
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functionalisation of ligands through click chemistry: long-lived NIR emission from organic Er(III) complexes with a perfluorinated core and a hydrogen-containing shell†
Y. Penga,
J. X. Hub,
H. Lua,
R. M. Wilsonb,
M. Motevallib,
I. Hernández*c,
W. P. Gillin*bd,
P. B. Wyatt*b and
H. Q. Ye*e
aDepartment of Macromolecular Science, State Key Laboratory of Molecular Engineering of Polymers, State Key Laboratory of ASIC and System, SIST Fudan University, Shanghai, 200433, P. R. China
bMaterials Research Institute and School of Physics and Astronomy, Materials Research Institute and School of Biological and Chemical Sciences, Queen Mary University of London, London E1 4NS, UK. E-mail: w.gillin@qmul.ac.uk; p.b.wyatt@qmul.ac.uk
cDepartamento CITIMAC, Facultad de Ciencias, Universidad de Cantabria, Santander 39005, Spain. E-mail: ignacio.hernandez@unican.es
dCollege of Physical Science and Technology, Sichuan University, Chengdu, 610064, P. R. China
eDivision of Physics and Applied Physics, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: hqye@ntu.edu.sg
First published on 22nd December 2016
Abstract
Erbium complexes with a fluorinated organic core shell linked to a hydrogen-containing shell, have been synthesized using the click reaction between erbium(III) bis(perfluoro-4-azidophenyl)phosphinate and a series of alkynes. The erbium 1.5 μm emission lifetimes in the hydrogen-containing erbium complexes exceed 140 μs, the longest ever reported in hydrogenated organic erbium systems. The visible sensitisation for erbium emission indicates a successful strategy that broadens the usage of non-fluorinated chromophores in organic erbium systems and allows more choices for ligand functionalization with exceptional efficiency for erbium emission.
Introduction
Organic erbium containing materials have been of great interest as the optical properties of erbium can be optimized by incorporating erbium ions into organic chromophores; these can efficiently harvest light and the energy-transfer can induce intense erbium emission through sensitization.1–3 However, the presence of high-energy oscillators (O–H or C–H units) should be avoided to minimize the vibrational quenching for near-infrared (NIR) emission in order to obtain a long 1.5 μm emission lifetime.4,5 Both experimental estimation and theoretical computation have suggested the need for a separation distance of >20 Å between NIR-emitting Er3+ ions and quenchers to overcome the quenching effect.6,7 The perfluorination of organic ligands has been well documented and the erbium emission lifetimes of up to hundreds of microseconds have been obtained due to the lower vibrational energy of fluorinated chemical units.8–10Although perfluorinated chromophores are preferred, the synthesis of these species is complex because the most efficient chromophores are highly conjugated multi-ring systems that are difficult to synthesis. Also, the availability of highly fluorinated functional chromophores is limited. If non-fluorinated chromophores are tolerable in the ligands through special confinement, it will greatly expand the scope of usable moieties. In this study, we proposed a pseudo-core–shell model (Fig. 1) with ‘central fluorination’ for the perfluorinated moiety carrying a binding site that is connected to the non-fluorinated moiety through a linkage. These ligands bind to an Er3+ ion with a perfluorinated protective core for the Er3+ ion in order to keep C–H bonds located in the relatively distant linkage and non-fluorinated shell/sheath. The resultant erbium 1.5 μm emission lifetimes reach over 140 μs, the longest ever reported for hydrogen-containing organic erbium species and even longer than those reported in some perfluorinated erbium systems.11 With the evidence that these ligands provide visible-range sensitization for erbium emission, this strategy allows the potentiality for functionalization and high efficiency NIR emission simultaneously.
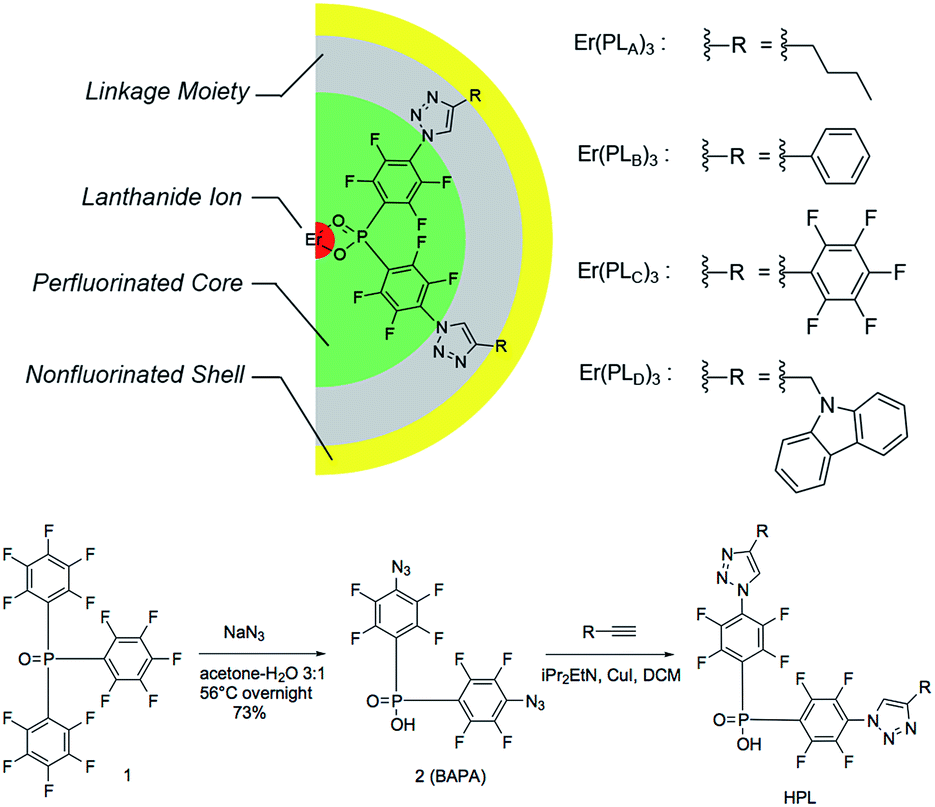 | ||
| Fig. 1 Pseudo-core–shell model of central-fluorinated erbium complexes and general route for ligand preparation. | ||
Experimental methods
The detailed ligand design chose P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bonds for the binding site (Fig. 1) because they have shown exceptional photoluminescence (PL) lifetimes for erbium complexes in previously reported systems.8 The perfluorinated core is derived from bis(perfluorophenyl)phosphinic acid. The electron-withdrawing PO bond and fluorine atoms on the phenyl ring are activated towards aromatic nucleophilic substitution, particularly at the para-position of the phenyl ring, providing a convenient way of functionalizing the system to introduce a linkage. Azide groups were chosen as precursors to the linkage because (1) the azide ion is a strong nucleophile and (2) the azide group is widely used in Hüisgen cycloaddition to form 1,2,3-triazoles, which is the preeminent example of a highly efficient and clean ‘click reaction’.12 According to this design, a large variety of non-fluorinated moieties can be incorporated into the ligands if appropriate alkynyl derivatives can be used in 1,2,3-triazole formation.
O double bonds for the binding site (Fig. 1) because they have shown exceptional photoluminescence (PL) lifetimes for erbium complexes in previously reported systems.8 The perfluorinated core is derived from bis(perfluorophenyl)phosphinic acid. The electron-withdrawing PO bond and fluorine atoms on the phenyl ring are activated towards aromatic nucleophilic substitution, particularly at the para-position of the phenyl ring, providing a convenient way of functionalizing the system to introduce a linkage. Azide groups were chosen as precursors to the linkage because (1) the azide ion is a strong nucleophile and (2) the azide group is widely used in Hüisgen cycloaddition to form 1,2,3-triazoles, which is the preeminent example of a highly efficient and clean ‘click reaction’.12 According to this design, a large variety of non-fluorinated moieties can be incorporated into the ligands if appropriate alkynyl derivatives can be used in 1,2,3-triazole formation.
The general route for ligand preparation is shown in Fig. 1. Tris(pentafluorophenyl)-phosphine oxide 1 was prepared following the literature procedure.13 Aromatic nucleophilic substitution by sodium azide in aqueous acetone replaced all of the p-fluorine atoms on the rings with azide groups. Hydrolysis of a P-aryl bond simultaneously occurred with the substitution, thus giving bis(4-azido-2,3,5,6-tetrafluorophenyl)phosphinic acid 2 (BAPA). The reactions of four terminal alkynes RnC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH (R1 = n-Bu; R2 = Ph; R3 = C6F5; R4 = carbazol-9-yl-CH2) with BAPA were performed through copper assisted azide–alkyne cycloaddition (CuAAC) to provide the products HPLA–D, respectively (see ESI for detailed synthesis procedures†). These four ligands were then mixed with ErCl3 solutions and ErL3 (L = PLA, PLB, PLC, PLD, respectively) complexes were formed as precipitates. The low solubility, coupled with existing work in the literature on similar materials,13 possibly suggests a structure involving bridging –O–P–O– groups.14
CH (R1 = n-Bu; R2 = Ph; R3 = C6F5; R4 = carbazol-9-yl-CH2) with BAPA were performed through copper assisted azide–alkyne cycloaddition (CuAAC) to provide the products HPLA–D, respectively (see ESI for detailed synthesis procedures†). These four ligands were then mixed with ErCl3 solutions and ErL3 (L = PLA, PLB, PLC, PLD, respectively) complexes were formed as precipitates. The low solubility, coupled with existing work in the literature on similar materials,13 possibly suggests a structure involving bridging –O–P–O– groups.14
The excitation spectra of the erbium emission were measured using a homemade continuous monochromic system. A xenon lamp was filtered and focused into a Jobin-Yvon Horiba Triax 180 spectrometer equipped with 1200 lines per mm gratings. The monochromatic light from the output of Triax 180 was focused into the sample. The erbium luminescence was focused into a Jobin-Yvon Horiba Triax 550 equipped with 600 lines per mm gratings. The illumination intensity on the sample was measured directly by a calibrated Newport 918D-UV-OD3R silicon photodetector to normalize the excitation spectra for variations in illumination intensity. The luminescence intensity was detected using a Hamamatsu R5509-72 nitrogen-cooled detector and measured using a 5029 single-phase lock-in amplifier. For the PL spectra at visible wavelength region the excitation was from a 375 nm laser diode. For the time-resolved PL measurements the excitation was from a Continuum Panther Optical Parametric Oscillator (OPO) laser pumped by a Continuum Surelite (SLI-10) Nd:YAG. The laser pulse is 5 ns. The time-resolved PL decay curves were recorded using a digital oscilloscope. The PL spectra were corrected by the response of the PMT detector. All samples were dried in vacuo prior to PL and excitation measurements.
Results and discussion
Diffuse reflectance measurements (ESI, Fig. S3†) were performed for solid-state complexes, due to the low solubility of the complexes, giving the absorption maxima of the solids over the UV range and weak fluorescence of the solid samples. The erbium PL spectra were explored using ∼5 ns laser pulse at 520 nm wavelength for direct excitation into the 2H11/2 level of Er3+ ions. The NIR photoluminescence of these complexes shows the approximate independence of 4I13/2 → 4I15/2 transitions on the composition, and the fine structures of emission peaks only slightly affected by local crystal fields (ESI, Fig. S2†).Excitation spectra, recorded at 1536 nm emission, for Er(PLA–D)3 over excitation wavelengths from 400 nm to 700 nm are shown in Fig. 2. It can be seen that excitation bands extend over the region of ∼400 nm to ∼500 nm, matching the tail of the reflectance spectra (see ESI†). It can be seen that the excitation for Er[(C6F5)2PO2]3 is solely the direct excitation from Er3+ ions, indicating the organic ligands provides no visible sensitization. Interestingly, the broad excitation for Er(PLA–D)3 over the visible region is identical in shape for each of the ligands. Given that the shell moieties used in this work lack extensive conjugation it is unlikely that they significantly contribute to the sensitization in the observed wavelength range. Hence, given the similarity in shape, it is believed that the sensitization in this case comes from the triazole linker. To estimate the sensitization effect for each complex, we took the excitation at 522 nm as comparison since the 522 nm absorption peak is due to the hypersensitive 4I15/2 → 2H11/2 transition, the intensity of which varies strongly with the local environment of the ion. Taking the ratio of the integrated size of the area (400 nm to 500 nm) relative to that integrated for 2H11/2 excitation (500 nm to 530 nm), we are able to evaluate the visible sensitization ratios as 4, 5, 3.5 and 11 for Er(PLA–D)3, respectively, which are comparable with or even larger than those for some perfluorinated organo–erbium complexes.11
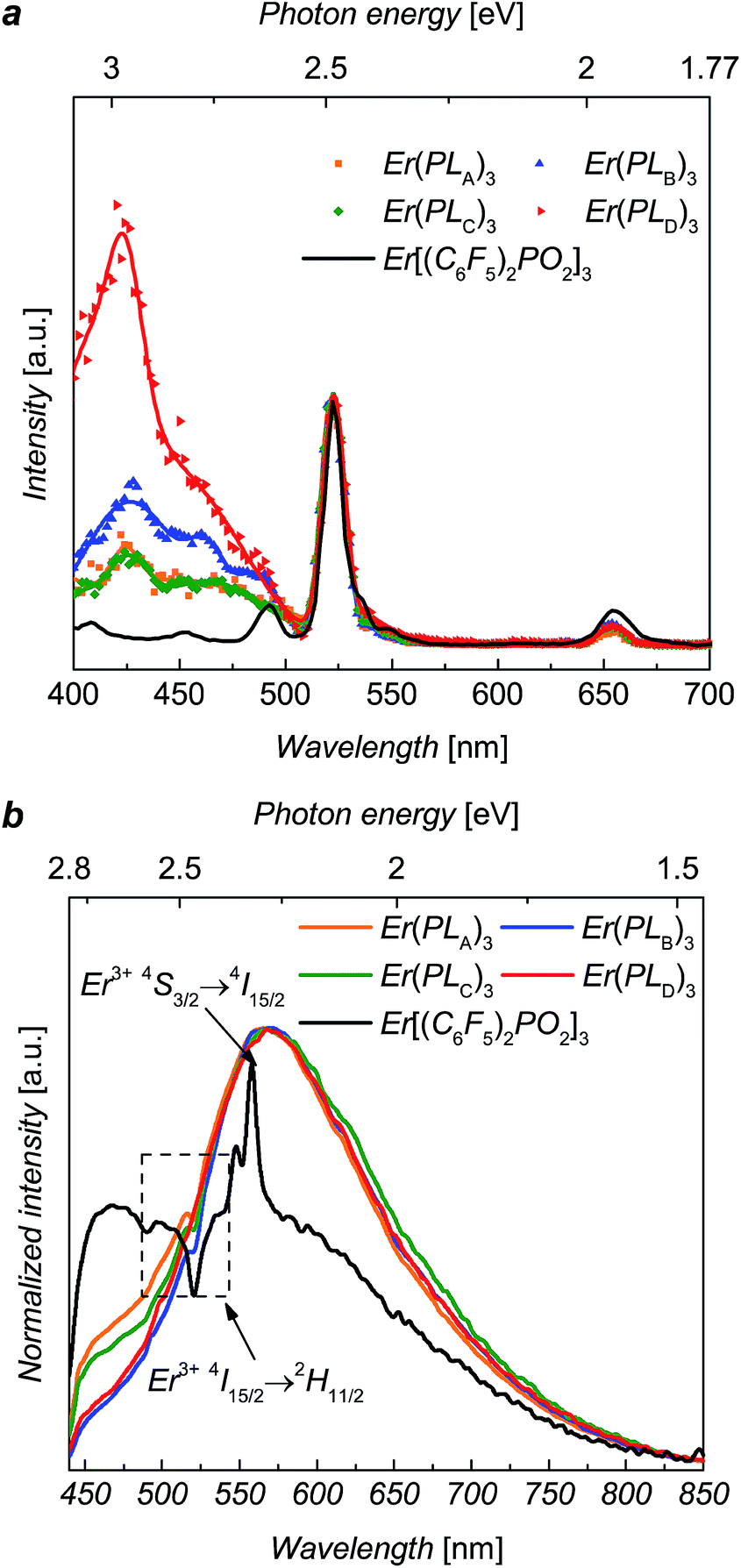 | ||
| Fig. 2 (a) Excitation spectra for Er(PLA–D)3 and recorded at 1536 nm emission at excitation wavelengths from 350 nm to 700 nm with continuous wave monochromatic lights. (b) PL spectra of Er(PLA–D)3 and over the visible region with 375 nm excitation. | ||
The visible PL spectra for the complexes, with 375 nm excitation, are shown in Fig. 2. For Er[(C6F5)2PO2]3 it is difficult to determine the peak accurately as it is weak and dominated by Er absorption (2H11/2) and emission (4S3/2 → 4I15/2) bands. Despite this it is clear that it is significantly blue shifted in comparison to the complexes Er(PLA–D)3. This suggests that the ligand emission is strongly dependent of the triazole linker, which supports the view that this group is contributing to the sensitization. These visible PL spectra clearly suggest that the emissive states of the ligands (1.5 eV to 2.8 eV) might be responsible for the sensitization as they can be resonant14,15 with the several intermediate levels of Er3+ ion such as the 4F7/2, 2H11/2, 4S3/2 and 4F9/2 states.
The erbium emission lifetimes of complexes Er(PLA–D)3 at 1536 nm wavelength were measured for a series of each complex where the Er3+ ions was diluted with Y3+ ions in order to explore the intrinsic erbium emission lifetimes without inter-ion interactions.13,16,17 As the Y3+ ion has approximately the same ionic radius as Er3+ ion, the Y(PLR)3 complexes are of the same phase as the corresponding Er(PLR)3 counterparts, which was indicated by X-ray powder diffraction (ESI, Fig. S4†). Consequently, it is unlikely that the mixture of the inactive yttrium complex will change the coordination structure around Er3+ ions. The addition of a mixture of ErCl3 and YCl3 solution to a ligand solution was assumed to produce ion mixing at an atomic level in ErxY1−x(PLR)3 complexes. The PL decays of ErxY1−x(PLB)3 complexes are shown in Fig. 3 as an example. The PL decay is clearly non-exponential for the 100% Er sample. As the erbium concentration decreases, the decay process tends to approach mono-exponential and becomes slower, suggesting gradual elimination of the concentration quenching due to inter-ion interactions of the erbium ions, likely cross-relaxation as observed in analogue compounds.17
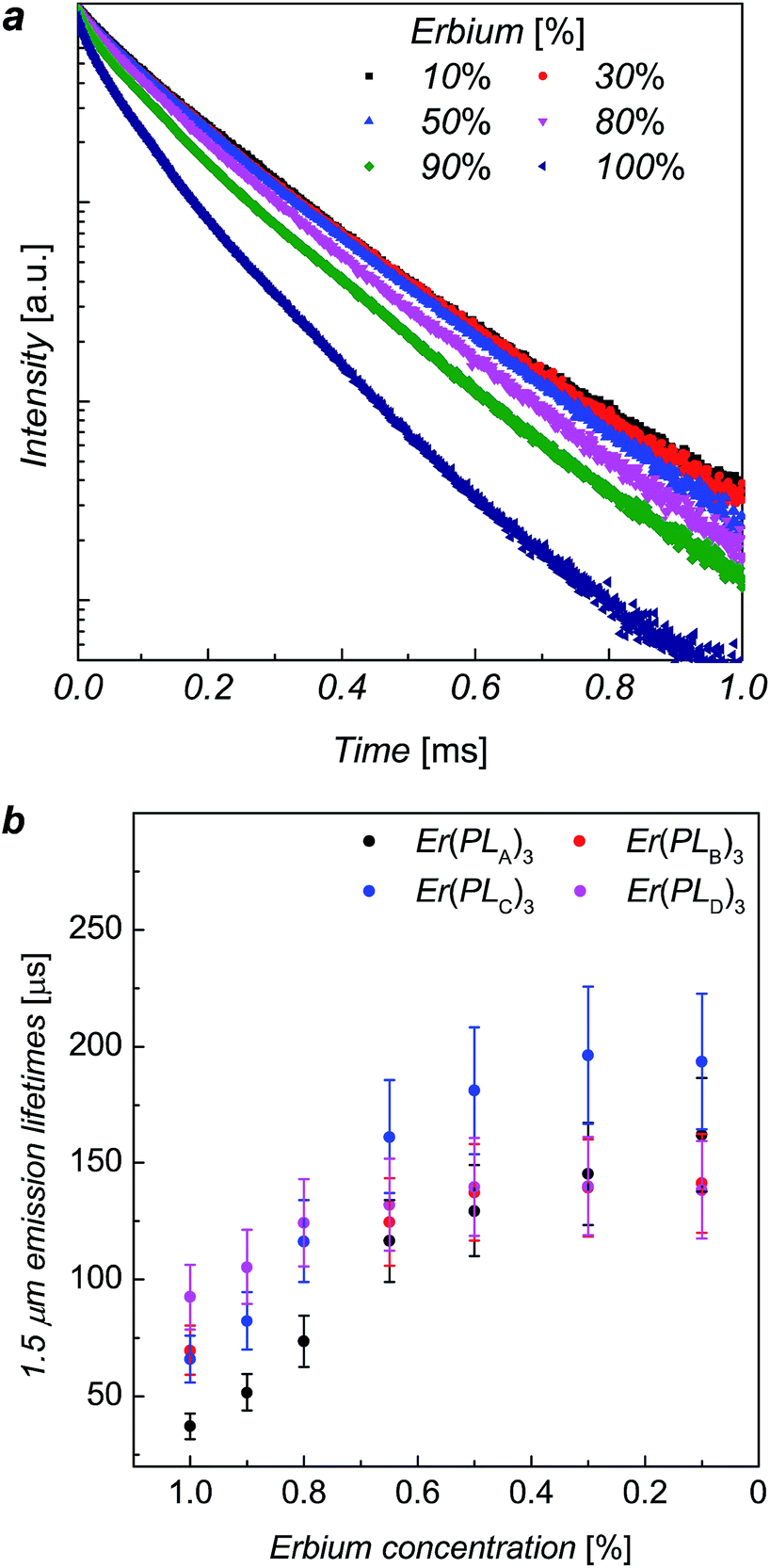 | ||
| Fig. 3 (a) The erbium 1536 nm emission decay for ErxY1−x(PLB)3, at 520 nm excitation. (b) Combined average lifetime versus Er3+ ion concentration plots for ErxY1−x(PLA–D)3 at room temperature with error bar of ±15%. | ||
The erbium emission lifetimes were obtained by fitting the PL decays with a stretched exponential function (see ESI†), which can give an average lifetime for both multi-exponential decay process and mono-exponential decay process.18,19 For each series the stretching factor, β, tends to increase with the decrease of the erbium concentration and β tends to reach a constant when the erbium concentration gets lower than 50%, whilst the average lifetimes increase with the decrease of erbium concentration (see details in ESI†). The lifetime increases faster when the erbium concentration is higher (>80%) while the increase trends slower when the erbium concentration less than 50%, gradually approaching the intrinsic lifetime limit. The change in β, lifetimes and erbium concentration means inter-ion interactions are suppressed when the average distance between neighbouring ions exceeds a critical value.
The average erbium emission lifetimes as a function of erbium concentration for each series of diluted complexes are plotted in Fig. 3b. The intrinsic erbium emission lifetimes without concentration quenching are estimated to be 157 μs, 141 μs, 195 μs and 140 μs for Er(PLA)3, Er(PLB)3, Er(PLC)3 and Er(PLD)3 respectively. Apart from the concentration quenching, the vibrational quenching due to O–H and C–H are mainly responsible to the short erbium 1.5 μm emission lifetimes in organic complexes. It can be expected that the reduction of erbium emission lifetimes of Er(PLA,B,D)3 compared with Er(PLC)3 is due to the stronger vibrational quenching caused by more hydrogen atoms on the shell moieties. It is extraordinary that the Er(PLC)3 gives an intrinsic erbium emission lifetime of 195 μs despite having six hydrogen atoms per formula unit. Surprisingly, Er(PLD)3, which contains 66 hydrogen atoms per formula unit, gives an exceptionally long intrinsic erbium emission lifetime of 140 μs. This demonstrates that it is possible to have highly conjugated chromophores introduced into these core–shell structures without significantly degrading the Er lifetime. These are by far the longest lifetimes reported for hydrogen-containing organic erbium systems highlighting the advantage of ‘central fluorination’. Potentially, application of the same strategy to sublimable complexes could result in even longer lifetimes as desired since vacuum sublimation is one of the common techniques to obtain the purity level that exceptional optical quality of erbium emission requires (e.g. by removing all traces of residual solvents).
Conclusions
We have proposed a new strategy, ‘central fluorination’, for ligand design. The shell moieties can have variations and then be easily attached to the core via click cycloaddition. We have demonstrated that the protection of perfluorinated shell could prevent vibrational quenchers on conjugated non-fluorinated organic chromophores from degrading the radiative lifetime of the erbium ions. This strategy provides a novel route to design organic sensitized erbium material systems by broadening the usage of non-fluorinated functional groups including chromophores.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge the EPSRC UK National Mass Spectrometry Facility at Swansea University. J. H. is financially supported by the China Scholarship Council and Queen Mary University of London. I. H. acknowledges financial support from the EU FP7 (Marie Curie-CIG-Grant 303535). Y. P., H. L. and H. Q. Y. acknowledge the China Scholarship Council and Queen Mary University of London, WPG acknowledges financial support from EPSRC EP/K004484/1 and National Science Foundation of China (61574095).References
- J.-C. G. Büenzli, Coord. Chem. Rev., 2015, 293, 19–47 CrossRef.
- Y. Hasegawa, Y. Wada and S. Yanagida, J. Photochem. Photobiol., C, 2004, 5, 183–202 CrossRef CAS.
- H. Q. Ye, Z. Li, Y. Peng, C. Wang, T. Li, Y. Zheng, A. Sapelkin, G. Adamopoulos, I. Hernández, P. Wyatt and W. Gillin, Nat. Mater., 2014, 13, 382–384 CrossRef CAS PubMed.
- R. H. Tan, M. Motevalli, I. Abrahams, P. B. Wyatt and W. P. Gillin, J. Phys. Chem. B, 2006, 110, 24476–24479 CrossRef CAS PubMed.
- A. Monguzzi, A. Milani, A. Mech, L. Brambilla, R. Tubino, C. Castellano, F. Demartin, F. Meinardi and C. Castiglioni, Synth. Met., 2012, 161, 2693–2699 CrossRef.
- L. Winkless, R. Tan, Y. Zheng, M. Motevalli, P. Wyatt and W. Gillin, Appl. Phys. Lett., 2006, 89, 1115 CrossRef.
- L. Ning, M. I. Trioni and G. P. Brivio, J. Mater. Chem., 2007, 17, 4464–4470 RSC.
- G. Mancino, A. J. Ferguson, A. Beeby, N. J. Long and T. S. Jones, J. Am. Chem. Soc., 2005, 127, 524–525 CrossRef CAS PubMed.
- P. B. Glover, A. P. Bassett, P. Nockemann, B. M. Kariuki, R. Van Deun and Z. Pikramenou, Chem.–Eur. J., 2007, 13, 6308–6320 CrossRef CAS PubMed.
- H. Q. Ye, Y. Peng, Z. Li, C. C. Wang, Y. X. Zheng, M. Motevalli, P. B. Wyatt, W. P. Gillin and I. Hernández, J. Phys. Chem. C, 2013, 117, 23970–23975 CAS.
- Y. Peng, H. Ye, Z. Li, M. Motevalli, I. Hernández, W. P. Gillin and P. B. Wyatt, J. Phys. Chem. Lett., 2014, 5, 1560–1563 CrossRef CAS PubMed.
- H. C. Kolb, M. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- L. Song, J. Hu, J. Wang, X. Liu and Z. Zhen, Photochem. Photobiol. Sci., 2008, 7, 689–693 CAS.
- G. H. Dahl and B. P. Block, Inorg. Chem., 1967, 6, 1439–1443 CrossRef CAS.
- G. L. Law, D. Parker, S. L. Richardson and K. L. Wong, Dalton Trans., 2009, 8481–8484 RSC.
- R. Tan, J. Pearson, Y. Zheng, P. Wyatt and W. Gillin, Appl. Phys. Lett., 2008, 92, 103303 CrossRef.
- I. Hernández, R. H. C. Tan, J. M. Pearson, P. B. Wyatt and W. P. Gillin, J. Phys. Chem. B, 2009, 113, 7474–7481 CrossRef PubMed.
- S. V. Eliseeva, D. N. Pleshkov, K. A. Lyssenko, L. S. Lepnev, J.-C. G. Bünzli and N. P. Kuzmina, Inorg. Chem., 2010, 49, 9300–9311 CrossRef CAS PubMed.
- C. Lindsey and G. Patterson, J. Chem. Phys., 1980, 73, 3348–3357 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25494b |
| This journal is © The Royal Society of Chemistry 2017 |