
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Readily switchable one-pot 5-exo-dig cyclization using a palladium catalyst†
Jaishree K. Mali,
Balaram S. Takale and
Vikas. N. Telvekar *
*
Department of Pharmaceutical Sciences and Technology, Institute of Chemical Technology, Mumbai 400 019, India. E-mail: vikastelvekar@rediffmail.com
First published on 12th January 2017
Abstract
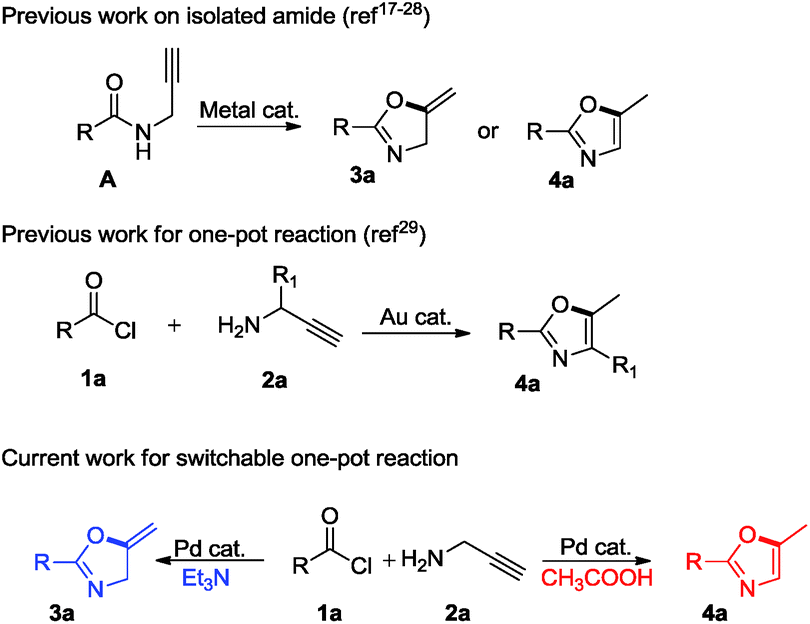
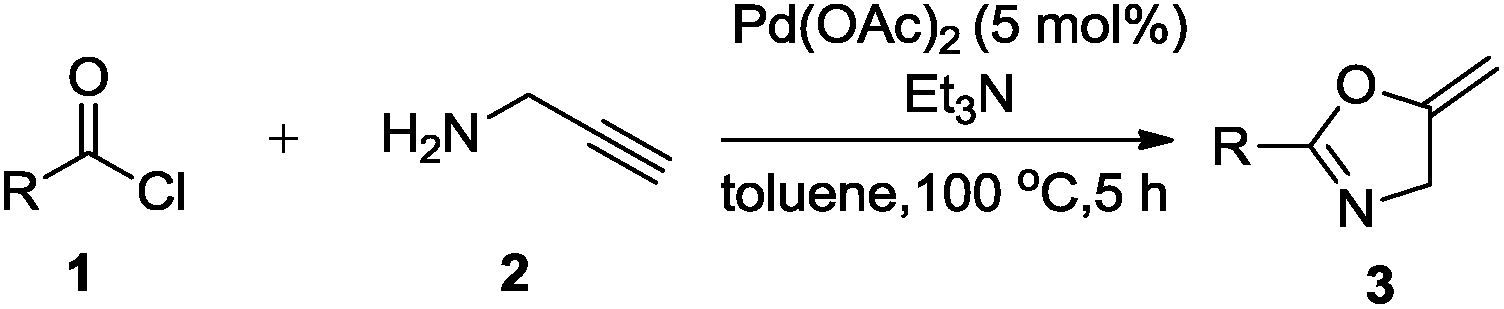
A convenient, ligand-free, Pd(OAc)2-catalyzed one-pot reaction has been developed for the synthesis of oxazolines and oxazoles from easily available acid chlorides and propargylamine. The reaction pathways could be easily modulated to selectively obtain oxazolines or oxazoles by merely changing the additives. This method proceeds via in situ sequential nucleophilic addition/elimination reactions followed by an intramolecular 5-exo-dig cycloisomerization reaction. An interesting observation in this case is the effect of an additive: a basic additive such as triethylamine resulted in the formation of 5-methylene oxazolines, while an acidic additive such as acetic acid resulted in the formation of 5-methyloxazoles. With the current protocol we are able to obtain good to moderate yields of the desired product without the need for the isolation of intermediates.
Introduction
Oxazoles and oxazolines are ubiquitous heterocycles present in a variety of natural products, pharmaceuticals, agrochemicals, foods and polymers.1–4 Additionally, 2-oxazolines serve as synthetic intermediates,5 protecting groups in organic synthesis, and catalysts, ligands or auxiliaries in asymmetric synthesis.6,7Several synthetic methods reported in the literature for substituted oxazole use Robinson–Gabriel synthesis cyclodehydration of 2-acylamino carbonyl compounds,8 isomerisation of the oxazolines,9 base promoted reactions of primary aromatic amides and 2,3-dibromopropene,10 catalytic decomposition of diazocarbonyl compounds in nitriles,11 reductive desulfuration of 5-methyl-4-methylthio-1,3-oxazoles,12 functionalisation (via metallation) of readily available oxazole13 and various condensation reactions.14
Other straightforward methods for the synthesis of methylene oxazoline and oxazole include cycloisomerization of propargylamides.15 However, in this context cyclization of propargylamides to the corresponding oxazoles was achieved by strong base or strong acid using harsh conditions.16
On the other hand, Lewis acids involving not only gold17 but also ZnI2 and FeCl3,18 cerium chloride,19 ferric bromide,20 zinc triflate,21 Bronsted acid22 catalysts have emerged to afford oxazoles or oxazolines from corresponding propargylic amides. Besides these, metals such as silver,23 copper,24 palladium,25 mercury,26 ruthenium,27 tungsten and molybdenum28 have been explored. In fact, all of these strategies possess respective drawbacks such as low yield, harsh reaction conditions, use of expensive ligands, and use of excess amount of additive or non-commercially available starting materials. Nevertheless, cycloisomerization of propargylamides to corresponding oxazole and oxazoline require synthesis, isolation and purification of propargylamides. Therefore, the development of single step protocol is highly desirable to minimize solvent waste and to maximize the yield of product. Taking this into consideration, Tran-DubéM et al.29 explored one-pot synthesis of oxazoles from acid chlorides and propargylamines using gold-catalyst. Our curiosity was increased to replace expensive gold catalyst with well known palladium catalyst.30
Gratifyingly, we found an interesting result while performing optimization studies, use of basic additive selectively resulted in oxazoline, while use of acidic additive selectively resulted in oxazole. We believe, such kind of readily switchable protocol which has wide applicability has not been explored previously (Scheme 1).
 | ||
| Scheme 1 Approach for the synthesis of dihydrooxazoles and oxazoles. | ||
Results and discussion
We initiated the present study with benzoyl chloride and propargylamine as model substrates. Accordingly, propargylamine (2a, 1.82 mmol), benzoyl chloride (1a, 1.81 mmol), triethylamine (TEA) (2.17 mmol) and 1 mol% of Pd(OAc)2 were heated in toluene as a solvent, in a sealed tube at 100 °C for 24 h, oxazoline 3a was formed in 20% yield (Table 1, entry 1). This results gave us a hint that Pd(OAc)2 is compatible with reaction conditions of one-pot synthesis. Further, we screened for the loading of Pd(OAc)2 from 2 to 10 mol% (Table 1, entries 2–13). Remarkably, the desired product was obtained in 85% yield with 5 mol% of the catalyst (Table 1, entry 3). We also tried to replace TEA with inorganic or organic bases such as K2CO3 and diisopropylethylamine but resulted in 20% and 10% yield, respectively. In the absence of Pd(OAc)2 and TEA, no product was formed (Table 1, entry 4). Only Pd(OAc)2 without any additive resulted in the trace amount of product 4a (confirmed by 1H NMR) (Table 1, entry 5). These blank reactions suggest that Pd(OAC)2 and TEA combination is crucial for selective formation of oxazoline 3a.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Additive | Solvent | Temp (°C)/time (h) | 3ac yield (%) | 4ac yield (%) |
| a Reaction conditions: acid chloride (1.82 mmol), propargylamine (1.81 mmol), catalyst Pd(OAc)2 (mol%), additive TEA (triethylamine) 2.17 mmol or acetic acid 1.81 mmol in 2 mL of dry solvent in a sealed tube.b Detected by 1H NMR.c Isolated yield. | ||||||
| 1 | 1 | TEA | Toluene | 100/24 | 20 | 0 |
| 2 | 2 | TEA | Toluene | 100/24 | 45 | 0 |
| 3 | 5 | TEA | Toluene | 100/24 | 85 | 0 |
| 4 | — | — | Toluene | 100/24 | 0 | 0 |
| 5 | 1 | — | Toluene | 100/24 | 0 | <2%b |
| 6 | 5 | TEA | CH2Cl2 | 40/24 | 30 | 0 |
| 7 | 5 | TEA | 1,2-DCE | 80/24 | 45 | 0 |
| 8 | 5 | TEA | CH3CN | 80/24 | 35 | 0 |
| 9 | 5 | TEA | THF | 65/24 | 32 | 0 |
| 10 | 5 | TEA | 1,4-Dioxane | 100/24 | 35 | 0 |
| 11 | 5 | TEA | Toluene | 100/12 | 85 | 0 |
| 12 | 5 | TEA | Toluene | 100/5 | 85 | 0 |
| 13 | 10 | TEA | Toluene | 100/5 | 88 | 0 |
| 14 | 5 | — | Toluene | 100/24 | 0 | 25 |
| 15 | 5 | Acetic acid | Toluene | 100/24 | 0 | 88 |
| 16 | 5 | Acetic acid | Toluene | 100/12 | 0 | 65 |

A variety of solvents were tested for this reaction, from which no product was observed in DMF and DMSO. However, toluene gave the best result (Table 1, entries 3, 6–10). Remarkably, shortening of the reaction time from 24 h to 5 h resulted in the same yield of the desired product (Table 1, entries 11 and 12). Further, putting 10 mol% of the catalyst with reaction time of 5 h did not show significant increment in the yield (Table 1, entry 13).
Surprisingly, we were able to obtain oxazole 4a exclusively when TEA was not used, although in very low amount (Table 1, entry 14). This strongly suggests that acidity is important for the formation of oxazole over oxazoline. To this end, a variety of acids including organic as well as inorganic solid acid catalysts were screened. Use of Amberlite IR 120-H, Amberlyst 15 (wet) and Montmorillonite led to the formation of the desired product in 15%, 30% and 50% yields respectively. Use of organic acids such as pivalic acid, trifluoroacetic acid and acetic acid resulted in 35%, 65% and 88% yield of the desired product, respectively. According to these results, an additive, TEA was replaced with 1.81 mmol of acetic acid to give 88% yield of the desired product when the reaction was carried out with 5 mol% of Pd(OAc)2 in toluene as a solvent at 100 °C for 24 h (Table 1, entry 15). Shortening of the time from 24 h to 12 h resulted in lower yield of the product (Table 1, entry 16). It was observed that the reaction did not proceed towards the completion when less than 1 equivalents of TEA and acetic acid were used respectively, while their excess amount did not affect the yield of the desired product.
Finally, we had optimized conditions in hands, that is for oxazoline 3a; 1a (1.82 mmol), 2a (1.81 mmol), 5 mol% of Pd(OAc)2, TEA (2.17 mmol) in toluene as solvent at 100 °C for 5 h, and for oxazole 4a; 1a (1.82 mmol), 2a (1.81 mmol), 5 mol% of Pd(OAc)2,acetic acid (1.81 mmol) in toluene as solvent at 100 °C for 24 h. Under these optimized reaction conditions, firstly we examined the scope of various acid chlorides for the synthesis of oxazolines (Table 2). Aroyl chlorides with either electron donating group on ortho-, meta- or para-position or electron withdrawing groups gave higher yields of the desired product (Table 2, entries 2–6).
|
|||
|---|---|---|---|
| Entry | Acid chlorides; 1 | Product; 3 | Yield of 3b (%) |
| a Reaction conditions: acid chloride (1.82 mmol), propargylamine (1.81 mmol), catalyst Pd(OAc)2 (5 mol%), TEA (triethylamine) 2.17 mmol in 2 mL of dry toluene in a sealed tube at 100 °C for 5 h.b Isolated yield. | |||
| 1 |  |
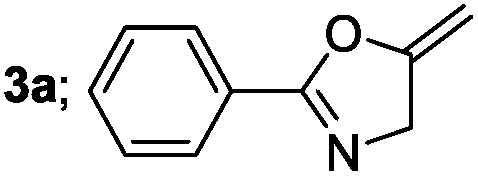 |
85 |
| 2 | 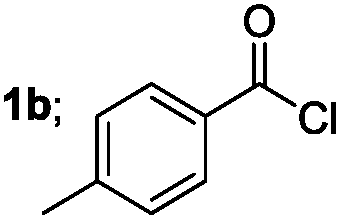 |
 |
85 |
| 3 | 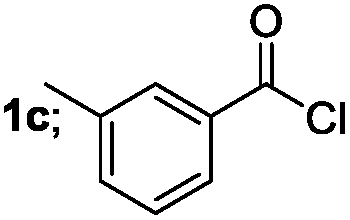 |
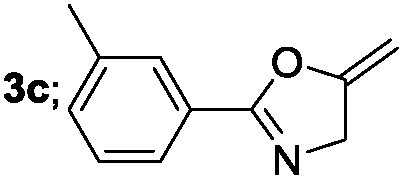 |
70 |
| 4 | 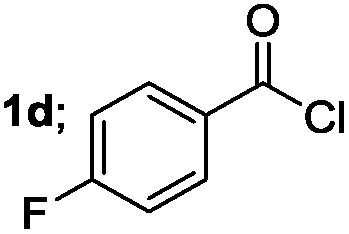 |
 |
92 |
| 5 |  |
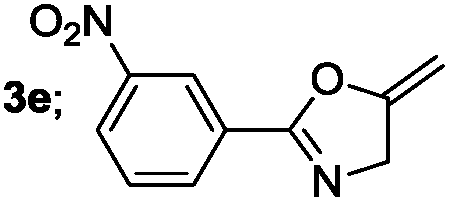 |
84 |
| 6 |  |
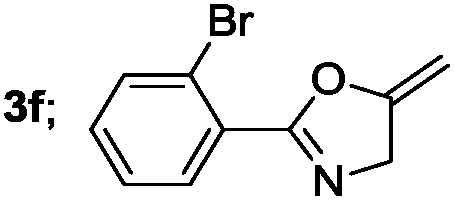 |
70 |
| 7 |  |
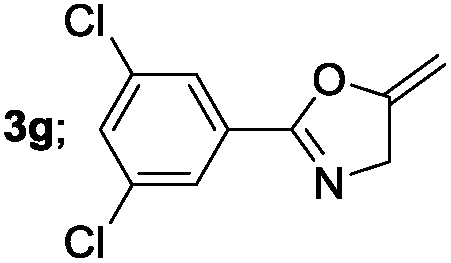 |
82 |
| 8 |  |
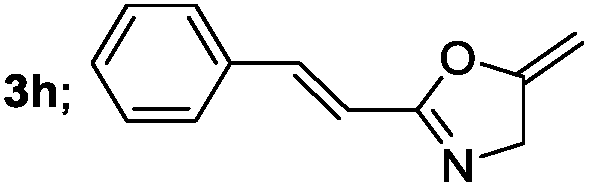 |
72 |
| 9 |  |
 |
70 |
| 10 |  |
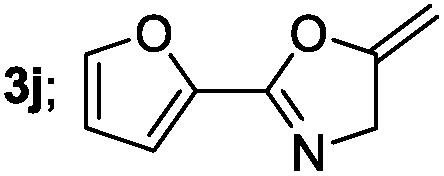 |
45 |
Aliphatic derivatives also furnished the desired product in moderate to good yield (Table 2, entries 8 and 9). Heterocyclic derivatives gave slightly lower yield (Table 2, entry 10). Noteworthy to mention, multiple substituent on the ring were also tolerated (Table 2, entry 7).
Secondly, using optimized reaction conditions available for oxazoles, we investigated the scope for variety of substrate (Table 3). Mono- and disubstituted aroyl chlorides were easily transformed to give good yields of the desired products (Table 3, entries 2–7). Both electron donating and electron withdrawing substrates were readily converted into the desired products (Table 3, entries 2–7). Furthermore, substituents at different positions on the aryl group (para-, meta-, ortho-positions), did not affect the reaction efficiency (Table 3, entries 2–6). Heteroaryl derivatives gave lower yield (Table 3, entry 10). Aliphatic acid chlorides also gave the desired products in good to moderate yields (Table 3, entries 8 and 9).
|
|||
|---|---|---|---|
| Entry | Acid chlorides; 1 | Product; 4 | Yield of 4b (%) |
| a Reaction conditions: acid chloride (1.82 mmol), propargylamine (1.81 mmol), catalyst Pd(OAc)2 (5 mol%), acetic acid 1.81 mmol in 2 mL of dry toluene in a sealed tube at 100 °C for 24 h.b Isolated yield. | |||
| 1 |  |
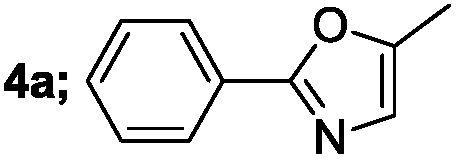 |
88 |
| 2 | 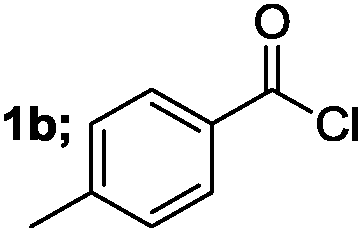 |
 |
90 |
| 3 | 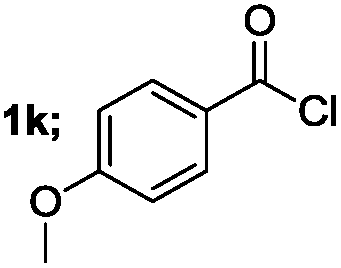 |
 |
70 |
| 4 |  |
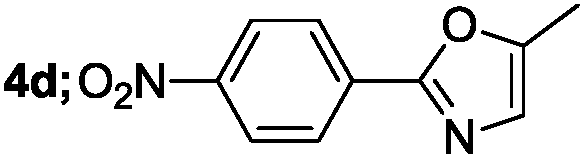 |
71 |
| 5 | 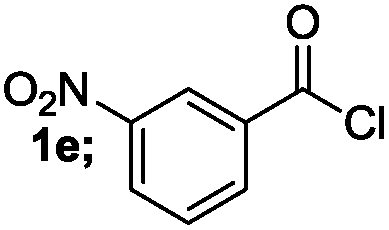 |
 |
84 |
| 6 |  |
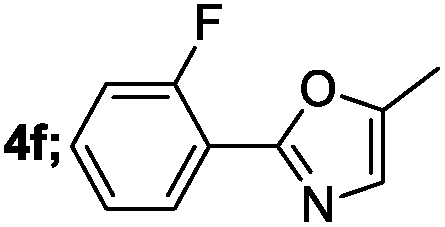 |
70 |
| 7 |  |
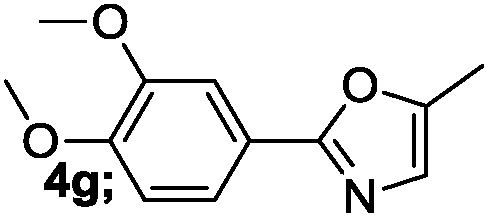 |
82 |
| 8 | 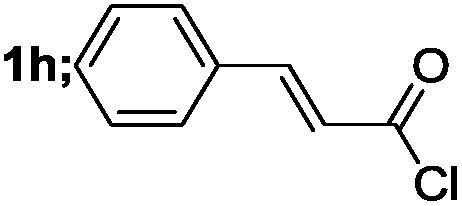 |
 |
82 |
| 9 | 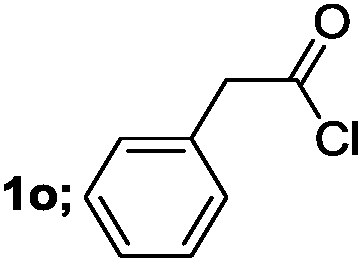 |
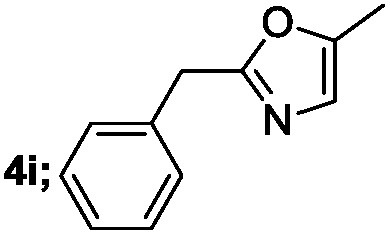 |
65 |
| 10 | 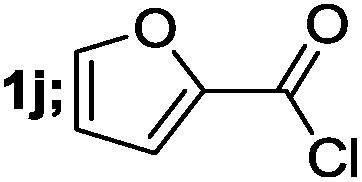 |
 |
40 |
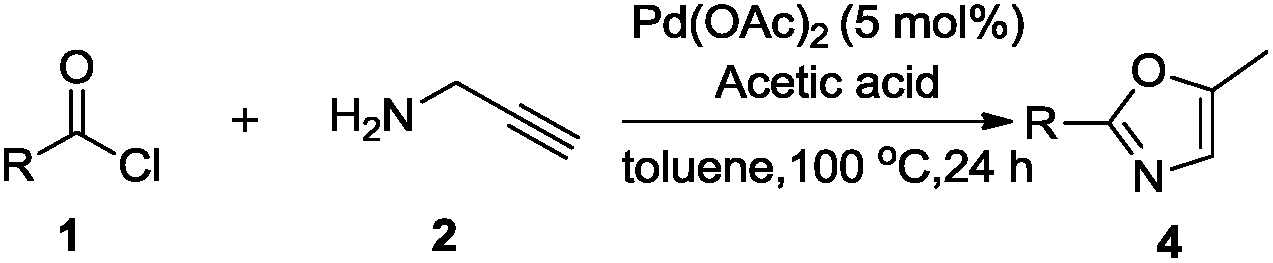
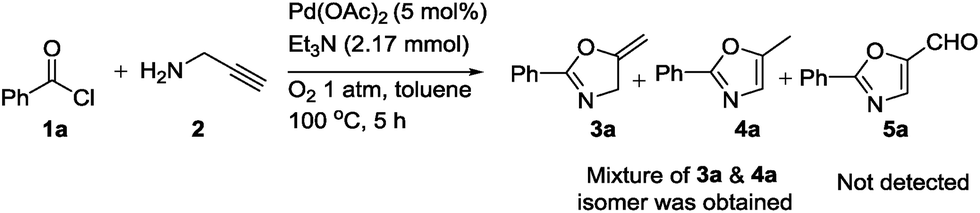
After this successful study, we performed one control experiment to check effect of atmospheric oxygen on the reaction (Scheme 2). It is reported in the literature25b that metal-vinylidene formed as an intermediate could be easily converted to aldehyde 5a. However, in our reaction condition we were not able to obtain such oxidized product 5a, and rather mixture of 3a and 4a could be obtained. It was noted that in the case of synthesis of oxazolines, small quantities of inactive palladium black were observed in the solution after the completion of reaction. On the other hand, in case of oxazoles, no formation of palladium black was observed. It could be hypothesized that acetic acid significantly helped in the regeneration of palladium acetate. In addition, to extend the scope of our methodology, we carried out reaction using secondary amine such as N-methylpropargylamine, but unidentified mixture of products was isolated.
 | ||
| Scheme 2 Control experiments. | ||
Based on the literature and experimental details, we proposed a plausible reaction pathway (Scheme 3). The reaction proceeds via in situ formation of propargylamide by nucleophilic addition/elimination reactions of acid chlorides and propargylamine. Palladium acetate coordinates to the carbon–carbon triple bond of in situ formed propargylamide A to form organo-palladium species B followed by triethylamine assisted intramolecular nucleophilic attack of enolate on alkyne to produce vinyl palladium C in 5-exo-dig manner. Intermediate C can either undergo proto-demetallation to give oxazoline 3 or it can undergo aromatization in the presence of excess of acetic acid to give oxazole 4. The path towards the formation of 4 includes initial protonation of double bond of vinyl palladium complex C to give D, (this complex is in resonance with complex D′) which undergoes aromatization by losing neighboring proton to form E before giving the final product 4 by proto-demetallation.
 | ||
| Scheme 3 Plausible reaction mechanism. | ||
Conclusions
In conclusion, we have developed one pot synthesis of 2-oxazolines and 2-oxazoles, catalyzed by palladium acetate via sequential nucleophilic addition/elimination reactions followed by intramolecular cycloisomerization reaction via 5-exo-dig mode. Moreover, the reaction pathway could be easily tuned by merely changing additives such as triethylamine or acetic acid to exclusively furnish 2-oxazolines or 2-oxazoles respectively. These protocols exhibit tolerance towards wide range of substrates to afford the desired product in moderate to good yield, except heteroaromatics, which furnished both the product oxazolines and oxazoles in slightly lower yield. Aliphatic acid chlorides also offered exclusively oxazolines and oxazoles in good yield without formation of complex mixture of the isomerised product. Being one-pot these reactions offer high step economy and does not require solvent exchanges or isolation of intermediates. In future, manipulation at the 5-methylene position of oxazolines could provide an avenue to wide variety of functionalized oxazoles. This protocol circumvents the use of expensive ligands. Synthetic application of the present work is underway in our laboratory.Acknowledgements
The author JKM is greatly thankful to the UGC (University Grant Commissions), New Delhi, India for providing a Basic Science Research (BSR) fellowship and VNT thanks the Science and Engineering Research Board (SERB), New Delhi, India, for providing financial support.References and notes
- (a) Z. Jin, Nat. Prod. Rep., 2011, 28, 1143–1191 RSC; (b) M. Tsuda, M. Yamakawa, S. Oka, Y. Tanaka, Y. Hoshino, Y. Mikami, A. Sato, H. Fujiwara, Y. Ohizumi and J. Kobayashi, J. Nat. Prod., 2005, 68, 462–464 CrossRef CAS PubMed; (c) L. J. Perez, D. J. Faulkner and W. Fenical, J. Nat. Prod., 2003, 66, 247–250 CrossRef CAS PubMed; (d) Z. Jin, Nat. Prod. Rep., 2013, 30, 869–915 RSC; (e) P. Wipf, Chem. Rev., 1995, 2115–2134 CrossRef CAS; (f) Z. Jin, Nat. Prod. Rep., 2006, 23, 464–496 RSC; (g) R. N. Misra, H. Xiao, K. S. Kim, S. Lu, W. Han, S. A. Barbosa, J. T. Hunt, D. B. Rawlins, W. Shan, S. Z. Ahmed, L. Qian, B. Chen, R. Zhao, M. S. Bednarz, K. A. Kellar, J. G. Mulheron, R. Batorsky, U. Roongta, A. Kamath, P. Marathe, S. A. Ranadive, J. S. Sack, J. S. Tokarski, N. P. Pavletich, F. Y. F. Lee, K. R. Webster and S. D. Kimball, J. Med. Chem., 2004, 47, 1719–1728 CrossRef CAS PubMed.
- (a) D. C. Palmer and S. Venkatraman, The Chemistry of Heterocyclic Compounds, A Series of Monographs, Oxazoles: Synthesis, Reactions, and Spectroscopy, Part A, ed. D. C. Palmer, John Wiley & Sons, NJ, 2003, vol. 60 CrossRef; (b) Oxazoles: Synthesis, Reactions and Spectroscopy, Part B, ed. D. C. Palmer, John Wiley & Sons, Hoboken, NJ, 2004 Search PubMed; (c) Y. Kato, N. Fusetani, S. Matsunaga, K. Hashimoto, K. S. Fujita and T. Furuya, J. Am. Chem. Soc., 1986, 108, 2780–2781 CrossRef CAS; (d) G. Pattenden, J. Heterocycl. Chem., 1992, 29, 607 CrossRef CAS.
- (a) J. A. Frump, Chem. Rev., 1971, 71, 483 CrossRef CAS; (b) J. A. Maga, J. Agric. Food Chem., 1978, 26, 1049–1050 CrossRef CAS PubMed.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS PubMed.
- H. H. Wasserman, K. E. Mccarthy and K. S. Prowse, Chem. Rev., 1986, 86, 845–856 CrossRef CAS.
- H. Vorbrüggen and K. Krolikiewicz, Tetrahedron, 1993, 49, 9353–9372 CrossRef.
- (a) A. Gissibl, M. G. Finn and O. Reiser, Org. Lett., 2005, 7, 2325–2328 CrossRef CAS PubMed; (b) H. Nlshiyama, H. Sakaguchi, T. Nakamura, M. Horlhata, M. Kondo and K. Itoh, Organometallics, 1989, 8, 846–848 CrossRef; (c) H. A. Mcmanus and P. J. Guiry, Chem. Rev., 2004, 104, 4151–4202 CrossRef CAS PubMed; (d) G. C. Hargaden and P. J. Guiry, Chem. Rev., 2009, 109, 2505–2550 CrossRef CAS PubMed.
- H. H. Wasserman and F. J. Vinick, J. Org. Chem., 1973, 38, 2407–2408 CrossRef CAS.
- R. Lakhan and B. Ternai, Adv. Heterocycl. Chem., 1974, 17, 99 CrossRef CAS.
- N. Yasmin and J. K. Ray, Synlett, 2009, 2825–2827 CAS.
- M. P. Doyle, W. E. Buhro, J. G. Davidson, R. C. Elliott, J. W. Hoekstra and M. Oppenhuizenlc, J. Org. Chem., 1980, 45, 3657–3664 CrossRef CAS.
- A. Herrera, R. Martinez-Alvarez, P. Ramiro, D. Molero and J. Almy, J. Org. Chem., 2006, 71, 3026–3032 CrossRef CAS PubMed.
- C. Kashima and H. Arao, Synthesis, 1989, 873–874 CrossRef CAS.
- (a) I. J. Turchi and M. J. S. Dewar, Chem. Rev., 1975, 75, 389–432 CrossRef CAS; (b) B. H. Lipshutz, Chem. Rev., 1986, 86, 795–819 CrossRef CAS; (c) R. H. Wiley and L. L. Bennett, Chem. Rev., 1949, 44, 447–476 CrossRef CAS; (d) C. T. Brain and J. M. Paul, Synlett, 1999, 1642–1644 CrossRef CAS.
- Y. Hu, X. Xin and B. Wan, Tetrahedron Lett., 2015, 56, 32–52 CrossRef CAS.
- (a) B. M. Nilsson, H. M. Vargas, B. Ringdahl and U. Hacksell, J. Med. Chem., 1992, 35, 285–294 CrossRef CAS PubMed; (b) P. Wipf, L. T. Rahman and S. R. Rector, J. Org. Chem., 1998, 63, 7132–7133 CrossRef CAS PubMed; (c) B. D. Conny Bogentoft, Ö. Ericsson and P. Stenberg, Tetrahedron Lett., 1969, 10, 4745–4748 CrossRef; (d) H. Araki, T. Katoh and M. Inoue, Synlett, 2006, 555–558 CAS; (e) K. E. Schulte, J. Reisch and M. Sommer, Arch. Pharm., 1966, 299, 107–112 CrossRef CAS.
- (a) S. K. Hashmi, J. P. Weyrauch, W. Frey and J. W. Bats, Org. Lett., 2004, 6, 4391–4394 CrossRef PubMed; (b) J. P. Weyrauch, A. S. K. Hashmi, A. Schuster, T. Hengst, S. Schetter, A. Littmann, M. Rudolph, M. Hamzic, J. Visus, F. Rominger, W. Frey and J. W. Bats, Chem. - Eur. J., 2010, 16, 956–963 CrossRef CAS PubMed; (c) A. S. K. Hashmi, M. C. Blanco Jaimes, A. M. Schuster and F. Rominger, J. Org. Chem., 2012, 77, 6394–6408 CrossRef CAS PubMed; (d) O. A. Egorova, H. Seo, Y. Kim, D. Moon, Y. M. Rhee and K. H. Ahn, Angew. Chem., Int. Ed., 2011, 50, 11446–11450 CrossRef CAS PubMed; (e) H. Peng, N. G. Akhmedov, Y. F. Liang, N. Jiao and X. Shi, J. Am. Chem. Soc., 2015, 137, 8912–8915 CrossRef CAS PubMed; (f) D. Aguilar, M. Contel, R. Navarro, T. Soler and E. P. Urriolabeitia, J. Organomet. Chem., 2009, 694, 486–493 CrossRef CAS; (g) A. S. K. Hashmi, A. M. Schuster and F. Rominger, Angew. Chem., Int. Ed., 2009, 48, 8247–8249 CrossRef CAS PubMed; (h) A. S. K. Hashmi, A. M. Schuster, M. Schmuck and F. Rominger, Eur. J. Org. Chem., 2011, 4595–4602 CrossRef CAS; (i) C. L. Paradise, P. R. Sarkar, M. Razzak and J. K. De Brabander, Org. Biomol. Chem., 2011, 9, 4017 RSC.
- G. C. Senadi, W. Hu and J. J. Wang, Org. Lett., 2013, 14, 4478–4481 CrossRef PubMed.
- G. Bartoli, C. Cimarelli, R. Cipolletti, S. Diomedi, R. Giovannini, M. Mari, L. Marsili and E. Marcantoni, Eur. J. Org. Chem., 2012, 630–636 CrossRef CAS.
- X.-H. Gao, P.-C. Qian, X.-G. Zhang and C.-L. Deng, Synlett, 2016, 27, 1110–1115 CrossRef CAS.
- B. Wang, Y. Chen, L. Zhou, J. Wang, C. H. Tung and Z. Xu, J. Org. Chem., 2015, 80, 12718–12724 CrossRef CAS PubMed.
- E. Merkul and T. J. J. Muller, Chem. Commun., 2006, 4817–4819 RSC.
- (a) M. Harmata and C. Huang, Synlett, 2008, 1399–1401 CrossRef CAS; (b) V. H. L. Wong, A. J. P. White, T. S. Hor and K. K. Hii, Adv. Synth. Catal., 2015, 357, 3943–3948 CrossRef CAS.
- (a) J. Chunyang, J. P. Burgess, J. A. Kepler and C. E. Cook, Org. Lett., 2007, 9, 1887–1890 CrossRef PubMed; (b) A. Alhalib and W. J. Moran, Org. Biomol. Chem., 2014, 12, 795–800 RSC.
- (a) A. Saito, K. Iimura and Y. Hanzawa, Tetrahedron Lett., 2010, 51, 1471–1474 CrossRef CAS; (b) E. M. Beccalli, E. Borsini, G. Broggini, G. Palmisano, S. Sottocornola, C. Organica, V. Uni and V. Uni, J. Org. Chem., 2008, 73, 4746–4749 CrossRef CAS PubMed; (c) A. Bacchi, M. Costa, B. Gabriele, G. Pelizzi and G. Salerno, J. Org. Chem., 2002, 67, 4450–4457 CrossRef CAS PubMed; (d) A. Arcadi, S. Cacchi, L. Cascia, G. Fabrizi, F. Marinelli, D. L. Aquila, V. Vetoio, C. Due and I.-L. Aquila, Org. Lett., 2001, 3, 2501–2504 CrossRef CAS PubMed; (e) A. Bacchi, M. Costa, N. Della Cà, B. Gabriele, G. Salerno and S. Cassoni, J. Org. Chem., 2005, 70, 4971–4979 CrossRef CAS PubMed; (f) A. Saito, K. Iimura and Y. Hanzawa, Tetrahedron Lett., 2010, 51, 1471–1474 CrossRef CAS.
- L. J. Street, J. R. Baker, J. L. Castro, M. S. Chambers, A. R. Guiblin, S. C. Hobbs, V. G. Matassa, A. J. Reeve, M. S. Beer, D. N. Middlemiss, A. J. Noble, J. A. Stanton, K. Scholey and R. J. Hargreaves, J. Med. Chem., 1993, 36, 1529–1538 CrossRef CAS PubMed.
- M. D. Milton, Y. Inada, Y. Nishibayashi and S. Uemura, Chem. Commun., 2004, 2712–2713 RSC.
- X. Meng and S. Kim, Org. Biomol. Chem., 2011, 9, 4429–4431 CAS.
- M. Tran-Dubé, S. Johnson and I. McAlpine, Tetrahedron Lett., 2013, 54, 259–261 CrossRef.
- (a) V. Michelet, P. Y. Toullec and J. P. Genêt, Angew. Chem., Int. Ed., 2008, 47, 4268–4315 CrossRef CAS PubMed; (b) G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285–2309 CrossRef CAS PubMed; (c) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127–2198 CrossRef CAS PubMed; (d) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS PubMed; (e) G. Zeni and R. C. Larock, Chem. Rev., 2007, 107, 303 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25857c |
| This journal is © The Royal Society of Chemistry 2017 |