DOI:
10.1039/C6RA25264H
(Paper)
RSC Adv., 2017,
7, 15397-15407
Bovine serum albumin as a nanocarrier for the efficient delivery of ginsenoside compound K: preparation, physicochemical characterizations and in vitro biological studies
Received
14th October 2016
, Accepted 24th January 2017
First published on 8th March 2017
Abstract
Ginsenosides are triterpenoids that are found in P. ginseng; they have numerous important structural, functional and pharmacological properties. In this work, a desolvation method was used to entrap ginsenoside CK within bovine serum albumin (BSA) to form BSA–CK nanoparticles (NPs), which enhance its aqueous solubility and stability. Following purification, the BSA–CK NPs were characterized by several physico-chemical techniques, including high pressure liquid chromatography (HPLC), electrophoresis, 1H NMR spectroscopy, Fourier transform infrared spectroscopy (FT-IR), field emission transmission electron microscopy (FE-TEM), zeta potential, particle size analysis by dynamic light scattering (DLS), and thermogravimetric analysis (TGA); the results confirm that the as-prepared BSA–CK NPs are spherical, highly monodispersed and stable in aqueous systems. In addition, the time-dependent and pH stabilities of the BSA–CK NPs were analyzed over a period of 8 days; the results suggest that the nanoparticles are stable in physiological buffer (pH 7.4), whereas they are readily degraded under acidic conditions (pH 5.0) which mimic intracellular pH conditions. Furthermore, comparative water solubility analysis of the BSA–CK NPs and standard CK showed that the BSA carrier enhances the water solubility of ginsenoside CK. In vitro cytotoxicity assays of the BSA–CK NPs and standard CK revealed that the BSA–CK NPs demonstrate greater in vitro therapeutic efficacy in the HaCaT skin cell line, the A549 lung cancer cell line, the HepG2 liver carcinoma cell line and the HT29 colon cancer cell line in comparison with standard CK. Moreover, RAW264.7 cells treated with BSA–CK NPs exhibited decreased lipopolysaccharide-induced NO production. Collectively, these results suggest that the BSA–CK NPs may be useful as a delivery vehicle in cancer cell lines and may also possess anti-inflammatory effects.
Introduction
Cancer is a major worldwide diesase which causes maximum number of death. However, various pharmacological anticancer drugs are available or are currently the subject of research and clinical trials; their major drawbacks include poor distribution throughout the body due to poor water solubility, poor tumor targeting, systemic toxicity which causes severe adverse effects, and short half-life in blood circulation. Another major limitation is that the commercial formulations of these drugs can induce hypersensitivity reactions; for example, Taxol® is formulated in a high concentration in Cremophor EL®, which limits its use in current cancer therapy due to the inducement of side effects.1 Additionally, poor water solubility leads to erratic absorption and poor oral bioavailability, which is a key limiting factor in the development of volume-limited formulations of drugs, such as ocular, intramuscular, pulmonary and nasal dosage forms. Thus, water solubility, which minimizes side effects, and site-specific delivery of cancer drugs, which helps to achieve high drug concentrations at target sites, are the major challenges of cancer therapy. To answer this challenge, biodegradable carrier systems have been found to be an ideal potential solution. In recent years, biomacromolecule proteins have emerged as potential drug delivery systems, overcoming various limitations of conventional therapy such as poor solubility, lack of bioavailability and poor therapeutic efficacy; thus, they are ideal candidates for drug delivery. These protein-based systems have the inherent properties of preferential uptake in tumor and inflamed tissues, lower toxicity, immunogenicity and biodegradability.2,3 Furthermore, the charged groups present in proteins assist in the physical entrapment of various drug molecules.
Among biomacromolecule proteins, albumin-based nanoparticles have recently been explored for their clinical applications due to their ready availability, biodegradability, preferential uptake in tumor and inflamed tissues, and low toxicity and immunogenicity. One major innovation is the development of paclitaxel–albumin nanoparticles (Abraxane), an example of nanometer albumin-bound technology (nab™), for the treatment of advanced non-small-cell lung cancer and metastatic breast cancer; Abraxane has been approved by the U.S. Food and Drug Administration (FDA).4 Studies have shown that nab-paclitaxel is significantly more effective, with double the response rate and increased time to disease progression, than paclitaxel formulated as Cremophor EL® (CrEL, Taxol®, CrEL–paclitaxel).4 Subsequently, albumin has been recognized as a prominent nanocarrier and thus has received much attention; around seven albumin-based drugs or imaging agents have been developed and are available on the market, and around ten such products are undergoing clinical trials for various applications.5 This has increased interest in the use of BSA as a nanocarrier for various other biomedical applications, including oncology, diabetes, hepatitis C and rheumatoid arthritis. Notably, albumin, with a molecular weight of 66.5 kDa, is the most abundant plasma protein, constituting more than half of human plasma proteins; it remains stable over a wide pH range from 4 to 9 and is thermally stable when heated at 60 °C for up to 10 hours without deleterious effects.6 These plasma proteins constitute crucial components in various biological processes, such as maintaining colloidal osmotic pressure, delivery of nutrients to cells, balancing plasma pH and solubilizing long chain fatty acids.2 Moreover, the unique ligand-delivery property of serum albumin imparts enhanced solubility for serum albumin-conjugated hydrophobic drugs in plasma and helps improve the pharmacokinetic properties of drug molecules in biological environments.7 Thus, we considered BSA as a carrier to load CK for delivery and enhanced efficacy.
Panax ginseng is traditionally used as a medicinal herb in oriental medicine in Korea, China and Japan.8 Ginsenosides are triterpene saponins and are the main active components in P. ginseng. Ginsenosides can be classified broadly as protopanaxadiols (PPD) or protopanaxatriols (PPT) and, more specifically, as major and minor ginsenosides based on the steroidal saponins and the number of sugar chains and linkages, all of which account for the diverse bioactivities of this class of compounds.9,10 Ginsenosides exhibit a vast array of pharmacological effects, such as anti-tumor, anti-aging and anti-inflammatory; they also show protective effects against many diseases, such as Alzheimer's disease, and anti-diabetic activity.11–13 In recent years, ginsenoside CK has gained special attention owing to its unique pharmacological properties. CK is the minor ginsenoside, which obtained from intestinal enzymes, after the oral administration of various major ginsenosides.14 The important pharmacological effects of CK include inhibition of proliferation and induction of apoptosis in human cancer cells.15 In addition, ginsenoside CK inhibits pro-inflammatory cytokines in lipopolysaccharide (LPS)-stimulated murine peritoneal macrophages.16
Although CK has been reported to have many pharmacological benefits, its lack of aqueous solubility and the need for target-specific delivery into tumor sites remain major drawbacks for its use in clinical trials.17 In this work, we attempted to load CK ginsenoside within BSA carrier, to form BSA–CK NPs which was expected to increase the solubility and stability of CK and enable its use as a potential anti-tumor and anti-inflammatory agent. In this study, BSA was chosen as a carrier to protect the CK active agent from proteolytic digestion and provide long circulation life in the bloodstream. The physicochemical characteristics of the BSA–CK NPs were determined using HPLC, 1H NMR, FT-IR, FE-TEM, particle size analysis, and zeta potential and TGA measurements. In addition, the time-dependent stability and pH stability of the nanoparticles were analyzed. Furthermore, the water solubilities of the BSA–CK NPs and standard CK were compared. Moreover, the in vitro cytotoxicity of the BSA–CK NPs was evaluated in HaCaT skin cell, A549 lung cancer, HepG2 liver cancer, HT-29 colon cancer and RAW264.7 (murine macrophage) cell lines in order to evaluate their anticancer and anti-inflammation potential.
Materials and methods
Materials
BSA (Mw = 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) was purchased from Sigma Aldrich Co. (St. Louis, MO, USA). Ginsenoside CK was purchased from the Ginseng Genetic Resource Bank (Kyung Hee University, Yongin, South Korea). The different cell lines, HaCaT skin cancer, A549 lung cancer, HepG2 liver carcinoma, HT29 colon cancer and RAW264.7, were purchased from the Korean Cell Line Bank (Seoul, South Korea). All other chemicals and solvents were of analytical grade and were used as received.
000 Da) was purchased from Sigma Aldrich Co. (St. Louis, MO, USA). Ginsenoside CK was purchased from the Ginseng Genetic Resource Bank (Kyung Hee University, Yongin, South Korea). The different cell lines, HaCaT skin cancer, A549 lung cancer, HepG2 liver carcinoma, HT29 colon cancer and RAW264.7, were purchased from the Korean Cell Line Bank (Seoul, South Korea). All other chemicals and solvents were of analytical grade and were used as received.
Preparation of BSA–CK NPs
In a quintessential preparation, the BSA–CK NPs were prepared via a desolvation method. Specifically, BSA was dissolved in water and sonicated for 5 to 10 minutes. Afterwards, the BSA water mixture was maintained under vigorous magnetic stirring at room temperature. Stirring was continued for 10 min; then, CK ginsenoside dissolved in ethanol was very slowly added dropwise to the BSA–water solution. The reaction mixture was maintained with further stirring for 24 hours at room temperature. The resulting mixture was transferred to a dialysis membrane (Mw cutoff: 3000); the mixture was then dialyzed against an excess amount of methanol/distilled water (75:25, v/v) for one day and against distilled water for 2 days. Finally, the dialyzed solution was lyophilized to obtain the BSA–CK NPs, which were used for further analytical characterizations and in vitro applications.
Characterization of BSA–CK NPs
The purity and amount of ginsenoside CK loaded in the BSA–CK-NPs was calculated using high pressure liquid chromatography (HPLC). For PAGE electrophoresis, samples in loading buffer (10 μL) were applied onto 4–12% native PAGE gels at the following concentrations: standard BSA (1 mg mL−1), BSA–CK NPs (1 mg mL−1 and 5 mg mL−1). Electrophoresis was performed for 2 hours at 120 V. The gels were stained for 30 min using solution I and then were de-stained for 1 hour with solution II and overnight with solution III. Loading buffer: TRIS (0.05 M, pH = 6.8), 2% (v/v) glycerol; running buffer: TRIS (0.125 M, pH = 8.3), 0.96 M glycine; solution I: 8% (w/v) Coomassie in methanol:acetic acid 5:1 (v/v); solution II: H2O:methanol:acetic acid 65:25:10 (v/v); solution III: H2O:methanol:acetic acid 88:5:7 (v/v).
The structure of the BSA–CK NPs was confirmed by nuclear magnetic resonance (1H NMR) and Fourier transform infrared (FT-IR) spectroscopy. The 1H NMR spectra were recorded at 300 MHz (JEOL, Tokyo, Japan); the samples were dissolved in D2O or CD3OD.17 The FT-IR spectra of the NPs were recorded with an FT-IR spectrophotometer (PerkinElmer Inc., Waltham, MA, USA) over the range of 4000 to 450 cm−1 using KBr pellets. The spectra recorded were plotted as transmittance (%) versus wavenumber (cm−1). The morphology and shape of the dried BSA–CK NPs were measured by field emission transmission electron microscopy (FE-TEM) using a JEM-2000F (JEOL) instrument operated at an acceleration voltage of 200 kV.18 To prepare the FE-TEM samples, a drop of a dilute dispersion of the as-prepared BSA–CK NPs in water was placed onto holey carbon film supported on a 200 mesh copper grid and allowed to air dry.19,20 FE-TEM was used to analyze the SEAD and FTT patterns of the BSA–CK NPs.18,21 The hydrodynamic particle size distribution, polydispersity index (PDI) and zeta potential were analyzed by dynamic light scattering (DLS) at 25 °C (Photal, Otsuka Electronics, Japan).22 The BSA–CK NPs sample was dispersed in water; as a reference, a dispersive medium of pure water with a refractive index of 1.3328, a viscosity of 0.8878, and a dielectric constant of 78.3 was used.23
For the time-dependent and pH stability studies, DLS was conducted to measure the sizes of the nanoparticles at regular time intervals. The time-dependent stability of the BSA–CK NPs was studied up to 8 days at regular time intervals by measuring the sizes of the nanoparticles using DLS. For the pH stability tests, similarly, the samples were dispersed in either phosphate-buffered saline (PBS, pH 7.4) or acetate buffer (pH 5.0), and the sizes of the nanoparticles were measured at regular time intervals.24 Solubility tests were conducted by dissolving the same concentrations of standard CK and CK entrapped in BSA–CK NPs in water at 0 and 24 hours; visual analysis, corresponding microscopic analysis (×400, Optinity, Korean Labtech, South Korea), and HPLC analysis were performed to measure the dissolved CK concentration. We confirmed that at each concentration, sufficient BSA–CK NPs were used such that the concentration of free CK was comparable to that of the CK entrapped in albumin nanoparticles, i.e. BSA–CK NPs, thus enabling thorough evaluation. Thermogravimetric analysis (TGA) was performed on a TGA machine (SDT Q600, TA Instruments, New Castle, DE, USA). For TGA analysis, the product was placed in an alumina pan and heated from 20 °C to 700 °C at a ramping time of 10 °C minute−1.
In vitro cell culture and cell cytotoxicity of BSA–CK NPs
All the cell lines were purchased from the American Type Culture Collection and the Korean Cell Line Bank (Seoul, South Korea). A549 (human lung cancer) and HT-29 (human colon cancer) cells were grown in RPMI 1640 culture medium (GenDEPOT Inc., Barker, TX), and HepG2 cells were grown in DMEM media (Welgene, Gyeongsangbuk-do, South Korea); both media were supplemented with 10% fetal bovine serum (FBS) and 1% penicillin G and streptomycin (Gibco-BRL, Gaithersburg, MD) at a temperature of 37 °C in a humidified incubator with a 5% CO2 atmosphere. Cell viability was determined by MTT (3,4,5-dimethylthiazol-2-yl-2-5-diphenyletrazolium bromide) assay. Cells were seeded in 96-well plates at a density of 1 × 105 cells mL−1. After 24 hours of incubation, the cells were treated with various concentrations of BSA–CK NPs and standard CK for 24 hours. After the incubation time was complete, 10 μL of MTT stock solution (5 mg mL−1) was added to each well and incubated for 4 hours. Then, the supernatants were removed and replaced with 100 μL of DMSO. The amount of formazan formed by the viable cells was measured using a multi-model plate reader (Bio-Tek Instruments, USA) at a test wavelength of 570 nm with a reference wavelength of 630 nm. In addition, cytotoxicity studies were conducted on HaCaT skin cell lines to analysis the viability of the cells.25
Hoechst nuclear staining
Hoechst 33258 staining was performed to capture the apoptotic induction of BSA–CK NPs on A549 cells. Cells were seeded into a 6-well plate at a density of 1 × 105 cells well−1 in 2 mL medium and incubated at 37 °C with 5% CO2 overnight. The cells were then treated with 15 μM of standard CK and BSA–CK NPs. After the treatment time was complete, the cells were washed twice with 1× PBS, fixed with 3.7% (v/v) formaldehyde for 5 min at room temperature, and washed twice with PBS. To dye the nucleus, the cells were stained with Hoechst 33258 solution (2 μg mL−1) for 20 min in dark conditions at room temperature. The nuclear morphologies of the Hoechst-positive cells were observed and photographed under a fluorescence microscope (×400, Optinity, Korean Labtech, South Korea) for further analysis.25 The scale bar (10 μm) was added using ImageJ software.
Cell migration assay
A 12-well plate was used to grow A549 cells. The cells were incubated with 5% CO2 in a 37 °C humidified atmosphere until they reached complete confluence. Next, the A549 cells were subjected to serum starvation by replacing the old medium with serum-free growth medium during a period of 24 hours. Prior to treatment, a scratch was made in each well using a 10 μL plastic pipette tip, and the wells were washed twice with 1× PBS to remove floating cells. Then, the cells were exposed to 15 μg mL−1 BSA–CK NPs in the presence or absence of EGF (epithelial growth factor) stimulation. Two random areas were photographed at the moment of treatment and after 24 hours under an optical microscope (Eclipse ME600L, Nikon Instruments, Melville, NJ). The images were analyzed with the T-scratch program.25
Anti-inflammatory activity of BSA–CK NPs
Murine macrophage cells (RAW 264.7) were seeded at a density of 5 × 103 cells well−1 in a 96-well microplate in RPMI-1640 medium containing 10% (v/v) fetal bovine serum (FBS) and 1% (V/V) P/S; the plate was incubated at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air for 24 hours. To determine the toxic effects of the BSA–CK NPs and standard ginsenoside CK on cell viability, the cells were treated with various concentrations of samples for 24 hours. After 1 day of treatment, the cell viability was measured by MTT assay. 10 μL MTT solution (5 mg mL−1) was added to each well; the plates were then incubated for an additional 3 to 4 hours, and the formed formazan was dissolved in DMSO; then, the absorbance of each well was recorded on a synergy 2 multi-mode microplate reader at 570 nm (BioteK, Vermont, USA). The optical density of formazan formed in the control (untreated) cells was taken as 100%. To measure the nitric level, RAW 264.7 cells were pretreated with BSA–CK NPs and standard ginsenoside CK for 1 hour and then stimulated with 1 μg μL−1 lipopolysaccharide (LPS) in the presence of the samples. Cells were incubated for 24 hours, after which time the Griess reagent was used to determine the nitrite levels in the medium. Briefly, 100 μL of supernatant was mixed with an equal volume of the Griess reagent. The resulting absorbance at 540 nm was measured using a microplate reader (Bio-Tek Instruments, Inc.). A standard sodium nitrite curve was included for each experiment.26
Statistical analysis
The statistical significance of the differences between the control group vs. the standard CK and BSA–CK NPs groups was determined using Student's t-test. P < 0.05 was considered to be significant, as indicated in the figures with asterisks (***).
Results and discussion
Preparation and physicochemical characterization of BSA–CK NPs
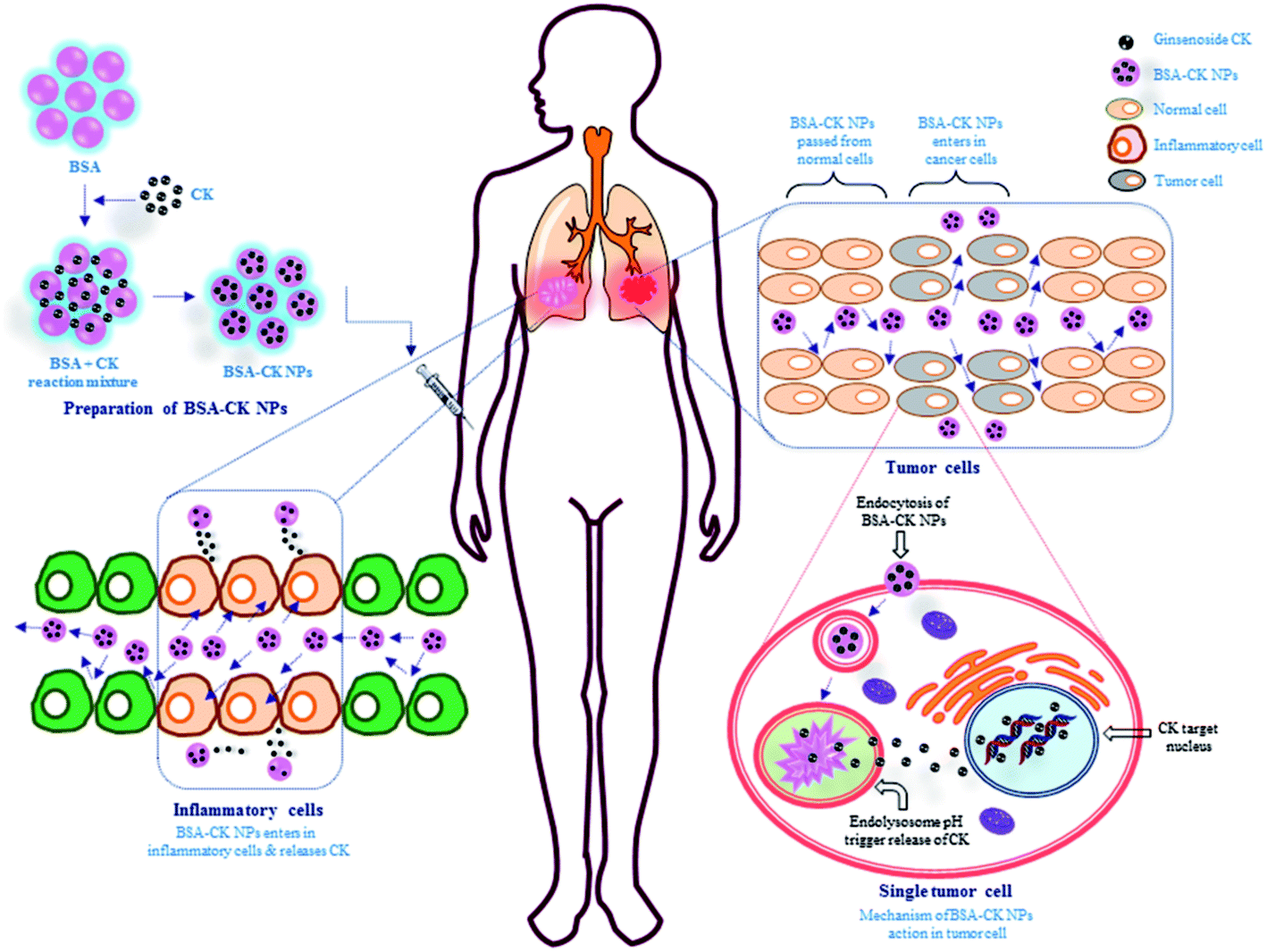
BSA, a common commercially available protein which has wide uses in fields such as nanocarrier development, drug delivery, sensing, self-assembly, and imaging, was selected as a carrier material due to its excellent biocompatibility; especially, the abundant functional groups on its surface can be conjugated easily with numerous molecules.27 Importantly, serum proteins serve as apt materials for nanoparticle formation; since they are the most naturally abundant protein found in blood and are chiefly responsible for the maintenance of blood pH, also possess non-toxic, biodegradable and non-immunogenic properties. BSA is a transport protein that is capable of binding reversibly with many exogenous and endogenous drugs. Various drugs with albumin nanoparticles have been synthesized by the desolvation method; BSA endows the drug with high dispersibility and biocompatibility, and it also has a multifunctional surface for conjugation with other targeting molecules. Similarly, the current study showed the preparation of BSA–CK NPs by a desolvation method, using ethanol as the desolvating agent.7,28 Fig. 1 exhibits the preparation of BSA–CK NPs for enhancing the solubility as well as the anticancer and anti-inflammatory effects of CK ginsenoside. This method endorses the creation of few aggregated particles and a homogeneous distribution; the particles remain stable in water and cell culture medium, as evidenced by in vitro analysis.
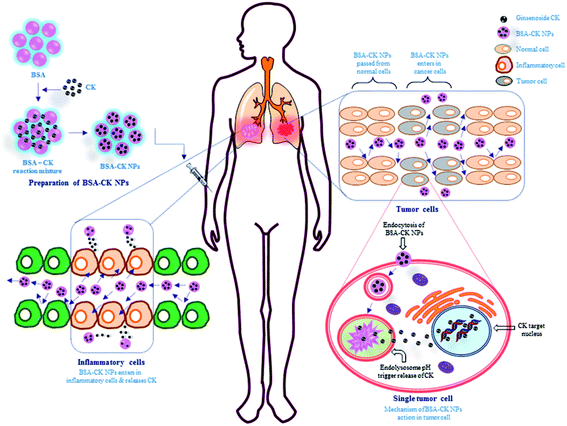 |
| | Fig. 1 Schematic illustration of the preparation of the BSA–CK NPs and their applications. The figure shows the stepwise preparation of the BSA–CK NPs by a desolvation method and the application of the synthesized NPs in inflammatory cells (RAW 264.7 cell) and cancer cells (A549 lung cancer cells), followed by the possible action mechanism. | |
HPLC and PAGE electrophoresis analysis of BSA–CK NPs
The amount of ginsenoside loaded in the BSA–CK NPs was calculated using HPLC. As shown in Fig. 2A, the BSA–CK NPs showed a clear peak of ginsenoside CK; the amount of loading was 0.4 mg of CK/mg of BSA–CK NPs. The samples were further analyzed by native PAGE electrophoresis; standard BSA and BSA–CK NPs were run in an electrophoresis gel, and the results clearly demonstrated the presence of BSA–CK NPs in comparison to BSA, while no free protein could be detected (Fig. 2B). The broadness of the bands reflected the high concentrations of the BSA–CK NP samples (1 mg mL−1 and 5 mg mL−1).
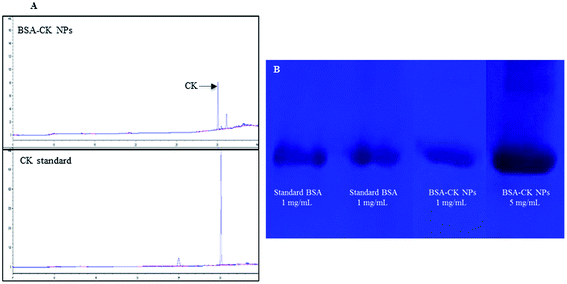 |
| | Fig. 2 HPLC analysis of the BSA–CK NPs compared with standard CK, revealing the CK peak and concentration in the synthesized BSA–CK NPs (A). Native PAGE analysis of standard BSA and purified BSA–CK NPs at low and high concentrations (B). | |
1H NMR and FT-IR analysis of BSA–CK NPs
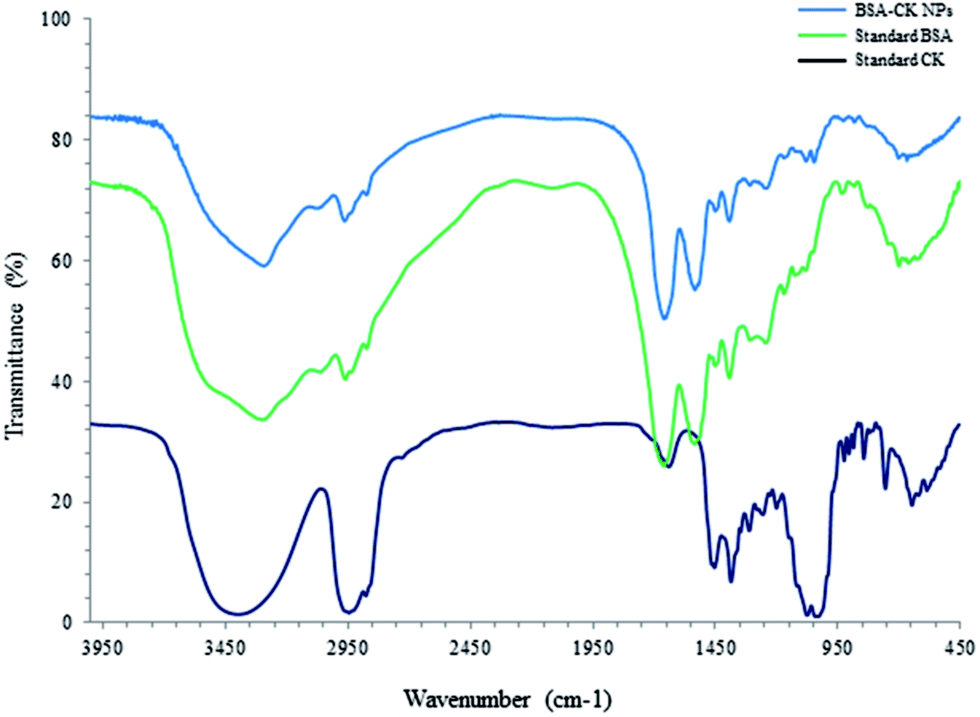
The 1H NMR spectra of the BSA–CK NPs, standard BSA and standard CK are shown in Fig. 3, which reveals the characteristic peak of CK protons in the spectrum of the BSA–CK NPs.17 Several interactions, including electrostatic interactions, hydrogen bonds, hydrophobic forces and van der Waals interactions, play key roles in the binding process of proteins with small molecules such as drugs. The FT-IR results of the BSA–CK NPs, standard BSA and standard CK are shown in Fig. 4, which exhibits the characteristic peak of CK in the spectrum of the BSA–CK NPs. The FT-IR results demonstrate a major shift in the peaks, which may be accredited to crosslinking between the positively charged amino groups of the proteins and the interaction of ginsenoside CK with residual amide species. Ginsenoside CK moves into the hydrophobic microenvironment of serum proteins, including human serum albumin (HSA), and interacts with the hydrophobic pockets of the protein. The interaction of ginsenoside with serum proteins mainly involves hydrophobic interactions, hydrogen bonding and van der Waals interactions. Like many other drug molecules, ginsenosides interact with a tryptophan residue (Trp 212) located inside the hydrophobic pocket of the BSA.29
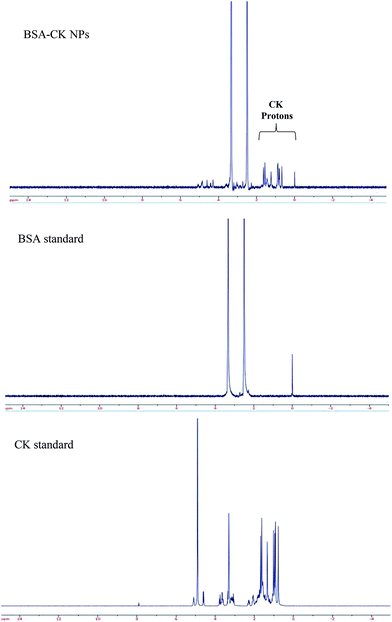 |
| | Fig. 3 1H NMR analysis of the BSA–CK NPs, standard BSA, and standard CK, revealing the presence of CK protons in the spectrum of the BSA–CK NPs. | |
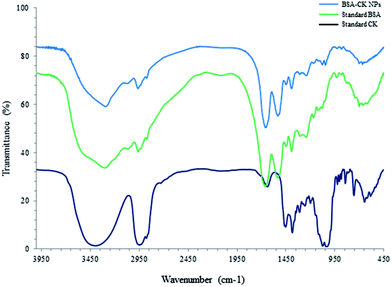 |
| | Fig. 4 FT-IR patterns of BSA–CK NPs, standard BSA and standard CK. | |
Size and morphology of BSA–CK NPs
The size of the nanocarrier system plays a crucial role in drug delivery, as nanoparticles up to 400 nm preferentially accrue in the tumor microenvironment via the enhanced permeability and retention (EPR) effect.30 Fig. 5 shows the shape and surface morphology of the BSA–CK NPs; a size of 30 to 50 nm was determined by FE-TEM.24 The images of the CK-loaded albumin nanoparticles demonstrate a spherical morphology. In addition, the SEAD and FFT patterns of the BSA–CK NPs were also demonstrated using FE-TEM.29
 |
| | Fig. 5 FE-TEM analysis, revealing the spherical shape of the synthesized BSA–CK NPs, with SEAD and FFT characterization of the BSA–CK NPs. | |
Particle size and zeta potential analysis of BSA–CK NPs
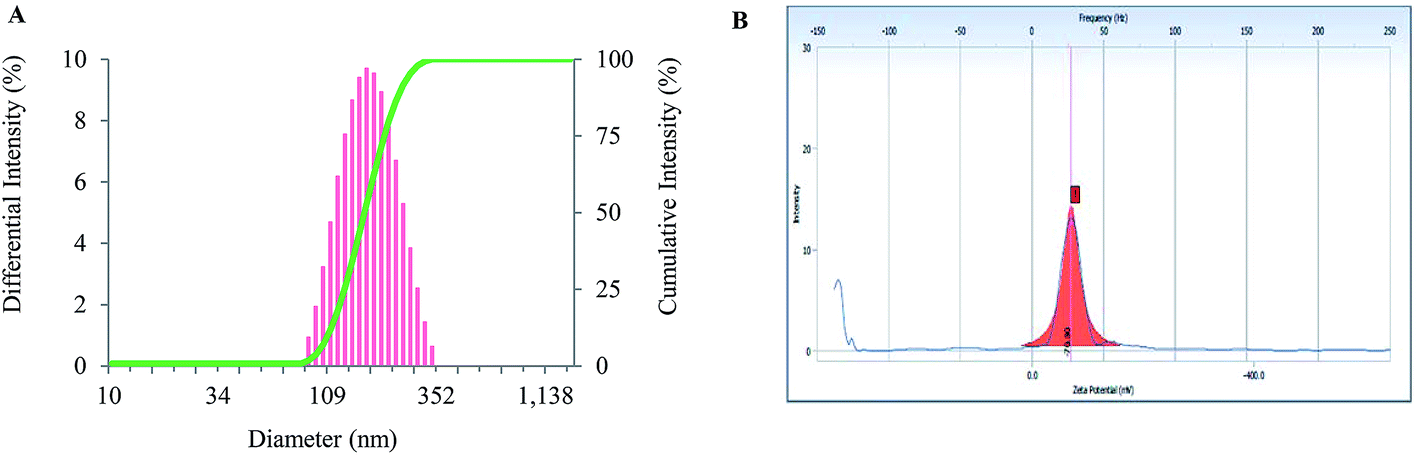
The size distribution of the prepared nanoparticles was acquired using DLS. The average hydrodynamic size of the BSA–CK NPs was found to be 157.2 nm (Fig. 6A). According to previous reports, nanoparticles smaller than 200 nm preferentially accumulate in tumor tissues due to the EPR effect and may exhibit long blood circulation effects by avoiding recognition by the reticuloendothelial system (RES). The hydrodynamic diameter is relatively higher than the diameter indicated from the FE-TEM results; this may be due to the presence of water surrounding the BSA–CK NPs during DLS in comparison with the sizes measured in the dried state by FE-TEM.31
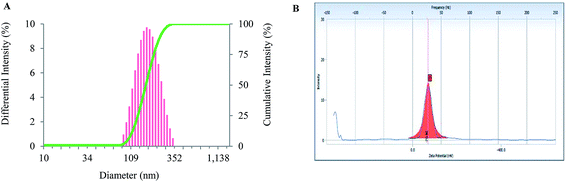 |
| | Fig. 6 Particle size and zeta potential analysis. Characterization of BSA–CK NPs by particle size analysis, showing the size-dependent distribution of the nanoparticles (A); zeta potential analysis showing the surface charge of −70.80 mV for the BSA–CK NPs, which corresponds with the stability of the nanoparticles (B). | |
The stability of the nanoparticles can be ascribed to their remarkably high zeta potential. The zeta potential of the BSA–CK NPs was found as −70.80 mV (Fig. 6B); which is quit lower then the previously reported, niclosamide-encapsulated BSA nanoparticle −34.2 mV, while that of pristine-encapsulated BSA nanoparticle −17.9 mV, thus showed higher stability.31 Thus, the BSA–CK NPs generated in the current study are highly stable. The value lies within the stable range, signifying that the nanoparticle formation led to the formation of a stable system. Moreover, the electrostatic repulsive force between the negatively charged surfaces of the nanoparticles affords high stability to the colloidal solution by preventing the nanoparticles from agglomerating in the colloid state.
Time-dependent and pH stability of BSA–CK NPs
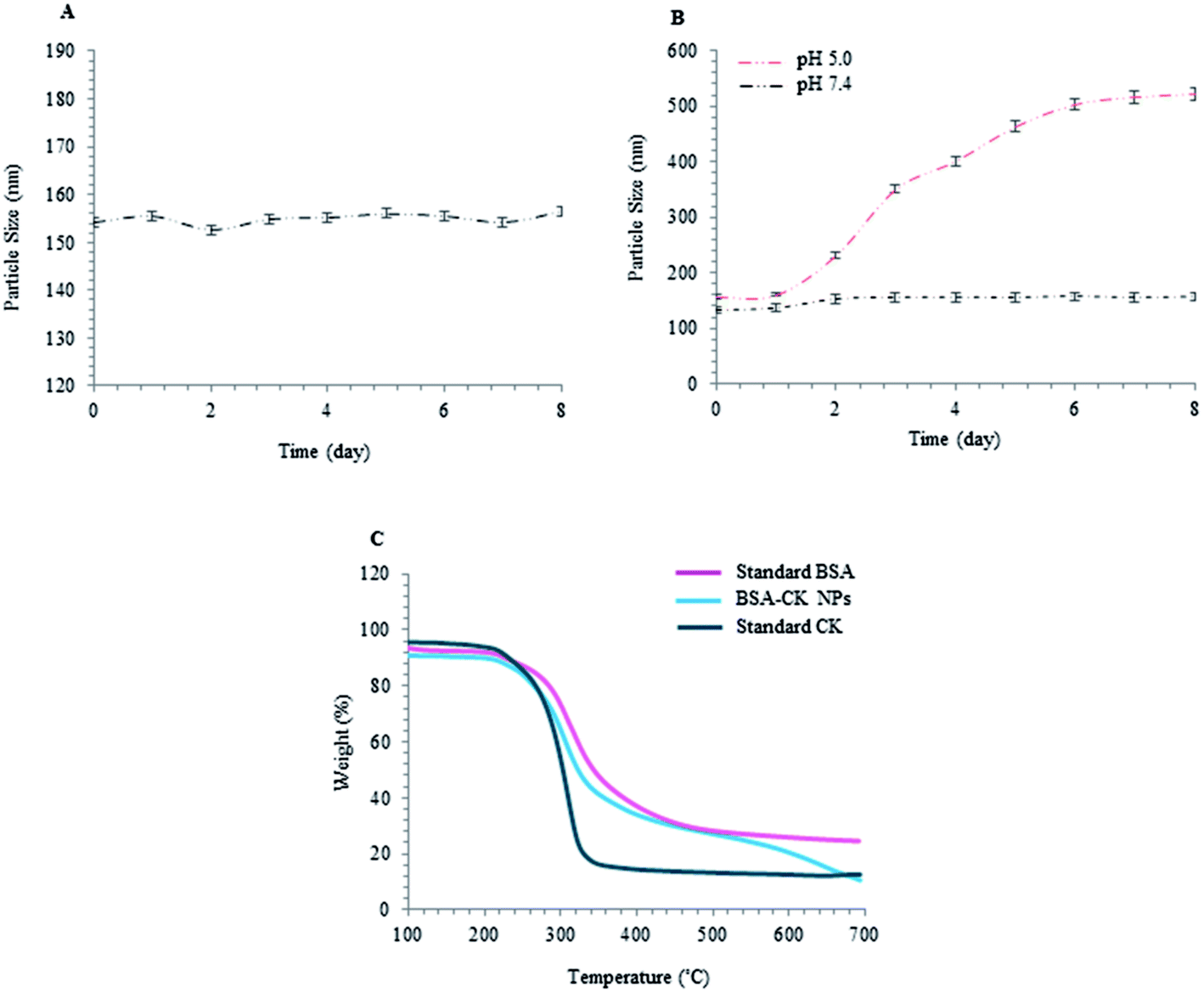
Various features, such as molecular weight, particle size, blood circulation time and stability, are very important factors that contribute to the accumulation of nanoparticles at tumor sites. Thus, the stabilities of the prepared BSA–CK NPs were investigated at different time intervals, up to 8 days, to analyze the increase in size of the BSA–CK NPs due to degradation of the nanoparticles under these conditions. As shown in Fig. 7A, over 8 days, there was no drastic change in the size of the BSA–CK NPs, which suggests the BSA–CK NPs are stable at room temperature in aqueous systems. Furthermore, for pH dependency analysis, the degradation of the nanoparticles was observed by measuring the particle size at 37 °C as a function of time in PBS (pH 7.4) and in acidic conditions (pH 5.0), which mimics tumor pH conditions (Fig. 7B). The stability of BSA–CK NPs was maintained for 8 days at pH 7.4, indicating its thermodynamic stability in aqueous media. On the other hand, the nanoparticle size increased in acidic conditions (pH 5.0), primarily due to the hydrolysis of CK from the BSA–CK NPs, resulting in the formation of large aggregates of the hydrolyzed CK through hydrophobic interactions. The most significant finding from our stability analyses was the high stability of the nanoparticles in physiological buffer, which indicates that the nanoparticles may exhibit prolonged circulation in vivo.24 From these observations, it is expected that the prepared BSA–CK NPs will be stable during blood circulation, whereas the BSA–CK NPs trigger CK release after reaching the tumor site in tumoral acidic pH conditions; this can be effective for cancer therapy (Fig. 1). Specifically, prolonged circulation of higher molecular weight nanoparticles in the blood consequently enhances tumor targeting by the EPR effect and by minimizing macrophage uptake.
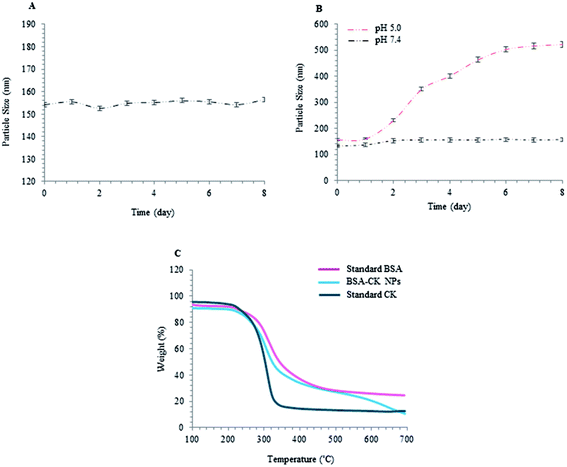 |
| | Fig. 7 Stability analysis. Time-dependent stability of the BSA–CK NPs using particle size analysis over a period of 8 days, with respect to different time intervals; (A) pH stability at different pH conditions, first in physiological buffer (pH 7.4) and then in acidic conditions (pH 5.0) which mimic the intracellular pH conditions of tumor cells (B); TGA analysis demonstrating the temperature stability of BSA–CK NPs (C). | |
Thermal stability of BSA–CK NPs
To analyze the ginsenoside loading in BSA, TGA was applied. Thermogravimetric (TG) analysis of standard ginsenoside CK and standard BSA as controls and of the BSA–CK NPs were conducted; the results depicted a slower degradation rate of the protein drug nanoformulation, indicating its improved stability compared to standard CK. The TGA profile shows the temperature stability and the degradation of BSA–CK NPs with respect to increasing temperature (Fig. 7C).29 From the results, it is clear that the particles start degrading at 250 °C; beyond 300 °C, an abrupt decrease in weight loss was found, which may be due to the loss of small molecules such as carbon dioxide and ammonia. At 350 °C to 550 °C, there is a considerable difference in weight loss; 9.39%, 4.58% and 7.50% weight losses were observed for BSA–CK NPs and the standard CK and BSA controls, respectively. Beyond 600 °C, a faster rate of degradation for the BSA–CK NPs was observed compared to BSA; this may be due to the crystalline nature of the entrapped CK molecules in the BSA–CK NPs.29 Meanwhile, no significant change was observed in BSA due to char formation in nitrogen atmosphere; an additional step was observed, related to the combustion of the char product in air atmosphere. The results clearly showed that the loading of CK in BSA–CK NPs enhances its stability compared to that of standard CK. Additionally, the temperature stability profile shows that the products are successfully degraded at higher temperatures.
Solubility of BSA–CK NPs
Aqueous solubility is one of the most important physicochemical factors for drug absorption. Furthermore, good aqueous solubility has been reported to play a major role in enhancing the pharmacokinetic properties of drugs, including their bioavailability, membrane flux and therapeutic efficacy. Importantly, serum albumin plays the major role of an “in vivo solubilizing agent” by enabling the solubility of a wide range of biomolecules and drugs in hydrophilic media, i.e. plasma. The solubility-enhancing properties of albumin correspond to its ability to form reversible binding complexes with ligands, which further allows the bound molecules (drug or ginsenosides) to flow in the blood at higher concentrations than their initial solubility would allow. Thus, to enhance the solubility of CK, ginsenoside CK was entrapped in BSA, leading to the formation of BSA–CK NPs. BSA considerably enhances the solubility, bioavailability and anti-tumor activity of various anticancer drugs. Owing to the hydrophilic nature of BSA, the prepared BSA–CK NPs were readily soluble in PBS (pH 7.4) or water, whereas free CK was insoluble at the same or even at much lower concentrations (Fig. 8). The synthesized BSA–CK NPs exhibited excellent stability in deionized water without obvious precipitation, even for up to a month, suggesting that the water dispersion ability of CK was significantly improved by the BSA coating. The enhanced solubility of CK with biocompatible materials has been previously reported.17 For example, Ji et al. reported that RGD-conjugated albumin nanoparticles remain soluble and disperse in aqueous systems without precipitation for up to 4 weeks.32 Thus, the entrapment of CK in the BSA nanocarriers contributed to the improved water dispersibility and biocompatibility of the BSA–CK NPs.
 |
| | Fig. 8 Aqueous solubility tests. Solubility of free ginsenoside CK and BSA–CK NPs in water, their corresponding microscopic images, and HPLC graphs of the supernatants. | |
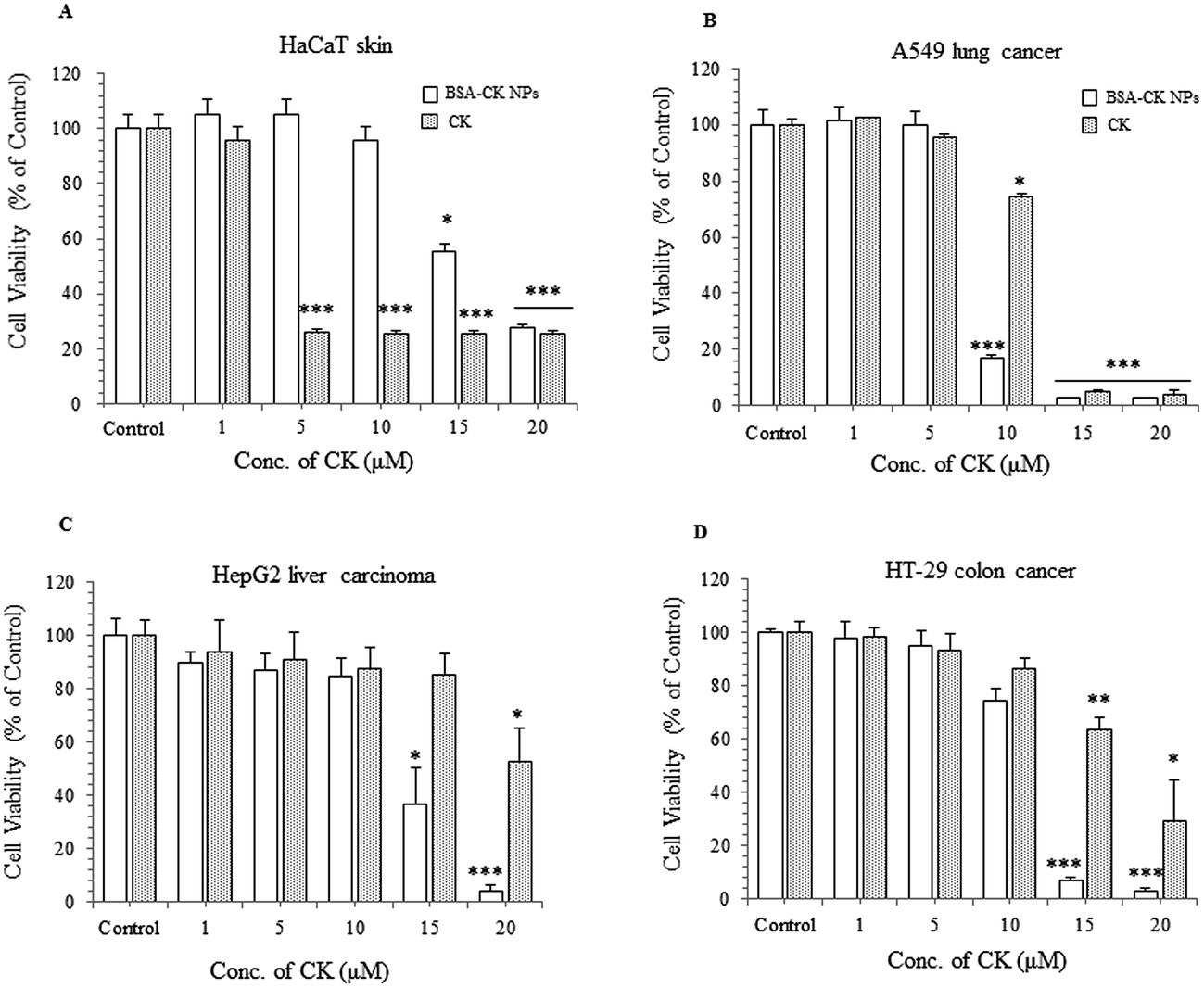
In vitro cytotoxicity of the BSA–CK NPs
To observe the in vitro cell viability and anticancer activity of the BSA–CK NPs, we examined the cytotoxicity of BSA–CK NPs and standard CK in the HaCaT skin cell, A549 lung cancer cell, HepG2 liver carcinoma, and HT-29 colon cancer cell lines by MTT assay. We confirmed that at each concentration, sufficient BSA–CK NPs were used such that the concentration of CK in the free drug was comparable to that in the entrapped drug, thus enabling thorough evaluation. As shown in Fig. 9A, as expected, the BSA–CK NPs showed very low cytotoxicity against HaCaT skin cells at 5 and 10 μg mL−1; however, at the same concentrations, standard CK showed cytotoxicity in HaCaT skin cells, which is considered in this study to be a normal cell line. This suggests that the BSA–CK NPs are not as toxic as standard CK in the HaCaT cell line; the cytotoxicity of CK is reduced due to the biocompatible nature of BSA. For cell cytotoxicity in cancer cell lines, the BSA–CK NPs exhibited significantly higher cytotoxicity in A549 lung cancer cells compared with the standard CK at 10 μg mL−1 (Fig. 9B); this suggests that the BSA–CK NPs at a concentration of 10 μg mL−1 are toxic to cancer cell lines but nontoxic in normal cell lines (HaCaT skin cell line). In addition, in HepG2 liver carcinoma cells, the BSA–CK NPs showed cytotoxicity at a concentration of 15 μg mL−1, whereas at the same concentration, standard CK is not greatly effective (Fig. 9C). The results are much more significant at a concentration of 20 μg mL−1. Similarly, in HT-29 colon cancer cells, the BSA–CK NPs showed cytotoxicity at a concentration of 15 μg mL−1 (Fig. 9D), which is significantly lower than that of CK; however, the cell viability in HaCaT cells was maintained at the same concentration. Thus, the results showed that the BSA–CK NPs showed higher toxicity than standard CK in these cancer cell lines.
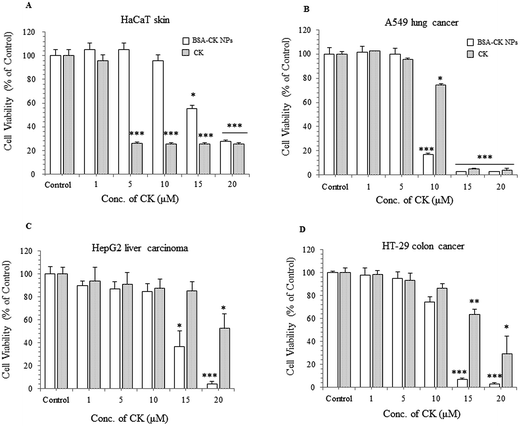 |
| | Fig. 9 In vitro effects of the BSA–CK NPs: cell viability in HaCaT skin cells (A); cell cytotoxicity in A549 lung cancer cells (B), HepG2 liver carcinoma cells (C), and HT-29 colon cancer cells (D). | |
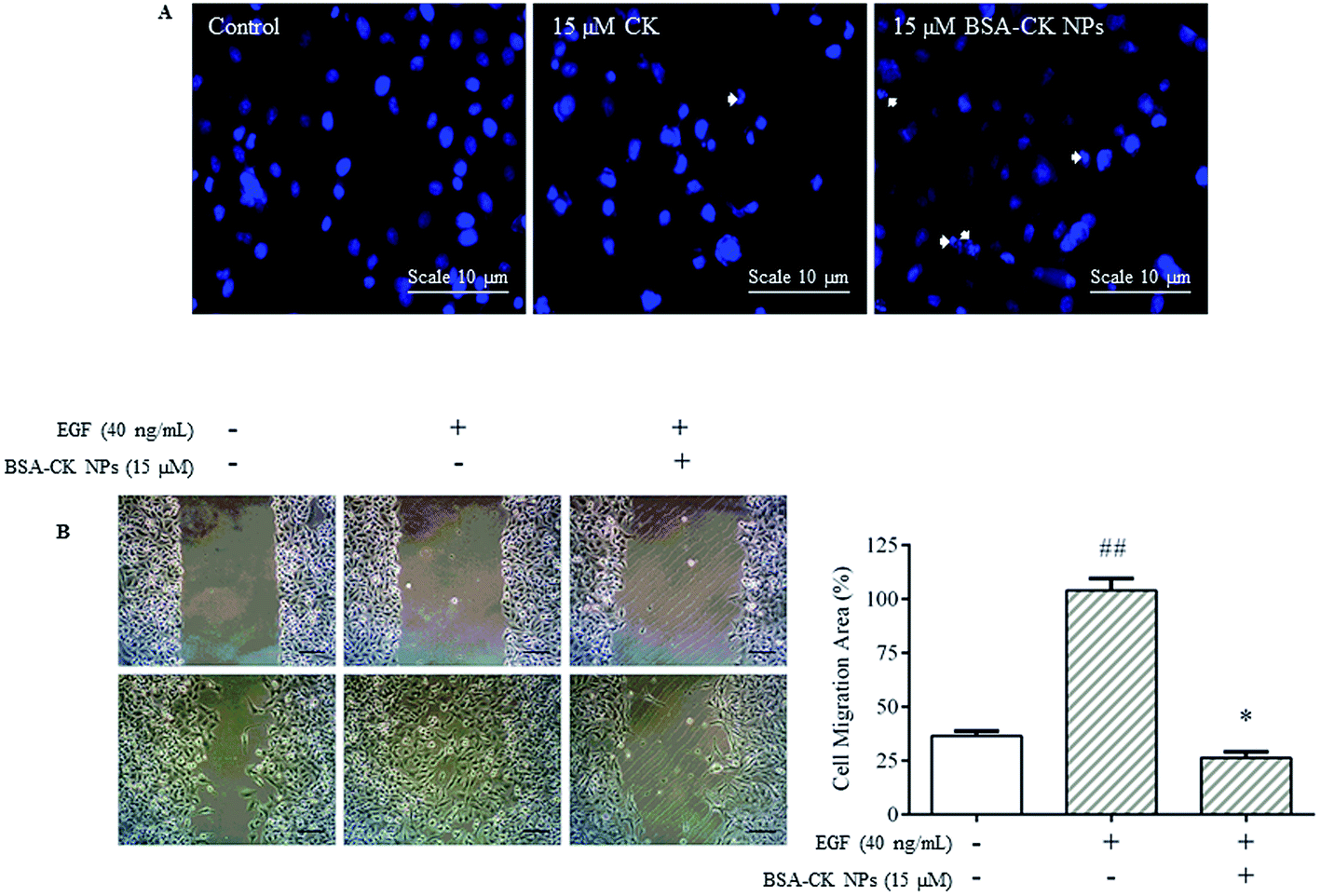
Hoechst nuclear staining and cell migration assay
All the MTT results for BSA–CK NPs and standard CK in different cell lines proclaimed that the BSA–CK NPs possessed higher cytotoxicity then standard CK in A549 lung cancer, HepG2 liver cancer, and HT-29 colon cancer cell lines. Among all the cancer cell lines, the BSA–CK NPs showed significant results in A549 lung cancer cell lines at a concentration of 10 μg mL−1. Thus, the A549 lung cancer cell line was considered for further study using more in vitro analysis methods. The results were further evidenced by staining the nuclei of A549 cells with Hoechst 33258 dye and quantifying the apoptotic effects of the BSA–CK NPs by HT Titer TACS™. Our data showed an induction of apoptosis through changes in the nucleus and also through the percentage of apoptotic cells induced by BSA–CK NPs, which was higher with 15 μM BSA–CK NPs than with standard CK (Fig. 10A). In addition, the BSA–CK NPs decreased cell migration; to confirm this, a wound healing assay was performed to observe the effects of the BSA–CK NPs on cell migration. The level of wound healing was measured by the average decrease in distance between the edges of the wounds at different time points in the presence or absence of EGF (40 ng mL−1) stimulation. The wound healing assay revealed that A549 cells that were not treated with BSA–CK NPs effectively healed the wounds, with a decrease to ∼90% of the original distance after 24 hours. However, treatment with 15 μg mL−1 BSA–CK NPs significantly repressed wound healing in A549 cells (Fig. 10B). Previously, the anticancer efficacy of niclosamide-encapsulated BSA nanoparticles was explored on A549 lung cancer and MCF-7 breast cancer cell lines; it was found that BSA enhanced the anticancer efficacy of niclosamide at a concentration of 5 μM compared with niclosamide alone by enhancing its stability and water solubility. The study suggests that the nanoformulation increases the solubility of niclosamide in aqueous medium so that it can be easily taken up by cells, resulting in enhanced anticancer activity in contrast to the free drug.31 Moreover, CK dissolved in DMSO also showed anticancer and anti-inflammatory effects; however, use of the organic solvent DMSO as a solubilizing agent leads to severe complications, as DMSO itself produces cytotoxic effects and is therefore not suitable for future clinical applications. Comparatively, the BSA–CK NPs avoid the use of organic solvents by increasing the water solubility and stability of ginsenoside CK.
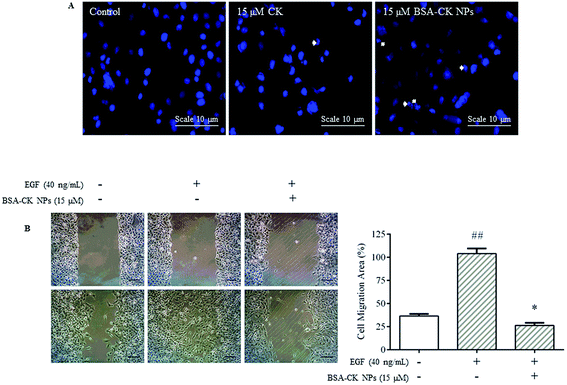 |
| | Fig. 10 BSA–CK NPs induce apoptosis and EGF-enhanced migration in A549 lung cancer cells. Hoechst 33258 staining in A549 cells. Apoptotic cells are indicated with white arrows; scale bar, 10 μm (A). The images correspond to the control, standard CK and BSA–CK NPs samples. The BSA–CK NPs reduce EGF-enhanced migration of A549 lung cancer cells. Effects of the BSA–CK NPs on wound healing (A). Analysis of migration ratio (%) within 24 hours in the wound healing assay (B). Data are shown as mean ± SEM (n = 3). ##p < 0.01 vs. control; **p < 0.01 vs. EGF-control. | |
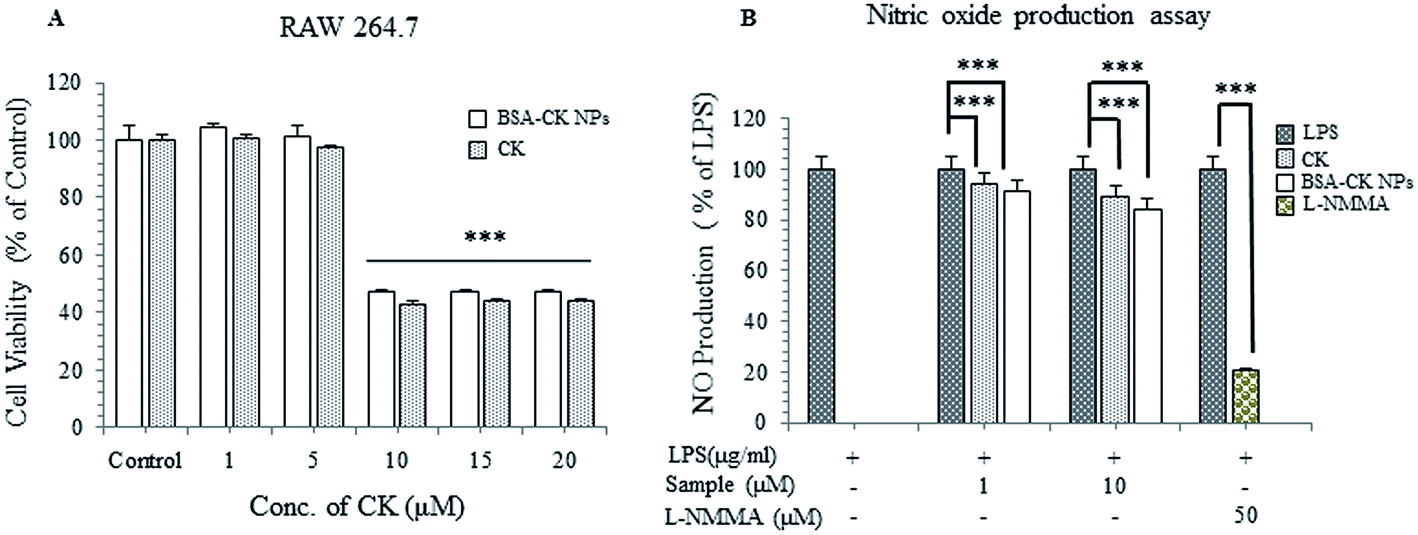
In vitro anti-inflammatory activity of BSA–CK NPs
To further investigate the anti-inflammatory effects of the BSA–CK NPs, an MTT assay was performed with the murine macrophage cell line (RAW 264.7); the cells were incubated with different concentrations of BSA–CK NPs and standard CK. The results showed that the BSA–CK NPs showed similar cell viability to standard CK on RAW 264.7 cells; however, the NPs showed higher inhibition of LPS-induced NO production compared with standard CK (Fig. 11A and B). The findings suggest that the BSA–CK NPs have enhanced anti-inflammatory efficacy compared with standard CK. Collectively, based on the anticancer and anti-inflammatory in vitro efficacy assays, the BSA–CK NPs are found to have more potent anticancer and anti-inflammatory efficacies then the standard CK drug.
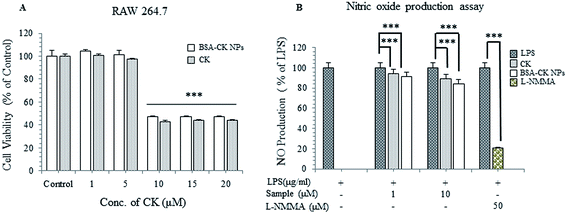 |
| | Fig. 11 In vitro anti-inflammatory analysis of the BSA–CK NPs on RAW 264.7 (murine macrophage) cell lines (A) and inhibition of LPS-induced NO production (B). | |
Consequently, the BSA–CK NPs formed by a desolvation method, in which CK was loaded in the backbone of hydrophilic BSA, further enhanced the solubility and stability of ginsenoside CK. In addition to this, loading BSA with CK helped to achieve high stability and excellent biocompatibility, which eliminates the use of toxic organic compounds such as cremophor and DMSO to solubilize CK for in vitro applications. The results support the excellent water dispersibility and biocompatibility of the BSA–CK NPs; furthermore, these NPs emerge as a potential candidate for anticancer and anti-inflammatory effects by enhancing the pharmacological effects of CK. Therefore, the BSA–CK NPs described in this study may be a potential candidate to deliver ginsenoside CK (drug) for the treatment of human cancer and inflammatory diseases. Based on the proposed principles, BSA, due to its biocompatible nature, can be considered for the loading of other pharmacologically important and water-insoluble drugs for medicinal applications, such as carrier systems or delivery vehicles; importantly, BSA could be replaced by HSA in future to create more promising and enhanced delivery systems.
Conclusion
We developed a novel anticancer ginsenoside CK-loaded delivery system based on a biodegradable nontoxic carrier, i.e. BSA. The particle size, morphology and stability analysis indicated that the BSA–CK NPs were quite stable, spherical in shape and nearly monodisperse in nature. This supported that the drug incorporation did not alter the size of the synthesized nanoparticles, whose sizes were within the optimal nanoparticle size range for drug delivery applications. The improved pharmacological efficacies of CK were demonstrated using in vitro cytotoxicity assays in various cell lines. The MTT results further support the nontoxic nature of the BSA nanocarriers; it was found that BSA sufficiently enhanced the anticancer ability of CK in cancer cells compared to the free CK in aqueous medium. Additionally, the BSA–CK NPs were found to possess good anti-inflammatory behavior compared to free CK. Overall, these results suggest that the BSA–CK NPs may be useful as vehicles for intracellular release of CK. However, it should be noted that this study utilizes BSA as a proof-of-concept investigation. In future, HSA would be considered instead of BSA to avoid probable immunologic consequences for in vivo trials. Thus, our studies propose that the chemotherapeutic potential of ginsenoside CK could be better utilized by loading it in a protein-based carrier for future clinical applications.
Acknowledgements
This research was supported by a grant from the Next-Generation BioGreen 21 Program (SSAC, grant#: PJ0120312016), Rural Development Administration, Republic of Korea.
References
- V. Hanusova, L. Skalova, V. Kralova and P. Matouskova, Curr. Cancer Drug Targets, 2015, 15, 35–52 CrossRef CAS PubMed.
- F. Kratz, J. Controlled Release, 2008, 132, 171–183 CrossRef CAS PubMed.
- W. Lohcharoenkal, L. Wang, Y. C. Chen and Y. Rojanasakul, BioMed Res. Int., 2014, 2014, 180549 Search PubMed.
- E. Miele, G. P. Spinelli, E. Miele, F. Tomao and S. Tomao, Int. J. Nanomed., 2009, 4, 99–105 CrossRef CAS.
- D. Zhao, X. Zhao, Y. Zu, J. Li, Y. Zhang, R. Jiang and Z. Zhang, Int. J. Nanomed., 2010, 5, 669–677 CAS.
- E. Neumann, E. Frei, D. Funk, M. D. Becker, H. H. Schrenk, U. Muller-Ladner and C. Fiehn, Expert Opin. Drug Delivery, 2010, 7, 915–925 CrossRef CAS PubMed.
- P. Saha and J. H. Kou, Eur. J. Pharm. Biopharm., 2002, 54, 319–324 CrossRef CAS PubMed.
- J. M. Lu, Q. Yao and C. Chen, Curr. Vasc. Pharmacol., 2009, 7, 293–302 CrossRef CAS PubMed.
- S. Kang and H. Min, J. Ginseng Res., 2012, 36, 354–368 CrossRef CAS PubMed.
- H. J. Kim, P. Kim and C. Y. Shin, J. Ginseng Res., 2013, 37, 8–29 CrossRef CAS PubMed.
- C. H. Lee and J. H. Kim, J. Ginseng Res., 2014, 38, 161–166 CrossRef CAS PubMed.
- S. Jiang, D. Ren, J. Li, G. Yuan, H. Li, G. Xu, X. Han, P. Du and L. An, Fitoterapia, 2014, 95, 58–64 CrossRef CAS PubMed.
- Y. C. Huang, C. Y. Lin, S. F. Huang, H. C. Lin, W. L. Chang and T. C. Chang, J. Agric. Food Chem., 2010, 58, 6039–6047 CrossRef CAS PubMed.
- J. Chen, H. Wu, Q. Wang, Y. Chang, K. Liu, S. Song, P. Yuan, J. Fu, W. Sun, Q. Huang, L. Liu, Y. Wu, Y. Zhang, A. Zhou and W. Wei, Inflammation, 2014, 37, 1608–1615 CrossRef CAS PubMed.
- Y. Li, T. Zhou, C. Ma, W. Song, J. Zhang and Z. Yu, J. Thorac. Dis., 2015, 7, 400–406 Search PubMed.
- H. Wu, J. Chen, Q. Wang, X. Jia, S. Song, P. Yuan, K. Liu, L. Liu, Y. Zhang, A. Zhou and W. Wei, Immunopharmacol. Immunotoxicol., 2014, 36, 124–129 CrossRef CAS PubMed.
- R. Mathiyalagan, S. Subramaniyam, Y. J. Kim, Y. C. Kim and D. C. Yang, Carbohydr. Polym., 2014, 112, 359–366 CrossRef CAS PubMed.
- P. Singh, Y. J. Kim and D. C. Yang, Artif. Cells, Nanomed., Biotechnol., 2015, 1–9, DOI:10.3109/21691401.2015.1115410.
- P. Singh, Y. J. Kim, H. Singh, C. Wang, K. H. Hwang, A. Farh Mel and D. C. Yang, Int. J. Nanomed., 2015, 10, 2567–2577 CAS.
- P. Singh, Y. J. Kim, H. Singh, R. Mathiyalagan, C. Wang and D. C. Yang, J. Nanomater., 2015, 2015, 10 Search PubMed.
- P. Singh, Y. J. Kim, C. Wang, R. Mathiyalagan and D. C. Yang, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 1150–1157 CAS.
- P. Singh, Y. J. Kim, C. Wang, R. Mathiyalagan, M. El-Agamy Farh and D. C. Yang, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 811–816 CAS.
- P. Singh, H. Singh, Y. J. Kim, R. Mathiyalagan, C. Wang and D. C. Yang, Enzyme Microb. Technol., 2016, 86, 75–83 CrossRef CAS PubMed.
- C. Chen, H. Hu, M. Qiao, X. Zhao, Y. Wang, K. Chen, X. Guo and D. Chen, Int. J. Pharm., 2015, 480, 116–127 CrossRef CAS PubMed.
- V. Castro-Aceituno, S. Ahn, S. Y. Simu, P. Singh, R. Mathiyalagan, H. A. Lee and D. C. Yang, Biomed. Pharmacother., 2016, 84, 158–165 CrossRef CAS PubMed.
- S. Ahn, P. Singh, V. Castro-Aceituno, S. Yesmin Simu, Y. J. Kim, R. Mathiyalagan and D. C. Yang, Artif. Cells, Nanomed., Biotechnol., 2016, 1–7, DOI:10.1080/21691401.2016.1228661.
- J. H. Choi, H. J. Hwang, S. W. Shin, J. W. Choi, S. H. Um and B. K. Oh, Nanoscale, 2015, 7, 9229–9237 RSC.
- A. Jithan, K. Madhavi, M. Madhavi and K. Prabhakar, Int. J. Pharm. Invest., 2011, 1, 119–125 CrossRef CAS PubMed.
- A. Gebregeorgis, C. Bhan, O. Wilson and D. Raghavan, J. Colloid Interface Sci., 2013, 389, 31–41 CrossRef CAS PubMed.
- K. H. Bae, H. J. Chung and T. G. Park, Mol. Cells, 2011, 31, 295–302 CrossRef CAS PubMed.
- A. B. Bhushan, P. Dubey, S. U. Kumar, A. Sachdev, I. Matai and P. Gopinath, RSC Adv., 2015, 5, 12078 RSC.
- S. Ji, J. Xu, B. Zhang, W. Yao, W. Xu, W. Wu, Y. Xu, H. Wang, Q. Ni, H. Hou and X. Yu, Cancer Biol. Ther., 2012, 13, 206–215 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Hina Singha,
Verónica Castro-Aceitunoa,
Sungeun Ahna,
Yeon Ju Kim*a and
Deok Chun Yang*ab
*ab,
Hina Singha,
Verónica Castro-Aceitunoa,
Sungeun Ahna,
Yeon Ju Kim*a and
Deok Chun Yang*ab