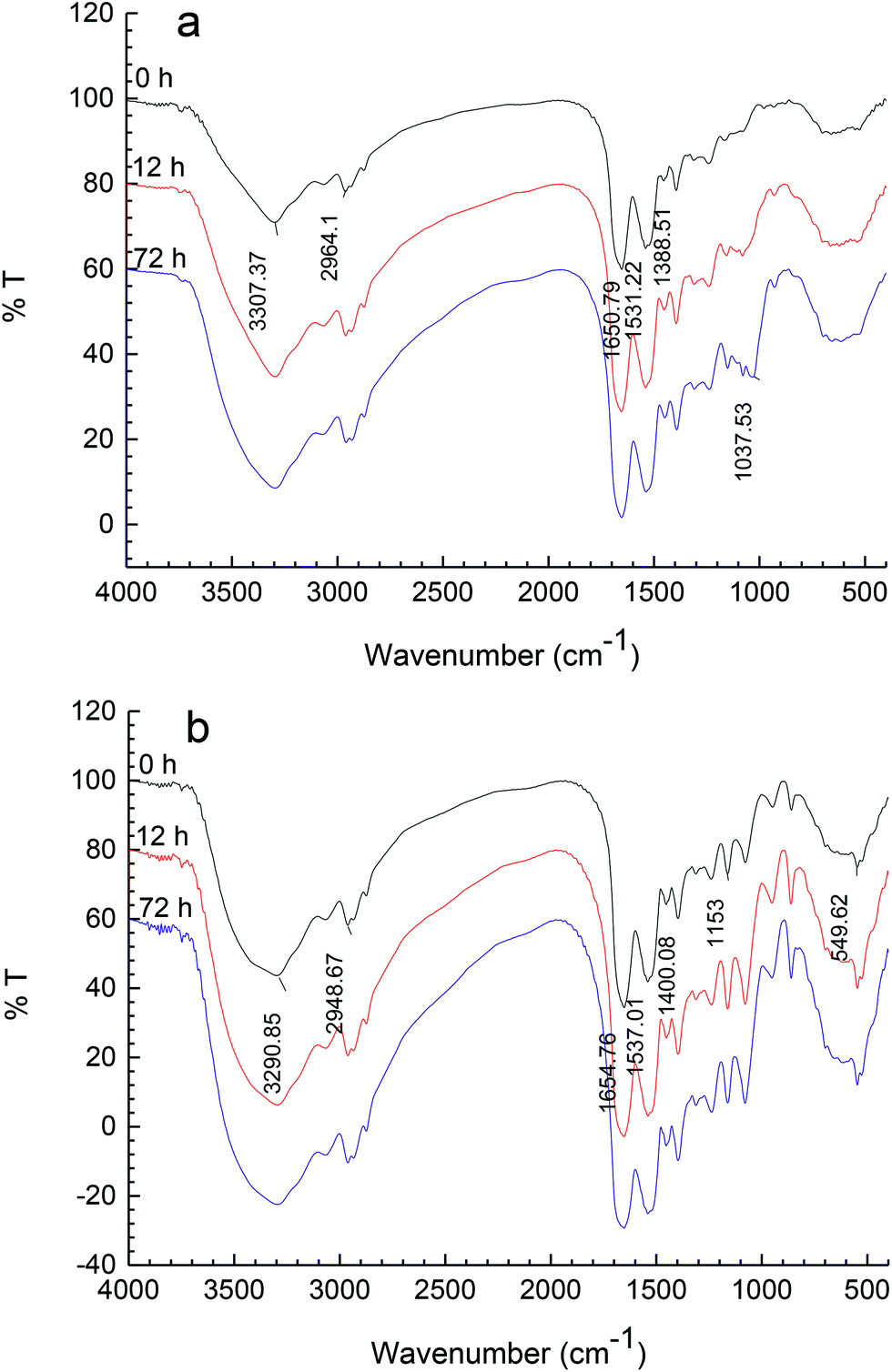 | ||
| Fig. 3 FT-IR spectra of MRPs. (a) Mf–glucose glycoconjugates, (b) Mf–maltodextrin glycoconjugates. | ||
In the spectra of M–M or M–G, it was observed that the absorptions of C–OH from 1015 cm−1 to 1200 cm−1 were stronger with the glycation progress, due to the saccharides increasingly attaching to Mf. The introduction of C–OH groups in the protein may increase the hydrophilicity of the system.24 There were obvious differences between M–M and M–G in the feature region of carbohydrates, because of the appearance of feature absorptions of glucose and maltodextrin.
3.4. Changes in solubility and heat stability
Solubility is one of the critical functional properties of proteins, which has great influence on other functionalities.25 Fig. 4a and b show the changes in solubility of MRPs in 0.1 M and 0.5 M NaCl (pH 7.5) with the progress of glycation. In low ionic strength medium (0.1 M NaCl, Fig. 4a), the total solubility of M–G in 0.1 M NaCl increased markedly with glycation. It reached a maximum at 8 hours of reaction. Such an increase was not yet observed in the protein with maltodextrin. This result conformed to the description of Saeki and Inoue,26 who significantly improved the solubility of Mf after conjugation with glucose. As described in another report,27 proteolysis of Mf often causes an increase in the solubility of Mf. However, almost no protein degradation was observed at the initial stage of the Maillard reaction, as shown in SDS-PAGE. Therefore, the increase in the total solubility in 0.1 M NaCl was mainly due to the glycation. This effect may be as a result of covalent linkage to carbohydrates, in which the net charge of the proteins changes and hydrophilic residues are produced.28 However, glucose is a reducing saccharide with small molecular weight, which has a faster reaction rate with Mf. Substitution of maltodextrin at the C-4 hydroxyl group of the glucosyl residues makes it less energetically prone to post-Amadori Maillard reactions.29 Thus, the enhancement of the solubility of M–M was weaker than for M–G. Meanwhile, it can be observed that a marked decrease in the amount of solubilized Mf occurred with further progression of glycation. This result indicated that the thermal denaturation of Mf during the later Maillard reaction had a great influence on the total solubility.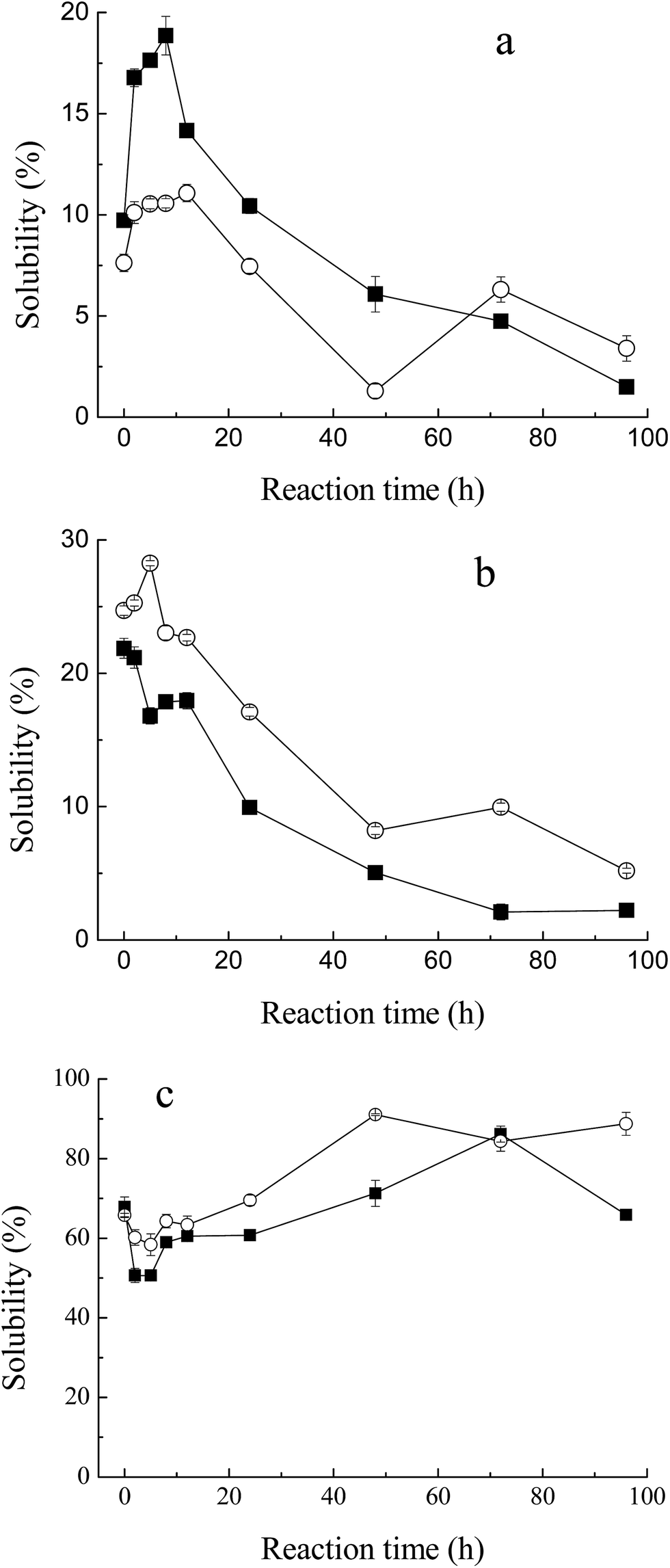 | ||
Fig. 4 Change in solubility of myofibrillar protein during glycation with sugars. (a) Low ionic strength medium (0.1 M NaCl, pH 7.5), (b) high ionic strength medium (0.5 M NaCl, pH 7.5), (c) effect of heat treatment (80 °C for 1 h) on heat stability of MRPs. Lyophilized myofibrillar proteins with glucose ( ), and maltodextrin ( ), and maltodextrin ( ). ). | ||
However, in high ionic strength medium (0.5 M NaCl, Fig. 4b), the solubility of M–M and M–G had a slight decrease at the early stage of the Maillard reaction and diminished more rapidly later. This depended on the heat denaturation of myofibrillar proteins, and glycation would have little effect on the total solubility in high ionic strength media. Protein is known to be stabilized in the presence of saccharide.30 As reported by Nishimura et al.,14 the difference in protein solubility with a decrease seemed to be caused by a shortage of saccharide as a thermal protectant. In this study, saccharides were only two times the weight of protein, which might be too small to defend Mf. Moreover, the solubility of M–M in high ionic strength solution was better than M–G in general, because maltodextrin was superior to glucose in inhibiting the protein denaturation during lyophilization and glycation,31 which was related to the stereospecific blockade and molecular weight of the carbohydrate.
A marked decrease in solubility in the process of glycation was observed. In addition to higher relative humidity and longer reaction time, lyophilization is a key factor that influences the total solubility, due to the denaturation of proteins. Saeki11 reported that, after lyophilization, the solubility of Mf decreased from 86% to 44%.
As shown in Fig. 4c, Mf itself has a relatively high heat stability at 68% solubility, when heated at 80 °C for 1 h. With the process of glycation, the heat stability of MRPs dropped at first, and then rose significantly to 90% at 48 h of reaction. As described in the solubility of MRPs, saccharides play an important role in protecting proteins from heat denaturation. However, at 2 h and 5 h of Maillard reaction, the influence of glycation was weaker than heat, which led to the reduction of heat stability. Simultaneously, with more and more saccharides attached to Mf, its stability against heating apparently improved. Besides, it can be observed that the heat stability of M–M was better than M–G in general. Enomoto et al.32 demonstrated that polysaccharide moieties inhibit the close approach and interaction of protein molecules in either their native or denatured state. Thus, the effect of maltodextrin in protecting Mf against heat is better than that of glucose. This conclusion is in agreement with the results of the solubility of M–M and M–G in 0.5 M NaCl.
3.5. Emulsifying properties
Fig. 5 shows the emulsifying properties of MRPs. Emulsion activity and stability in this study indicate the ability to absorb at the oil/water interface at the end of homogenization,33 and consequently, to stabilize emulsion. With the process of glycation, the turbidity of the emulsion of the glycated Mf increased from 0.9 to 1.3 (OD at 500 nm) within 12 h. At the later stage of the Maillard reaction, with the protein polymerization or browning, the emulsion activity decreased to 1.0, due to the loss of the binding spot of the protein for the O/W interface. This result was in agreement with the changes in solubility of MRPs. However, the enhancement of emulsion stability was not significant because the ratio of sugar and protein plays a critical role in the mechanism behind the observed increase in emulsifying properties. The increase in steric stabilization may endow greater emulsifying properties to the conjugate.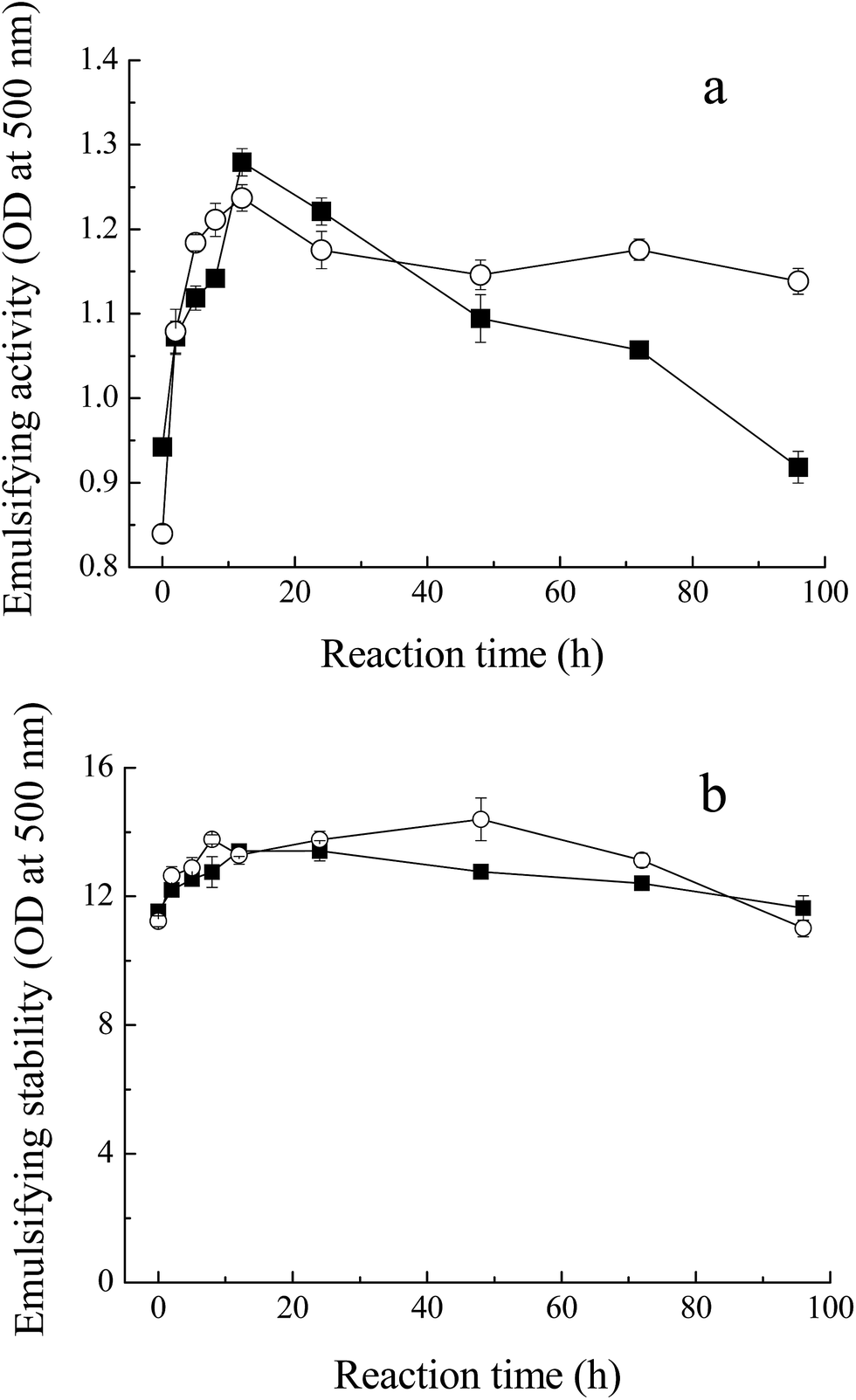 | ||
Fig. 5 Emulsifying properties of MRPs. (a) Emulsifying activity of Mf conjugated with sugars, (b) emulsifying stability of glycated Mf. Myofibrillar proteins with glucose ( ), and maltodextrin ( ), and maltodextrin ( ). ). | ||
In addition, unlike the conclusion of Li et al.,34 who measured the effects of saccharide structure on properties, the difference between M–M and M–G was not distinct. Former research proposed that branched polysaccharides could improve the emulsifying properties of Maillard conjugates more efficiently and provide more steric hindrance, preventing coalescence of the coated fat droplets.35,36 However, maltodextrin has few hydrophobic groups and branches, compared to other polysaccharides. Meanwhile, maltodextrin has the inclusion effect and glucose has a faster reaction with Mf. In conclusion, the emulsifying properties of M–M were slightly superior to M–G.
3.6. Foaming capacity and stability
The foaming capacity17 and foaming stability (FS) were measured for M–M and M–G through the height of foaming. As shown in Fig. 6a, the glycated Mf showed significant improvement compared to native Mf and even reached the highest FC of 80% at the incubation period of 8 h. The foaming capacity of both M–M and M–G dropped markedly with further reaction. The glycated proteins may increase the flexibility of the native protein structure, the conformational change, and the protein surface rearrangement, which lead to enhanced protein adsorption at the air–water interface by bubbling.37 In addition, Chevalier et al.38 proved that the properties of saccharides were the key factors that influence the foaming properties of glycated Mf. Moreover, protein solubility is the principal condition for proteins to have a good foaming activity, and the variations of FC within the process of the Maillard reaction were the same for solubility. Foaming stability, which was determined by the volume of liquid drained from the foam over time, reached the highest value of 60% (M–G) and 40% (M–M) at 8–12 h of reaction, as shown in Fig. 6b and c. Moreover, regardless of the phase of foaming disappearance (2, 10, 20 and 30 min), MRPs of 8 h and 12 h, on the whole, maintained the highest FS. Chevalier et al.38 indicated that the modification with sugar improved the foam stability of protein, as a result of the enhancement of solubility, hydration of protein and the amount of protein adsorbed at the air–water interface. | ||
Fig. 6 Foaming properties of MRPs. (a) Foaming capacity of MRPs, (b) foaming stability of Mf conjugated with glucose, (c) foaming stability of Mf conjugated with maltodextrin. Myofibrillar proteins with glucose ( ), and maltodextrin ( ), and maltodextrin ( ). Reaction time: 0 h ( ). Reaction time: 0 h ( ), 2 h ( ), 2 h ( ), 5 h ( ), 5 h ( ), 8 h ( ), 8 h ( ), 12 h ( ), 12 h ( ), 24 h ( ), 24 h ( ), 48 h ( ), 48 h ( ), 72 h ( ), 72 h ( ), 96 h ( ), 96 h ( ). ). | ||
Li et al.39 reported that glycated proteins with saccharides of higher molecular weight may increase the foaming properties of MRPs more efficiently. However, in this study, changes in the foaming activity of M–M and M–G were almost analogous. This may due to the faster reaction of Mf and glucose, and the special structure of maltodextrin. Glycation of glucose and Mf may change the surface properties and flexibility of protein to a larger extent, but maltodextrin may contribute to the better protein adsorption at the air–water interface by bubbling. Thus, the foaming capacity of M–G was slightly superior to that of M–M. However, the foaming stability of M–G was apparently better than that of M–M, probably because maltodextrin has a larger steric hindrance than glucose. The larger space structure of the saccharide may have prevented liquid running off the foam, and stabilized the formation of foam, which markedly improved the foaming stability of the MRPs.
4. Conclusion
The changes in physicochemical properties of M–G and M–M indicate that the reaction between Mf and glucose was faster than Mf and maltodextrin, while the protection of maltodextrin for Mf was better than glucose. The functional properties of Mf glycated with both glucose and maltodextrin changed significantly, compared to unglycated Mf, and the conjugates of M–G were superior to M–M in solubility and foaming properties; however, M–M had better heat stability and emulsifying properties. This may be ascribed to the difference in molecular weight and steric hindrance of sugars. Glucose may be prone to bind with Mf, while maltodextrin, with large molecular weight and steric hindrance, has advantage of protecting proteins from heat denaturation. Thus, the differences in functional properties of M–M and M–G are in agreement with the results of the physicochemical properties of MRPs.In conclusion, both glucose and maltodextrin are favorable sugar donors for glycation with Mf. These results may provide a theoretical basis for further research on glycation of Mf, such as the management of the reaction stage and choice of sugars. In addition, the improvement of functional properties resulting from glycation will contribute to better utilization of fish myofibrillar proteins in processed food.
Acknowledgements
This work was supported financially by the National Natural Science Foundation of China (No. 31301437) and Natural Science Foundation of Zhejiang (No. LQ13C200003).References
- Y. Yamashita, K. Arai and K. Nishita, Bulletin of the Japanese Society for the Science of Fish, 1978, 44, 485–489 CrossRef CAS.
- T. Gill, J. Chan, K. Phonchareon and A. Paulson, Food Res. Int., 1992, 25, 333–341 CrossRef CAS.
- M. Friedman, J. Agric. Food Chem., 1996, 44, 631–653 CrossRef CAS.
- J. Liu, Q. Ru and Y. Ding, Food Res. Int., 2012, 49, 170–183 CrossRef CAS.
- C. M. Oliver, L. D. Melton and R. A. Stanley, Crit. Rev. Food Sci. Nutr., 2006, 46, 337–350 CrossRef CAS PubMed.
- R. Sato, S. Katayama, T. Sawabe and H. Saeki, J. Agric. Food Chem., 2003, 51, 4376–4381 CrossRef CAS PubMed.
- H. Saeki and M. Tanabe, Fish. Sci., 1999, 65, 967–968 CAS.
- K. Fujiwara, T. Oosawa and H. Saeki, J. Agric. Food Chem., 1998, 46, 1257–1261 CrossRef CAS.
- K. Nishimura, M. Murakoshi, S. Katayama and H. Saeki, Biosci., Biotechnol., Biochem., 2011, 75, 247–254 CrossRef CAS PubMed.
- J. Liu, X. Wang and Y. Ding, Carbohydr. Polym., 2013, 92, 484–489 CrossRef CAS PubMed.
- H. Saeki, J. Agric. Food Chem., 1997, 45, 680–684 CrossRef CAS.
- M. Dubois, K. A. Gilles, J. K. Hamilton, P. Rebers and F. Smith, Anal. Chem., 1956, 28, 350–356 CrossRef CAS.
- H. Frister, H. Meisel and E. Schlimme, Fresenius' Z. Anal. Chem., 1988, 330, 631–633 CrossRef CAS.
- K. Nishimura, M. Murakoshi, S. Katayama and H. Saeki, Food Sci. Technol. Res., 2010, 17, 69–75 CrossRef.
- K. N. Pearce and J. E. Kinsella, J. Agric. Food Chem., 1978, 26, 716–723 CrossRef CAS.
- C. C. Sánchez and J. M. R. Patino, Food Hydrocolloids, 2005, 19, 407–416 CrossRef.
- E. Huff-Lonergan, T. Mitsuhashi, F. Parrish and R. M. Robson, J. Anim. Sci., 1996, 74, 779–785 CrossRef CAS PubMed.
- S. Katayama, J. Shima and H. Saeki, J. Agric. Food Chem., 2002, 50, 4327–4332 CrossRef CAS PubMed.
- R. Sato, T. Sawabe, H. Kishimura, K. Hayashi and H. Saeki, J. Agric. Food Chem., 2000, 48, 17–21 CrossRef CAS PubMed.
- I. A. Farhat, S. Orset, P. Moreau and J. M. Blanshard, J. Colloid Interface Sci., 1998, 207, 200–208 CrossRef CAS PubMed.
- J. A. Turner, L. R. Sivasundaram, M.-A. Ottenhof, I. A. Farhat, R. S. Linforth and A. J. Taylor, J. Agric. Food Chem., 2002, 50, 5406–5411 CrossRef CAS PubMed.
- C. M. Oliver, A. Kher, D. McNaughton and M. A. Augustin, J. Dairy Res., 2009, 76, 105–110 CrossRef CAS PubMed.
- F.-L. Gu, J. M. Kim, S. Abbas, X.-M. Zhang, S.-Q. Xia and Z.-X. Chen, Food Chem., 2010, 120, 505–511 CrossRef CAS.
- J. C. Cardoso, R. L. C. Albuquerque Jr, F. F. Padilha, F. O. Bittencourt, O. de Freitas, P. S. Nunes, N. L. Pereira, M. J. V. Fonseca and A. A. S. Araújo, J. Therm. Anal. Calorim., 2010, 104, 249–254 CrossRef.
- J. Yongsawatdigul and B. O. Hemung, J. Food Sci., 2010, 75, C251–C257 CrossRef CAS PubMed.
- H. Saeki and K. Inoue, J. Agric. Food Chem., 1997, 45, 3419–3422 CrossRef CAS.
- R. Sato, T. Sawabe and H. Saeki, J. Food Sci., 2005, 70, C58–C62 CrossRef CAS.
- E. Sanmartín, J. C. Arboleya, M. Villamiel and F. J. Moreno, Compr. Rev. Food Sci. Food Saf., 2009, 8, 332–344 CrossRef.
- A. Kato, K. Murata and K. Kobayashi, J. Agric. Food Chem., 1988, 36, 421–425 CrossRef CAS.
- D. Christ, K. P. Takeuchi and R. L. Cunha, J. Food Sci., 2005, 70, E230–E238 CrossRef CAS.
- K.-I. Izutsu, N. Aoyagi and S. Kojima, Chem. Pharm. Bull., 2004, 52, 199–203 CrossRef CAS PubMed.
- H. Enomoto, C.-P. Li, K. Morizane, H. R. Ibrahim, Y. Sugimoto, S. Ohki, H. Ohtomo and T. Aoki, J. Agric. Food Chem., 2007, 55, 2392–2398 CrossRef CAS PubMed.
- A. Kato, T. Fujishige, N. Matsudomi and K. Kobayashi, J. Food Sci., 1985, 50, 56–58 CrossRef CAS.
- Y. Li, F. Zhong, W. Ji, W. Yokoyama, C. F. Shoemaker, S. Zhu and W. Xia, Food Hydrocolloids, 2013, 30, 53–60 CrossRef CAS.
- A. Kato, K. Minaki and K. Kobayashi, J. Agric. Food Chem., 1993, 41, 540–543 CrossRef CAS.
- B. Zhang, X. Guo, K. Zhu, W. Peng and H. Zhou, Carbohydr. Polym., 2015, 127, 168–175 CrossRef CAS PubMed.
- L. Were, N. Hettiarachchy and U. Kalapathy, J. Food Sci., 1997, 62, 821–824 CrossRef CAS.
- F. Chevalier, J.-M. Chobert, Y. Popineau, M. G. Nicolas and T. Haertlé, Int. Dairy J., 2001, 11, 145–152 CrossRef CAS.
- R. Li, N. Hettiarachchy, S. Rayaprolu, M. Davis, S. Eswaranandam, A. Jha and P. Chen, J. Food Sci. Technol., 2015, 52, 6067–6072 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |