
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Retracted Article: Reduction of neuronal damage and promotion of locomotor recovery after spinal cord injury by early administration of methylprednisolone: possible involvement of autophagy pathway
Yichao Jin†
,
Shaofeng Yang† and
Xiaohua Zhang*
Department of Neurosurgery, Shanghai Renji Hospital, Shanghai Jiaotong University, School of Medicine, Shanghai 200127, People's Republic of China. E-mail: zhangxh1969@hotmail.com; Fax: +86-21-58394262; Tel: +86-21-34506561
First published on 12th January 2017
Abstract
Autophagy is involved in the secondary damage associated with spinal cord injury (SCI), however, the role of autophagy remains to be elucidated. There are no reports regarding changes in autophagy after methylprednisolone (MP) treatment after acute SCI. In the present study, we examined changes in autophagy and apoptosis and explored the possible relationship between autophagy and apoptosis. We found that SCI produced by clamp force in rats caused apoptotic cell death in the injured area at different time points and MP treatment could significantly decrease the number of apoptotic cells; we also found that SCI could induce an increase in autophagy in the injured area and both neurons and astrocytes were co-stained with autophagic biomarkers, including microtubule-associated protein light chain-3 and Beclin-1, and an apoptotic biomarker, cysteinyl aspartate specific proteinase-3; MP treatment could also promote the locomotor recovery after SCI. The results suggest an increase in autophagy after MP treatment may be a neuroprotective mechanism via inhibition of apoptosis.
1. Introduction
Spinal cord injury (SCI) often results in permanent neurologic deficits, whose outcome depends on the extent of secondary damage produced by a series of cellular and molecular events initiated by the primary trauma, including inflammation, edema, ischemia hemorrhage, electrolyte imbalance, arachidonic acid release, glutamate excitotoxicity, apoptosis and lipid peroxidation leading to membrane lysis.1–6 Apoptosis, or programmed cell death type-I, induced through multiple mechanisms such as oxidative stress, activation of death receptor tumor necrosis factor-α, expression of death receptor ligand FAS and caspase activation, may play a pivotal role in secondary SCI.2,3,7–13 Another type of programmed cell death-autophagic cell death is also involved in the secondary damage associated with SCI.14–20 However, whether the function of autophagy is protective or detrimental in SCI remains to be elucidated.One of the aims of current therapies is to reverse the post-traumatic cell death caused by SCI to preserve the remaining intact structure and function of spinal cord.15 Methylprednisolone (MP) has been used as the drug treatment of choice for more than a decade in humans suffering from an SCI, although some debates have recently arisen.21 J. Vaquero et al. found that early administration of a single bolus of MP decreased apoptotic cell death after SCI.21 As to changes in autophagy after administration of MP for treatment of acute SCI, Chen et al. adopted a contusion SCI model and observed an inhibition in autophagy expression with MP treatment in neurons soon after contusion injury.22 However, whether autophagic cell death is beneficial or detrimental after SCI remain to be determined.
This study used an animal model of SCI produced by clamp force in adult rats to determine the effects of MP treatment on apoptosis and autophagy expression.
2. Experimental
2.1 Ethics statement
All animal procedures were approved by the animal care and experimental committee of the school of medicine, Shanghai Jiaotong University (Shanghai, China). All animal experiments were carried out at Shanghai Jiaotong University School of Medicine following national institutes of Health guidelines and local Institutional Animal Care and Use Committee regulations. Adult male Sprague-Dawley rats were used in present study. Rats were housed in individual cages in a temperature- and humidity-controlled animal facility with a 12 h light/dark cycle. Rats were housed in the animal facility for at least 7 days before surgery and were given free access to food and water during this period.2.2 Experimental design
A total of 216 adult male Sprague-Dawley rats weighing 280–320 g were divided into three groups randomly: SCI (SCI, n = 72), SCI with intraperitoneal injection of saline (saline, n = 72) and SCI with intraperitoneal injection of MP (MP, 30 mg kg−1 body weight, n = 72). All rats, except those used in the behavioral tests, were sacrificed at 6 h, 24 h or 72 h after SCI. For hematoxylin and eosin (HE) staining, terminal deoxyribonucleotidyl transferase (TdT)-mediated biotin-16-dUTP nick-end labeling (TUNEL) staining and immunohistochemical analysis, 54 (18 for each group) rats were perfused transcardially with 4% paraformaldehyde in phosphate-buffered saline (PBS) at 6 h, 24 h or 72 h after SCI. Specimens were removed, further fixed at 4 °C overnight; for western blotting, 54 (18 for each group) rats were perfused transcardially with saline and then specimens were removed and preserved in −80 °C freezer; for EM, 54 (18 for each group) rats were perfused transcardially with 2.5% glutaraldehyde and 2.5% paraformaldehyde in 0.1 mmol L−1 cacodylate buffer (pH 7.4) and then specimens were post-fixed in the same fixatives for 24 h; 54 (18 for each group) rats were used for behavioral assessment.2.3 SCI induction
Rats were anaesthetized with an intraperitoneal injection of 10% chloral hydrate (3.3 ml kg−1 body weight) and then fixed in the prone position. A T5–T9 midline skin incision was made, and the paravertebral muscles were dissected. From T6 to T8, the spinous processes were removed, and a laminectomy was performed. The dura was left intact. An aneurysm clip with a 70 g closing force (Yasargil FE 721, Aesculap, Germany) was applied to the T7 level of the spinal cord for 1 min.23,24 At the end of the procedure, the clip was removed, and the surgical wound was closed in layers with silk sutures. The drugs were administered intraperitoneally immediately after the wound was closed. Bladders were expressed twice a day until spontaneous voiding began.2.4 HE staining
Rats were subjected to deep anesthesia by 10% chloral hydrate. At 6 h, 24 h and 72 h after SCI, all rats were perfused transcardially with 4% paraformaldehyde in PBS. Specimens were removed, further fixed at 4 °C overnight, and immersed in 30% sucrose/PBS at 4 °C overnight. Specimens were mounted in OCT. Serial sections were obtained using a cryostat and stained with toluidine blue for 30 min and then 2–3 drops of glacial acetic acid. Once the nucleus and granulation were clearly visible, sections were mounted in Permount or Histoclad.Sections were cut in a microtome and adhered to glass slides with polylysine. Images of the ipsilateral hippocampus were captured at 200× by using a microscope (Nikon Labophot; Nikon USA, Melville, NY).
2.5 TUNEL staining
TUNEL staining was used to detect cell death of injured spinal cord. A TUNEL assay was performed according to the manufacturer's directions by using an In Situ Cell Death Detection Kit (Roche, USA). In brief, sections were washed for 30 min, 10 min each. Then, sections were incubated in 0.1% Triton-X and 0.1% sodium citrate, and rinsed three times with PBS for 10 min each time. Sections were incubated in 0.3% H2O2 in PBT for 30 min to inhibit endogenous peroxidase activity and then rinsed with PBS. Sections were then incubated with 50 μl TUNEL reaction mixture (In Situ Cell Death Detection Kit, Fluorescein, Roche, USA) in a humidified atmosphere for 60 min at 37 °C in the dark. Then, sections were rinsed three times with PBS. Sections were then observed under a fluorescence microscope (Nikon TE300, Japan). Negative control was carried out by incubating sections in 50 μl of label solution without terminal transferase instead of TUNEL reaction mixture per well. 4,6-Diamino-2-phenyl indole (DAPI) staining was used for total cell count. At least ten randomly selected microscopic fields were used for counting the TUNEL-positive cells (400× magnifications). Cell counting was conducted by an investigator blinded to the group conditions. The average number of TUNEL-positive cells of total cell number in 10 microscopic fields per section was evaluated as cell death index.2.6 Immunohistochemical analysis
The 4 μm-thick formalin-fixed OCT-embedded sections were subjected to immuno-fluorescence analysis to determine the immunoreactivity of cysteinyl aspartate specific proteinase-3 (Caspase-3), microtubule-associated protein light chain-3 (LC3) and Beclin-1. Endogenous peroxidase was blocked with 3% H2O2 for 5 min followed by a brief rinse in distilled water and a 15 min wash in PBS. Sections were cooled at room temperature for 20 min and rinsed in PBS. Nonspecific protein binding was blocked with an incubation in 5% horse serum for 40 min. Sections were incubated with primary antibodies (anti-LC3 [CST3868], anti-Beclin-1 [CST3738], anti-Caspase-3 [CST9662] and anti-IgG, all diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200; from Suzhou Ard Biological Co., Ltd.) for 1 h at room temperature, followed by a 15 min wash in PBS. Sections were incubated with FITC/Cy3-conjugated IgG (1:500 dilution; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 60 min at room temperature. For negative controls, sections were incubated in the absence of a primary antibody. From each brain, ten microscopic fields per section from five to eight brain sections separated by at least 150 μm were photographed using a random number generator for counting the LC3 and Beclin-1-positive cells (400× magnifications, Nikon TE300, Japan).
:200; from Suzhou Ard Biological Co., Ltd.) for 1 h at room temperature, followed by a 15 min wash in PBS. Sections were incubated with FITC/Cy3-conjugated IgG (1:500 dilution; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 60 min at room temperature. For negative controls, sections were incubated in the absence of a primary antibody. From each brain, ten microscopic fields per section from five to eight brain sections separated by at least 150 μm were photographed using a random number generator for counting the LC3 and Beclin-1-positive cells (400× magnifications, Nikon TE300, Japan).
2.7 Immunofluorescent double labeling
Double immunofluorescence was used to identify expression of cell type markers in autophagic biomarker-positive and apoptotic biomarker-positive cells. The expression of neuronal nuclei (NeuN) and glial fibrillary acidic protein (GFAP) were determined for neurons and astrocytes, respectively. The primary antibodies for immunofluorescence were mouse anti-rat NeuN (1:200, Santa Cruz Biotechnology), mouse anti-rat GFAP (1:100, Santa Cruz Biotechnology), rabbit anti-rat LC3 (1:200, Santa Cruz Biotechnology), rabbit anti-rat Beclin-1 (1:200, Santa Cruz Biotechnology) and Caspase-3 (1:200, Cell Signaling Technology). The sections were incubated with a primary antibody in PBS with 1% bovine serum albumin for 30–40 min at room temperature followed by washing and application of secondary antibodies. The secondary antibodies were Alexa Fluor 568 goat anti-mouse for NeuN and GFAP (Invitrogen, Carlsbad, CA, USA), and Alexa Fluor 488 goat anti-rabbit for LC-3, Beclin-1 and Caspase-3 (Invitrogen). We performed double labeling for NeuN and GFAP/LC-3, Beclin-1 or Caspase-3 to localize expression of LC-3, Beclin-1 and Caspase-3. After final wash, sections were protected with cover slips with anti-fading mounting medium sealed with nail polish and stored at 4 °C for preservation.
2.8 Western blot analysis
The frozen spinal cord was mechanically lysed in 20 mM Tris (pH 7.6), containing 0.2% sodium dodecyl sulfate (SDS), 1% Triton X-100, 1% deoxycholate, 1 mM phenylmethylsulfonyl fluoride, and 0.11 IU ml−1 aprotinin (all purchased from Sigma-Aldrich, Inc.). The lysates were centrifuged at 12000g for 20 min at 4 °C. The protein concentration was estimated by the Bradford method. The samples (60 μg per lane) were separated by 12% SDS polyacrylamide gel electrophoresis and electro-transferred onto a polyvinylidene-difluoride membrane (Bio-Rad Lab, Hercules, CA). The membrane was blocked with 5% skimmed milk for 1 h at room temperature and incubated with primary antibodies against LC-3 and Beclin-1 (1:1000 dilutes, both from CST), Caspase-3 (1:500, Abcom), Bcl-2 (1:1000, Santa Cruz Biotechnology), Bax (1:1000, Santa Cruz Biotechnology) and p62 (1:1000, CST) at 4 °C overnight. β-Actin (diluted in 1:50000, Sigma-Aldrich) was used as the loading control. After the membrane was washed three times in TBS + Tween-20 (TBST) for 10 min each, it was incubated in the appropriate horseradish peroxidase-conjugated secondary antibody (diluted 1:10000 in TBST) for 2 h. Then, the membrane was washed three times in TBS + Tween-20 (TBST) for 10 min each. The blotted protein bands were visualized by enhanced chemiluminescence western blot detection reagents (Millipore, IL) and exposed to X-ray film. The developed films were digitized using an Epson Perfection 2480 scanner (Seiko Corp, Nagano, Japan). The results were quantified by Quantity One Software (BioRad) and normalized to a loading control, β-actin. Values from SCI group were used as 100%.
2.9 Transmission electron microscopy
For electron microscopy, injured spinal cords were fixed in phosphate-buffered glutaraldehyde (2.5%) and osmium tetroxide (1%). Dehydration of the samples was accomplished in acetone solutions at increasing concentrations. The tissue was embedded in an epoxy resin. Semi-thin (1 μm) sections of the samples were then made and stained with toluidine blue. Then, 600 Å-thin sections were made from a selected area of tissue defined by the semi-thin section, and these sections were stained with lead citrate and uranyl acetate. The ultrastructure of the sections was observed under a transmission electron microscope (JEM-1200X). From each brain, five to eight 13500× fields from five to eight brain sections were chosen for analysis.
2.10 Behavioral testing
Two weeks before surgery, all rats were trained to adapt to a standardized open field (a molded plastic tube with a smooth floor, 900 mm in diameter with a wall height of 70 mm).25 At different time points (ctrl, 3 d, 7 d, 21 d) after SCI and administration of MP or vehicle, Basso, Beattie and Bresnahan (BBB) scoring protocol was used to analyze behavior as described previously.25,26 Experienced handlers placed the rats in the center of the field, where they were observed for 4 min, and then placed back in their cages. All experiments were performed in a double-blind manner.2.11 Statistical analysis
All data are presented as the mean ± standard error of mean (SEM). SPSS 17.0 (SPSS Inc., Chicago, IL) was used for statistical analysis of the data. All data, including biochemical changes in autophagy and apoptosis and the TEM results, were subjected to a one-way analysis of variance (ANOVA) followed by Bonferroni's post hoc test. The behavioral outcomes, evaluated with BBB scores, were subjected to a two-way ANOVA. Statistical significance was inferred at p < 0.05.3. Results
3.1 HE staining
In injured rats, paraplegia was observed immediately after SCI and there were no signs of complete functional recovery throughout the entire follow-up period. Zones of microhemorrhages, tissue edema and signs of neurodegeneration were detected throughout the entire follow-up period. However, in the MP-treated animals, a lesser degree of tissue edema was observed in the zone of lesion (Fig. 1).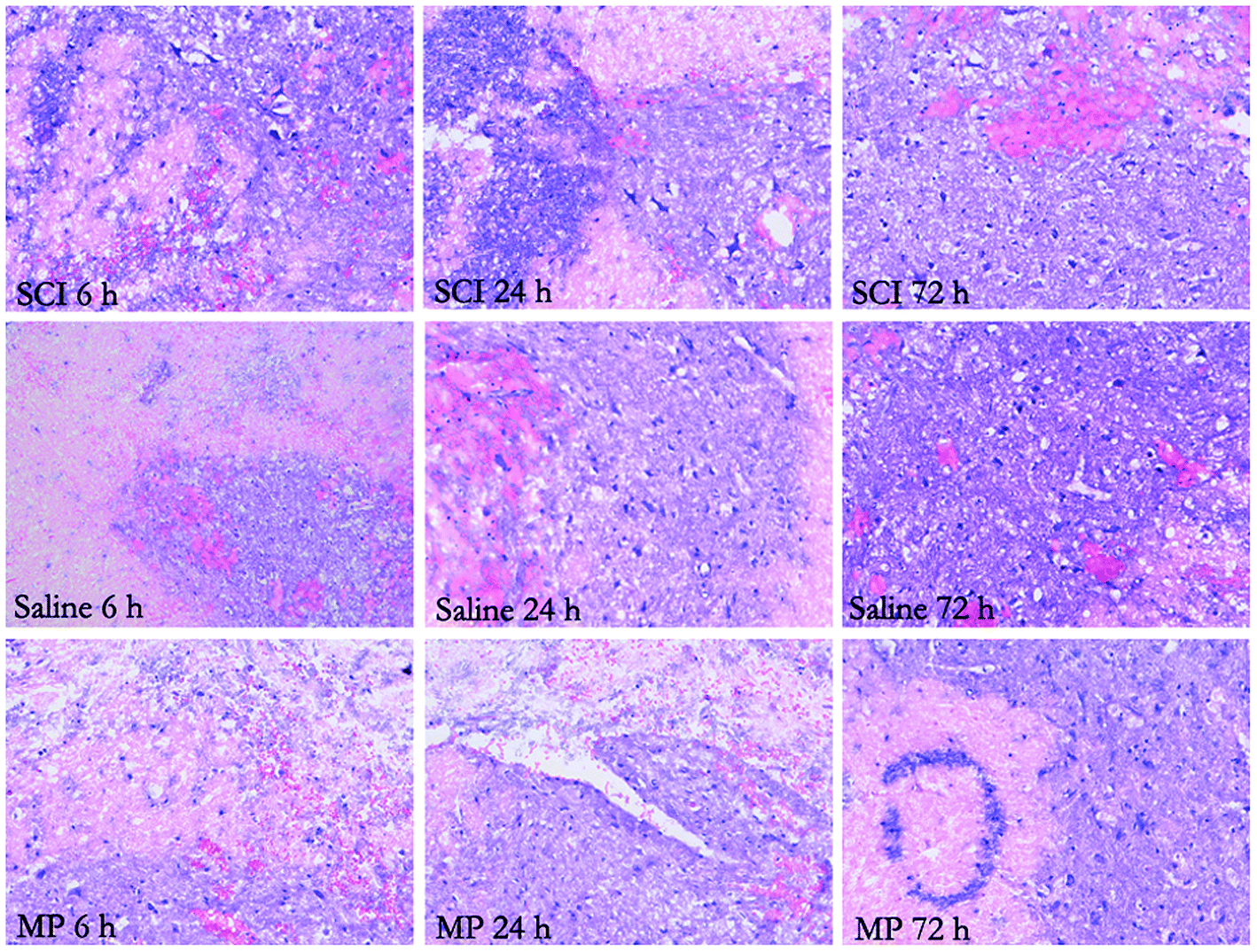 | ||
| Fig. 1 Hematoxylin and eosin staining of the injured spinal cord at 6 h, 24 h and 72 h after SCI. Zones of microhemorrhages, tissue edema and signs of neurodegeneration were detected. A lesser degree of tissue edema was observed in the zone of lesion of the MP-treat animals. | ||
3.2 MP treatment could decrease apoptotic cell death after SCI
 | ||
| Fig. 2 Terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) staining of the injured spinal cord at different time points. Arrows indicate TUNEL-positive cells (green: nucleus; blue: nucleus). The average number of TUNEL-positive cells of total cell number was evaluated as cell death index. Magnification, 100×. (**p < 0.01, ***p < 0.001; MP vs. saline). | ||
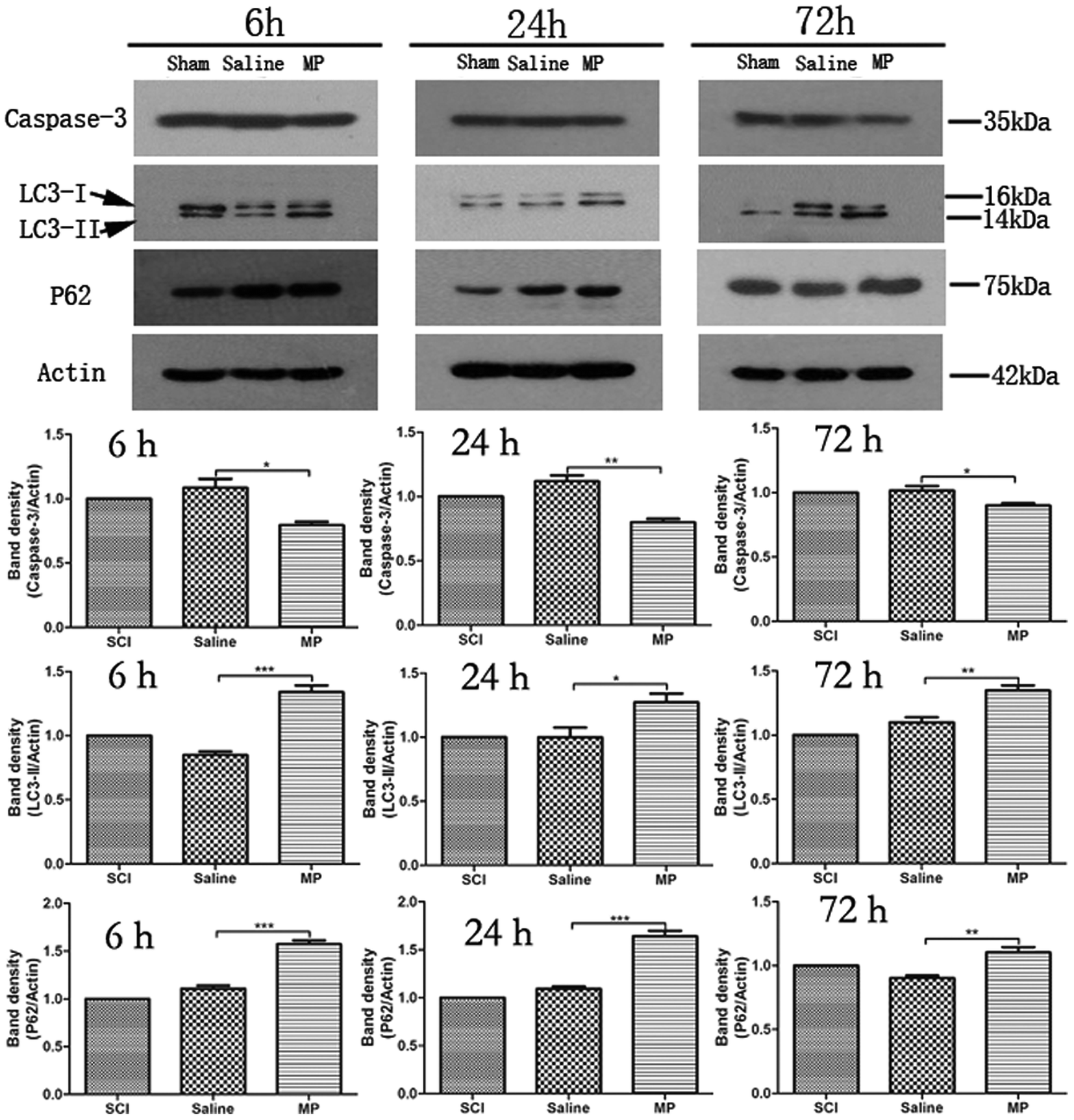 | ||
| Fig. 3 Immunoblots of Caspase-3, LC3-II and p62 in injured spinal cord. β-Actin was used as load control. MP treatment could significantly down-regulate expression of Caspase-3 and up-regulate the expression of LC3-II and p62. (*P < 0.05, **P < 0.01, ***p < 0.001; MP vs. saline). | ||
3.3 MP treatment activates autophagy
Immunohistochemical staining of LC3 and Beclin-1 was adopted to assess the influence of MP treatment on autophagy at each time point. Saline administration had no effect on the expression of LC3 and Beclin-1. The number of cells expressing LC3 and Beclin-1 were increased in MP group compared with the SCI group and saline group at each time point (Fig. 4 and 5). | ||
| Fig. 4 Immunofluorescent analysis of LC3 expression in injured spinal cord. The average number of LC3-positive cells of total cell number was evaluated. MP treatment could significantly increase the LC3-positive cells in the injured spinal cord. Arrows indicate LC3-positive cells (green: cytoplasm; blue: nucleus). **p < 0.01; ***p < 0.001. Magnification, 400×. | ||
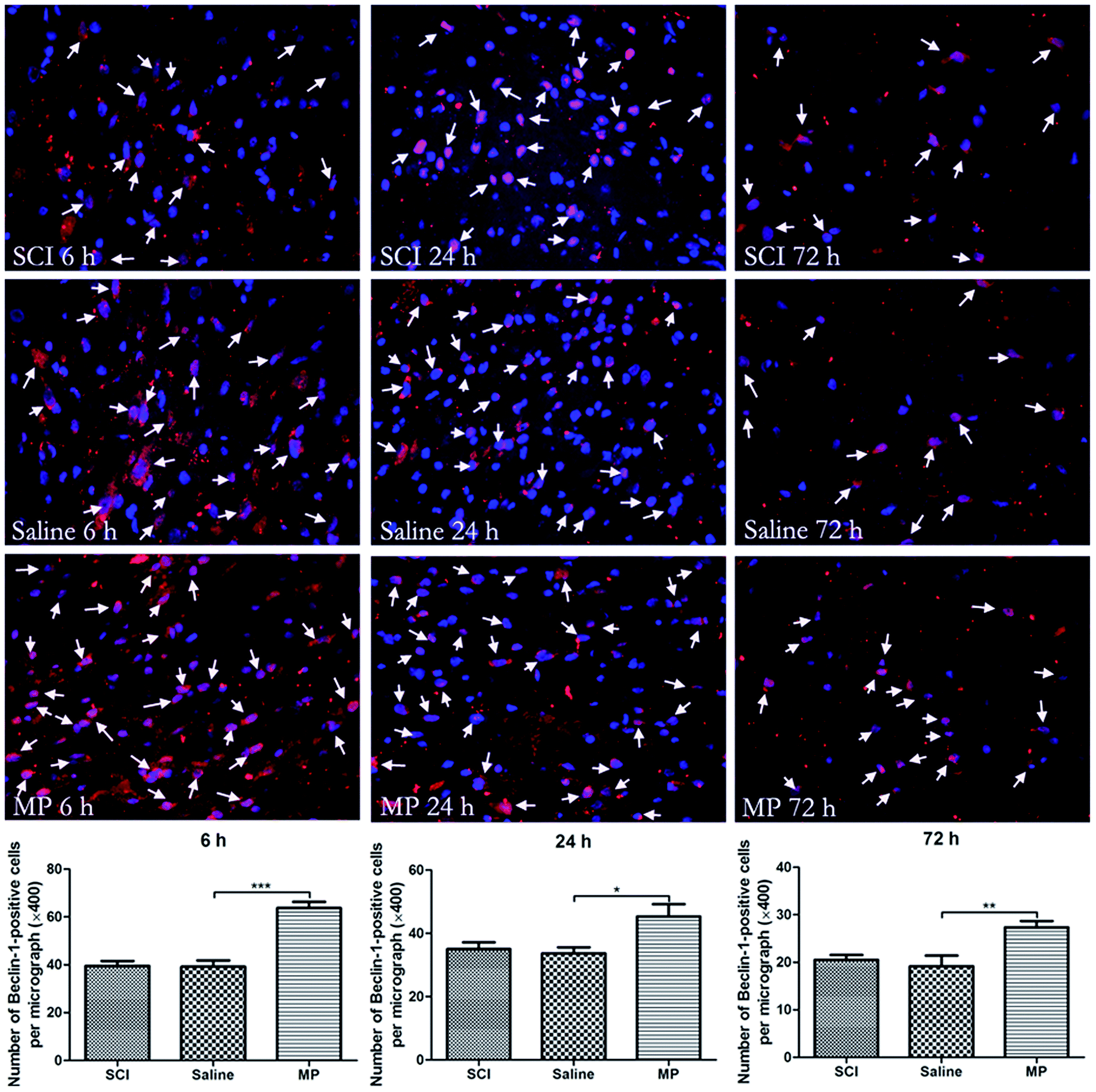 | ||
| Fig. 5 Immunofluorescent analysis of Beclin-1 expression in injured spinal cord. The average number of Beclin-1-positive cells of total cell number was evaluated. MP treatment could significantly increase the number of Beclin-1-positive cells in the injured spinal cord. Arrows indicate Beclin-1-positive cells (green: cytoplasm; blue: nucleus). *p < 0.05, **p < 0.01; ***p < 0.001. Magnification, 400×. | ||
To investigate the role of MP treatment on autophagy, western blots of LC3-II expressions were performed. Saline administration had no effect on the expression of LC3-II. Compared with saline group, MP treatment could significantly increase the LC3-II expression at each time point (6 h: 135.8% ± 4.2% vs. 79.5% ± 3.4%; 24 h: 125.6% ± 2.9% vs. 105.8% ± 3.1%; 72 h: 138.6% ± 3.6% vs. 109.6% ± 4.1%; MP vs. saline). (Fig. 3).
The appearance of autophagosomes (APs) and autolysosomes (ALs) under transmission electron microscopy is a morphologic hallmark unique to autophagy. Acute SCI could induce ultrastructural changes including accumulation of APs, ALs and mitochondrial swelling. Saline administration had no effect on the accumulation of APs and ALs. Number of APs and ALs was significantly increased after MP treatment compared with SCI group and saline group (6 h: 7.8 ± 0.8 vs. 4.2 ± 0.7; 24 h: 6.5 ± 0.9 vs. 3.5 ± 0.4; 72 h: 4.5 ± 0.6 vs. 2.8 ± 0.3; MP vs. saline) (Fig. 6)
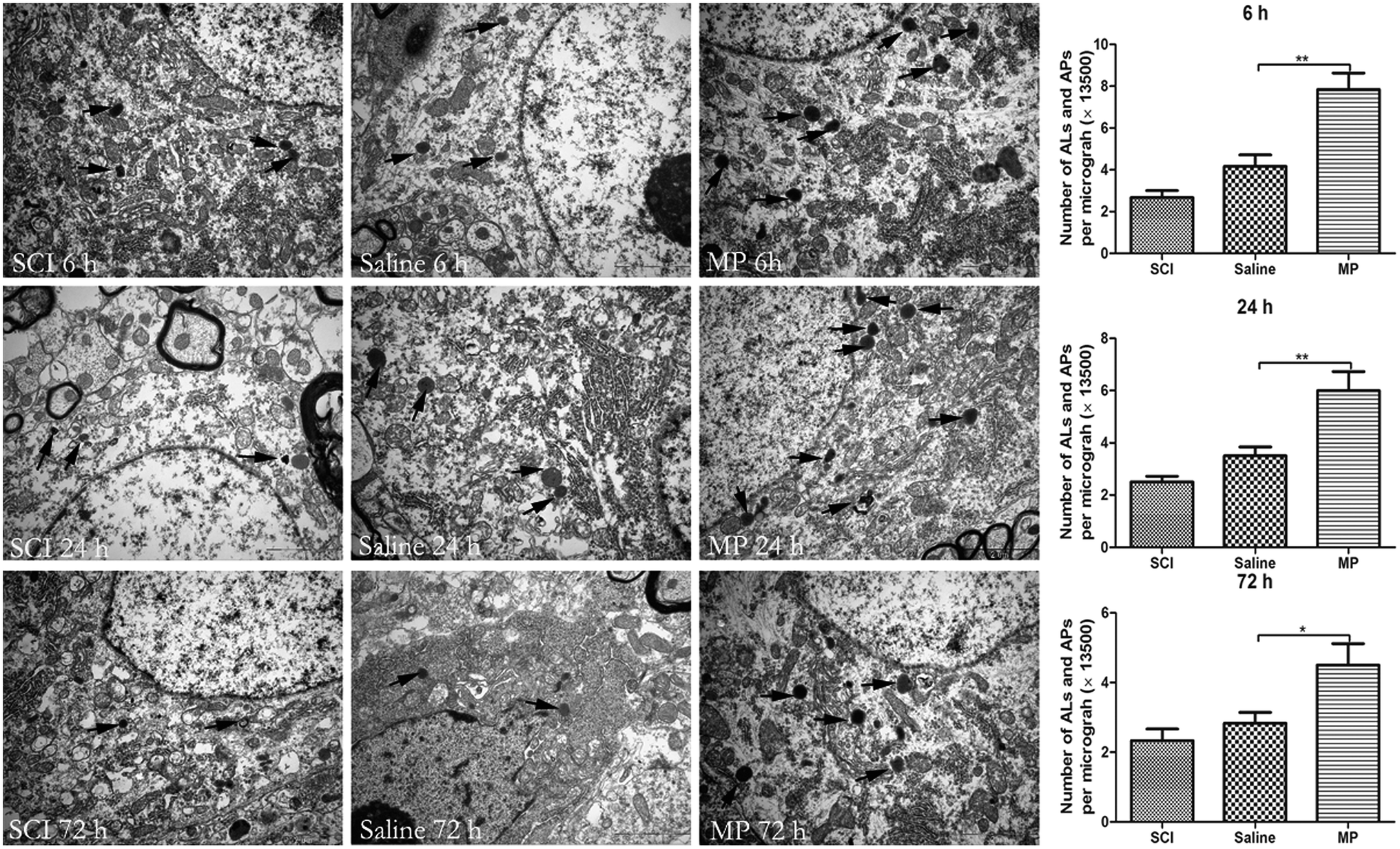 | ||
| Fig. 6 Electron micrographs of autophagy-related vesicular compartments. Scale bar is 2 μm. Arrows indicate the autophagosomes and autolysosomes. *p < 0.05; **p < 0.01. | ||
3.4 MP treatment activates “autophagic flux”
The protein p62 is a substrate of the autophagic process, and its expression level is a marker of autophagic flux in vivo.27 Western blot of p62 was used to analyze the degree of autophagic flux. Saline administration had no effect on autophagic flux after SCI. Compared with saline group, MP treatment could significantly increase the p62 expression at each time point (6 h: 152.7% ± 5.2% vs. 109.8% ± 3.4%; 24 h: 110.6% ± 1.9% vs. 162.5% ± 3.8%; 72 h: 113% ± 3.2% vs. 93.8% ± 2.9%; MP vs. saline). (Fig. 3).3.5 Autophagy and apoptosis occur in both neurons and astrocytes
To study the location of autophagy and apoptosis, immunohistochemical staining of Beclin-1, LC3, Caspase-3 and other markers (such as NeuN, which indicates neurons and GFAP, which indicates astrocytes) was performed. Both autophagy and apoptosis were co-localized with neurons and astrocytes. (Fig. 7, 8 and 9).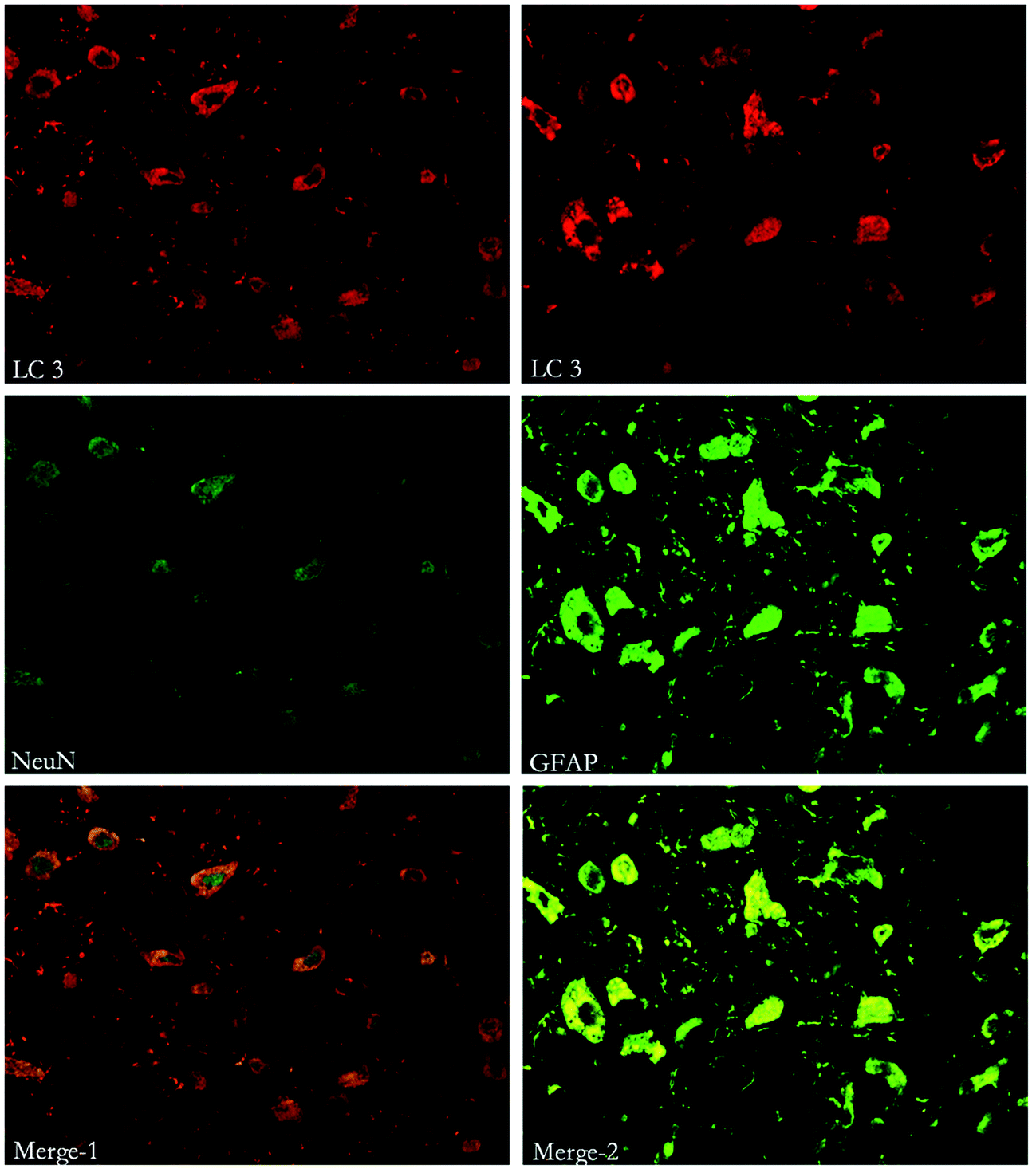 | ||
| Fig. 7 Immunofluorescent double labeling of LC3 and NeuN or GFAP. The results indicate that cells expressing LC3 were co-stained with both NeuN and GFAP. | ||
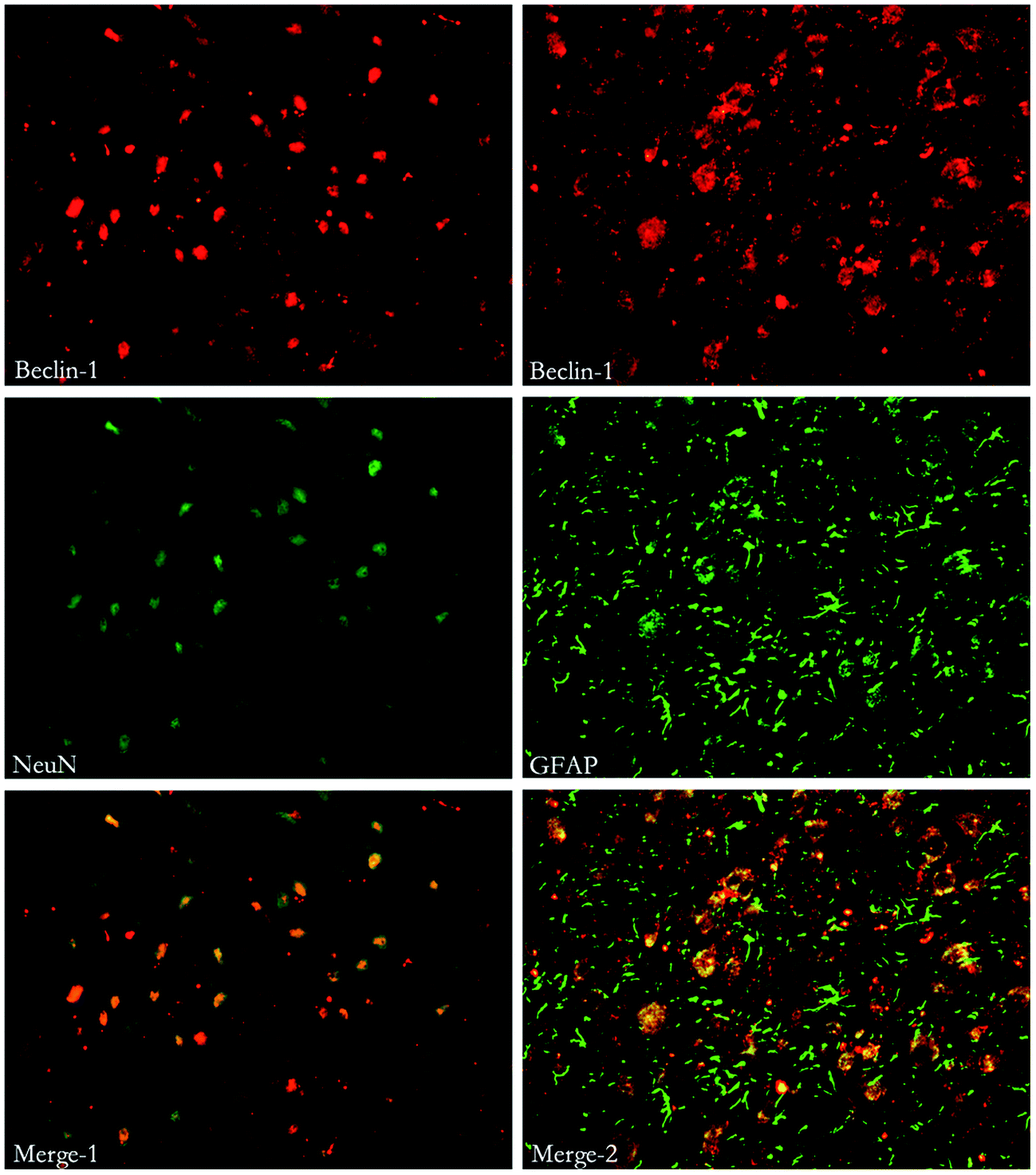 | ||
| Fig. 8 Immunofluorescent double labeling of Beclin-1 and NeuN or GFAP. The results indicate that cells expressing Beclin-1 were co-stained with both NeuN and GFAP. | ||
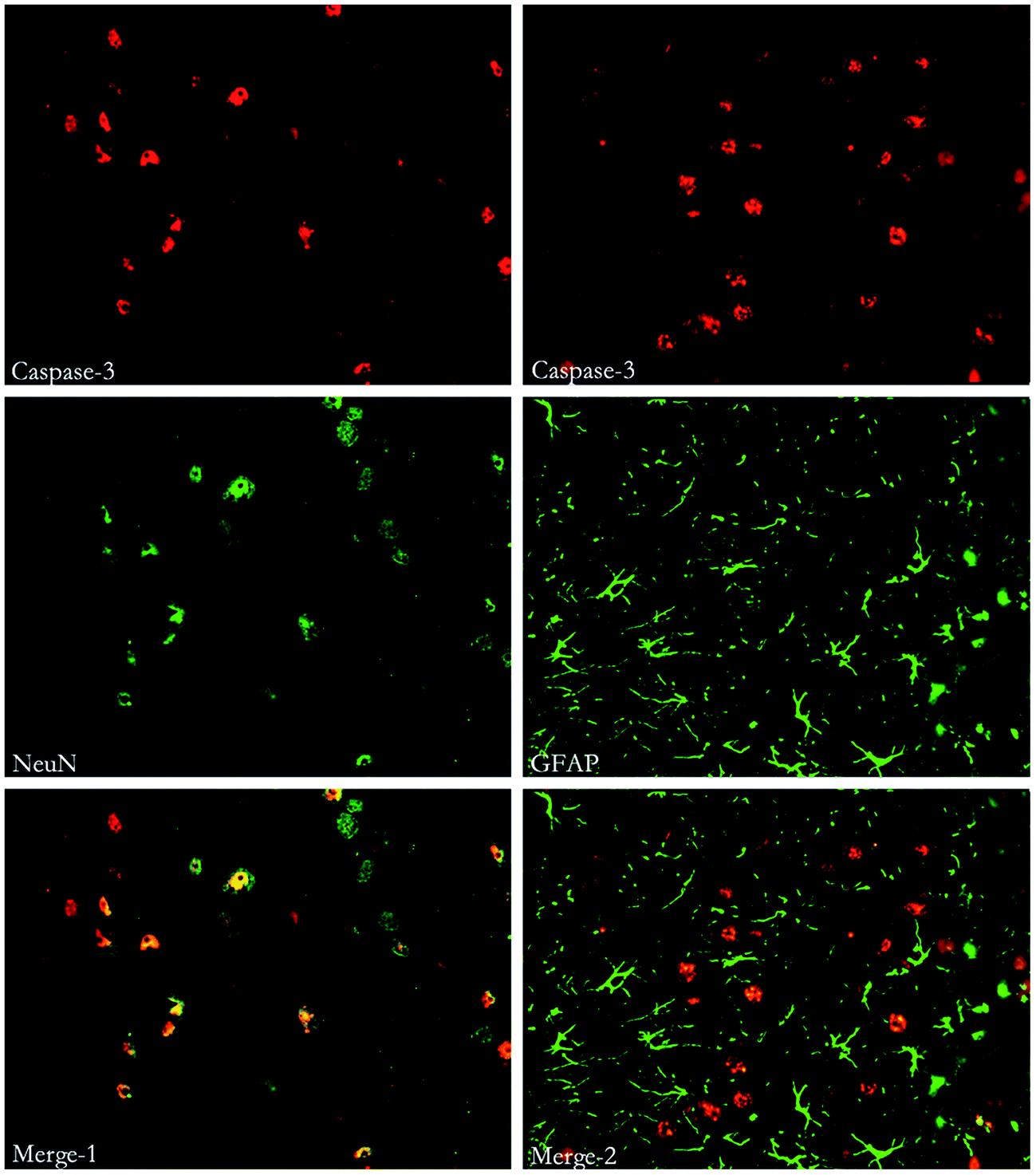 | ||
| Fig. 9 Immunofluorescent double labeling of Caspase-3 and NeuN or GFAP. The results indicates that cells expressing Caspase-3 were co-stained with both NeuN and GFAP. | ||
3.6 Relationship between autophagy and apoptosis
To investigate the relationship between autophagy and apoptosis, western blots were performed to assess the ratio of Beclin-1/Bcl-2 and Bcl-2/Bax. Belin-1/Bcl-2 indicates the activation of autophagy and Bcl-2/Bax reflects the apoptosis. Saline administration had no effect on the changes of autophagy and apoptosis. MP could significantly up-regulate the ratio of Beclin-1/Bcl-2 and down-regulate the ratio of Bcl-2/Bax at each time point. (Fig. 10).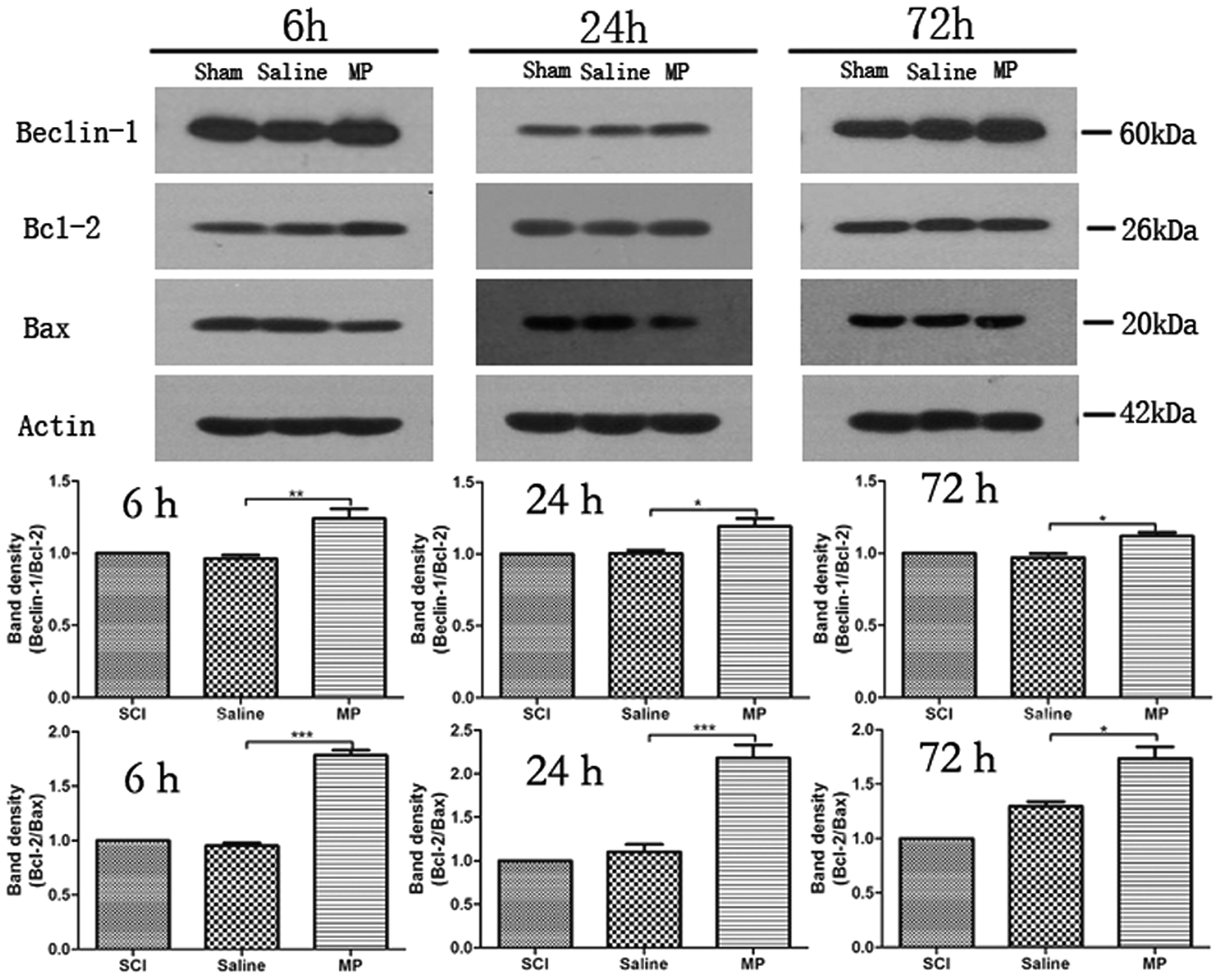 | ||
| Fig. 10 Immunoblots of Beclin-1, Bcl-2 and Bax in injured spinal cord. β-Actin was used as a loading control. Data in the bar graph represent mean ± SEM from four rats in each group. Up-regulation of Beclin-1/Bcl-2 and down-regulation of Beclin-1/Bcl-2 was observed. | ||
3.7 MP treatment activates the recovery of behavioral function
BBB score was used to evaluate the effects of MP treatment in behavioral function of rats after SCI at different time points (control, 3 d, 7 d, 14 d, 21 d). Saline administration had no effect on the recovery of behavioral function. In the MP group, the averages of total BBB scores were significantly higher than those in the SCI group and saline group. SCI could induce the severe impairments in the behavioral function of rats. At 3 d after SCI, BBB score of rats in MP group was 1.5 ± 0.3, significantly higher than those in SCI group and saline group (p < 0.05). Behavioral function of rats gradually recovered in the follow-up period. At 21 d after SCI, BBB score of rats in MP group increased to 6.5 ± 0.3, still significantly higher than those in other two groups (p < 0.001). (Fig. 11).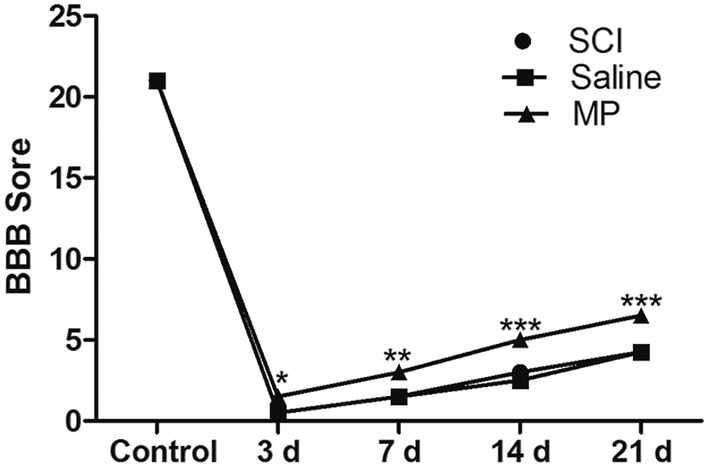 | ||
| Fig. 11 BBB score of rats at different time points after SCI (control, 3 d, 7 d, 14 d, 21 d). In the MP group, the averages of total BBB scores were significantly higher than those in the SCI group and saline group. (*p < 0.05, **p < 0.01, ***p < 0.001; MP vs. saline.) | ||
3.8 Discussion
In present study, we found that SCI produced by clamp force in rats caused apoptotic cell death in the injured area at different time points after SCI and MP treatment could significantly decreased the number of apoptotic cells; we also found that SCI could induce an increase in autophagy in the injured area and both neurons and astrocytes were co-stained with autophagic biomarkers, including LC3 and Beclin-1, and an apoptotic biomarker, Caspase-3. MP treatment could promote the locomotor recovery after SCI. The results suggest that an increase in autophagy after MP treatment may be a neuroprotective mechanism.In a previous study, apoptosis following SCI was shown in neurons and glial cells in the zone of lesion 1 h after trauma; between 4 and 8 h post-injury, the number of apoptotic cell increased.28 TUNEL assay, a biochemical assessment, is commonly used to detect DNA fragmentation resulting from cell death. This assay relies on the presence of nicks in the DNA of apoptotic (and some necrotic) cells and the nicks can be identified through catalyzation of the added dUTP (fluorescein) by terminal transferase; Caspase-3 plays a central role in the apoptotic cascade as an executioner caspase, and its activation marks a point-of-no-return in the complicated cascade of apoptosis induction.29,30 In addition, Bax–Bax homodimers can promote apoptosis and Bcl-2/Bax heterodimers can restrain apoptosis.31 The Bcl-2/Bax ratio plays an important role in determining whether apoptosis is promoted or inhibited.32 In accordance with previous studies, SCI produced by clamp force could induce significant apoptosis both in neurons and glial cells, as demonstrated by these different biochemical assessments.
The mechanisms of induction of apoptosis after SCI are elusive. Springer J. E. et al. found that upstream and downstream components of the Caspase-3 apoptotic pathway are activated after traumatic spinal cord injuries in rats and occur early in the neurons and astrocytes adjacent to and distant from the injury site.33 Inukai T. et al. suggested that overexpression of TNF-α, TNFR1 and TNFR2 participated in apoptosis of oligodendrocytes in the chronically compressed spinal cord.34 Satake K. et al. found that iNOS may contribute toward apoptosis in damaged area following SCI.35 Expression of pro-inflammatory cytokines also promotes neuronal apoptosis.36 However, the specific mechanisms of apoptosis following acute SCI remain elusive.
Another type of programmed cell death, autophagy, is also involved in the pathological changes after SCI. It is a highly regulated process which is involved not only in the balance between protein synthesis and degradation, but also in the execution of cell death.37 LC3 is one of the most reliable markers of autophagy induction, and can be linked with ubiquitinated substrates by p62, a marker of autophagic flux in vivo, thus regulating the selective autophagic clearance of protein aggregates.27,38 As a key protein involved in the regulation of autophagy, Beclin-1 is an indispensable protein for subsequent formation of the Beclin-1-Vps34–Vps15 core complex.39,40 Some studies have indicated that autophagy protects neurons, reduces neuronal damage and promotes locomotor recovery via inhibition of apoptosis.14,15 This perspective was confirmed by the results that rapamycin, which enhances expression of autophagy by inhibiting mTOR, was neuroprotective at the lesion site following SCI.16,41,42 There are also counterviews. Kanno H. et al. found that autophagic cell death was induced in the damaged tissue after SCI and clearly contribute to neuronal tissue damage after SCI.17,18 Drugs including valproic acid and bisperoxovanadium which can reduce autophagy, promoted functional recovery following SCI.19,20 Whether the function of autophagy is protective or detrimental for neural tissue in SCI remains to be elucidated.
As to the location of autophagy, we found that autophagy was expressed in both neurons and astrocytes. There are also counterviews. Chen H. C. et al. found that LC3-positive cells were co-localized with neuronal nuclei, but not with glial fibrillary acidic protein.22 However, Tang P. et al. demonstrated that both neurons and astrocytes were co-stained with LC3B-II with different start time.14 Different results may be due to the different types of damage.
SCI treatment strategies targeting apoptosis have been long sought.43 In the past, MP has been considered beneficial after SCI because of its abilities to inhibit lipid oxidation and hydrolysis, and form active oxygen and free radicals.44–46 First report concerning the anti-apoptotic effect of MP was conducted by Ray, which concluded that co-administration of calpeptin and MP in rats immediately after SCI could reduce apoptosis in the lesion site and adjacent areas via inhibition of calpain activity and inflammatory process.47 In 2006, J. Vaquero et al. also found that early administration of MP could decrease apoptotic cell death, possible mechanisms include changes in the expression of certain genes implicated in apoptosis such as Bcl-2, Caspases, Bcl-x, Bax and p-53.21 In the present study, we have found that MP treatment could not only decrease apoptotic cell death but also increase autophagy. Negative modulation of apoptosis may be one possible mechanism of the cytoprotective function of autophagy.
Possible mechanisms for the effects of MP treatment include the interactions between Beclin-1 and Bcl-2/Bcl-xL. Beclin-1 is a novel Bcl-2-homology (BH)-3 domain only protein, mainly located in the cytoplasmic structures, including the ER, mitochondria, and the peri-nuclear membrane.48 Under nutrient-sufficient conditions, Beclin-1 is bound by Bcl-2 or Bcl-xL, inhibiting its ability to initiate autophagy. Anti-apoptotic Bcl-2 family members interact with the BH-3 domain of Beclin-1.49 During starvation or other stress conditions, however, Bcl-2 and Bcl-xL must be displaced from Beclin-1 to permit autophagy. Previous studies have explored the possible mechanisms of dislocation of Bcl-2 and Beclin-1, which is necessary for induction of autophagy. Possible mechanisms include competitive displacement of Beclin-1 BH3 domain by other Bcl-2 family proteins or translocation of the nuclear protein high-mobility group box 1 (HMGB1) to the cytosol, JNK-mediated phosphorylation of Bcl-2, death-associated protein kinase (DAPK)-mediated phosphorylation of Beclin-1, nutrient-deprivation factor-1 dysfunction, Beclin-1 self-interaction and tumor necrosis factor receptor-associated factor 6-mediated ubiquitination of Beclin-1.49–56 In addition, we also found that MP treatment could regulate the interaction between endogenous Bcl-2 and Beclin-1 by altering their total protein levels.
4. Conclusion
In the present study, we found that the BBB scores were significantly higher for rats in MP group compared with those in SCI and saline groups. A possible mechanism for this effect includes activation of autophagy induced by MP treatment, which could decrease apoptosis, protect neuronal locomotor function and enhance the behavioral recovery of rats.Acknowledgements
This work was supported by grants from National Science and Nature Grant (No. 81271381, 81471333, 81601061, 81300996), and Shanghai Shenkang hospital development center project (No. SHDC12013906).References
- H. M. Bramlett and W. D. Dietrich, Brain Res., 2007, 161, 125–141 Search PubMed.
- B. K. Kwon, W. Tetzlaff, J. N. Grauer, J. Beiner and A. R. Vaccaro, Spine J., 2004, 4, 451–464 CrossRef PubMed.
- J. Lu, K. W. Ashwell and P. Waite, Spine, 2000, 25, 1859–1866 CrossRef CAS PubMed.
- L. H. Sekhon and M. G. Fehlings, Spine, 2001, 26, S2–S12 CrossRef CAS PubMed.
- C. H. Tator, Brain Pathol., 1995, 5, 407–413 CrossRef CAS.
- C. H. Tator and M. G. Fehlings, J. Neurosurg., 1991, 75, 15–26 CrossRef CAS PubMed.
- T. Haider, R. Hoftberger, B. Ruger, M. Mildner, R. Blumer, A. Mitterbauer, T. Buchacher, C. Sherif, P. Altmann, H. Redl, C. Gabriel, M. Gyöngyösi, M. B. Fischer, G. Lubec and H. J. Ankersmit, Exp. Neurol., 2015, 267, 230–242 CrossRef CAS PubMed.
- A. L. Bowes and P. K. Yip, J. Neurotrauma, 2014, 31, 1753–1766 CrossRef PubMed.
- E. D. Hall and J. E. Springer, NeuroRx, 2004, 1, 80–100 CrossRef PubMed.
- C. A. Oyinbo, Acta Neurobiol. Exp., 2011, 71, 281–299 Search PubMed.
- P. G. Popovich, P. Wei and B. T. Stokes, J. Comp. Neurol., 1997, 377, 443–464 CrossRef CAS PubMed.
- J. W. Rowland, G. W. Hawryluk, B. Kwon and M. G. Fehlings, Neurosurg. Focus, 2008, 25, E2 CrossRef PubMed.
- K. T. Wright, E. I. W. Masri, A. Osman, J. Chowdhury and W. E. Johnson, Stem Cells, 2011, 29, 169–178 CrossRef CAS PubMed.
- P. Tang, H. Hou, L. Zhang, X. Lan, Z. Mao, D. Liu, C. He, H. Du and L. Zhang, Mol. Neurobiol., 2014, 49, 276–287 CrossRef CAS PubMed.
- Z. Y. Wang, J. H. Lin, A. Muharram and W. G. Liu, Apoptosis, 2014, 19, 933–945 CrossRef CAS PubMed.
- A. Sekiguchi, H. Kanno, H. Ozawa, S. Yamaya and E. Itoi, J. Neurotrauma, 2012, 29, 946–956 CrossRef PubMed.
- H. Kanno, H. Ozawa, A. Sekiguchi, S. Yamaya and E. Itoi, Spine, 2011, 36, E1427–E1434 CrossRef PubMed.
- H. Kanno, H. Ozawa, A. Sekiguchi and E. Itoi, Neurobiol. Dis., 2009, 33, 143–148 CrossRef CAS PubMed.
- C. L. Walker, M. J. Walker, N. K. Liu, E. C. Risberg, X. Gao, J. Chen and X. M. Xu, PLoS One, 2012, 7, e30012 CAS.
- H. H. Hao, L. Wang, Z. J. Guo, L. Bai, R. P. Zhang, W. B. Shuang, Y. J. Jia, J. Wang, X. Y. Li and Q. Liu, Neurosci. Bull., 2013, 29, 484–492 CrossRef CAS PubMed.
- J. Vaquero, M. Zurita, S. Oya, C. Aguayo and C. Bonilla, Histol. Histopathol., 2006, 21, 1091–1102 CAS.
- H. C. Chen, T. H. Fong, A. W. Lee and W. T. Chiu, Spine, 2012, 37, 470–475 CrossRef PubMed.
- B. Cemil, K. Topuz, M. N. Demircan, G. Kurt, K. Tun, M. Kutlay, O. Ipcioglu and Z. Kucukodaci, Acta Neurochir., 2010, 152, 1583–1590 CrossRef PubMed , discussion 1590.
- G. Kurt, E. Ergun, B. Cemil, A. O. Borcek, P. Borcek, O. Gulbahar and N. Ceviker, Surg. Neurol., 2009, 71, 332–336 CrossRef PubMed , discussion 336.
- D. M. Basso, M. S. Beattie and J. C. Bresnahan, Exp. Neurol., 1996, 139, 244–256 CrossRef CAS PubMed.
- D. M. Basso, M. S. Beattie, J. C. Bresnahan, D. K. Anderson, A. I. Faden, J. A. Gruner, T. R. Holford, C. Y. Hsu, L. J. Noble, R. Nockels, P. L. Perot, S. K. Salzman and W. Young, J. Neurotrauma, 1996, 13, 343–359 CrossRef CAS PubMed.
- R. Mathew, C. M. Karp, B. Beaudoin, N. Vuong, G. Chen, H. Y. Chen, K. Bray, A. Reddy, G. Bhanot, C. Gelinas, R. S. Dipaola, V. Karantza-Wadsworth and E. White, Cell, 2009, 137, 1062–1075 CrossRef CAS PubMed.
- A. Aslan, M. Cemek, M. E. Buyukokuroglu, K. Altunbas, O. Bas and Y. Yurumez, Food Chem. Toxicol., 2012, 50, 2554–2559 CrossRef CAS PubMed.
- S. E. Logue and S. J. Martin, Biochem. Soc. Trans., 2008, 36, 1–9 CrossRef CAS PubMed.
- B. A. Eldadah and A. I. Faden, J. Neurotrauma, 2000, 17, 811–829 CrossRef CAS PubMed.
- D. Liu, C. Lu, R. Wan, W. W. Auyeung and M. P. Mattson, J. Cereb. Blood Flow Metab., 2002, 22, 431–443 CrossRef CAS PubMed.
- C. Zhou, X. Li, W. Du, Y. Feng, X. Kong, Y. Li, L. Xiao and P. Zhang, Chemotherapy, 2010, 56, 393–402 CrossRef CAS PubMed.
- J. E. Springer, R. D. Azbill and P. E. Knapp, Nat. Med., 1999, 5, 943–946 CrossRef CAS PubMed.
- T. Inukai, K. Uchida, H. Nakajima, T. Yayama, S. Kobayashi, E. S. Mwaka, A. R. Guerrero and H. Baba, Spine, 2009, 34, 2848–2857 CrossRef PubMed.
- K. Satake, Y. Matsuyama, M. Kamiya, H. Kawakami, H. Iwata, K. Adachi and K. Kiuchi, Mol. Brain Res., 2000, 85, 114–122 CrossRef CAS PubMed.
- K. L. Wu, S. H. Chan, Y. M. Chao and J. Y. Chan, Neurobiol. Dis., 2003, 14, 19–31 CrossRef CAS PubMed.
- Y. Tsujimoto and S. Shimizu, Cell Death Differ., 2005, 12(suppl. 2), 1528–1534 CrossRef CAS PubMed.
- Y. Ichimura, T. Kumanomidou, Y. S. Sou, T. Mizushima, J. Ezaki, T. Ueno, E. Kominami, T. Yamane, K. Tanaka and M. Komatsu, J. Biol. Chem., 2008, 283, 22847–22857 CrossRef CAS PubMed.
- C. He and B. Levine, Curr. Opin. Cell Biol., 2010, 22, 140–149 CrossRef CAS PubMed.
- R. S. Clark, H. Bayir, C. T. Chu, S. M. Alber, P. M. Kochanek and S. C. Watkins, Autophagy, 2008, 4, 88–90 CrossRef CAS PubMed.
- H. C. Chen, T. H. Fong, P. W. Hsu and W. T. Chiu, J. Surg. Res., 2013, 179, e203–210 CrossRef CAS PubMed.
- Z. Y. Wang, W. G. Liu, A. Muharram, Z. Y. Wu and J. H. Lin, NeuroImmunoModulation, 2014, 21, 257–267 CrossRef CAS PubMed.
- V. Cavallucci and M. D'Amelio, Curr. Pharm. Des., 2011, 17, 215–229 CrossRef CAS PubMed.
- T. B. Ducker and S. M. Zeidman, Spine, 1994, 19, 2281–2287 CrossRef CAS PubMed.
- E. D. Hall, J. Neurosurg., 1992, 76, 13–22 CrossRef CAS PubMed.
- E. D. Hall, Cell. Mol. Neurobiol., 1993, 13, 415–432 CrossRef CAS PubMed.
- S. K. Ray, G. G. Wilford, D. C. Matzelle, E. L. Hogan and N. L. Banik, Ann. N. Y. Acad. Sci., 1999, 890, 261–269 CrossRef CAS PubMed.
- A. Oberstein, P. D. Jeffrey and Y. Shi, J. Biol. Chem., 2007, 282, 13123–13132 CrossRef CAS PubMed.
- R. Kang, H. J. Zeh, M. T. Lotze and D. Tang, Cell Death Differ., 2011, 18, 571–580 CrossRef CAS PubMed.
- S. Luo and D. C. Rubinsztein, Cell Death Differ., 2007, 14, 1247–1250 CrossRef CAS PubMed.
- M. C. Maiuri, G. Le Toumelin, A. Criollo, J. C. Rain, F. Gautier, P. Juin, E. Tasdemir, G. Pierron, K. Troulinaki, N. Tavernarakis, J. A. Hickman, O. Geneste and G. Kroemer, EMBO J., 2007, 26, 2527–2539 CrossRef CAS PubMed.
- Y. Wei, S. Pattingre, S. Sinha, M. Bassik and B. Levine, Mol. Cell, 2008, 30, 678–688 CrossRef CAS PubMed.
- E. Zalckvar, H. Berissi, M. Eisenstein and A. Kimchi, Autophagy, 2009, 5, 720–722 CrossRef CAS PubMed.
- E. Zalckvar, H. Berissi, L. Mizrachy, Y. Idelchuk, I. Koren, M. Eisenstein, H. Sabanay, R. Pinkas-Kramarski and A. Kimchi, EMBO Rep., 2009, 10, 285–292 CrossRef CAS PubMed.
- R. Kang, K. M. Livesey, H. J. Zeh, M. T. Loze and D. Tang, Autophagy, 2010, 6, 1209–1211 CrossRef CAS PubMed.
- D. Tang, R. Kang, K. M. Livesey, C. W. Cheh, A. Farkas, P. Loughran, G. Hoppe, M. E. Bianchi, K. J. Tracey, H. J. Zeh and M. T. Lotze, J. Cell Biol., 2010, 190, 881–892 CrossRef CAS PubMed.
Footnote |
| † These two authors contribute equally to the study. |
| This journal is © The Royal Society of Chemistry 2017 |