
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cichoric acid regulates the hepatic glucose homeostasis via AMPK pathway and activates the antioxidant response in high glucose-induced hepatocyte injury†
Di Zhu,
Ni Zhang,
Xuelian Zhou,
Mengying Zhang,
Zhigang Liu and
Xuebo Liu*
Laboratory of Functional Chemistry and Nutrition of Food, College of Food Science and Engineering, Northwest A&F University, 28. Xinong Road, Yangling 712100, China. E-mail: xueboliu@aliyun.com; Fax: +86-29-87092325; Tel: +86-29-87092325
First published on 5th January 2017
Abstract
Cichoric acid (CA), a plant-based nutraceutical, is extracted from Echinacea purpurea and other edible plants and vegetables and exhibits multiple biological functions, including antioxidant and hypoglycemic effects. The objective of this study was to determine the effect of CA on the energy regulation pathways and the antioxidant response system in insulin resistance and diabetes-induced hepatic injury and the underlying molecular mechanisms. The streptozotocin-induced diabetic C57BL/6J mice and glucosamine-induced HepG2 cells were observed to evaluate the hepatic protective effects of CA. Compared to the control, CA (60 mg kg−1 d−1, in drinking water for 4 weeks) inhibited hepatic injury and chronic inflammation in diabetic mice via antioxidant defence and regulated the balance of gluconeogenesis and glycolysis. CA (100 μM) regulated glucose metabolism and activated antioxidant response in glucosamine-induced HepG2 cells. CA increased the phosphorylation of the AMP-activated protein kinase and glycogen synthase kinase-3β and stimulated glycogen synthesis and glucose uptake. CA also activated the Nrf2-Keap1 pathway and increased the antioxidant enzyme expression. CA is a potential natural nutraceutical for regulating hepatic glucose homeostasis and antioxidant response, which improved insulin resistance and hepatic injury in diabetes.
1. Introduction
Chronic hyperglycemia plays a critical role in the development of diabetes, which leads to an intracellular oxidative stress, insulin resistance, and a series of cellular responses.1,2 As the center of peripheral glucose homeostasis and sensitivity to insulin,3 the liver plays an essential role in the systemic insulin resistance and the regulation of blood sugar levels. Furthermore, the liver is also responsible for oxidative and detoxifying processes.4 Oxidative stress contributes to mitochondrial dysfunction, insulin resistance, and diabetes complications.5,6 Oxidative stress in diabetes attenuates the antioxidant response and tissue injury.7 Antioxidant/detoxifying enzyme genes, such as quinone oxidoreductase-1 (NQO-1), glutathione peroxidase (GPx), heme oxygenase-1 (HO-1), glutathione-S-transferase (GST), superoxide dismutase (SOD), and catalase, generally defend against oxidative stress-induced damage,8,9 which is mediated and activated by the Nrf2-Keap1 pathway.10,11 Therefore, we hypothesize that the antioxidant response plays a vital role in the remission of the occurrence and development of diabetes.Disorders of energy metabolism, such as glucose and lipid metabolism, are another crucial contributing factor in the development of diabetes. A previous study demonstrated that the energy metabolism of mice fed a high-fat diet was disorganized and insulin resistance was generated in the liver.12,13 AMP-activated protein kinase (AMPK) functions as an energy sensor for energy homeostasis. AMPK is expressed in all eukaryotic cells,14 and it controls the metabolism of glucose and lipids in the liver.15,16 Many reports demonstrated that AMPK was activated by an increase in the AMP/ATP ratio. The activation of AMPK stimulates the catabolic pathways, which enhance glucose uptake and glycolysis,16 reduce the hepatic gluconeogenic enzyme expressions of phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G-6-Pase) genes,17,18 inhibit hepatic lipogenesis and cholesterol production15 and accelerate fatty acid oxidation14 in the key tissues of type 2 diabetes pathogenesis, such as adipose tissues, and liver. Sirtuin type 1 (SIRT1) is a member of the mammalian sirtuin family that is upstream of liver kinase B1 (LKB1), which is upstream of AMPK.15,19 The interaction of SIRT1 and AMPK in the control of energy homeostasis of hepatocytes is a hot topic in diabetic mechanism research.20 Nevertheless, there were few reports about the effect of CA on the regulation of SIRT1 and AMPK in liver tissue.
Cichoric acid (CA) is a phenolic acid extracted from Echinacea purpurea, chicory root and other edible plants and vegetables.21,22 The CA content in Echinacea purpurea roots is 16.80–24.30 mg g−1.23 As a potential natural nutraceutical, CA exhibits various biological activities, such as anti-inflammatory and antioxidant response,24 and it improves insulin resistance.13,25,26 Our previous study investigated the anti-inflammatory effect of CA and the promotion of glucose uptake of glucosamine-mediated HepG2 cells. However, the underlying mechanisms of CA in the relationship between antioxidant response and insulin resistance improvement (i.e., anti-diabetic properties) have not been explored.
The present study explores whether CA improves hepatic injury and regulates glucose metabolism in STZ-induced diabetes in vivo. We also examine whether CA regulates hepatic glucose metabolism via AMPK-SIRT1 energy regulation systems, and antioxidant response in glucosamine-induced HepG2 cells in vitro.
2. Materials and methods
2.1. Reagents and antibodies
Cichoric acid (purity ≥ 99%), which was extracted from Echinacea purpurea, was purchased from Chengdu Pufei De Biotech Co., Ltd (Chengdu, Sichuan, China). Insulin and N-acetyl-L-cysteine22 were obtained from Sigma Chemical Co. (St. Louis, MO, USA). All inhibitors and activators were purchased from Selleck (Shanghai, China). Glucosamine (purity ≥ 99%) was provided by Beyotime Institute of Biotechnology (Haimen, Jiangsu, China). Streptozotocin (STZ) was purchased from MP Biomedicals (Shanghai, China). The Glucose Oxidase Method Kit was purchased from Applygen (Beijing, China). A catalase assay kit (A007-1), malondialdehyde assay kit (A003-1), glutathione peroxidase assay kit (A005), glutathione-S-transferase assay kit (A004), reduced glutathione assay kit (A006-1), superoxide dismutase assay kit (A001-3) and liver/muscle glycogen assay kit (A043) were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, Jiangsu, China). The periodic acid-Schiff stain assay kit was obtained from Solarbio (Beijing, China). The Amplex® Red Hydrogen Peroxide/Peroxidase Assay Kit was purchased from Invitrogen (California, USA). ECL was obtained from Thermo Scientific (Waltham, MA, USA). All other chemicals were analytical grade. Primary antibodies against HO-1, NQO-1, GAPDH, NRF2, KEAP1, Lamin B, Na+/K+-ATPase α1, GLUT2, IRS-1, PTP1B, p-CREB and horseradish peroxidase-conjugated secondary antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The p-GSK, SIRT1, AMPK, p-AMPK, p-IRS-1 S307, p-IκB, IκB, p-IKKβ, NF-κB, p-AKT and AKT antibodies were purchased from Cell Signaling Technology Company (Shanghai, China). The p-IRS-1 Y612 antibody was purchased from Millipore (Bedford, MA, USA).2.2. Cell culture and treatment
HepG2 cells were obtained from the Fourth Military Medical University (Xi'an, Shaanxi, China) and cultivated in RPMI 1640 medium containing 10% FBS, 100 units of penicillin, and 100 μg ml−1 streptomycin at 37 °C with 5% CO2 and 95% air atmosphere. An insulin resistant cell model was constructed according to a previous method.25 Cells at 80–90% confluence were pretreated with or without 18 mM glucosamine (dissolved in phosphate buffer) for 18 h to create insulin resistance. The cells were starved for 2 h using a serum-free medium, prior to drug treatment. Different concentrations of CA dissolved in dimethylsulfoxide (DMSO) were added for 24 h with or without pretreatment with inhibitors or activators; the same amount of DMSO was added to the control group.2.3. Glucose uptake
The supernatant was collected and measured at 570 nm using an enzyme standard from a glucose diagnostic kit. Glucose content was calculated from a standard curve based on the optical density of each sample. The glucose content of the experimental group medium was subtracted from the glucose content of the original RPMI-1640 medium to obtain the glucose uptake.2.4. H2O2 measurements
Briefly, HepG2 cells were cultured in a 96-well plate at 1 × 105 cells per well. The cells were cultured with 18 mM glucosamine for 18 h, followed by incubation with different concentrations of CA for 24 h. The cells were lysed using cell lysis buffer for western blotting and IP. H2O2 generation was determined using the Amplex Red hydrogen peroxide/peroxidase assay kit (Invitrogen), according to the manufacturer's instructions.2.5. Antioxidant enzymes and glycogen measurements
HepG2 cells were collected after treatment and centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 10 min at 4 °C. The liver tissue samples were collected through homogenization and centrifugation at 3000g for 10 min at 4 °C. The enzymatic activity of MDA, SOD, CAT, GSH, GST, GPx and glycogen contents in the supernatant were measured using commercial kits.
000g for 10 min at 4 °C. The liver tissue samples were collected through homogenization and centrifugation at 3000g for 10 min at 4 °C. The enzymatic activity of MDA, SOD, CAT, GSH, GST, GPx and glycogen contents in the supernatant were measured using commercial kits.
2.6. Western blotting
HepG2 cells were lysed using lysis buffer (Nuclear and Cytoplasmic Protein Extraction Kit or cell lysis buffer for western blotting and IP) containing 1% PMSF and 20 mM NaF for 15 min. Lysates were centrifuged for 10 min at 15000 rpm at 4 °C. Hepatocyte proteins were separated using SDS/PAGE and transferred to PVDF membranes. 5% nonfat milk was used to block the membranes for 2.5 h at room temperature. The membranes were incubated with primary antibodies overnight at 4 °C. The results of western blotting were visualized using ECL. A Molecular Imager Chemidoc XRS System (Bio-Rad, Shanghai, China) and Quantity One software were used to expose protein bands and perform semiquantitative analysis, respectively.
2.7. Animals
Eight-week-old male C57BL/6J mice were purchased from the medical department of Xi'an Jiaotong University. The mice were housed individually under standard conditions (12 h light–dark cycle, humidity at 50 ± 15%, temperature of 22 ± 2 °C) with free access to water and an AIN93M standard diet (NMF, 3601 kcal kg−1, 10% of energy as fat and 14.1% of energy as protein) (Trophic Animal Feed High-tech Co. Ltd, China). All of the experimental procedures followed the Guide for the Care and Use of Laboratory Animals: eighth edition, ISBN-10: 0-309-15396-4, and conformed to the Administration Rule of Laboratory Animals (016000291/2011-8494). The housing facility was a barrier housing facility, and it was in keeping with the National Standard of China, Laboratory Animal-Requirements of Environment and Housing Facilities (GB 14925-2010/XG1-2011). The ethical animal protocol was approved by the animal ethics committee of Xi'an Jiaotong University and the compliance to the Helsinki Declaration.2.8. Generation of the diabetic mouse model and CA treatment
Mice received adaptive feeding for one week, followed by intraperitoneal injections of STZ (50 mg kg−1 body weight) or vehicle (0.1 mol L−1 sodium citrate buffer, pH 4.5) after an overnight fast for five days. The animals developed hyperglycemia (blood glucose levels ≥ 13.9 mmol L−1) one week after STZ injection, and these mice were considered diabetic, as previously described.27 Diabetic mice were divided into STZ and CA groups. The CA group received CA in drinking water at a dose of 60 mg kg−1 d−1, and the control group and STZ groups were given distilled water for 4 weeks. The animal protocol was approved by the Animal Ethics Committee of Xi'an Jiaotong University. Mice were anesthetized using trichloroethane, and all efforts were made to minimize suffering. The livers were immediately resected and washed in a PB solution to remove excess blood, fixed in 4% paraformaldehyde at 4 °C for morphology or stored at −80 °C for gene testing.2.9. Liver histopathology and glycogen stain
Fixed livers in 4% paraformaldehyde were dried, embedded in paraffin and cut into 5 μm sections using a microtome. After deparaffinizing and rehydrating the sections, the tissues were stained with hematoxylin and eosin (H&E) dyestuff or the periodic acid-Schiff (PAS) dyestuff, and they were imaged under an optical microscope at a 400× magnification (Olympus, Tokyo, Japan).2.10. RNA isolation and RT-PCR
Total mRNA was extracted from liver samples using an mRNA extraction kit (9767, Takara, Dalian, Liaoning, China). A 1 μg sample of total mRNA was reverse transcribed into cDNA in 20 μL of reaction mixture using the PrimeScript™ RT Master Mix RR036A. Real-time PCR was performed on cDNA samples using SYBR® Premix Ex Taq™ II RR036A. ESI Table S1† lists the sequences for the PCR primers. Primer sequences are available upon request. The following RT-PCR thermal cycle was performed: pre-incubation, 10 min at 95 °C; amplification, 40 cycles of 15 s at 95 °C, 1 min at 60 °C. Each sample was analyzed in triplicate. The transcript levels of target genes were calculated using the comparative method (2−ΔΔCt). Results were normalized to the housekeeping gene Gapdh.2.11. Statistics
All data are expressed as means ± standard deviation (S.D.) or standard error of mean (SEM) for three or more independent experiments. Statistical analyses were performed using one-way analysis of variance (one-way ANOVA) followed by Tukey's multiple-range test in SPSS 19.0. Differences were considered statistically significant at p < 0.05.3. Results
3.1. Effect of CA on the alleviation of liver inflammation via activating antioxidant response in diabetic mice
CA (the chemical structure of CA is shown in Fig. 1A) was added to the drinking water of STZ induced diabetic mice to verify the effect of CA on the alleviation of liver inflammation in vivo. Normal liver cells were arranged in order, compactly and radially around a central vein and exhibit a large, round nucleus in the center of the hepatocyte and a uniform cytoplasm. The hepatic lobules are normal and clearly visible. The liver architecture was distorted in diabetic mice and exhibited lobular structural damage, focal necrosis, and inflammatory cell infiltration. Hepatic cells became pyknotic and fractured. In contrast, these changes described above were markedly alleviated in the CA group (Fig. 1B).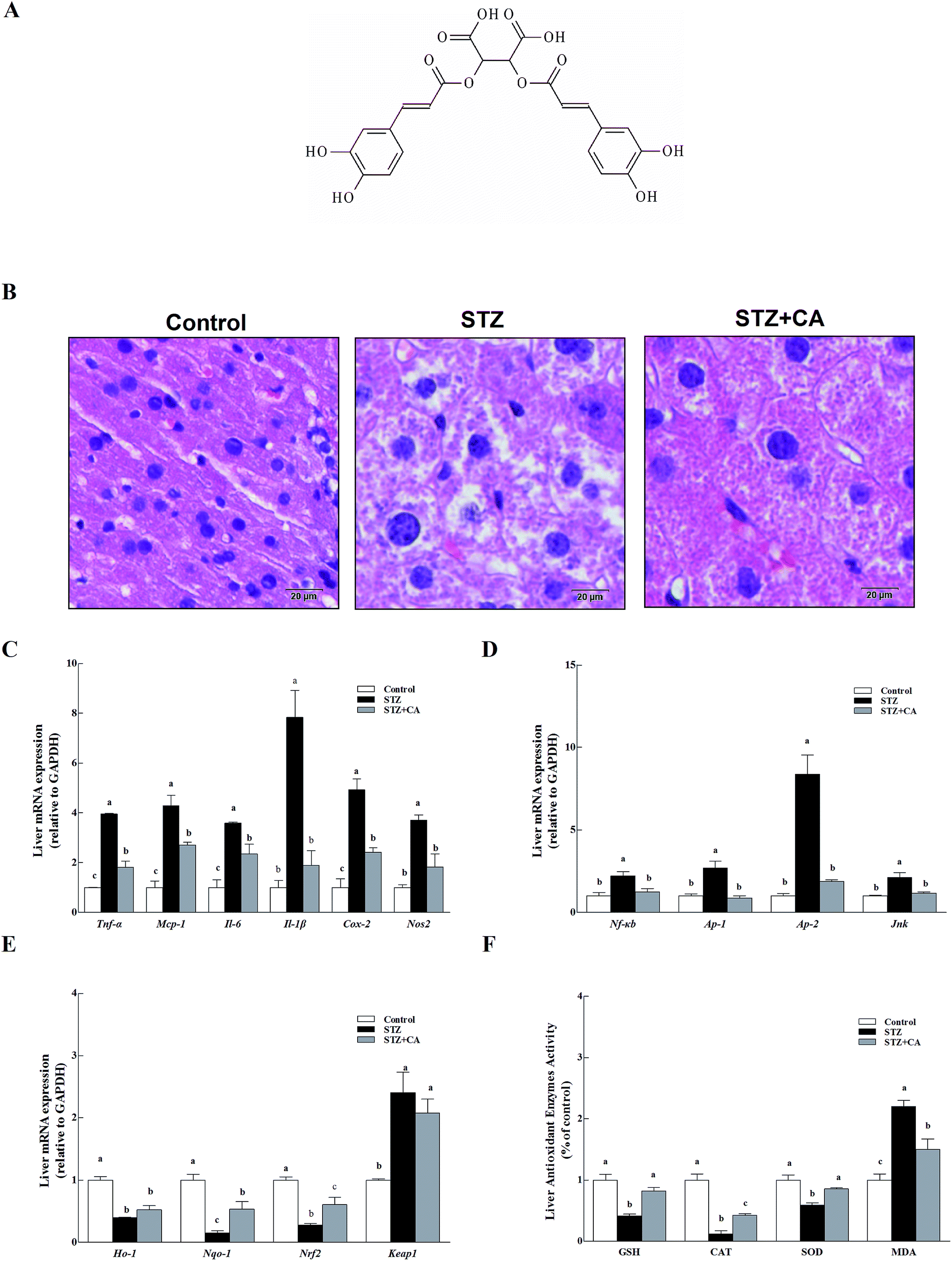 | ||
| Fig. 1 Effect of CA on the alleviation of liver inflammation via activating antioxidant response in diabetic mice. (A) Chemical structure of CA; (B) liver tissues were stained with H&E. Scale bar = 20 μm, magnification 400×. Levels of (C) inflammatory cytokines (Tnf-α, Mcp-1, Il-6, Il-1β, Nos2 and Cox-2), (D) anti-inflammatory pathway (Nf-κb, Ap-1, Ap-2, and Jnk), and (E) the antioxidant response (Ho-1, Nqo-1, Nrf2, Keap1) in the liver were assessed using RT-PCR. The mRNA for these factors was quantified using Gapdh as the internal standard. (F) The expression of antioxidant enzymes (SOD, CAT, and GSH) and MDA were detected in the liver homogenate. The results are expressed as the means ± SEM. Values with different superscripts are significantly different, p < 0.05. | ||
Inflammatory cytokines mRNA, including tumor necrosis factor (Tnf-α), monocyte chemotactic protein 1(Mcp-1), interleukin-6/1β (Il-6, and Il-1β), inducible nitric oxide synthase (Nos2) and cyclooxygenase-2 (Cox-2) in the livers of STZ-diabetic mice were significant, compared to normal mice (p < 0.01) (Fig. 1C). Members of the anti-inflammatory pathway (nuclear transcription factor-κB (Nf-κb), transcription activator-1/2 (Ap-1/2), and c-Jun N-terminal kinase (Jnk)) were also remarkably activated in the STZ group (Fig. 1D). However, these changes were markedly alleviated in the CA group. The antioxidant response was partially attenuated in the STZ group. CA increased the expression of antioxidant enzymes Ho-1 and Nqo-1 mRNA to improve antioxidant response via activation of the Nrf2-Keap1 pathway (Fig. 1E). After tissue homogenization, the enzymatic activities of antioxidant enzymes (GSH, CAT and SOD) and MDA in liver tissues were detected. CA also significantly increased the expression of antioxidant enzymes and suppressed the MDA levels (Fig. 1F), compared to STZ-induced diabetic mice.
3.2. Effect of CA on the alleviation of liver dysfunction in diabetic mice
The gluconeogenesis genes G-6-Pase and Pepck were significantly up-regulated and glycolysis genes glucokinase (Gck), pyruvate kinase (Pk) and phosphofructokinase (Pfk) were significantly down-regulated in the liver tissue of STZ-diabetic mice, compared to normal mice, and these changes were markedly alleviated in the CA group. However, there were no significant changes in G-6-Pase, Gck, Pk or Pfk mRNA copies, compared with the normal control group (Fig. 2A). CA treated mice had a significant increase in glycogen synthesis by 43.4%, compared to the STZ-induced diabetic mice (Fig. 2B), as shown in the PAS glycogen stain (Fig. 2C). To further explore whether CA regulated hepatic glucose homeostasis by promoting the activation of AMPK pathway from the cytoplasm to the membrane via the PI3K/AKT pathway, the expression of AMPK and the downstream protein expression were detected. In STZ-induced diabetic mice, the phosphorylation of AMPK was significantly down-regulated and the phosphorylation of ACC was significantly up-regulated, which were remarkably reversed by the CA supplement (Fig. 2D and E). | ||
| Fig. 2 Effect of CA on the alleviation of liver dysfunction in diabetic mice. Levels of (A) gluconeogenesis genes (G-6-Pase and Pepck) and glycolysis genes (Gck, Pk and Pfk) in the liver were assessed using RT-PCR. The mRNA for these factors was quantified using Gapdh as the internal standard. After liver homogenization, (B) the hepatic glycogen was detected by the PAS glycogen assay kit and was read at 620 nm. (C) The hepatic glycogen storage in STZ-diabetic mice was stained with PAS. Scale bar = 20 μm, magnification 400×. (D) The impact of CA on the phosphorylation of AMPK and ACC were analyzed using western blot. AMPK served as controls. (E) The protein bands were quantified using densitometry. Values are expressed as the fold change compared to the control, which was arbitrarily set to 1. The results are expressed as the means ± SEM (n = 3). Values with different superscripts are significantly different, p < 0.05. | ||
3.3. CA activated the AMPK-Sirt1 pathway and hepatic glucose metabolism pathway
Previous research established that glucosamine induced insulin resistance, and CA accelerated glucose uptake and promoted the translocation of glucose transporter 2 (GLUT2) from the cytoplasm to the membrane in HepG2 cells.25 We investigated the phosphorylation of IRS-1 and AKT in HepG2 cells to determine whether CA could be directly concerned with the regulation of the insulin/IGF-1 signaling pathway (IIS). Protein tyrosine phosphatase 1B (PTP1B) is a negative regulator of glucose and insulin homeostasis.28 PTP1B expression was up-regulated in the glucosamine group. Glucosamine pre-treatment for 18 h impaired insulin signaling. In contrast, CA increased the phosphorylation of p-IRS-1 Y612 and AKT by 33.5% and 23.4%, and reduced the phosphorylation of p-IRS-1 S307 and the expression of PTP1B by 40.1% and 30.4% in HepG2 cells, respectively. The addition of CA and insulin increased the phosphorylation of p-IRS-1 Y612 and AKT by 23.8% and 19.7%, and decreased the phosphorylation of p-IRS-1 S307 and the expression of PTP1B by 20.5% and 28.0%, respectively, compared to the insulin group. Rosiglitazone (ROG) is an insulin sensitizer that was used as a positive control (10 μM) (Fig. 3A and B). | ||
| Fig. 3 CA activated the AMPK-Sirt1 pathway and hepatic glucose metabolism pathway. To construct the insulin resistance model, HepG2 cells were treated with 18 mM glucosamine for 18 h. Cells were starved with serum-free medium for 2 h and incubated with CA (100 μM) for 24 h. Insulin (100 nM) was added during the last 30 min. ROG was used as a positive control (10 μM). (A) The total PTP1B, IRS-1 and AKT, and the phosphorylation of p-IRS-1 S307, p-IRS-1 Y612 and AKT were detected in cells. (C) The phosphorylation of AMPK and Sirt1 was detected in cells using the western blot. (E) The impact of CA on the phosphorylation of CREB and GSK-3β were analyzed using the western blot. AMPK and GAPDH served as controls. (B, D, and F) The protein bands were quantified using densitometry. Values are expressed as the fold change compared to the control, which was arbitrarily set to 1. The results are expressed as the means ± SD of at least three replicates. Values with different superscripts are significantly different, p < 0.05. | ||
AMPK and SIRT1 interacted with each other and played a critical role in energy metabolism regulation.20 High glucosamine inhibited AMPK phosphorylation and SIRT1 expression (Fig. 3C and D). CA treatment of glucosamine-induced HepG2 cells enhanced p-AMPK levels and Sirt1 expression in a concentration-dependent manner (0, 25, 50, and 100 μM). CA treatment activated glycogen synthase kinase-3 (GSK-3β) in HepG2 cells, which reduced liver gluconeogenesis via inhibition of cAMP-response element binding protein (CREB), nuclear translocation and the phosphorylation of downstream substrates (Fig. 3E and F).
3.4. CA regulated glucose metabolism via activation of the AMPK pathway
To further investigate whether CA regulated hepatic glucose homeostasis via the AMPK pathway, the effect of CA on glycogen synthesis and glucose uptake were explored with the AMPK activator AICAR (5-aminoimidazole-4-carboxamide-1-b-D-ribofuranoside) and the AMPK inhibitor compound C. AMPK phosphorylation was profoundly increased by AICAR and decreased by compound C (Fig. 4A and B). CA exhibited a synergistic effect with AICAR on AMPK phosphorylation, and this effect was reversed in the presence of compound C. Compared to glucosamine-induced HepG2 cells, the CA and AICAR accelerated GSK-3β phosphorylation and glycogen synthesis (Fig. 4A–C), and increased the membrane translocation of GLUT2 and glucose uptake (Fig. 4A, B and D), which were also reversed by compound C.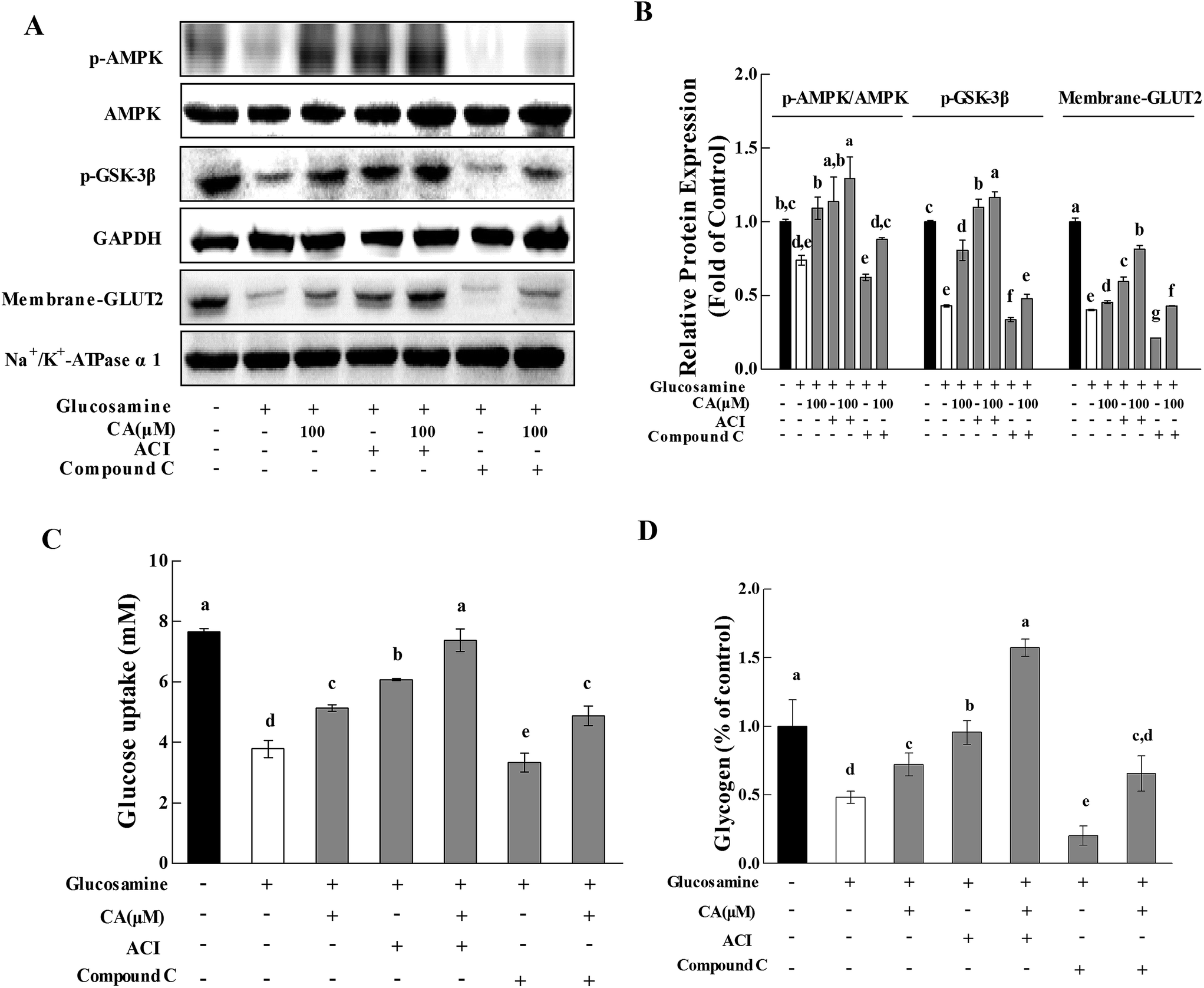 | ||
| Fig. 4 CA regulated glucose metabolism via activation of the AMPK pathway. Cells were incubated with 18 mM glucosamine for 18 h and starved for 2 h with serum-free medium. Compound C (20 μmol L−1) or AICAR (2 mmol L−1) was added to the cells for 30 min before and during the incubation with CA for 24 h. (A) The phosphorylation of AMPK and GSK-3β and the expression of membrane-GLUT2 were detected in cells using the western blot. AMPK, GAPDH and Na+/K+-ATPase α1 served as controls. (B) The protein bands were quantified using densitometry. (C) The glucose content in the culture medium was measured using a glucose uptake analysis kit, which presents the glucose uptake of cells. (D) The glycogen content in the cell lysis buffer was measured using a glycogen analysis kit. Values are expressed as the fold change compared to the control, which was arbitrarily set to 1. The results are expressed as the means ± SD of at least three replicates. Values having different superscripts are significantly different, p < 0.05. | ||
3.5. Effect of CA on H2O2 release and antioxidant enzymes
H2O2 exhibited a 41.3% increase in the high glucosamine group, compared to the control group (Fig. 5A). H2O2 in the mitochondria of the high glucosamine group was also significantly increased by 58%, compared to the control group. H2O2 concentration increased significantly (12.2%, 18.7%, and 26.3% at 25, 50, and 100 μM CA, respectively) in a concentration dependent manner in the CA groups, compared to the high glucosamine group. Glucosamine treatment caused an obvious reduction in GSH levels in HepG2 cells (Fig. 5B). However, CA treatment (25, 50, and 100 μM) significantly increased in GSH enzymatic activity by 12.8%, 21.3% and 29.0%, respectively, compared to the glucosamine group.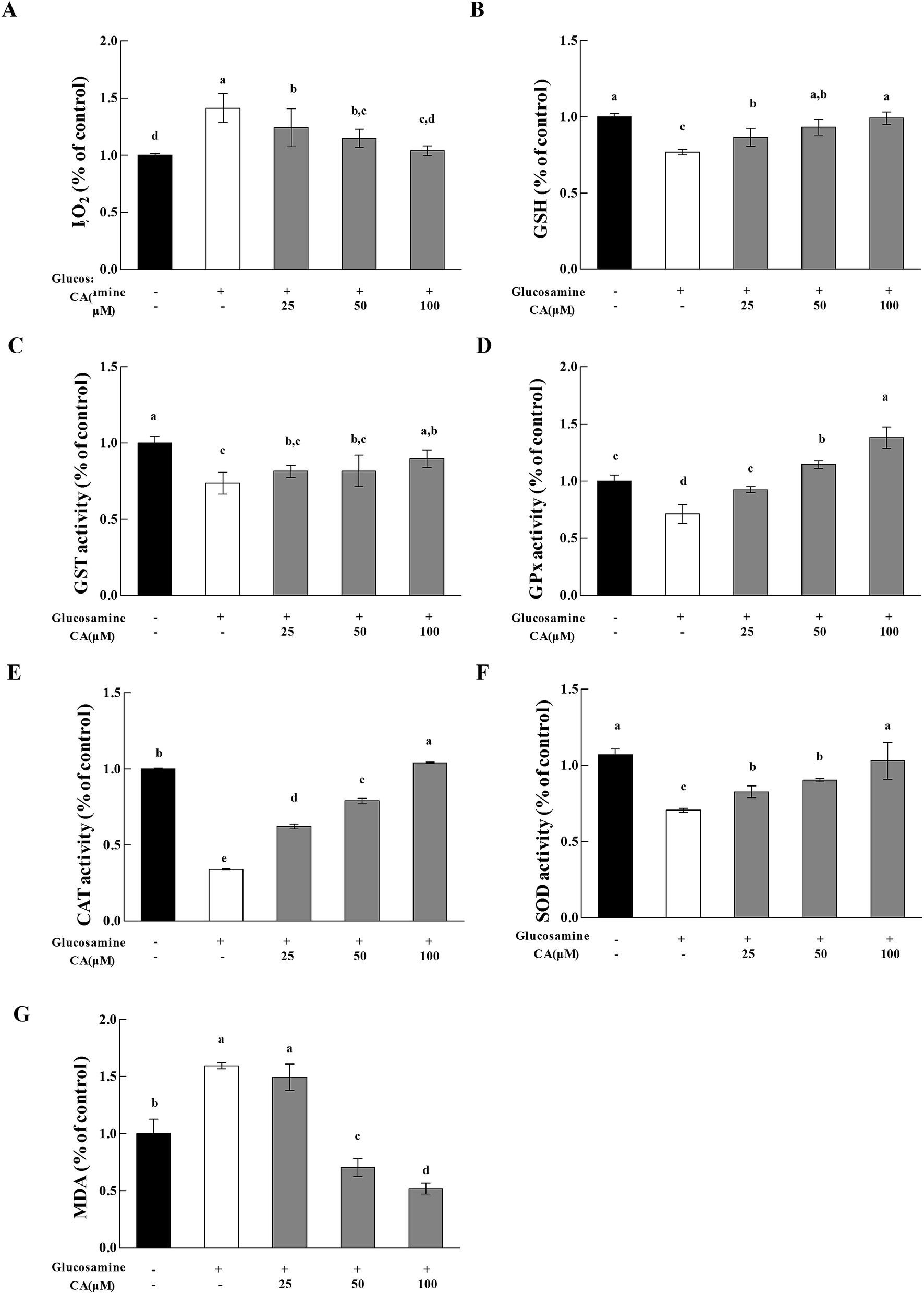 | ||
| Fig. 5 Effect of CA on H2O2 release and antioxidant enzymes. Cells were incubated with 18 mM glucosamine for 18 h and starved for 2 h with serum-free medium. CA (100 μM) was added for 24 h. (A) H2O2 release in cell extracts was immediately collected in 96-well plates, and absorbance was read at 560/590 nm. (B) Cells were lysed, and proteins were reacted. GSH levels was read at 420 nm. (C) GST activity was read at 412 nm. (D) GPx activity was read at 412 nm. (E) CAT activity was read at 405 nm. (F) SOD activity was read at 450 nm. (G) MDA levels was read at 532 nm. Values are expressed as the fold change compared to a vehicle control that was arbitrarily set to 1. The results are expressed as the means ± SD of at least three replicates. Values having different superscripts are significantly different, p < 0.05. | ||
We measured GPx, GST, and SOD activities and MDA levels to evaluate the effects of CA administration on the antioxidant response of high glucosamine-treated HepG2 cells. High glucosamine treatment caused an obvious attenuation in GST levels in HepG2 cells, compared to the control group (p < 0.05). However, CA administration significantly increased GST (Fig. 5C) activities in high glucosamine-treated HepG2 cells in a concentration-dependent manner. In contrast, CA treatments (25, 50 and 100 μM) significantly increased GPx activities by 40.7%, 60.7%, and 93.7%, respectively (Fig. 5D), CAT activities by 2.20%, 2.90%, and 9.20%, respectively (Fig. 5E), and SOD activities by 17.2%, 28.0%, and 46.1%, respectively (Fig. 5F), compared to the glucosamine-treated group. Moreover, CA administration significantly attenuated the elevated MDA levels in high glucosamine-treated HepG2 cells in a concentration-dependent manner (Fig. 5G).
3.6. CA restored glucosamine-induced impairment of the insulin signaling pathway via elimination of H2O2
After incubation of glucosamine for 18 h and the following treatment of 100 μM CA for 24 h in HepG2 cells, CA treatment significantly facilitated NF-E2-related factor 2 (Nrf2) nuclear translocation and decreased kelch-like ECH-associated protein-1 (Keap-1) expression in HepG2 cells (p < 0.05). CA treatment remarkably increased the nuclear translocation of NRF2 and decrease in KEAP-1, compared to the high glucosamine incubation group in HepG2 cells (p < 0.05) (Fig. 6A and B). CA also significantly increased the expression of HO-1 and NQO-1 in a concentration-dependent manner, compared to the high glucosamine group (Fig. 6C and D). The insulin signaling pathway was investigated using the antioxidant N-acetyl-cysteine22 to verify that CA reversed the inhibition of the insulin signaling pathway via suppression of oxidative stress. CA and NAC synergistically increased the phosphorylation of p-IRS-1 Y612 and AKT by 33.8% and 10.4%, respectively, and reduced the phosphorylation of p-IRS-1 S307 by 21.2% in the HepG2 cells. CA and NAC treatment promoted GLUT2 membrane translocation by 22.8%, compared to the NAC group (Fig. 6E and F).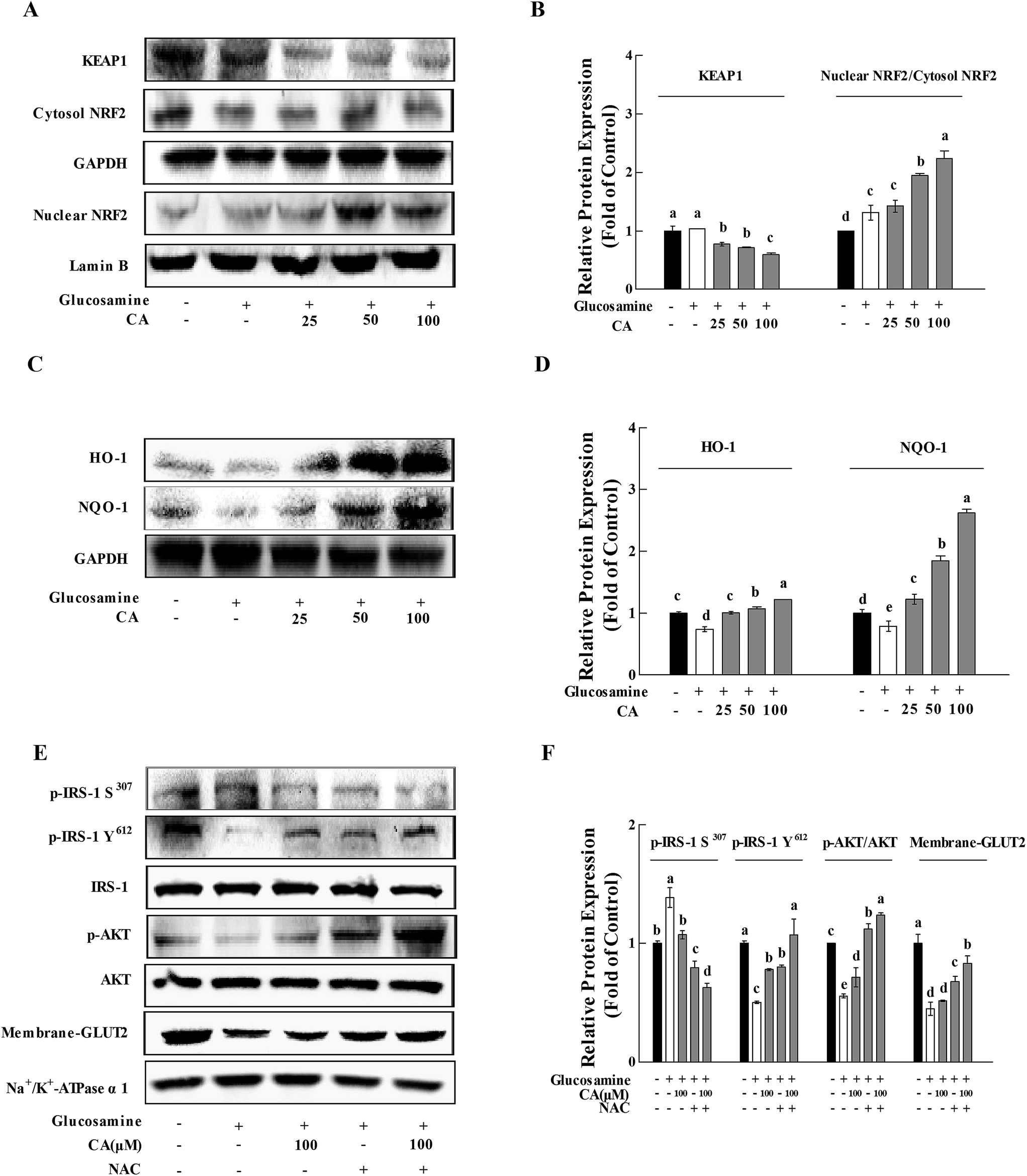 | ||
| Fig. 6 CA restored glucosamine-induced impairment of the insulin signaling pathway via the elimination of H2O2. Cells were incubated with 18 mM glucosamine for 18 h and starved for 2 h with serum-free medium. CA (100 μM) was added for 24 h. (A) KEAP1, NRF2 and nuclear NRF2 in cells were detected using the western blot. GAPDH and Lamin B served as controls. (C) HO-1, NQO-1 and GAPDH were detected in cells using the western blot. (E) NAC (5 mM) was added to the cells for 30 min before and during the incubation with CA for 24 h. Insulin (100 nM) was added during the last 30 min. The phosphorylation of p-IRS-1 S307, p-IRS-1 Y612, AKT and membrane-GLUT2 were detected in cells using the western blot. IRS-1, AKT and Na+/K+-ATPase α1 served as controls. The protein bands (B, D, and F) were quantified using densitometry. Values are expressed as the fold change compared to control, which was arbitrarily set to 1. The results are expressed as the means ± SD of at least three replicates. Values having different superscripts are significantly different, p < 0.05. | ||
4. Discussion
Liver tissue is the center of peripheral glucose homeostasis, and it accounts for more than 80% of glucose release during the fasting state.3 Two major mechanisms contribute to hyperglycemia in diabetes. One mechanism is the increase of hepatic glucose output, and the other mechanism is the decrease of glucose uptake, which leads to the generation of insulin resistance and glucose metabolic disorder in the key glucose-metabolizing organs.12,17 HepG2 cells are human hepatocellular carcinoma cells that are similar to normal human hepatocytes, and these cells are generally used to investigate hypoglycemic effects in hepatocytes.25,29Blood glucose homeostasis is maintained by gluconeogenesis and glycolysis.30 Gluconeogenesis and glycogen synthesis are increased in diabetes and hyperglycemia livers, and glycolysis enzymes are attenuated.31 PEPCK, and G-6P-ase are the key rate limiting enzymes in the regulation of gluconeogenesis,17,32 and these enzymes are suppressed by AMPK via an increase in the orphan nuclear receptor small heterodimer partner (SHP) expression in the liver.33 CA suppressed Pepck and G-6-Pase gene expression and increased the gene expression of glycolysis enzymes, including Gck, Pk, and Pfk, in the present study (Fig. 2A). The effect of CA on glycogen synthesis was also observed with quantitative and qualitative research techniques (Fig. 2B and C).
Our previous studies investigated whether CA stimulated GLUT2 membrane translocation and glucose uptake and inhibited the generation of reactive oxygen species (ROS),25 but the underlying mechanism and relevance of these effects were not explored; as ESI,† it was validated that CA reversed insulin resistance through activating the IIS signaling pathway and antioxidant response. Like carnosic acid,34 CA promoted the phosphorylation of p-IRS-1 Y612 and AKT and activated the IIS signaling pathway; moreover, CA remarkably suppressed the p-IRS-1 S307 (Fig. 3A and B). During exploring the IIS signaling pathway, it was found that the activation of p-IRS-1 S307 down-regulated PI3K/AKT and the downstream pathway in the high glucose-induced cells, leading to insulin resistance.35
Furthermore, it was reported that PTP1B, IκB, and MAPKs are important proteins that influence the IIS signaling pathway via activation of serine phosphorylated IRS-1.17 The negative influence of PTP1B on glucose and insulin homeostasis contributes to the development of diabetes, obesity28 and other metabolic syndromes.18 PTP1B is a potential screening target for anti-diabetic compounds.36 The allosteric inhibition of CGA and CHA of PTP1B produced favorable results.18 CA inhibited PTP1B (Fig. 3A and B) because of its extreme affinity for the PTP1B molecule. The NF-κB signaling pathway also mediates inflammation, and IIS signaling pathway impairment was linked to the phosphorylation of p-IRS-1 S307.12 The inhibitor of nuclear factor kappa-B kinase β (IKKβ) is a key enzyme in NF-κB activation, and it induces serine phosphorylation of IRS-1. Subsequently, the phosphorylation of p-IRS-1 S307 reduces the functional interaction between IRS-1 tyrosine phosphorylation and downstream PI3K signaling in response to insulin.37 This study also examined the effect of CA on IKKβ. ESI Fig. S1† shows that CA inhibited IKKβ phosphorylation via suppression of the oxidative stress process.
It was reported that AMPK stimulated AKT phosphorylation.32 AMPK and SIRT1 cooperated within the cell to maintain the homeostasis of glucose and lipid metabolism,14,15,20 and this cooperation has become an attractive target for anti-diabetic natural products discovery, such as cocoa flavonoids17 and resveratrol.38 Furthermore, an impairment of the AMPK-Sirt1 pathway is associated with insulin resistance and diabetes.20 AMPK, activated by the AMP/ATP ratio, accelerates the fatty acid oxidation and inhibits enzyme activities of acetyl CoA carboxylase.22,39 In this study, it was proved that CA enhanced the phosphorylation of AMPK in vitro (Fig. 3C and D) and in vivo (Fig. 2D and E). In fact, to regulate the glucose metabolism, multiple pathways were involved in regulating glucose homeostasis, such as the LXR signaling pathway,40 leptin signaling pathway41 and FOXO families.42,43 Besides the AMPK pathway, the regulation of glucose homeostasis by CA is probably involved in other pathways. For example, the LepR-STAT3 signaling pathway has been involved in the regulation of hepatic gluconeogenesis, which can be inhibited by PTP1B.41 It is speculated that CA regulated the glucose homeostasis via the inhibition of PTP1B. GSK-3 is downstream of Akt and AMPK, and it is inhibited by phosphorylation, which leads to the subsequent phosphorylation and inactivation of glycogen synthase.17,38 GSK-3β also plays a vital role in the nuclear translocation of CREB via TORC2 activation (a co-activator of CREB), which regulates gluconeogenesis.44 Consistent with the present research, CA treatment of glucosamine-induced HepG2 cells enhanced GSK-3β phosphorylation and decreased CREB phosphorylation in a concentration-dependent manner (Fig. 3E and F). This study also proved that CA expedited glycogen synthesis, which was enhanced by AICAR and reversed by compound C (Fig. 4). Therefore, CA decreased the production of glucose, increased the glycogen synthesis and suppressed gluconeogenesis via an AMPK signaling pathway. Notably, AMPK activation promoted the membrane translocation of GLUT16 and glucose uptake independently of insulin.45 This study also demonstrated that CA expedited the membrane translocation of GLUT2 and glucose uptake via an AMPK signaling pathway. Taken together, our findings indicate that CA regulated hepatic glucose homeostasis via the AMPK signaling pathway.
The increase of oxidative stress induced by hyperglycemia may contribute to the progressive deterioration of peripheral insulin sensitivity.1,5 We established a STZ-induced diabetes model and administered CA through drinking water to further investigate the role of the CA on the alleviation of hyperglycemia-induced liver damage. Previous studies demonstrated that the liver tissue of diabetic mice exhibits oxidative stress and inflammatory responses12 induced by hyperglycemia. CA treatment decreased the mRNA expression levels of the inflammatory cytokines (Fig. 1C) and members of the anti-inflammatory pathway (Fig. 1D), and activated the expression of antioxidant enzymes (Fig. 1E and F). These pathways are implicated in inflammation and insulin resistance. Previous studies suggested that inflammation confuses normal metabolism and damage in the insulin signaling pathway, which increases the risk for the development of insulin resistance, diabetes and diabetic complications.5,6 Therefore, the inhibition of inflammation development is crucial to slowing down the process of diabetes.
CA is a phenolic acid that exhibits remarkable antioxidant activity.24 In addition, the liver is the vital tissue for distribution of CA after gavage administration.46 This study demonstrated that high-dose glucosamine increased H2O2 generation, which was reversed by CA (Fig. 5A). Previous studies demonstrated that Nrf2 was the primary transcription factor for numerous detoxifying and regulating genes of redox homeostasis in the liver,10 and it has been evaluated as a target of the anti-oxidative stress effect. Nrf2 normally combines with the negative regulator Keap1 in the cytoplasm, as an inactive form. Nevertheless, Nrf2 separates from Keap1 under oxidant conditions, enters into the nucleus, and binds to an antioxidant response element.11 The GSH and related enzymes protect cells from free radical-mediated damage in the liver; CAT and SOD activities represent the antioxidant response;8 the MDA free radical reaction reflects the effect of lipid peroxides.9 Plant extracts, such as resveratrol, protect cells via activation of Nrf2 expression.4,47 CA dramatically increased HO-1 and NQO-1 expression via the inhibition of Keap-1 and activation of Nrf2 nuclear translocation in the present study, which regulated the expression of redox enzymes in glucosamine-induced HepG2 cells and livers of STZ-induced diabetic mice (Fig. 1, 5 and 6).
5. Conclusion
In summary, we observed that CA markedly activated the antioxidant response to inhibit hepatic injury and chronic inflammation, and regulated the balance of gluconeogenesis and glycolysis in STZ-induced diabetic mice. It was further found that CA regulated hepatic glucose homeostasis via the AMPK pathway in glucosamine-induced HepG2 cells. Moreover, CA down-regulated the expression of PTP1B and IRS-1 serine phosphorylation and activated the PI3K/AKT pathway to reverse the insulin resistance via activation of the antioxidant response in vitro. These results strongly suggest that CA may be used as a functional food-derived compound for the regulation of hepatic glucose metabolism and prevention of liver damage in diabetes.Abbreviations used
| AICAR | 5-Aminoimidazole-4-carboxamide-1-b-D-ribofuranoside |
| AMPK | AMP-activated protein kinase |
| AP-1/2 | Transcription activator |
| ARE | Antioxidant response element |
| CA | Cichoric acid |
| CREB | cAMP-response element binding protein |
| COX | Cyclooxygenase |
| GCK | Glucokinase |
| G6Pase | Glucose-6-phosphatase |
| GLUTs | Glucose transporters |
| GPx | Glutathione peroxidase |
| GS | Glycogen synthase |
| GSK-3 | Glycogen synthase kinase-3 |
| GST | Glutathione-S-transferase |
| HO-1 | Heme oxygenase-1 |
| IIS | Insulin/IGF-1 signaling pathway |
| IKKβ | Inhibitor of nuclear factor kappa-B kinase β |
| I-κB | α inhibitory kappa B |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| JNK | c-Jun N-terminal kinase |
| Keap-1 | Kelch-like ECH-associated protein-1 |
| LKB1 | Liver kinase B1 |
| MCP-1 | Monocyte chemotactic protein 1 |
| NAC | N-Acetyl-cysteine |
| NF-κB | Nuclear transcription factor-κB |
| Nrf2 | NF-E2-related factor 2 |
| NQO-1 | Quinone oxidoreductase-1 |
| PEPCK | Phosphenolpyruvate carboxykinase |
| PFK | Phosphofructokinase |
| PK | Pyruvate kinase |
| PTP1B | Protein tyrosine phosphatase 1B |
| ROG | Rosiglitazone |
| SHP | Orphan nuclear receptor small heterodimer partner |
| SIRT1 | Sirtuin type 1 |
| STZ | Streptozotocin |
| TNF-α | Tumor necrosis factor |
Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2016YFD0400601), Science and Technology Coordination Project of Innovation in Shaanxi province, (No. 2014KTCL02-07) and Scientific Startup Funds for Doctors of Northwest A&F University (Z109021611).References
- C. M. Palmeira, A. P. Rolo, J. Berthiaume, J. A. Bjork and K. B. Wallace, Toxicol. Appl. Pharmacol., 2007, 225, 214–220 CrossRef CAS PubMed.
- M. Brownlee, Nature, 2001, 414, 813–820 CrossRef CAS PubMed.
- O. Wang, J. Liu, Q. Cheng, X. Guo, Y. Wang, L. Zhao, F. Zhou and B. Ji, PLoS One, 2015, 10, e0118135 Search PubMed.
- A. L. Eggler, K. A. Gay and A. D. Mesecar, Mol. Nutr. Food Res., 2008, 52(suppl. 1), S84–S94 Search PubMed.
- L. Rochette, M. Zeller, Y. Cottin and C. Vergely, Biochim. Biophys. Acta, 2014, 1840, 2709–2729 CrossRef CAS PubMed.
- M. El Assar, J. Angulo and L. Rodriguez-Manas, Free Radical Biol. Med., 2013, 65, 380–401 CrossRef CAS PubMed.
- J. L. Evans, I. D. Goldfine, B. A. Maddux and G. M. Grodsky, Endocr. Rev., 2002, 23, 599–622 CrossRef CAS PubMed.
- S. Y. Ou, G. M. Jackson, X. Jiao, J. Chen, J. Z. Wu and X. S. Huang, J. Agric. Food Chem., 2007, 55, 3191–3195 CrossRef CAS PubMed.
- I. Cordero-Herrera, M. Á. Martín, L. Goya and S. Ramos, Food Res. Int., 2015, 69, 194–201 CrossRef CAS.
- S. Wang, W. Zheng, X. Liu, P. Xue, S. Jiang, D. Lu, Q. Zhang, G. He, J. Pi, M. E. Andersen, H. Tan and W. Qu, Environ. Sci. Technol., 2014, 48, 13478–13488 CrossRef CAS PubMed.
- D. D. Zhang, Drug Metab. Rev., 2006, 38, 769–789 CrossRef CAS PubMed.
- Z. Liu, I. Y. Patil, T. Jiang, H. Sancheti, J. P. Walsh, B. L. Stiles, F. Yin and E. Cadenas, PLoS One, 2015, 10, e0128274 Search PubMed.
- H. Xiao, G. Xie, J. Wang, X. Hou, X. Wang, W. Wu and X. Liu, Food Res. Int., 2013, 54, 345–353 CrossRef CAS.
- G. R. Steinberg and B. E. Kemp, Physiol. Rev., 2009, 89, 1025–1078 CrossRef CAS PubMed.
- B. Viollet, R. Mounier, J. Leclerc, A. Yazigi, M. Foretz and F. Andreelli, Diabetes Metab., 2007, 33, 395–402 CAS.
- D. G. Hardie, Nat. Rev. Mol. Cell Biol., 2007, 8, 774–785 CrossRef CAS PubMed.
- I. Cordero-Herrera, M. A. Martin, L. Goya and S. Ramos, Food Chem. Toxicol., 2014, 64, 10–19 CrossRef CAS PubMed.
- S. K. Baskaran, N. Goswami, S. Selvaraj, V. S. Muthusamy and B. S. Lakshmi, J. Chem. Inf. Model., 2012, 52, 2004–2012 CrossRef CAS PubMed.
- D. R. Alessi, K. Sakamoto and J. R. Bayascas, Annu. Rev. Biochem., 2006, 75, 137–163 CrossRef CAS PubMed.
- L. E. Nelson, R. J. Valentine, J. M. Cacicedo, M. S. Gauthier, Y. Ido and N. B. Ruderman, Am. J. Physiol.: Cell Physiol., 2012, 303, C4–c13 CrossRef CAS PubMed.
- M. Landmann, G. Kanuri, A. Spruss, C. Stahl and I. Bergheim, Nutrition, 2014, 30, 882–889 CrossRef CAS PubMed.
- M. Innocenti, S. Gallori, C. Giaccherini, F. Ieri, F. F. Vincieri and N. Mulinacci, J. Agric. Food Chem., 2005, 53, 6497–6502 CrossRef CAS PubMed.
- P. Molgaard, S. Johnsen, P. Christensen and C. Cornett, J. Agric. Food Chem., 2003, 51, 6922–6933 CrossRef CAS PubMed.
- L. Dalby-Brown, H. Barsett, A. K. Landbo, A. S. Meyer and P. Molgaard, J. Agric. Food Chem., 2005, 53, 9413–9423 CrossRef CAS PubMed.
- D. Zhu, Y. Wang, Q. Du, Z. Liu and X. Liu, J. Agric. Food Chem., 2015, 63, 10903–10913 CrossRef CAS PubMed.
- D. Tousch, A. D. Lajoix, E. Hosy, J. Azay-Milhau, K. Ferrare, C. Jahannault, G. Cros and P. Petit, Biochem. Biophys. Res. Commun., 2008, 377, 131–135 CrossRef CAS PubMed.
- J. Thomas, M. L. Garg and D. W. Smith, J. Nutr. Biochem., 2014, 25, 313–318 CrossRef CAS PubMed.
- D. D. Song, Y. Chen, Z. Y. Li, Y. F. Guan, D. J. Zou and C. Y. Miao, Biochim. Biophys. Acta, 2013, 1831, 1368–1376 CrossRef CAS PubMed.
- J. F. Williams and J. M. Olefsky, Endocrinology, 1990, 127, 1706–1717 CrossRef CAS PubMed.
- L. Agius, Best Pract. Res., Clin. Endocrinol. Metab., 2007, 21, 587–605 CrossRef CAS PubMed.
- J. Zhou, G. Xu, Z. Bai, K. Li, J. Yan, F. Li, S. Ma, H. Xu and K. Huang, Toxicol. Appl. Pharmacol., 2015, 289, 409–418 CrossRef CAS PubMed.
- I. Cordero-Herrera, M. A. Martin, L. Bravo, L. Goya and S. Ramos, Mol. Nutr. Food Res., 2013, 57, 974–985 CAS.
- Y. D. Kim, K. G. Park, Y. S. Lee, Y. Y. Park, D. K. Kim, B. Nedumaran, W. G. Jang, W. J. Cho, J. Ha, I. K. Lee, C. H. Lee and H. S. Choi, Diabetes, 2008, 57, 306–314 CrossRef CAS PubMed.
- C. W. Tsai, K. L. Liu, Y. R. Lin and W. C. Kuo, Mol. Nutr. Food Res., 2014, 58, 654–664 CAS.
- G. Sabio, M. Das, A. Mora, Z. Zhang, J. Y. Jun, H. J. Ko, T. Barrett, J. K. Kim and R. J. Davis, Science, 2008, 322, 1539–1543 CrossRef CAS PubMed.
- K. A. Kenner, E. Anyanwu, J. M. Olefsky and J. Kusari, J. Biol. Chem., 1996, 271, 19810–19816 CrossRef CAS PubMed.
- D. Cai, M. Yuan, D. F. Frantz, P. A. Melendez, L. Hansen, J. Lee and S. E. Shoelson, Nat. Med., 2005, 11, 183–190 CrossRef CAS PubMed.
- J. A. Baur, K. J. Pearson, N. L. Price, H. A. Jamieson, C. Lerin, A. Kalra, V. V. Prabhu, J. S. Allard, G. Lopez-Lluch, K. Lewis, P. J. Pistell, S. Poosala, K. G. Becker, O. Boss, D. Gwinn, M. Wang, S. Ramaswamy, K. W. Fishbein, R. G. Spencer, E. G. Lakatta, D. Le Couteur, R. J. Shaw, P. Navas, P. Puigserver, D. K. Ingram, R. de Cabo and D. A. Sinclair, Nature, 2006, 444, 337–342 CrossRef CAS PubMed.
- N. Henin, M. F. Vincent, H. E. Gruber and G. Van den Berghe, FASEB J., 1995, 9, 541–546 CAS.
- S. W. Beaven, A. Matveyenko, K. Wroblewski, L. Chao, D. Wilpitz, T. W. Hsu, J. Lentz, B. Drew, A. L. Hevener and P. Tontonoz, Cell Metab., 2013, 18, 106–117 CrossRef CAS PubMed.
- A. Bravard, G. Vial, M. A. Chauvin, Y. Rouille, B. Bailleul, H. Vidal and J. Rieusset, Cell Commun. Signaling, 2014, 12, 4 CrossRef PubMed.
- M. Lu, M. Wan, K. F. Leavens, Q. Chu, B. R. Monks, S. Fernandez, R. S. Ahima, K. Ueki, C. R. Kahn and M. J. Birnbaum, Nat. Med., 2012, 18, 388–395 CrossRef CAS PubMed.
- X. Xiong, R. Tao, R. A. DePinho and X. C. Dong, PLoS One, 2013, 8, e74340 CAS.
- H. D. Yuan and G. C. Piao, Biosci., Biotechnol., Biochem., 2011, 75, 1079–1084 CrossRef CAS PubMed.
- Y. Zhang, L. Jin, Q. Chen, Z. Wu, Y. Dong, L. Han and T. Wang, Fitoterapia, 2015, 102, 7–14 CrossRef CAS PubMed.
- Y. Wang, G. Xie, Q. Liu, X. Duan, Z. Liu and X. Liu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1031, 139–145 CrossRef CAS PubMed.
- Z. Ungvari, Z. Bagi, A. Feher, F. A. Recchia, W. E. Sonntag, K. Pearson, R. de Cabo and A. Csiszar, Am. J. Physiol.: Heart Circ. Physiol., 2010, 299, H18–H24 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The attenuation of CA on NF-κB signaling pathway via suppressing oxidative stress, and the primer sequences used for semi-quantitative RT-PCR analysis. See DOI: 10.1039/c6ra25901d |
| This journal is © The Royal Society of Chemistry 2017 |