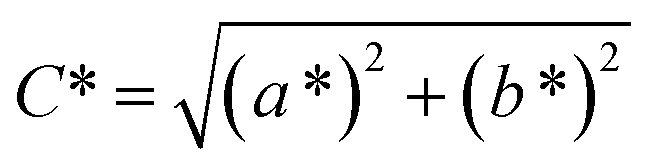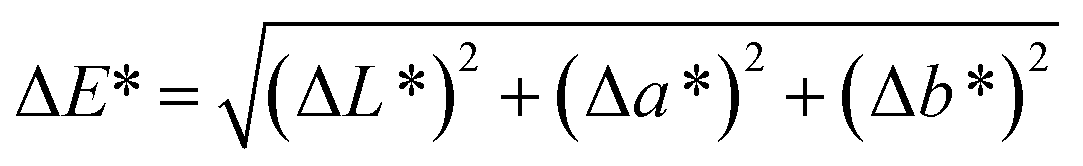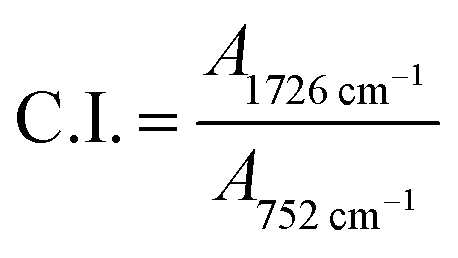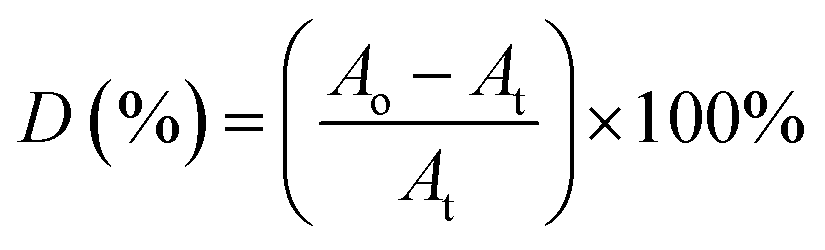DOI:
10.1039/C6RA25313J
(Paper)
RSC Adv., 2017,
7, 112-120
Degradation of ultra-high molecular weight poly(methyl methacrylate-co-butyl acrylate-co-acrylic acid) under ultra violet irradiation†
Received
16th October 2016
, Accepted 5th December 2016
First published on 22nd December 2016
Abstract
A new acrylic terpolymer, poly(methyl methacrylate-co-butyl acrylate-co-acrylic acid) [P(MMA-co-BA-co-AA)] of ultra-high molecular weight (UHMW) was synthesized via seeded emulsion polymerization. This polyacrylic showed good film properties; high transparency, water resistance and mechanical flexibility that may suitable for many environmental based applications such as coating, packaging, label sensors etc. In order to access the photo-stability of this material for environmental application, studies were conducted under UV illumination of a short-wavelength (λ = 254 nm) in air. The responses were collected at different irradiation times by using several characterization techniques: infrared/UV-visible spectroscopy (FTIR/UV-Vis), gel permeation chromatography (GPC), atomic force microscopy (AFM) and thermogravimetric analysis (TGA). Two distinguishable structures, cross-linked and fragmented chains, were formed under photo-irradiation at different times of exposure. The formation of cross-linked structures at short irradiation times (t < 60 min) increases the chain length as validated from the increase in average-molecular weight (Mw), whilst at longer irradiation time the fragmentation causes a decrease in the chain length (decrease in Mw). Only the chain scission at longer irradiation time (t > 60 min) causes the copolymer to degrade. The centre of reaction was identified at the pendent group and no effect of main chain destabilization was observed throughout the experimental condition. The occurrence of chain cleavage during photo-degradation causes chain–chain separation, as visually seen under the imaging technique and this coincides with the observed drop in thermal stability. Photo-oxidation was also proposed to occur simultaneously with photo-degradation as the irradiation was performed in air.
Introduction
Photo-degradation is generally a destructive reaction undergone by polymers that alters material properties in undesirable ways in the presence of sunlight and/or air. The alternation can be visualized from a loss in mechanical strength, formation of cracks/holes and discolorations in the material at prolonged duration.1–3 This process is commonly observed in large molecules, particularly in polymers during their service lifetime. The long molecular chain of polymer makes the material susceptible to degradation and the level of deteriorations shows linear dependence to the polymers molecular weight, as reported by Rebollar et al.4 The strength of reacting ultra-violet (UV) photons (e.g. UVA, UVB, UVC etc.) with the polymer chains is indeed a concern. The higher the energy of the reacting photons, the greater the deterioration imposes to polymer structure. Thus, it is crucial to perform a preliminary study on the effect of UV-irradiation (including short-wavelength rays) and air towards polymer properties before deciding on the potential application. This is to ensure that the material durability is sustained at the course of an application at prolong usage time.
Vast information is available in literature reporting the photo-behavior of polyacrylates under UV exposure. The most commonly reported polyacrylates are poly(methyl methacrylate) (PMMA) and its derivative copolymers. Polyacrylates show high demand in vast design material development for interior and exterior use. The degradation of PMMA has been studied in a wide range of UV-wavelengths (248 nm,5 193 nm and 157 nm (ref. 6 and 7)) at different exposure time. Generally, all the studied polymers show a pronounce degradation with increasing irradiation time successive to high numbers of chain cleavage.8 The effect is rather significant under the illumination of short UV wavelength. This hypothesis was proven true in many case studies involving homopolymer and similar trends were reported for copolymers such as poly(methyl methacrylate-co-methyl acrylate) P[MMA-co-MA]9 and poly(vinyl acetate-co-butyl acrylate) P[VA-co-BA].10
The degradation of polymer, forming shorter chains, was initiated by the chain scission process due to the photo-excitation of atoms that bearing an unpaired electron or double bond.11,12 This electronic transition (during photo-excitation) generates short-lived radicals that participate incessantly in photochemical reaction and cause major structural deterioration. In some cases, a crosslink structure can be observed during the initial time of irradiation. This structural alternation shows no negative effect on the polymer properties. But prime deterioration in material properties arises as consequent of the chain scission reaction. Generally, the weakest bond in the polymer chain, usually pendant group, is susceptible to chain cleavage. While for the main chain scission, it occurs only if the reacting UV-photons have sufficiently high energy (short-wavelength UV) to cause the cleavage. The degradation is more pronounced if cleavage happens at the main chain as compared to side chain scission as this may lead to photo-ablation.4 In order to suppress the polymer sensitivity towards light, UV-inhibitors from the classes of benzotriazoles (e.g. 2-(2′-hydroxyphenyl) benzotriazoles), metallic complexes (e.g. nickel dibutyldithiocarbamate), pigments (e.g. carbon black) are incorporated into the polymer matrix.13 This alternative is identified as an economically favored way to improve the UV shielding of polymer.
In this paper, the photo-behavior of P(MMA-co-BA-co-AA) with ultra-high molecular weight (UHMW) was studied under the illumination of UV ray with wavelength of 254 nm and 365 nm at different time of exposure. Only the result for 254 nm irradiation was presented as the UV of λ = 365 nm (produces low energy photon) is incapable of inducing chemical changes in the copolymer structure. This agrees well to the discussion reported by Shirai et al. (1999), stating the absence of photo-degradation in PMMA under the illumination of UV wavelength longer than 320 nm.14 Although much studies on the photo-degradation have been reported in the literature, those studies focused on polyacrylic with less emphasis on UHMW polyacrylic. Therefore, this paper could shed some light on the degradation behavior of polymer with huge molecular weight that ranges in millions. The chemical changes resulted by the UV irradiation were assessed from the FTIR study and supplementary data were obtained from UV-Vis-NIR spectroscopy. Other techniques such as AFM and TGA were also used to understand the changes in copolymer morphology and thermal stability, respectively. The reason for degradation study conducted at 254 nm – UV is owing to the wide utilization of this wavelength in many fields such as in label tracking, optical sensors, forensic analysis etc. Thus, the suitability of the copolymer to be used in various applications can be accessed from the present study.
Experimental
Polymer material
UHMW-P(MMA-co-BA-co-AA) film with average molecular weight (Mw) of 2 × 106 g mol−1 was used in this study. The copolymer was synthesized via emulsion polymerization according to the method reported in Ramesh et al. (2016).15 The emulsion was processed into thin film with complete drying at 60 °C prior to structural elucidation and irradiating with UV. The chemical structure of the final yield was confirmed using 13C-nuclear magnetic resonance spectroscopy (NMR, Bruker Avance III) at 500 MHz against tetramethylsilane as an internal reference standard. The dissolving solvent used was deuterated chloroform.
Irradiation procedure
The irradiation of copolymer film was performed in air under the dark condition. UV lamp (model no. UVL-56, intensity: 1.0 mW cm−2) emitting 254 nm ray was used in this study. The photo-response of the film was studied in variation of the UV exposure time at 30, 60, 120, 240, 360 and 480 minutes.
Characterization techniques
All irradiated samples were characterized using the techniques of spectroscopy, chromatography, imaging and thermal. The neat copolymer (or non-irradiated sample) was used as a comparison.
Spectroscopy studies. Attenuated total reflectance-Fourier transform infrared (ATR-FTIR, Model: Themoscientific Nicolet iS10 FTIR Spectrometer) and ultra violet-visible-near infrared (UV-Vis-NIR, Model: AvaSpec-ULS2048 StarLine) spectroscopy techniques were used to study the chemical changes in sample upon UV irradiating at different exposure time. The FTIR and UV-Vis-NIR spectra were recorded in the range of 4000–650 nm (in wavenumber, with 4 cm−1 resolution) and 1100–180 nm (in wavelength), respectively. Chromaticity study was also performed using UV-Vis-NIR spectrometer with an attached UV light source (Spectroline Model TVC-312A). The parameters of L* (lightness) and color coordinates (a* and b*) were measured. The color coordinates a* is comprised of the color directions from +a* (red direction) to −a* (green direction), whereas the b* coordinates the color direction is from +b* (yellow direction) to −b* (blue direction). Using the parameter of a* and b*, the C* (metric chroma-color intensity) and ΔE* (total color difference) values were respectively calculated using the eqn (1) and (2);| |
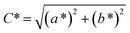 | (1) |
| |
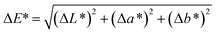 | (2) |
Chromatography study. Changes in the average molecular weight were identified using gel permeation chromatography (GPC, Waters 2414 GPC system) equipped with Waters 2414 refractive index detector, Waters 717 plus autosampler, Waters 600 controller and Waters Styrogel columns conditioned at 35 °C. Polystyrene standard was used for calibration. The tested P(MMA-co-BA-co-AA) samples were dissolved in tetrahydrofuran at a concentration of 2.0 mg mL−1, then filtered through a 0.22 μm PTFE filter. Similar solvent was used as the eluent. A total of 100 μL aliquot samples were injected in the system and the analysis was run for 55 min. The selected flow rate of the THF mobile phase is was 1.0 mL min−1.
Imaging study. The roughness and surface morphological changes due to UV exposure were studied using an atomic force microscope (AFM, Model: Ambios Q-Scope Series) at ambient condition. For AFM imaging, samples were periodically removed from the exposure cell and analyzed under the scanning probe microscope operated in contact mode. The frequency scan was 1.0 mHz. Scan atomic software was used for the surface viewing and to calculate the surface roughness. The mean root square (rms) roughness values reported here was the average of three measurements.
Thermal analysis. Thermogravimetric analyzer (TGA, Model: TA Instrument Q500) was used to study the thermal decomposition. The measurement was performed in nitrogen atmosphere over the recorded temperature range of 30–600 °C at 50 °C min−1 heating rate.
Results and discussion
Molecule structure of P(MMA-co-BA-co-AA)
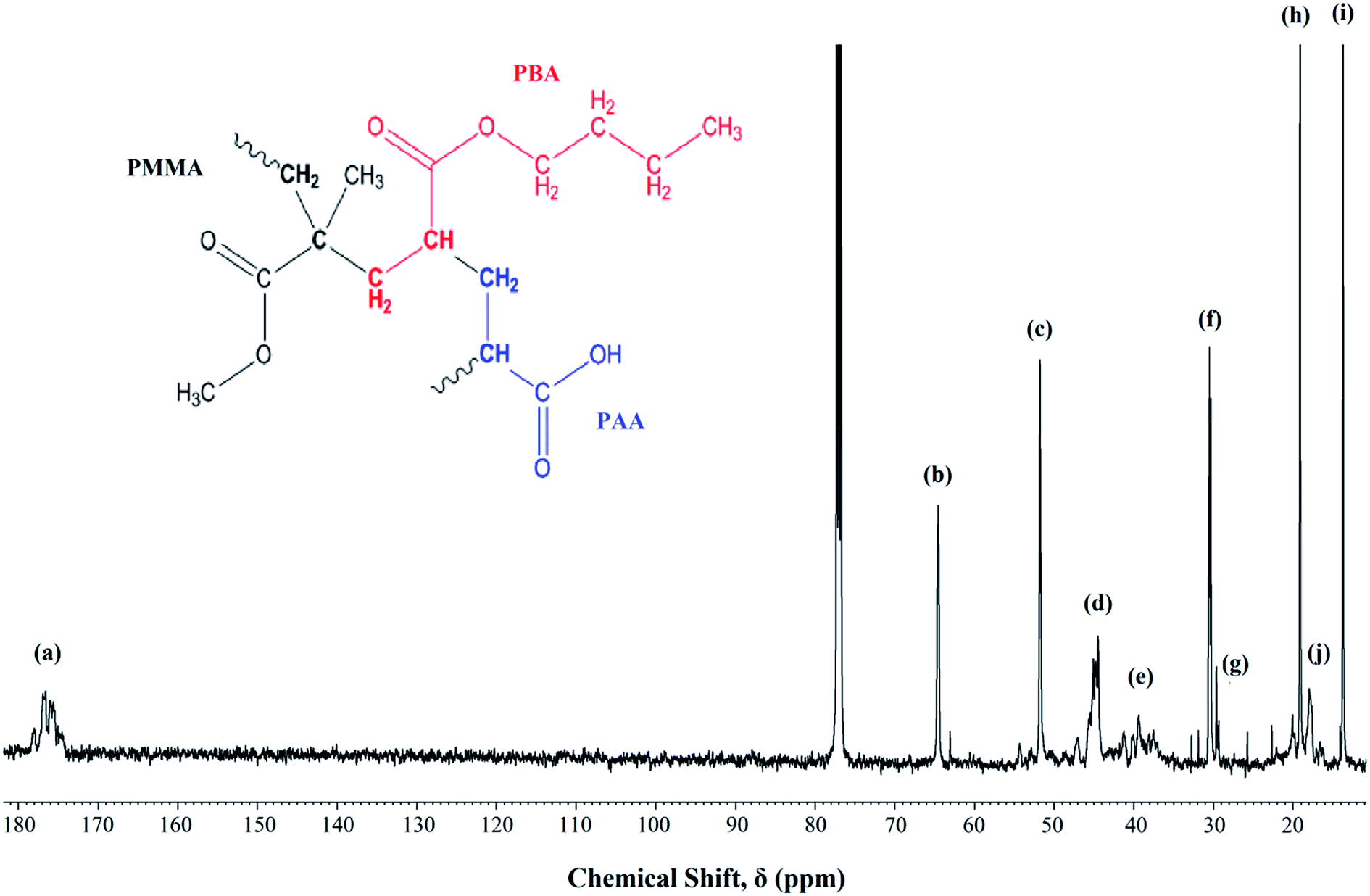
Fig. 1 shows the 13C-NMR spectrum of P(MMA-co-BA-co-AA). The signal assignments were labeled from the highest shielded carbon group to the lowest. The signals assignment are as follows; chemical sift, δ = 170–180 ppm (a) represents the carbonyl carbon (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the ester (in MMA and BA) and carboxylic (in AA) functional group, δ = 65 ppm (b) can be assigned to –OCH2 in PBA, δ = 52 ppm (c) corresponds to –OCH3 in PMMA, δ = 44 ppm (d) corresponds to CH2–C backbone carbon of PMMA, δ = 36–40 ppm (e) is signaled by –CH backbone carbon of PBA and PAA, δ = 31 ppm (f) corresponds to the carbon next to the carbon attached to the oxygen in –OCH2CH2CH2CH3 in PBA, δ = 29 ppm (g) represents the –CH2 backbone carbon in PMMA, PBA and PAA, δ = 19 ppm (h) represents the carbon next to the aforementioned carbon environment and the final peak at δ = 13 and 18 ppm [(i) and (j) respectively] represent the methyl carbon (–CH3) in PBA and PMMA respectively. The coexistence of all the carbon atoms signaling from all the added monomers confirms the successful polymerization.
O) of the ester (in MMA and BA) and carboxylic (in AA) functional group, δ = 65 ppm (b) can be assigned to –OCH2 in PBA, δ = 52 ppm (c) corresponds to –OCH3 in PMMA, δ = 44 ppm (d) corresponds to CH2–C backbone carbon of PMMA, δ = 36–40 ppm (e) is signaled by –CH backbone carbon of PBA and PAA, δ = 31 ppm (f) corresponds to the carbon next to the carbon attached to the oxygen in –OCH2CH2CH2CH3 in PBA, δ = 29 ppm (g) represents the –CH2 backbone carbon in PMMA, PBA and PAA, δ = 19 ppm (h) represents the carbon next to the aforementioned carbon environment and the final peak at δ = 13 and 18 ppm [(i) and (j) respectively] represent the methyl carbon (–CH3) in PBA and PMMA respectively. The coexistence of all the carbon atoms signaling from all the added monomers confirms the successful polymerization.
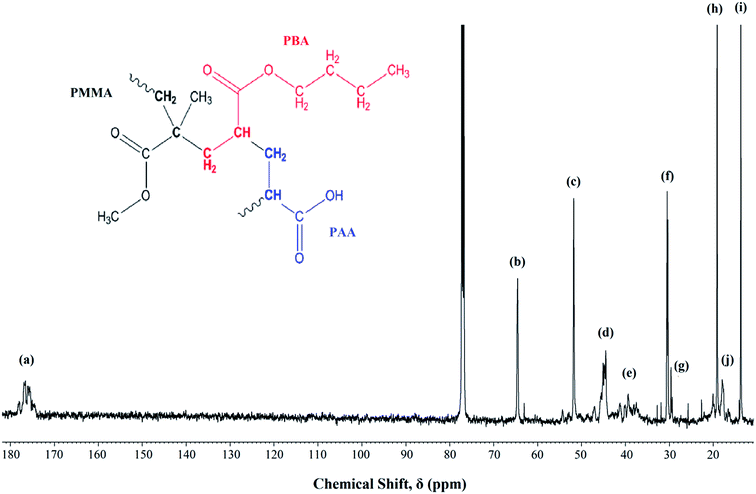 |
| | Fig. 1 13C NMR of UHMW-P(MMA-co-BA-co-AA) thin film measured at room temperature. Inset: copolymer structure. | |
Photo-response of P(MMA-co-BA-co-AA)
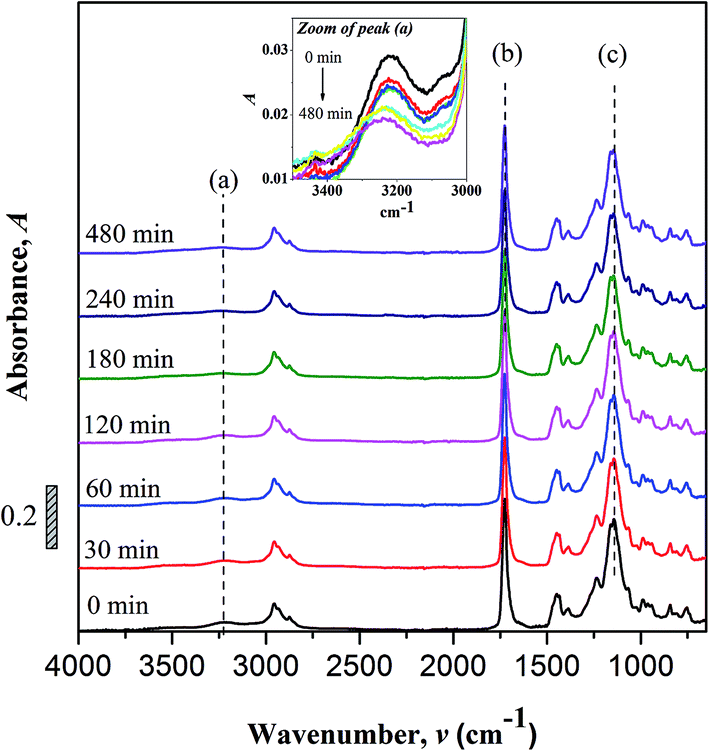
FTIR spectroscopy. Changes to the chemical structure (bond scission/forming) of copolymer after UV irradiation were monitor using FTIR. Fig. 2 shows the FTIR spectra of non-irradiated and photo-irradiation samples. The assignments of some important peaks of the copolymer are as summarized; –OH stretch (3225 cm−1), C–H stretch (2955 to 2874 cm−1), CO stretch (1726 cm−1), C–H bending (1350 and 1500 cm−1), C–C–O stretch (1237 cm−1), C–O–C stretch (1160 and 1142 cm−1) and skeletal C–C stretch (1064 and 755 cm−1).
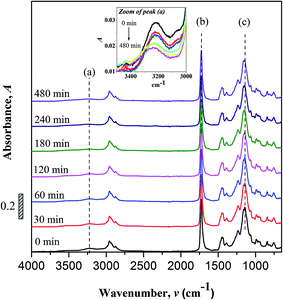 |
| | Fig. 2 FTIR spectra of non-irradiated and UV irradiated UHMW-P(MMA-co-BA-co-AA). Inset: zoomed area of peak (a) [FTIR characteristic peaks of (a) hydroxyl (O–H), (b) carbonyl (CO) and (c) ester (C–O–C)]. | |
No peak shifting was observed in the UV-irradiated sample with reference to the non-irradiated sample. However, minor changes in the peak intensities were observed evidencing chemical changes in the copolymer upon UV-irradiation. The bands that undergo prominent changes are the functionalities of hydroxyl (O–H), carbonyl (CO) and ester (C–O–C) moieties. The FTIR characteristic peaks of the fore mentioned functionalities are alphabetically labeled in the FTIR figure as; (a) hydroxyl (O–H), (b) carbonyl (CO) and (c) ester (C–O–C). The –OH characteristic peak, which represents the AA segments within the copolymer chain, decreases in the peak intensity upon irradiating [cf. Fig. 2 (inset)]. This may arise from the photo-oxidation reaction of –OH group, which results in the formation of oxidized products containing CO chromophores.16–19 For the functionalities of carbonyl and ester moieties, the changes exhibited are conversed based on the plotted carbonyl index (C.I.) and ester decomposition (D), which are respectively presented in Fig. 3 and 4. The C.I. value was obtained by normalizing the characteristic absorbance peak of CO with respect to the absorbance of ν(C–C) peak at 752 cm−1, shown in eqn (3):20
| |
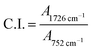 | (3) |
where
A represents the absorbance of peak. Whereas for the ester decomposition value (
D),
eqn (4) was employed:
| |
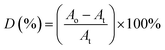 | (4) |
where
Ao and
At represent the absorbance value of C
O band at 1726 cm
−1 respectively before and after irradiation.
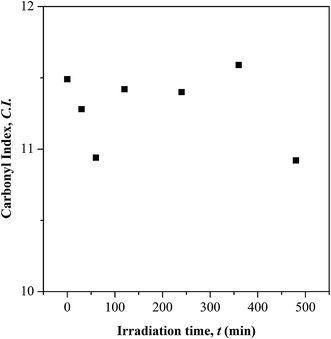 |
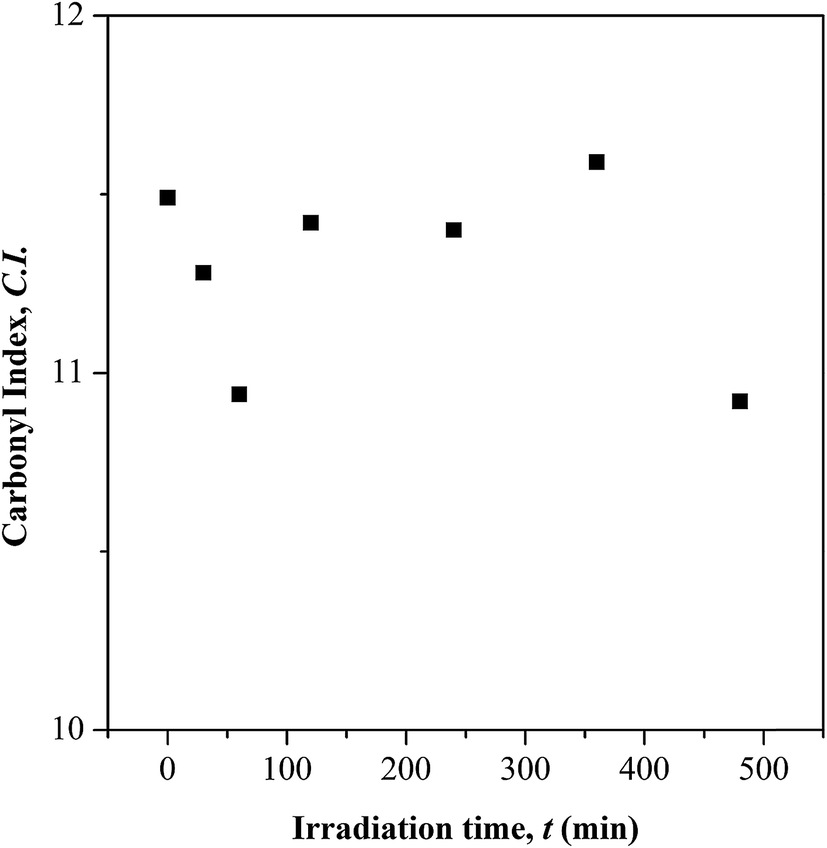
| | Fig. 3 Carbonyl index (C.I.) at different UV irradiation time. | |
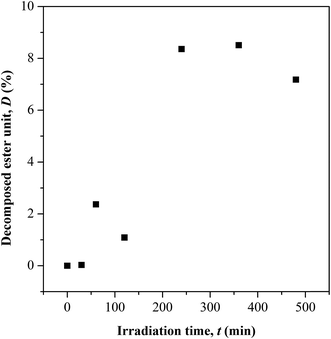 |
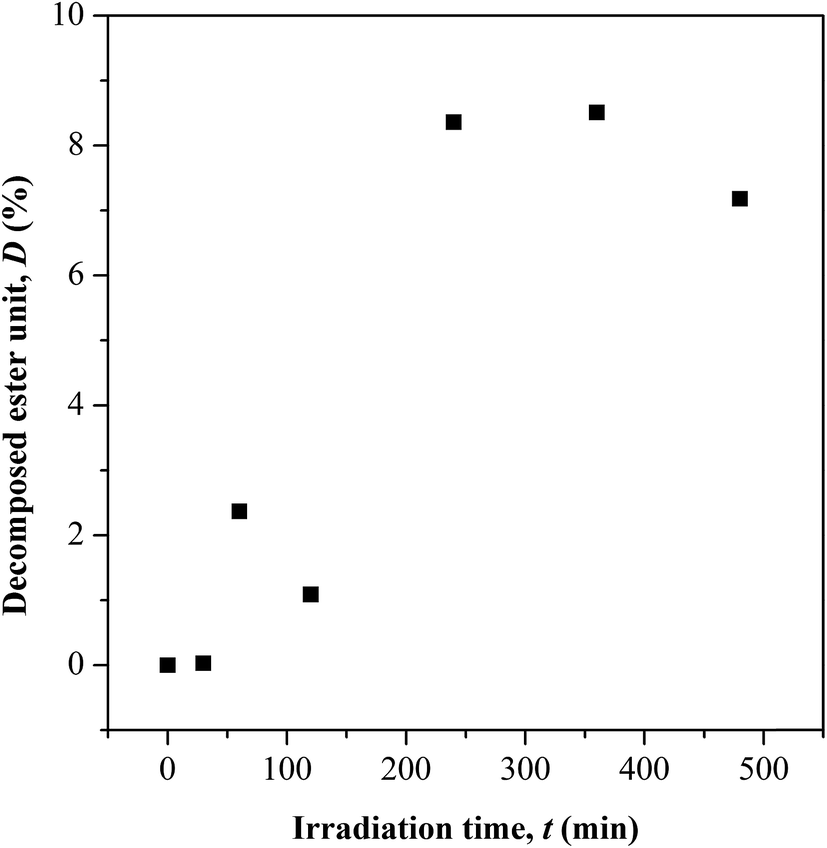
| | Fig. 4 Variation in ester chain cleavage at different UV irradiation time. | |
Based on the C.I. plot, it is understood that the CO concentration reduces at initial UV-irradiation and increases gradually with increase in irradiation time. The initial drop in the chromophore concentration is associated to the photo-destruction of π bond in CO, which later reacts with another radically active C–O˙ site that adjacent in coordination forming crosslinked structure rather than chain scission. The absence of chain scission at short irradiation time is well supported by the unchanged number of ester decomposition, as seen at 30 minutes of irradiation in Fig. 4. At prolong irradiation, an exponential increase in CO concentration was observed as subsequent of the cleavage of crosslinked bonds. Therefore, increase in ester decomposition units is accounted. The ester cleavage is found to be pronounced above 240 minutes of irradiation time, evidence from the abrupt increase in the decomposed ester unit. The product that potential to form upon photo-degradation is not possible to detect in this measurement since most of the characteristic absorption bands of the products overlap in the same region. The mechanism of bond destruction and forming in CO chromophore upon UV-irradiation is discussed in detail under UV-Vis-NIR spectroscopy subsection.
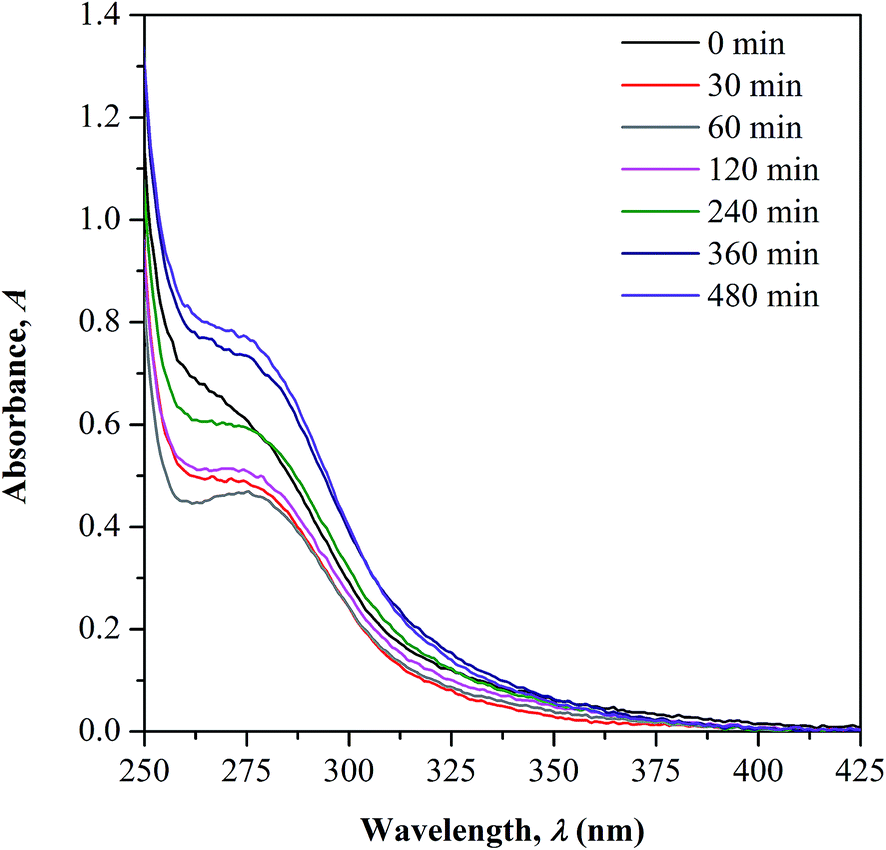
UV-Vis-NIR spectroscopy. UV-Vis-NIR spectroscopy is used as a supplementary tool to support the observed changes in CO concentration during irradiation as previously discussed in FTIR measurement. The overlaid UV-Vis-NIR spectra measured for both non-irradiated and irradiated samples are presented in Fig. 5. All the test samples show an appearance of two absorption bands in the UV region; peak at below 250 nm and within 250 to 350 nm that correspond to the characteristic bands of conjugated unsaturation and carbonyl chromophores, respectively. There is no occurrence of new absorption band observed upon UV-irradiation, implying the absence of new chromophore structure formation. The changes in the CO characteristic peak as function of the irradiation time is the only focus of the discussion.
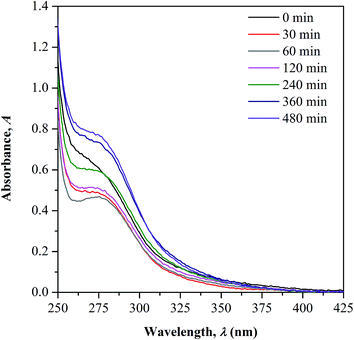 |
| | Fig. 5 UV-Vis-NIR absorption spectra of UHMW-P(MMA-co-BA-co-AA) – before and after UV irradiation. | |
In the spectrum of non-irradiated sample, a shoulder-like curve is found to appear in the wavelength region studied as the copolymer contains the CO functional group in its structure. As the sample was irradiated to 60 minutes, a more defined peak with reduced intensity can be observed. With further increase in the irradiation time, a broader shoulder with increasing intensities was observed. The initial decrease in the CO peak intensity is accounted due to the photochemical disruption of π bond in CO, which later causes reduction in the concentration. The electron in the π bond undergoes photo-excitation from ground to higher energy state (π → π* transition) and cleaves the bond.11,21,22 This consequently generates active radicals at the C–O˙ which then reacts further with the side chain of the adjacent monomer units forming crosslinked structure via oxygen bridge.
With increased irradiation time, the CO peak becomes broader and increases in intensity, evidencing the increase in CO concentration. This observation may arise upon the cleavage of the initially formed crosslinking bond as the result of the photo-excitation of sigma (σ) electron in C–O. As the consequence, free radicals are generated at the cleaved sites and rearranges forming stable structure. This gives high chances for the regeneration of CO group. The increment in CO concentration with irradiation time well supports the occurrence of photo-degradation on the surface of copolymer as more chain scission occurs randomly. Photo-oxidation process is also possible to take place simultaneously with the degradation as the irradiation was performed in air.
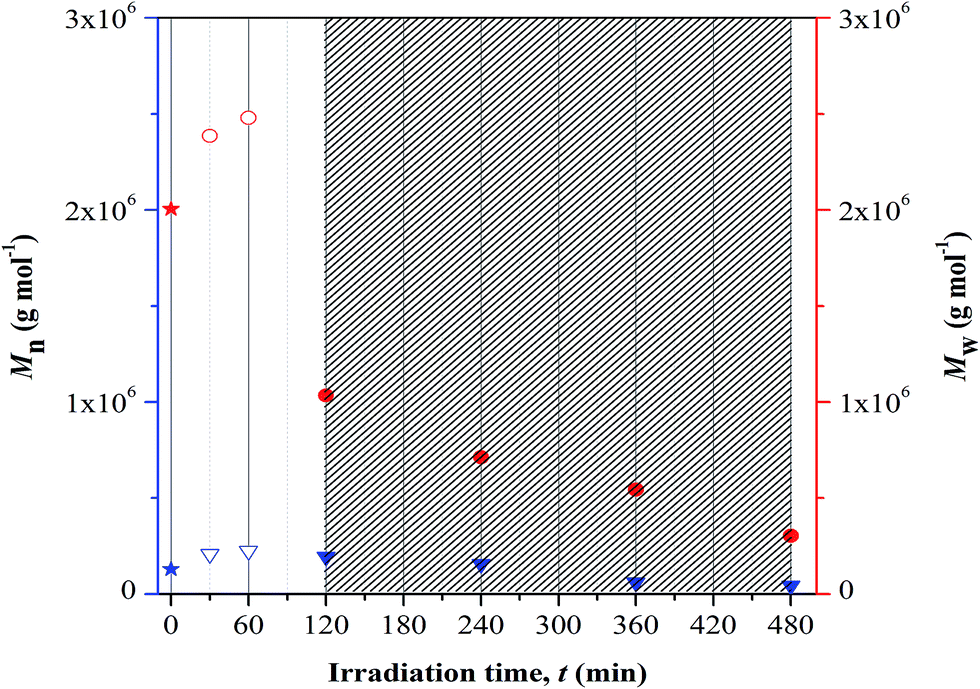
GPC analysis. GPC analysis was employed to study the changes in the polymer chain length upon photolytic and/or crosslinking reaction at different UV irradiation time. The correlation was studied based on the variation in the number-average (Mn) and weight-average (Mw) molecular weight, as presented in Fig. 6. Based on the plotted results, increase in both the average-molecular weights was visualized with increase in irradiation up to 60 min and at longer time of exposure these values drop rapidly. The increase in average-molecular weights corresponds to the characteristic feature of crosslinking process. The terpolymer chains increase in length during this reaction as quantitatively proven by the increase in Mn values. The reported Mn value represents the kinetic length of a chain. At longer irradiation time, there is a decrease in the average-molecular weights implying random chain scission. This process symbolizes degradation. The terpolymer chains are now cleaved and the kinetic length is reduced, as denoted by the decrease in the Mn values with irradiation time. This may progressively increase the degradation rate and consequently P(MMA-co-BA-co-AA) may lose its structural integrity (including properties).
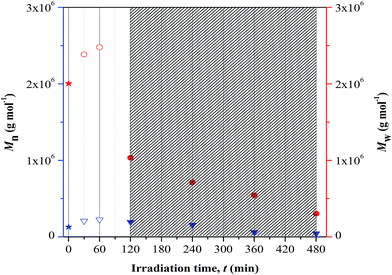 |
| | Fig. 6 Changes in average-molecular weights at different UV irradiation time. Note: blue and red color symbols/Y-axis are respectively represent Mn and Mw values. Symbols representation; star: non-irradiated sample, down-pointing triangle and circle: irradiated samples (open symbol-cross linked structure and filled symbol-fragmentation). Dense region showed degraded samples. | |
There are two types of photolysis mechanisms that well adopted under irradiation of UV; chain scission or unzipping. These mechanisms may occur simultaneously. In finding for the most favorable mechanism to take place during the irradiation, the theoretical view point reported by Wochnowski et al. was considered.12 It was reported that the decrease in the molecular weight that is associated to the unzipping process was observed to be slower than those observed for photo-degradation. Referring to this theoretical view, we proposed that the observed immense reduction in the Mw at irradiation of UV may be associated to be the effect of chain scission. This effect shows higher dominance at longer irradiation time. The immense reduction in the molecular mass of copolymer under UV exposure imposes negative impact on the chains properties; example as lost in the chemical and mechanical integrity. These changes then affect the ability of the materials to perform adequately in some applications and give rise to safety concern.
Chromaticity analysis. The chromaticity analysis was performed to study the color changes in sample upon UV irradiation. The color data are presented in Table 1 by the parameters of L*, a*, b*, C* and ΔE*. For the non-irradiated sample, it is slightly yellowish due to the presence of CO functional group. As the copolymer film is irradiated, the sample experiences a quick lightening and shifts towards blue coordination. The shifting is caused by the loss of yellowness (b*) and redness (a*) on the surface. The decrease in a* and b* values are consistent with the reduction in CO (yellow color reflecting chromophore23) concentration, as subsequent of its involvement in forming crosslinking structure at low irradiation time. Upon 60 minutes of irradiation, the bluish color gradually intensifies and the C* and ΔE* values increase. The observed improvement in the lightness (or brightness) of the non-irradiated sample at shorter UV irradiation time showed the materials applicability in optical- and packaging-based development.
Table 1 Chromaticity data of UHMW-P(MMA-co-BA-co-AA) collected at different UV irradiation time
| Irradiation time (t, min) |
L* |
a* |
b* |
C* |
ΔE* |
| 0 |
98.30 |
0.01 |
0.26 |
0.2602 |
0 |
| 30 |
100.54 |
−0.18 |
−1.93 |
1.9384 |
3.1384 |
| 60 |
105.06 |
−0.15 |
−3.54 |
3.543 |
7.7565 |
| 120 |
99.52 |
−0.10 |
−0.52 |
0.5295 |
1.4522 |
| 240 |
102.52 |
−0.13 |
−0.89 |
0.8994 |
4.4842 |
| 360 |
99.37 |
−0.23 |
−1.51 |
1.5274 |
2.0822 |
| 480 |
99.65 |
−0.08 |
−1.56 |
1.5620 |
2.2678 |
At 120 minutes of irradiation, the color shifts towards yellow coordination with reduced lightness. This observation illustrates the increase in CO concentration upon chain scission reaction. The reduction in the L* value may symbolize the occurrence of chain scission. With prolong irradiation time, non-linear changes in L*, a* and b* values were observed. The color change at each irradiation time is not noticeable owing to the low intensity of the color.
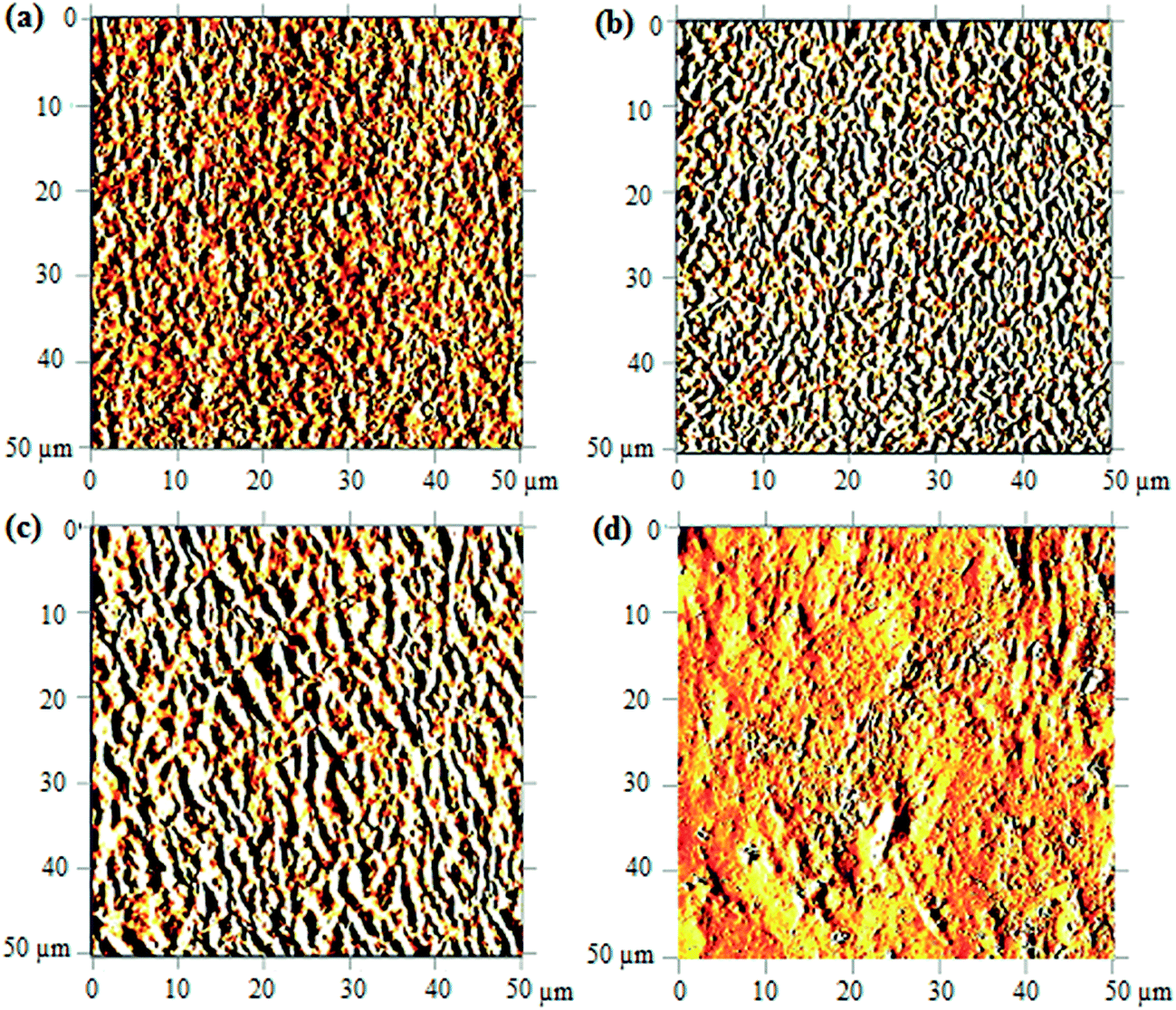
AFM study. The morphological of copolymer upon UV-irradiation can be visualized from the 2D-AFM images shown in Fig. 7. Only the images of degraded samples at 120 minutes, 240 minutes and 480 minutes of UV-irradiation were reported; the changes in morphology are understood using the non-irradiated copolymer image as the reference of comparison. In the non-irradiated copolymer, as depicted in Fig. 7(a), the surface exhibits a compact chain arrangement with uniform heights. This arrangement is found to be more diffusive upon UV-irradiation above 120 minutes, as shown in Fig. 7(b and c). Alternation on the surface architecture is caused by the scission of crosslinking bond (oxygen bridge) and ester/hydroxyl bond. This subsequently causes separation between the compactly joined polymer chains and the fraction of free volume increases.
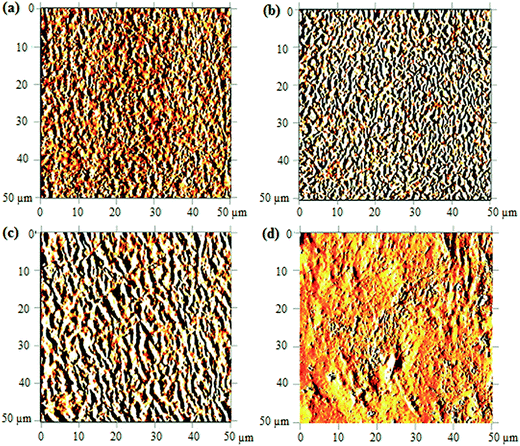 |
| | Fig. 7 AFM images of UHMW-P(MMA-co-BA-co-AA) captured before UV irradiation (a) and after irradiating at 120 min (b), 240 min (c) and 480 min (d). | |
As the irradiation time prolongs to 480 minutes, high amount of copolymer chains are cleaved. The resulted short segment chains are loosely bound on surface (due to massive free volume surrounded) and may possibly be etched. This results in major irregularities in the structure shown in Fig. 7(d). The increasing irregularities on the surface structure upon increasing UV-irradiation may also quantitatively be validated from the measured arithmetic mean roughness (Ra) values. The values increase from 8.4 nm (in reference sample) to 17.1 nm, 31.0 nm and 32.7 nm upon irradiation at 120, 240 and 480 minutes, respectively. The chain scission is the most probable cause of the increase in the morphological irregularities (or increase in Ra) at prolonged irradiation.24
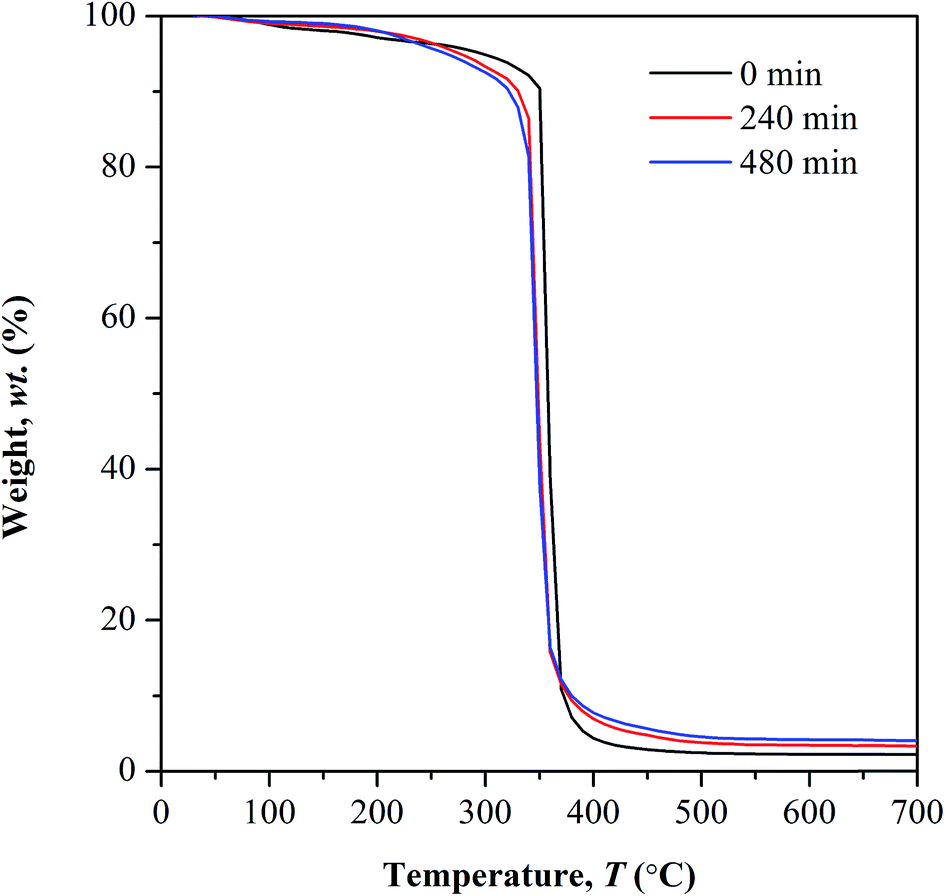
TGA study. Fig. 8 depicts the TGA curves of non-irradiated and photo-irradiated copolymer. The first stage of weight loss occurring at temperature below 100 °C corresponds to water desorption process. The weight lost exhibited at this stage decreases with increasing irradiation time. This happens as the subsequent of photo-oxidation involving –OH bond which in turn lowers the copolymer affinity towards water. This observation is supported by the reduction in FTIR peak intensity ranging at 3100 to 3400 cm−1.
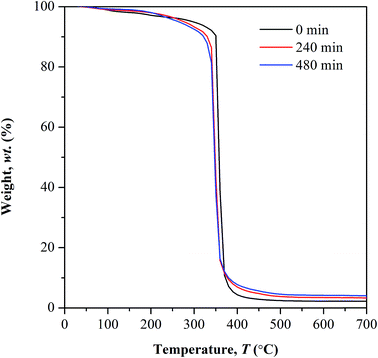 |
| | Fig. 8 Thermal analysis of UHMW-P(MMA-co-BA-co-AA) before and after UV irradiation. | |
Above 100 °C, lower weight loss was observed in all the irradiated samples until 250 °C. This owes to the decrease in the available water content that subject to evaporation, as the irradiated samples have low affinity towards water. At further elevated temperature above 250 °C, a distinct decomposition step with highest weight loss was observed in all the tested samples. This decomposition reaction is initiated by random chain scission. The degradation temperature (Td) at the highest weight loss shifts to lower values in photo-irradiated samples, from 360 °C (non-irradiated) to 347 °C (240 min irradiation) and 343 °C (480 min of irradiation). The Td is defined here as the onset temperature for degradation. The shifting of Td to lower temperature, at increasing irradiation time implies the increment in the concentration of weak linkages during photolytic reaction as the UV exposure time prolongs. Therefore low amount of heat is sufficient to cause degradation on weak linkages. The average residual mass in all the tested samples is lower than 5 wt% at 600 °C.
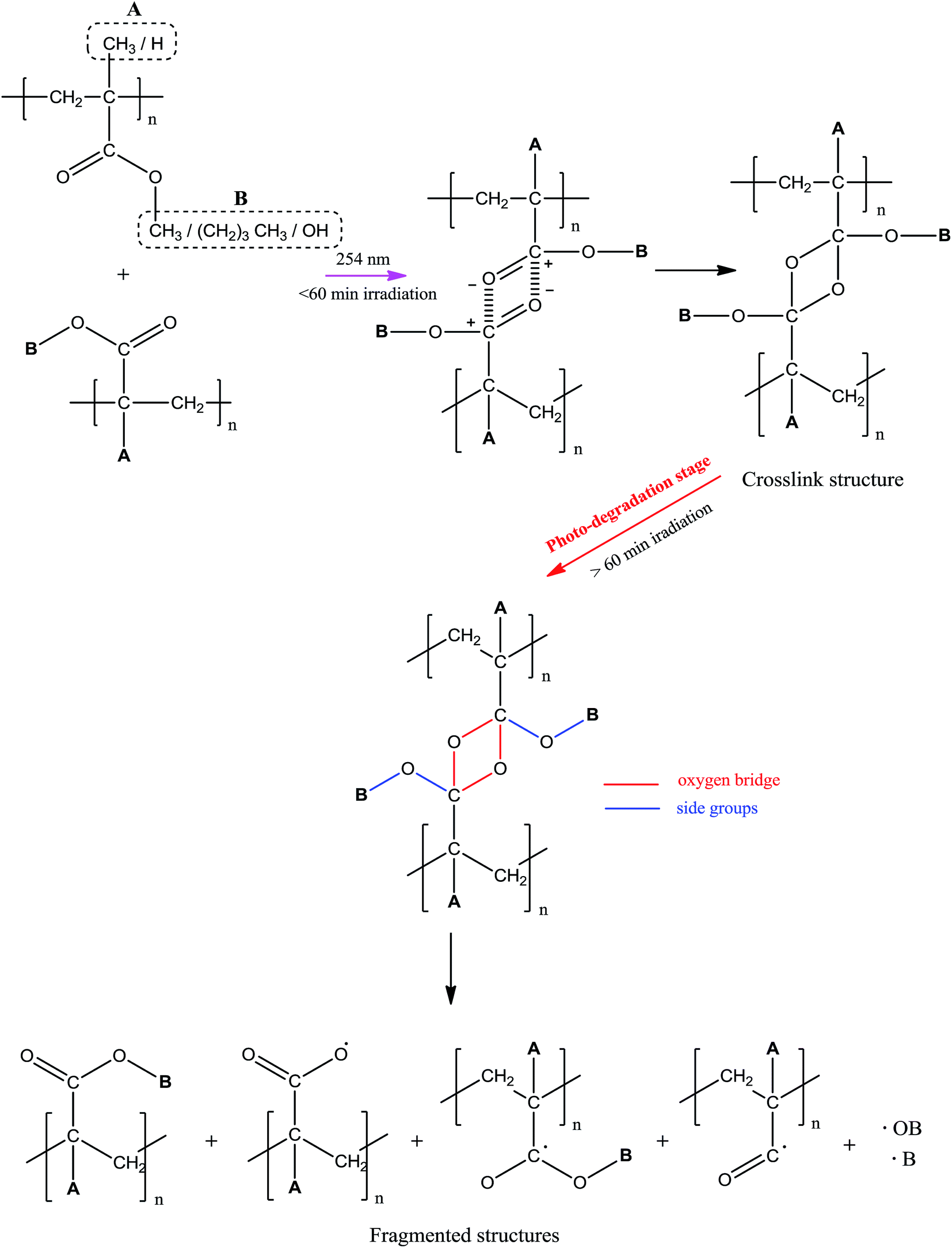
Proposed mechanism of photo-degradation. The photo-behavior of UHMW-P(MMA-co-BA-co-AA) at different irradiation time was proposed in reference to the molecular weight changes (cf. GPC result), supplemented with FTIR and UV-Vis results. The schematic diagram of the predicted copolymer photo-behavior was summarized in Scheme 1. Throughout the experimented irradiation time two different structures were formed; crosslink structure below 60 min of irradiation and fragmented (or short) structures formed at longer irradiation time. The formations of cross linkages at irradiation below 60 min were identified from the increase in Mw and the new linkages were formed between the adjacent carbonyl groups, according to the spectroscopy scrutiny. Cross-linking is prone to occur at low irradiation time due to ‘cage’ recombination of the primary radicals, as the effect of constrained chain mobility.12 At longer irradiation time, chains are actively being cleaved and the Mw of copolymer decreases. This causes major structural defect and contributes to degradation. The sites of bond scissions were identified at oxygen bridge (cross-linkages), ester (–OCH3) or/and methyl (CH3) groups. Only the side chains in the copolymer structure found to actively involved in bond forming/breaking reaction while main chains remained stable throughout irradiation, as evidence from the absence of CC peak in FTIR spectra at 1600–1650 nm. The possible photoproducts that are formed during degradation are therefore limited to the decomposition products of side chains. Photo-oxidation reaction is also likely to occur simultaneously with the degradation as the irradiation was performed in air.
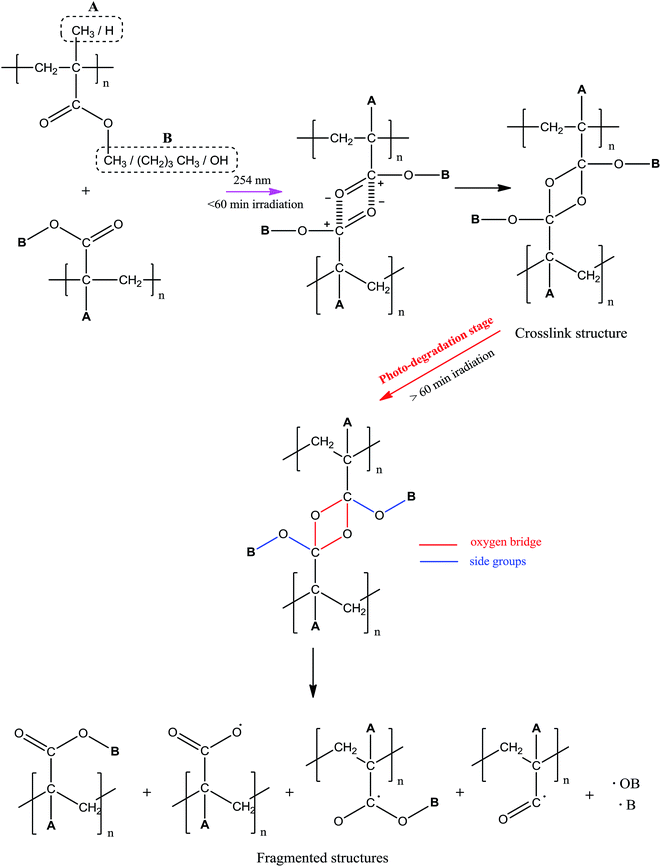 |
| | Scheme 1 Mechanism of photo-behavior of UHMW-P(MMA-co-BA-co-AA). | |
Conclusion
In summary, the UHMW-P(MMA-co-BA-co-AA) film undergoes photo-degradation under the illumination of 254 nm-UV at above 60 minutes of irradiation. At shorter irradiation time, crosslinked structure is prone to form due to high “cage” effect combination of radicals. All the exhibited changes in wise of chemical structure, film color, morphology and thermal degradation at different irradiation time signify the dependency of the copolymer degradation with irradiation time. The properties of the copolymer become poor upon photo-degradation and the effect is more pronounced with irradiation time. There is no tendency for the cleavage of copolymer main chain; instead all the changes are due to the cleavage of the side chains of the copolymer.
Acknowledgements
My sincere gratitude to “Scheme Bright Spark, University of Malaya” for awarding me with scholarship throughout the years of my studies. This work is financially supported by Fundamental Research Grant Scheme (FP012-2015A), from Ministry of Education, Malaysia. My special thanks to Assoc. Prof. Dr Roslan Bin Md Nor (Physics Department, UM) for allowing me to use the UV light facility in his lab which was supported by his UM RACE grant under the Project No: CR014-014.
References
- E. Yousif and R. Hadded, SpringerPlus, 2013, 2, 398 CrossRef PubMed.
- R. Geetha, A. Torikai, S. Nagaya and K. Fueki, Polym. Degrad. Stab., 1987, 19, 279 CrossRef CAS.
- B. Singh and N. Sharma, Polym. Degrad. Stab., 2008, 93, 561 CrossRef CAS.
- E. Rebollar, G. Bounos, M. Oujia, C. Domingo, S. Georgiou and M. Castillejo, J. Phys. Chem. B, 2006, 110, 14215 CrossRef CAS PubMed.
- S. Beauvois, D. Renaut, R. Lazzaroni, L. D. Laude and J. L. Bredas, Appl. Surf. Sci., 1997, 109–110, 218 CrossRef.
- A. Costera, J. M. Figuera, F. Florido, I. Garcia-Moreno, E. P. Collar and R. Sastre, J. Appl. Phys., 1995, 60, 261 Search PubMed.
- A. Costera, I. Garcia-Moreno, I. F. Florida, J. M. Figuera, R. Sastre, S. M. Hooker, J. S. Cashmore and C. E. Webb, J. Appl. Phys., 1995, 77, 2343 CrossRef.
- B. Ahmed, S. K. Raghuvanshi, Siddhartha, N. P. Sharma, J. B. M. Krishna and M. A. Wahab, Progress in Nanotechnology and Nanomaterials, 2013, 2, 42 Search PubMed.
- N. Grassie, A. Scotney, R. Jenkins and T. I. Davis, Chem. Zvesti, 1972, 26, 208 CAS.
- S. Duquesne, J. Lefebvre, R. Delobel, G. Camino, M. LeBras and G. Seeley, Polym. Degrad. Stab., 2004, 83, 19 CrossRef CAS.
- V. Kumar, R. G. Sonkawa de, Y. Ali and A. S. Dhaliwal, Int. J. Appl. Eng. Res., 2011, 2, 419 CAS.
- C. Wochnowski, M. A. Shams Eldin and S. Metev, Polym. Degrad. Stab., 2005, 89, 252 CrossRef CAS.
- S. D. Su’ad and I. M. I. Ismail, Orient. J. Chem., 2008, 24, 35 Search PubMed.
- M. Shirai, T. Yamamoto and M. Tsunooka, Polym. Degrad. Stab., 1999, 63, 481 CrossRef CAS.
- R. Shanti, F. Bella, Y. S. Salim, S. Y. Chee, S. Ramesh and K. Ramesh, Mater. Des., 2016, 108, 560 CrossRef CAS.
- S. G. Prasad, D. Abhijit and D. Udayan, Int. J. Spectrosc., 2011, 1 CrossRef CAS.
- A. Sionkowska, J. Photochem. Photobiol., B, 2006, 82, 9 CrossRef CAS PubMed.
- A. L. Andrady, A. Torikai and T. Kobatake, J. Appl. Polym. Sci., 1996, 62, 1465 CrossRef CAS.
- S.-M. Wang, Q.-Z. Huang and Q.-S. Wang, Carbohydr. Res., 2005, 340, 1143 CrossRef CAS PubMed.
- O. S. Kushwaha, C. V. Avadhani and R. P. Singh, Adv. Mater. Lett., 2014, 5, 272 CrossRef.
- L. Calcagno, G. Compagnini and G. Foti, Nucl. Instrum. Methods Phys. Res., Sect. B, 1992, 65, 413 CrossRef.
- M. F. Zaki, J. Phys. D: Appl. Phys., 2008, 41, 175 CrossRef.
- A. J. Domb, J. Kost and D. Wiseman, Handbook of Biodegradable Polymers, 1998, Retrieved from https://books.google.com.my/books?id=iLjhl6AvfIsC Search PubMed.
- E. C. Azevedo, E. M. Nascimento, G. O. Chierice, S. C. Neto and C. M. Lepienski, Polimeros, 2013, 23, 305 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25313j |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a and
K. Ramesha
*a and
K. Ramesha