DOI:
10.1039/C6RA25572H
(Paper)
RSC Adv., 2017,
7, 6259-6265
The proteomic profiling of calenduloside E targets in HUVEC: design, synthesis and application of biotinylated probe BCEA†
Received
20th October 2016
, Accepted 7th December 2016
First published on 18th January 2017
Abstract
We previously found the total saponins from the medicinal herb Aralia elata, which exhibited strong anti-oxidative and anti-apoptotic effects. calenduloside E (CE) is one of the major triterpenoid saponin compounds isolated from these total saponins. However, the endothelial protection effect and the probable protein targets of CE have not been fully characterized. In the present study, a biotin-conjugated CE analogue (BCEA) was employed as a molecular probe to research and analyse its protein targets and signaling pathways. Compared with its parental compound CE, BCEA exhibited a similar protective effect against ox-LDL induced HUVEC damage. A chemical proteomic approach identified 128 proteins that related to the cell survival signaling pathways as the targets for BCEA. Meanwhile some of these cell survival signaling pathways that showed a higher P-value in KEGG pathway analysis were associated with anti-apoptotic activity. Moreover, further evaluation with flow cytometry, JC-1 staining assays and cleaved caspase-3 activity confirmed the anti-apoptotic effect of BCEA. Taken together, these results suggested that CE can improve cell viability most likely through anti-apoptotic mechanisms, and provided the basis for the further optimization of the endothelial protection compounds.
Introduction
Molecular target identification is one of the most significant facets and major challenges for drug discovery and development. Although various pharmacological strategies have been developed to screen active compounds, there are still no direct effective methods for understanding the biological action mechanism of active small molecules due to their unclear targets.1–3 In recent years, increasing studies have demonstrated that chemical proteomics is one of the most powerful methods for identifying the targets of small molecules.4 Common approaches to identify small molecule targets involve tethering of the bioactive compounds with suitable functional groups (e.g. biotin) which serve as an affinity tag allowing the bound proteins to be pulled-down and isolated with streptavidin.5–7 The isolated proteins are then separated and identified by mass spectrometry.8,9 Furthermore, the functional validation and pathway analysis of target proteins should also be carried out by correlating binding site information with proteomics data for the sake of understanding the biological mechanism of active compounds.10,11
Bioactive natural products from medicinal herbs have played a major role in discovering novel drugs and formed a treasury that supplies candidate drugs with structure–function diversity. Aralia elata (Miq) Seem (AS), a kind of homologous plant food medicine, has been used as a tonic, antiarrhythmic, anti-arthritic, antihypertensive and anti-diabetic agent in traditional Chinese medicine.12,13 The total saponins extracted from AS, which are found to be the main pharmacological active ingredients of AS, have been proved to exhibit anti-myocardial ischemic, anti-hypoxic activities, anti-oxidative as well as anti-inflammatory and anti-apoptotic capacity.14–16 Moreover, as main components of A. elata Xinmaitong capsules (Clinical Trial Approval Number 2003L01111 by China Food and Drug Administration), AS was developed for the treatment of coronary heart disease, and have been successfully completed Phase III clinical trials in China.17 Various oleanane-type triterpene saponins were extracted and identified from AS.13,18 A previous report demonstrated that some triterpenoid saponin analogue from AS could tremendously protect the endothelial cells from injury and reduce apoptotic endotheliocytes.19–21 calenduloside E (CE, Fig. 1A) is one of the major natural pentacyclic triterpenoid saponins isolated from AS.22–24 However, the endothelial protection effect and protein targets analysis of CE have not been investigated.
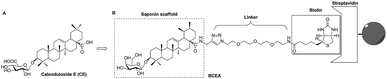 |
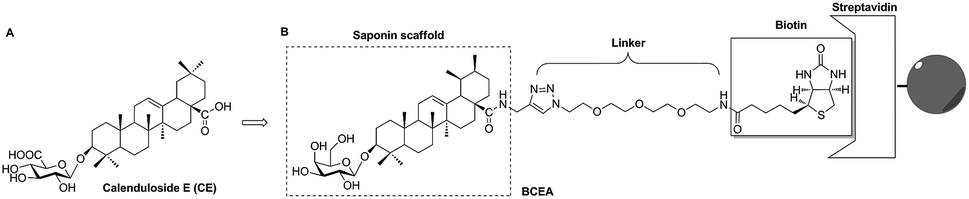
| | Fig. 1 The design of biotinylated probe BCEA from lead compound calenduloside E (CE). (A) The chemical structure of CE. (B) The construction of biotinylated probe BCEA. The saponin scaffold of CE was used as the active moiety. The PEG chain coupled 1,2,3-triazole which synthesized by a Cu(I) catalyzed Huisgen 1,3-dipolar cycloaddition reaction was employed as the linker. The biotin moiety was applied as the tag that bound to the streptavidin resin to immobilize and isolate the complexation of probe and target. | |
According to previous studies, the analogs of CE which derived from amidation at the C-28 position with propargylamine, glycosylation at the C-3 position with D-galactose or change of aglycone from oleanane to ursane, all kept the similar activity of parental compound CE.25 In this current study, we describe the design and construction of biotinylated-CE analogue (BCEA, Fig. 1B). Potential saponin scaffold of CE was chosen as the active moiety of probe, and the PEG chain coupled 1,2,3-triazole part which synthesized by a Cu(I) catalyzed Huisgen 1,3-dipolar cycloaddition reaction was appended to the C-28 carboxylic of saponin scaffold and employed as the hydrophilic linker. The biotin moiety was applied as the tag that bound to the streptavidin resin to immobilize and isolate the complexes of probe and target.26–28
Herein, we investigate whether the parent compound CE and its probe BCEA can protect endotheliocytes subjected to ox-LDL injury on human umbilical vein endothelial cells (HUVEC).29 BCEA was employed as small molecular probe for the proteomic profiling of the protein targets of CE. By the approach of chemical proteomics through liquid chromatography with tandem mass spectrometry (LC-MS/MS), the profile of the pull-down proteins bound to BCEA were analysed. Through functional validation and pathway analysis, our data reveal that CE may function by means of reducing apoptosis to increase endothelial cells survival.
Results and discussion
Chemistry
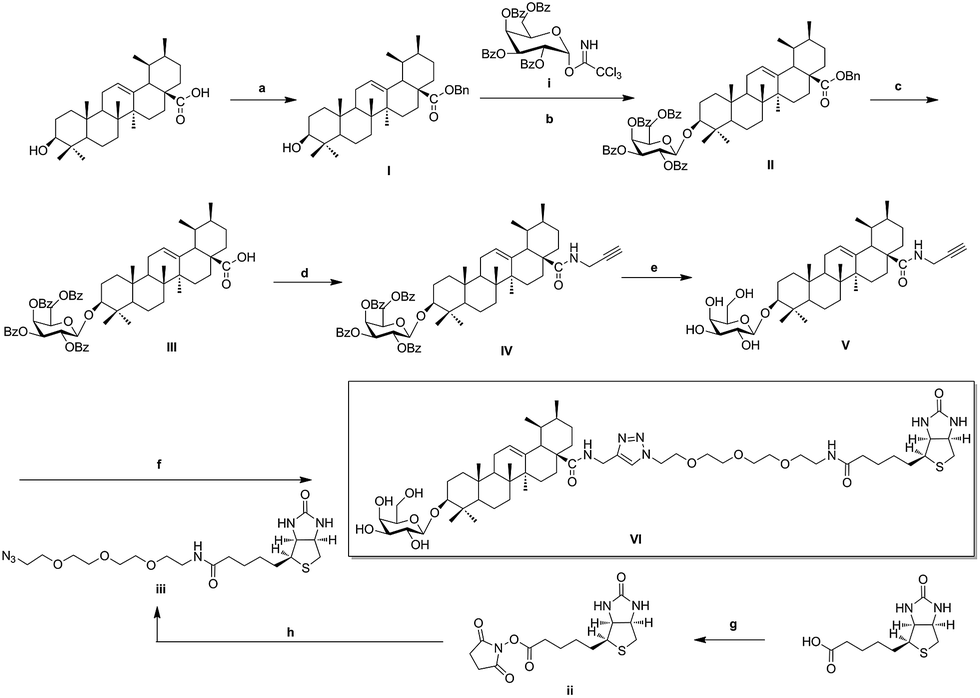
As illustrated in Scheme 1, naturally abundant ursolic acid was treated with benzyl bromide (BnBr), potassium carbonate solution (K2CO3), and tetrabutylammonium bromide (TBAB) in dry dichloromethane (DCM) to obtain a good yield of I. Glycosyl donor i was prepared from galactose, conditions reported previously by Schmidt.30 Compound I reacted with glycosyl donor i in Lewis acidic conditions trimethylsilyl trifluoromethanesulfonate (TMSOTf) to provided compound II, which was subjected to hydrogenation to obtain compound III in the presence of catalytic amount of 10% Pd–C at atmospheric pressure. Compound IV was attained via amidation with propargylamine of C-28 carboxyl group of saponin scaffold, and then followed by deprotection of glycosyl-groups in the presence of NaOMe/MeOH solution to gain Compound V. The above reaction conditions were reported previously by our previous patent.25 Biotinylated PEG chain iii with azide group exposed was synthesized according to the follow general procedures. D-Biotin reacted with N-hydroxysuccinimide (HOSu) in the 1-ethyl-3-(3-dimethyllaminopropyl)carbodiimide hydrochloride (EDCI) condition to get compound ii, which was amidated with amino-PEG3-azide to obtain biotinylated azides chain iii in 49% total yield. In the final step, Huisgen [3 + 2] cycloaddition of compound V with biotinylated azides chain iii in the presence of copper(I) thiophene-2-carboxylate (CuTC) catalyst in methanol solution resulted in the formation of 1,4 substituted triazolyl saponin derivatives VI (BCEA).31,32
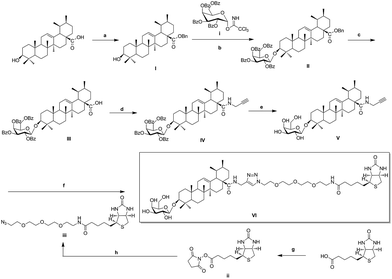 |
| | Scheme 1 Synthesis of biotinylated probe BCEA. Reagents and conditions: (a) BnBr, K2CO3, TBAB, DCM–H2O, rt, 18 h; (b) glycosyl donor i, TMSOTf, 4 Å MS, DCM, rt, 2–4 h; (c) H2, Pd–C (10%), EtOAc, reflux, 4–6 h; (d) HOBt, EDCI, propargylamine, rt, 6–8 h; (e) NaOMe, MeOH, rt, 2–3 h; (f) compound iii, CuTc, MeOH, rt, 12 h; (g) HOSu, EDCI, DMF, 12 h; (h) amino-PEG3-azide, DMF, TEA, 8 h. | |
In vitro protective activity of CE and biotinylated probe BCEA
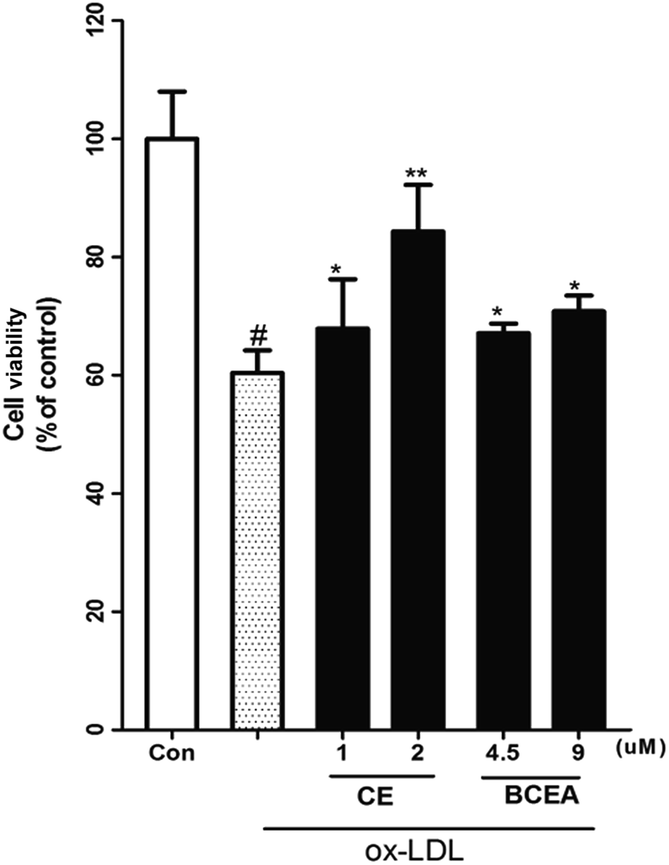
ox-LDL induces dysfunction and injury in vascular endothelial cells (VECs), which are the pathogenetic basis for atherosclerosis formation and normally used for estimation of compounds potential. Moreover, biotin coupled with linker must be attached to the active compound at a position that does not affect the potential. Here, we evaluated the endothelial protective effect of CE and its biotinylated probe (BCEA) on ox-LDL induced HUVEC damage. As shown in Fig. 2, pretreating the cells with CE or BCEA for 8 h followed by 24 h of ox-LDL exposure respectively resulted in remarkable protective effect. Although the activity is lower than that of the original compound CE (1, 2 μM), the probe BECA still showed acceptable protective activity on HUVEC (4.5, 9 μM).
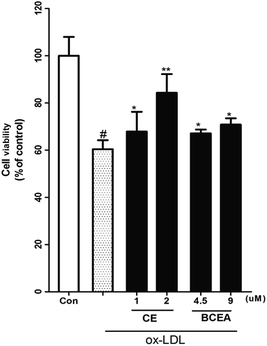 |
| | Fig. 2 Evaluation of endothelial protective effect of CE and BCEA on ox-LDL induced HUVEC damage. Cell viability of HUVEC cells incubated with different concentrations (1, 2 μM) of CE and (4.5, 9 μM) of BCEA for 8 h. Pretreatment with CE or BCEA significantly attenuated ox-LDL-induced cell injury. #P < 0.01 versus control group, **P < 0.01 versus ox-LDL treatment group, *P < 0.05 versus ox-LDL treatment group. | |
Labelling profile of BCEA by chemical proteomics approach
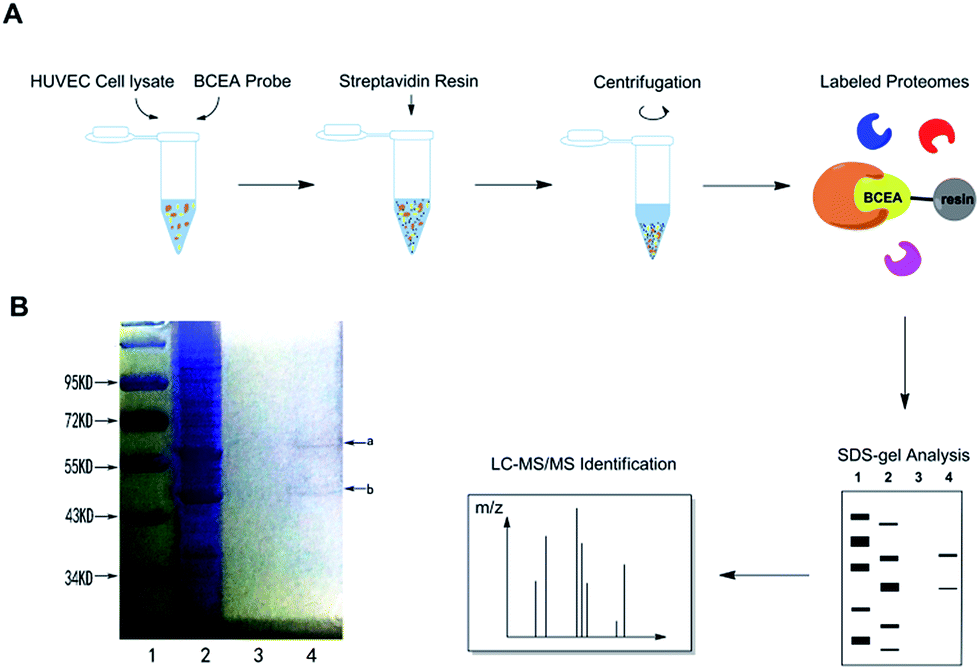
We hypothesized that CE bound to certain proteins to achieve its protective effect on ox-LDL induced HUVEC damage.29 We mixed BCEA probe with HUVEC cell lysate in order to allow specific binding proteins to attach to the active moiety of probe. For the protein isolation, the BECA–protein complexes were incubated with streptavidin resin to immobilize the proteins. After extensive washing with binding buffer, the nonbinding proteins were eluted and then the resin was boiled with SDS buffer.9 The main procedure is described in Fig. 3A. We next examined the binding proteins using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and stained the gel with coomassie blue staining reagent (Fig. 3B). BCEA binding proteins were separated by SDS-PAGE and collected by cutting the gel with the visible protein bands. Lane 1 is the marker, lane 2 is the whole cell lysate. The streptavidin resin beads were washed with binding buffer 5 times for eliminating the nonbinding proteins. The last wash elution solution was kept. Lane 3 is the last time elution solution. The resin beads were then denatured in SDS sample buffer. Lane 4 is the resin boiled with SDS buffer. There are two clear visible protein band a and band b in lane 4 which can be identified on the gel. Band a has a molecular weight between 55–72 kDa, and band b is between 43–55 kDa.
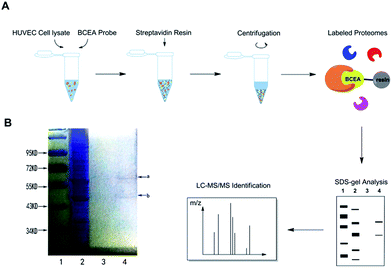 |
| | Fig. 3 Labelling profile of BCEA by chemical proteomics. (A) Schematic illustration of BCEA–protein interactions with proteomic profiling. HUVEC cell lysate incubated with BCEA probe (18 μM) for 1 h. (B) The binding proteins precipitation was separated by SDS-PAGE and stained by coomassie blue staining. | |
Identification of CE targets by LC-MS/MS and pathway analysis of CE targets
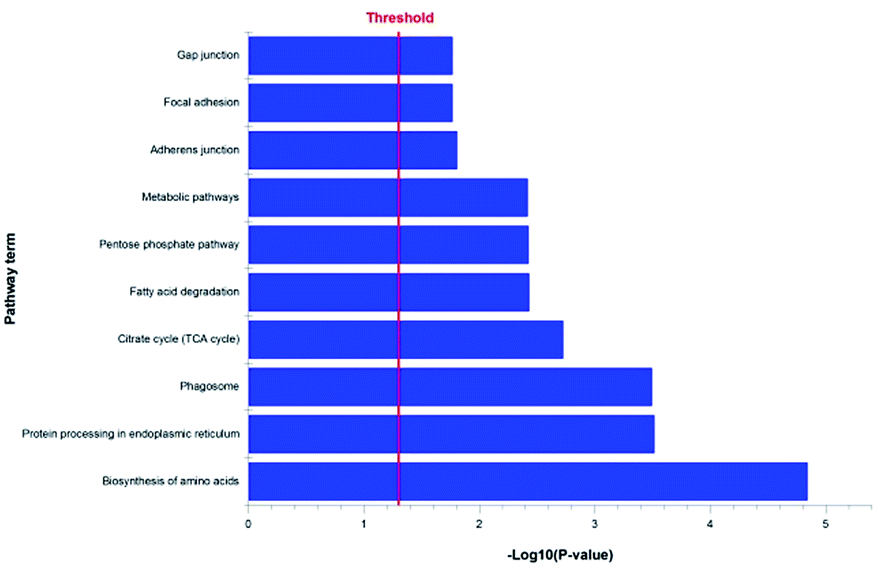
Our previous studies confirmed that some triterpenoid saponins isolated from AS could improve cell viability by means of reducing apoptotic cells. In order to explore the relevant mechanism of triterpenoid saponin CE by analyzing its protein targets and signaling pathways, the chemical proteomics has been employed to identify the probable binding targets of CE by determining the peptides molecular weight of digested protein. In our study, a total of 750 proteins were successfully identified from band a and band b using LC-MS/MS in our experiment.10,11 A unique peptide can act as a “protein tag” in protein identification.33 Only proteins identified with at least one unique peptide were considered statistically reliable hits, which resulted in a list of 744 proteins. To shrink the numbers of lower possibility of the potential targets, we then got rid of the protein with the score of zero. Consequently, we have identified 587 proteins as the more possibility targets of CE (the full list of the quantified proteins was shown in ESI Table 1†). Through the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis of these proteins, 128 proteins involved in cell survival pathways were found including protein processing in endoplasmic reticulum, citrate cycle (TCA cycle), adherens junction, gap junction and estrogen signaling pathway that associated with anti-apoptosis effect (Fig. 4). Meanwhile, we also found some else cell survival signaling pathways (PI3K-AKT, VEGF, MAPK and ER stress) that contributed to antiapoptotic activity. Taken together, these results suggested that CE could greatly improve the cell viability most likely through anti-apoptosis effect. However, it is too early to speculate which protein is the role player at this stage. We will identify all the highly scored proteins on the list in our future study.
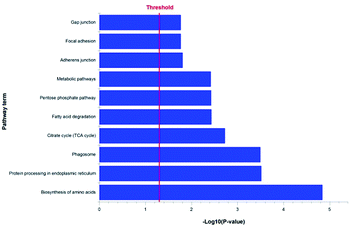 |
| | Fig. 4 The proteins function and pathway analysis of CE targets. | |
BCEA protected against ox-LDL-induced HUVEC apoptosis
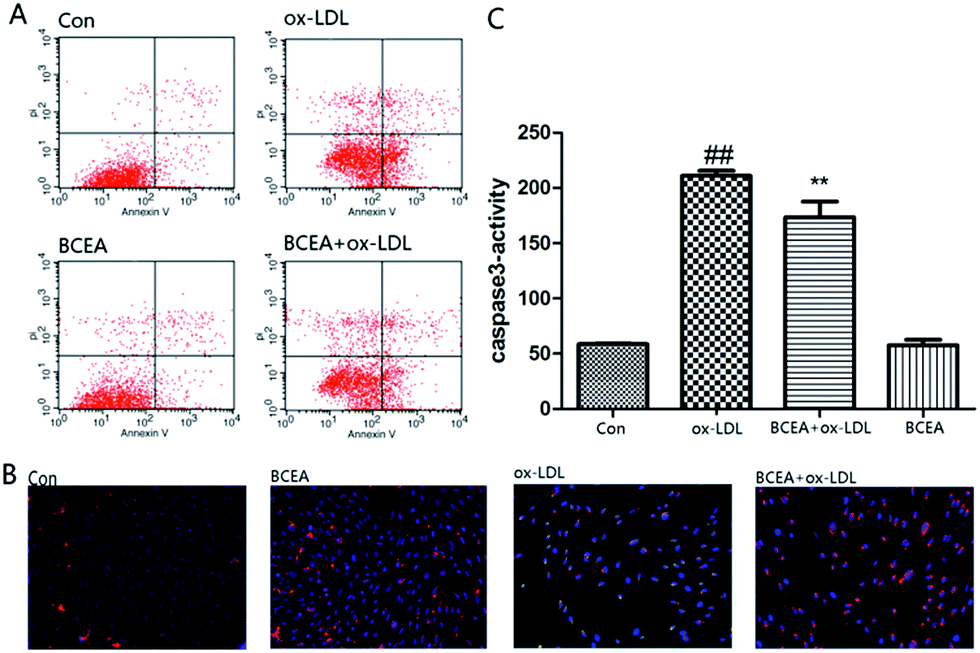
Phosphatidylserine (PS) exposure on the extra-cellular side of the cell membrane occurs during the early stage of apoptosis. PS exposure was investigated through flow cytometry under Annexin V–PI double staining. After the stimulation of ox-LDL for 24 h, the percentage of Annexin V(+)/PI(−) cells increased. In this study, the percent of Annexin V(+)/PI(−) cells was significantly reduced in the BCEA pretreatment group in Fig. 5A. Molecular function and cell component analysis found numerous mitochondrial proteins, this suggested CE may play an important role in mitochondria. The disruption of mitochondrial membrane potential is an early event in the apoptotic cascade. Mitochondrial depolarization was detected by JC-1 staining in Fig. 5B. ox-LDL group exhibited an increase in green fluorescence intensity indicating mitochondrial dissipation, while the BCEA treatment group showed clearly red fluorescence, indicating that BCEA can protect the cells against ox-LDL injury. Caspase-3 activity is the important part during apoptosis, we assessed caspase-3 activation using a fluorogenic substrate named Asp–Glu–Val–Asp–7-amino-4 trifluoromethylcoumarin (DEVD-AFC). As shown in Fig. 5C, pretreatment of BECA significantly inhibited active caspase-3 levels, which were increased in response to ox-LDL treatment.
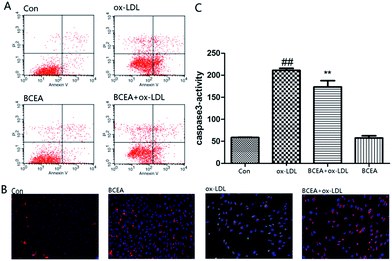 |
| | Fig. 5 BCEA protected against ox-LDL-induced HUVEC apoptosis. (A) Inhibitory effects of BCEA (9 μM, 8 h) on ox-LDL-induced HUVEC apoptosis were detected by using flow cytometry; (B) the protect effect of BCEA (9 μM, 8 h) on mitochondrial membrane potential were detected by JC-1 staining. (C) Analysis of cleaved caspase-3 activity of BCEA (9 μM, 8 h) by spectrophotometer. | |
Experimental
All the reagents were used without further purification unless otherwise specified. Solvents were dried and redistilled prior to use in the usual manner. Human umbilical vein endothelial cells (HUVECs) were isolated from several fresh human umbilical veins using 0.1% collagenase I. The neonate cords were donated by the Maternal and Child Care Service Centre in Beijing, China. Human ox-LDL (by copper ion-induced LDL oxidation, malondialdehyde >25 nmol mg−1) were obtained from Union-Bio Technology (Beijing, China). Annexin V(+)/PI(−) staining kit was obtained from Invitrogen (Eugene, OR, USA). MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] was the products of Sigma Chemical Co. (St. Louis, MO, USA). The fluorescent dye JC-1 was purchased from Sigma-Aldrich (St. Louis, MO, USA). Caspase-3 fluorometric assay kits were acquired from BioVision (CA, USA). Analytical TLC was performed using silica gel HF254. Preparative column chromatography was performed with silica gel H. 1H and 13C NMR spectra were recorded on a Bruker Advance III 600 MHz spectrometer. HRMS were obtained on a Thermofisher LTQ-Orbitrap XL.
The synthesis of compounds I–V and the glycosyl donor i were described in previous studies (Scheme 1).
Chemistry
Synthesis of compound ii. To a solution of D-biotin (500 mg, 2.05 mmol) in dry DMF (10 mL), HOSu (283 mg, 2.46 mmol) and EDCI (470 mg, 2.46 mmol) were added and the reaction mixture was stirred at room temperature for 12 h. Reaction was monitored by TLC and the suspension was filtered out and the filtrate was washed respectively by water and then methanol to give pure compound ii as white solid. (476 mg, 68% yield); 1H-NMR (600 MHz, DMSO-d6) δ: 6.40 (s, 1H, –CONH–CH–), 6.34 (s, 1H, –CONH–CH–), 4.32–4.30 (m, 1H, –CONH–CH–), 4.16–4.14 (m, 1H, –CONH–CH–), 3.12–3.09 (m, 1H, –CH–S–), 2.85–2.81 (m, 5H, –CO–CH2–CH2–CO–, –CH2–S–), 2.67 (t, J = 7.5 Hz, 2H, –CO–CH2–), 2.58 (d, J = 12.4 Hz, 1H, –CH2–S–), 1.67–1.39 (m, 6H, –CH2–CH2–CH2–CH–); 13C-NMR (150 MHz, DMSO-d6) δ: 170.2, 168.9, 162.6, 61.0, 59.2, 55.2, 39.9, 30.0, 27.8, 27.5, 25.4, 24.3.
Synthesis of compound iii. To a solution of compound ii (100 mg, 0.29 mmol) in DMF (1 mL) was dropped 11-azido-3,6,9-trioxaundecan-1-amine (63.3 mg, 0.29 mmol, 58 μmL) and TEA (58.7 mg, 0.58 mmol, 80 μL). The mixture was reacted at room temperature for 8 h. When complete, the solvent was evaporated and the crude product was subjected to column chromatography (eluent: CH2Cl2–CH3OH, 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to gain pure compound iii as white solid. (92.8 mg, 72% yield); 1H-NMR (600 MHz, MeOD) δ: 4.51–4.48 (m, 1H, –CONH–CH–), 4.32–4.30 (m, 1H, –CONH–CH–), 3.68–3.61 (m, 10H, –(OCH2CH2)2–OCH2–), 3.56–3.54 (m, 2H, –OCH2–CH2–N3), 3.39–3.35 (m, 4H, –CONH–CH2–, –COCH2–), 3.23–3.19 (m, 1H, –CH–S–), 2.93 (dd, J = 12.8 Hz, 5.0 Hz, 1H, –CH2–S–), 2.71 (d, J = 12.8 Hz, 1H, –CH2–S–), 2.24–2.21 (m, 2H, –OCH2–CH2–N3), 1.77–1.57 (m, 4H, –CH2–CH2–CH2–CH–), 1.48–1.41 (m, 2H, –CH2–CH2–CH2–CH–); 13C-NMR (150 MHz, MeOD) δ: 176.1, 166.0, 71.7, 71.6, 71.5, 71.3, 71.1, 70.6, 63.4, 61.6, 56.9, 51.8, 41.0, 40.4, 36.8, 29.7, 29.5, 26.8; HRMS calcd mass for C18H32N6O5SNa [M + Na]+ 467.2053, found 467.2051.
:1) to gain pure compound iii as white solid. (92.8 mg, 72% yield); 1H-NMR (600 MHz, MeOD) δ: 4.51–4.48 (m, 1H, –CONH–CH–), 4.32–4.30 (m, 1H, –CONH–CH–), 3.68–3.61 (m, 10H, –(OCH2CH2)2–OCH2–), 3.56–3.54 (m, 2H, –OCH2–CH2–N3), 3.39–3.35 (m, 4H, –CONH–CH2–, –COCH2–), 3.23–3.19 (m, 1H, –CH–S–), 2.93 (dd, J = 12.8 Hz, 5.0 Hz, 1H, –CH2–S–), 2.71 (d, J = 12.8 Hz, 1H, –CH2–S–), 2.24–2.21 (m, 2H, –OCH2–CH2–N3), 1.77–1.57 (m, 4H, –CH2–CH2–CH2–CH–), 1.48–1.41 (m, 2H, –CH2–CH2–CH2–CH–); 13C-NMR (150 MHz, MeOD) δ: 176.1, 166.0, 71.7, 71.6, 71.5, 71.3, 71.1, 70.6, 63.4, 61.6, 56.9, 51.8, 41.0, 40.4, 36.8, 29.7, 29.5, 26.8; HRMS calcd mass for C18H32N6O5SNa [M + Na]+ 467.2053, found 467.2051.
Synthesis of compound VI (BCEA). To a solution of compound V (10.0 mg, 15.2 mmol) in methanol (1 mL), CuTc (0.29 mg, 1.52 mmol) were added at room temperature. To this mixture, compound iii (6.8 mg, 15.2 mmol) was added and the reaction mixture was stirred until its completion. The methanol was evaporated and the crude product was purified through column chromatography (eluent: CH2Cl2–CH3OH–H2O, 50:10:1) to offer pure compound V as white solid. (13.5 mg, 81% yield). 1H-NMR (600 MHz, pyridine-d5) δ: 8.54 (s, 1H, N′-H), 8.14 (s, 1H, H-33), 8.07 (t, J = 5.3 Hz, 1H, N-H), 7.48 (s, 2H, N′′-H), 5.48 (t, J = 3.4 Hz, 1H, H-12), 4.89 (d, J = 7.7 Hz, 1H, H-1′), 4.82 (d, J = 5.3 Hz, 2H, H-31), 4.63–4.61 (m, 3H), 4.54–4.47 (4H, m), 4.37–4.35 (1H, m, H-49), 4.19 (dd, J = 3.4 Hz, 9.5 Hz, 1H), 4.14 (t, J = 6.0 Hz, 1H), 3.87 (t, J = 5.1 Hz, 2H, H-35), 3.70–3.68 (4H, m, H-36, 37), 3.63–3.58 (8H, m, H-38, 39, 40, 41), 3.42 (dd, J = 11.8 Hz, 4.4 Hz, 1H, H-3), 3.22–3.19 (m, 1H, H-47), 2.93 (dd, J = 12.5 Hz, 4.9 Hz, 1H, H-48-1), 2.87 (d, J = 12.5 Hz, 1H, H-48-2), 2.42 (d, J = 10.7 Hz, 1H, H-18), 2.38 (t, J = 7.4 Hz, 2H, H-43), 1.33 (s, 3H, CH3), 1.23 (s, 3H, CH3), 1.00 (s, 3H, CH3), 0.97 (d, J = 6.4 Hz, 3H, CH3), 0.95 (s, 3H, CH3), 0.91 (s, 3H, CH3), 0.80 (s, 3H, CH3); 13C-NMR (150 MHz, pyridine-d5) δ: 177.6, 173.0, 164.3, 145.6, 139.2, 126.1, 123.8, 107.5, 88.8, 76.7, 75.4, 73.1, 70.7, 70.6, 70.6, 70.5, 70.4, 70.3, 69.7, 62.4, 62.3, 60.5, 56.2, 55.8, 53.5, 50.2, 47.9, 47.7, 42.5, 41.0, 39.9, 39.8, 39.7, 39.5, 39.3, 38.9, 37.9, 36.8, 36.1, 35.6, 33.3, 31.1, 29.0, 28.9, 28.3, 28.2, 26.7, 26.0, 24.9, 23.8, 23.6, 21.3, 18.4, 17.4, 17.0, 16.9, 15.7; HRMS calcd mass for C57H93N7O12SNa [M + Na]+ 1122.6501, found 1122.6490.
Biological studies
Cell viability analysis. Cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as previously described.22 Briefly, HUVEC cells were plated on 96-well plates at a density of 8 × 104 cells per well. The cultures were grown at 37 °C for 24 h. Then the cells were pretreated respectively with CE (1, 2 μM) or BCEA (4.5, 9 μM) for 8 h, then flowed by ox-LDL (70 μg mL−1, 24 h). 20 μL MTT (5 mg mL−1) was added to each well and incubated for 4 h. The medium was then removed, and the formazan crystals were dissolved with dimethyl sulphoxide (DMSO). Absorbance was read at 570 nm on a microplate reader (TECAN Infinite M1000, Austria).
Biotin–neutravidin pull-down assay. HUVEC cells (1.0 × 107) were disrupted in RIPA lysis buffer and sonicated with freshly added protease inhibitor cocktail (CST). The cell lysate (200 μL) was incubated with 2 μL BCEA (18 μM) at room temperature for 1 h. The mixture was further incubated with equilibrated and packed streptavidin resin at room temperature for 1 h, which was followed by centrifugation (1 min, 2500 rpm) and five times repeated washing with 0.5 mL binding buffer to wash out nonbinding proteins. The pull-down assay was performed according to the protocol of streptavidin (20347 Pierce® Streptavidin Agarose Resins). The resin was boiled with 2× SDS loading buffer for 10 min and then electrophoresed on a 10% SDS-polyacrylamide gel. The resulting gel was visualized with coomassie blue staining.
In gel digestion. The CBB-stained SDS-PAGE gel lane was manually cut into 6 slices depending on protein molecular weight (MW). Each slice was diced into 1 mm × 1 mm pieces, and then subjected to in-gel tryptic digestion, essentially as described by Wilm.34 Briefly, the gel pieces were rinsed thrice using Milli-Q water (Millipore) and destained twice with 25 mM NH4HCO3 in 50% acetonitrile (ACN, Amer-sham) at 37 °C until the color depigmented completely. The dried gels were incubated with 50 mM tris(2-carboxyethyl)phosphine (TCEP, Sigma) in 25 mM NH4HCO3 at 56 °C for 1 h to reductively cleave the disulfide bonds of proteins and then the resulting sulfhydryl functional groups were alkylated by 100 mM iodoacetamide (IAA, Amersham) in 25 mM NH4HCO3 at room temperature in the dark for 0.5 h. Subsequently, the proteins were digested with 20 ng μL−1 porcine trypsin (modified proteomics grade, Sigma) overnight at 37 °C. The resulting tryptic peptides mixture was extracted twice from the gel pieces with 5% trifluoroacetic acid (TFA, Fluka) in 50% ACN solution. The pooled extracts were evaporated in a vacuum centrifuge (Labconco, Kansas, MO), and resuspended with 0.1% methanoic acid (Sigma) prior to the LC separation and MS detection.
LC-MS/MS analysis. All digested peptide mixtures were separated by reverse phase (RP) HPLC followed by tandem MS analysis. RP-HPLC was performed on a surveyor LC system (Thermo Finnigan, San Jose, CA). Samples were loaded into a trap column (Zorbax 300SB-C18 peptide traps, 300 μm × 65 mm, Agilent Technologies, Wilmington, DE) first at a 3 μL min−1 flow rate after the split for peptides enrichment and desalting. After flow-splitting down to about 1.5 μL min−1, peptides were transferred to the analytical column (RP-C18, 150 μm × 150 mm, Column Technology, Inc., Fremont, CA) for separation with a 195 min linear gradient from 96% buffer A (0.1% methanoic acid in water) to 50% buffer B (84% ACN, 0.1% methanoic acid in water) at a flow rate of 250 nL min−1. The analytical column was regenerated for 20 min with buffer A before loading the next sample. A thermo QE linear ion trap mass spectrometer equipped with a nanospray source was used for the MS/MS experiment in the positive ion mode. The temperature of the ion transfer capillary was set at 160 °C. The spray voltage was 3.0 kV and normalized collision energy was set at 35.0% for MS/MS. The MS analysis was performed with one full MS scan (m/z 400–1800) followed by 10 MS/MS scans on the 10 most intense ions from the MS spectrum with the dynamic exclusion settings: repeat count 2, repeat duration 30 s, exclusion duration 90 s. Data were acquired in data-dependent mode using Xcalibur software.
Bioinformatics analysis. Tandem mass spectra were searched against mascot 2.1 (Local Host) RAT protein database. The search results were then filtered using a cut off of 1% for peptide false identification rate. Peptides with Z score < 4 or delta-mass > 5 ppm were rejected. Furthermore, the minimum number of peptides to identify a protein was set to 1. The default parameters for the Quantitative software Profile Analysis 2.0 software were used throughout the analysis. Bioinformatics analysis was carried out to categorize proteins based on biological processes, cellular component and molecular function using annotations in Protein ANalysis THrough Evolutionary Relationships (PANTHER) database v 6.1 (http://www.pantherdb.org), which is in compliance with gene ontology (GO) standards. Signaling pathway analysis was performed with the tools on the Kyoto Encyclopedia of Genes and Genome (KEGG) database (http://www.genome.jp/kegg/pathway.html), respectively. Identified proteins in different groups were analyzed for molecular function, biological process or pathway term in PANTHER using the binomial test.
Assessments of cell apoptosis. The HUVECs were incubated with ox-LDL (70 μg mL−1, 24 h) and pretreated with BCEA (9 μM) 8 h before the apoptosis analysis. For the detection of early apoptosis and necrosis, double fluorescence staining was performed using an Annexin V-FITC/PI apoptosis staining kit according to the manufacturer's instructions. Cellular fluorescence was measured using flow cytometry analysis with a FACS Calibur Flow Cytometer (BD Biosciences, USA).
Determination of mitochondrial transmembrane potential (ΔΨm). We used 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1) (Sigma-Aldrich, St. Louis, USA) to analyse changes in mitochondrial transmembrane potential. HUVECs were cultured on cover slips. After removal of the ox-LDL, the cells were washed twice with warm PBS, and incubated with JC-1 (2 μM final concentration) for 30 min in the dark, finally washed twice with PBS, images were captured using a fluorescence microscope (Leica, Germany).
Analysis of caspase-3 activation. Caspase-3 activity was measured using a Fluorometric Assay Kit (BioVision, USA) according to the manufacturer's instructions. The samples were read in a Fluoroskan Ascent FL fluorometer (Thermo Fisher Scientific, USA) using 400 nm excitation and 505 nm emission wave-lengths. The results were expressed as fold change over the control.
Statistical analysis. The MTT assay was performed three times. Data are presented as mean ± SD. The groups were compared using one-way ANOVA followed by Tukey's multiple comparison tests using the statistics module of Graph Pad Prism 5.0. A value of P < 0.05 was considered statistically significant.
Conclusions
In summary, our present study found that CE possesses a profound endothelial protection effect against ox-LDL induced HUVEC damage, and reported for the first time the construction of biotinylated probe BCEA which displayed endothelial protective effect with similar efficacy as CE. The underlying mechanisms of CE-mediated endothelial protection appear to be largely dependent on the cell survival signaling pathways, which associated with anti-apoptotic activity. It would be interesting to understand these signaling pathways and identify the direct binding targets in the future. Our observations indicated that natural product CE might act as a potential lead compound, and provided a rationale for the further development of endothelial protection agent on the strength of anti-apoptotic signaling pathways.
Acknowledgements
This work was supported by the Natural Sciences Foundation of Beijing (Grant No. 7144225), the National Natural Sciences Foundation of China (Grant No. 81302656 and 81502929), and the National Science and Technology Major Project (Grant No. 2015ZX09501004-001-003), the CAMS Innovation Fund for Medical Science (CIFMS) (Grant No. 2016-I2M-1-012) and Peking Union Medical College Graduate Student Innovation Fund (Grant No. 2016-1007-06).
Notes and references
- F. Sams-Dodd, Drug Discovery Today, 2005, 10, 139–147 CrossRef CAS PubMed.
- J. P. Overington, B. AL-Lazikani and A. L. Hopkins, Nat. Rev. Drug Discovery, 2006, 5, 993–996 CrossRef CAS PubMed.
- J. Eder, R. Sedrani and C. Wiesmann, Nat. Rev. Drug Discovery, 2014, 13, 577–587 CrossRef CAS PubMed.
- K. Wierzba, M. Muroi and H. Osada, Curr. Opin. Chem. Biol., 2011, 15, 57–65 CrossRef CAS PubMed.
- D. Guiffant, D. Tribouillard, F. Gug, H. Galons, L. Meijer, M. Blondel and S. Bach, Biotechnol. J., 2007, 2, 68–75 CrossRef CAS PubMed.
- G. C. Terstappen, C. Schlüpen, R. Raggiaschi and G. Gaviraghi, Nat. Rev. Drug Discovery, 2007, 6, 891–903 CrossRef CAS PubMed.
- K. Markham, Y. Bai and G. SchmittUlms, Anal. Bioanal. Chem., 2007, 389, 461–473 CrossRef CAS PubMed.
- G. Z. Cui, L. Shan, I. K. Chu, G. H. Li, G. P. H. Leung, Y. Q. Wang, Y. W. Kwan, S. W. Chan, M. P. M. Hoi and S. M. Y. Lee, RSC Adv., 2015, 5, 74605–74610 RSC.
- X. Yi, B. Zhong, K. M. Smith, W. J. Geldenhuys, Y. Feng, J. J. Pink, A. Dowlati, Y. Xu, A. Zhou and B. Su, J. Med. Chem., 2012, 55, 3425–3435 CrossRef CAS PubMed.
- J. G. Wang, J. B. Zhang, C. J. Zhang, Y. K. Wong, T. K. Lim, Z. C. Hua, B. Liu, S. R. Tannenbaum, H. M. Shen and Q. S. Lin, Sci. Rep., 2016, 6, 22146–22153 CrossRef CAS PubMed.
- J. G. Wang, C. J. Zhang, J. B. Zhang, Y. K. He, Y. M. Lee, S. B. Chen, T. K. Lim, S. K. Ng, H. M. Shen and Q. S. Lin, Sci. Rep., 2015, 5, 7896–7907 CrossRef CAS PubMed.
- A. I. Baranov, J. Ethnopharmacol., 1982, 6, 339–353 CrossRef CAS PubMed.
- N. X. Nhiem, H. Y. Lim, P. V. Kiem, C. V. Minh, V. K. Thu, B. H. Tai, T. H. Quang, S. B. Song and Y. H. Kim, Bioorg. Med. Chem. Lett., 2011, 21, 6143–6147 CrossRef PubMed.
- G. B. Sun, H. B. Xu, F. C. Wen, W. Zhang, T. Ding and X. B. Sun, Chin. Pharmacol. Bull., 2006, 22, 1092–1095 Search PubMed.
- F. C. Wen, H. B. Xu, W. Zhang, T. Ding and X. B. Sun, World Science and Technology-Modernization of Traditional Chinese Medicine and Materia Medica, 2005, 7, 5–8 Search PubMed.
- H. W. Deng, Y. J. Li, N. Shen, X. Chen and Z. C. Zhou, Chin. J. Pharmacol. Toxicol., 1988, 2, 20–23 CAS.
- M. Wang, X. D. Xu, H. B. Xu, F. C. Wen, X. P. Zhang, H. Sun, F. Yao, G. B. Sun and X. B. Sun, J. Ethnopharmacol., 2014, 155, 240–247 CrossRef CAS PubMed.
- J. H. Lee, Y. W. Ha, C. S. Jeong, Y. S. Kim and Y. Park, Arch. Pharmacal Res., 2009, 32, 831–840 CrossRef CAS PubMed.
- M. Wang, X. B. Meng, Y. L. Yu, G. B. Sun, X. D. Xu, X. P. Zhang, D. Xin, J. X. Ye, H. B. Xu, Y. F. Sun and X. B. Sun, Apoptosis, 2014, 19, 1727–1735 CrossRef CAS PubMed.
- M. Wang, G. B. Sun, J. Y. Zhang, Y. Luo, Y. L. Yu, X. D. Xu, X. B. Meng, M. D. Zhang, W. B. Lin and X. B. Sun, Int. J. Cardiol., 2015, 185, 167–176 CrossRef PubMed.
- Y. Luo, X. Dong, Y. L. Yu, G. B. Sun and X. B. Sun, J. Ethnopharmacol., 2015, 163, 241–250 CrossRef CAS PubMed.
- Z. Wang, S. Song, H. Lu, G. Chen, S. Xu, Y. Sagara, N. Kitaoka, M. Manabe and H. Kodama, Clin. Chim. Acta, 2003, 336, 65–72 CrossRef CAS.
- M. Hu, K. Ogawa, Y. Sashida and P. G. Xiao, Phytochemistry, 1995, 39, 179–184 CrossRef CAS.
- M. Y. Shi, Y. Yang, L. M. Cheng, S. Zhao, Y. T. Sun, H. B. Xu, J. P. Fawcett, X. B. Sun and J. K. Gu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 951–952, 129–134 CrossRef CAS PubMed.
- X. D. Xu, Y. Tian, G. B. Sun, X. B. Sun and M. Wang, Faming Zhuanli Shenqing Gongkai Shuomingshu, 2015, CN105111272A Search PubMed.
- G. C. Zhou, F. Liu, J. T. Wan, J. Wang, D. C. Wang, P. Wei and P. K. Ouyang, Eur. J. Med. Chem., 2013, 70, 631–639 CrossRef CAS PubMed.
- M. J. Evans and B. F. Cravatt, Chem. Rev., 2006, 106, 3279–3301 CrossRef CAS PubMed.
- P. Y. Yang, K. Liu, M. H. Ngai, M. J. Lear, M. R. Wenk and S. Q. Yao, J. Am. Chem. Soc., 2010, 132, 656–666 CrossRef CAS PubMed.
- M. Qin, Y. Luo, X. B. Meng, M. Wang, H. W. Wang, S. Y. Song, J. X. Ye, R. L. Pan, F. Yao, P. Wu, G. B. Sun and X. B. Sun, Vasc. Pharmacol., 2015, 70, 23–34 CrossRef CAS PubMed.
- R. R. Schmidt and J. Michel, Angew. Chem., Int. Ed., 1980, 19, 731–732 CrossRef.
- Y. Tian, Y. H. Wang, H. Shang, X. D. Xu and Y. F. Tang, Org. Biomol. Chem., 2015, 13, 612–619 CAS.
- H. Shang, Y. Wang, Y. Tian, J. Feng and Y. Tang, Angew. Chem., Int. Ed., 2014, 53, 5662–5666 CrossRef CAS PubMed.
- Y. P. Zhao and Y. H. Lin, Genomics, Proteomics Bioinf., 2010, 8, 33–41 CrossRef CAS.
- M. Wilm, A. Shevchenko, T. Houthaeve, S. Breit, L. Schweigerer, T. Fotsis and M. Mann, Nature, 1996, 379, 466–469 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25572h |
| ‡ These two authors contributed equally to this paper. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Shan Wang‡,
Hai Shang,
Min Wang,
Guibo Sun*,
Xudong Xu* and
Xiaobo Sun*
,
Shan Wang‡,
Hai Shang,
Min Wang,
Guibo Sun*,
Xudong Xu* and
Xiaobo Sun*