
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insight into the structure and energy of Mo27SxOy clusters†
Xingchen Liua,
Dongbo Caoa,
Tao Yangb,
Hao Lib,
Hui Gea,
Manuel Ramosc,
Qing Pengd,
Albert K. Deardene,
Zhi Cao f,
Yong Yangag,
Yong-Wang Liag and
Xiao-Dong Wen*ag
f,
Yong Yangag,
Yong-Wang Liag and
Xiao-Dong Wen*ag
aState Key Laboratory of Coal Conversion, Institute of Coal Chemistry Chinese Academy of Sciences, Taiyuan, Shanxi, 03001 China. E-mail: wxd@sxicc.ac.cn
bState Key Laboratory of Heavy Oil Processing, China University of Petroleum, Beijing 102249, China
cDepartment of Physics and Mathematics, Universidad Autónoma de Cd. Juárez, 450 del Charro Ave. Cd, Juárez, Mexico 32310
dNuclear Engineering and Radiological Sciences, University of Michigan, Ann Arbor, MI 48109-2104, USA
eDepartment of Physics, Berea College, Berea, KY 40403, USA
fDepartment of Chemistry, University of California, Berkeley, California 94720, USA
gSynfuels China Co. Ltd., Huairou, Beijing 100195, China
First published on 31st January 2017
Abstract
Oxygen incorporated molybdenum sulfide (MoS2) nanoparticles are highly promising materials in hydrodesulfurization catalysis, mechanical, electric, and optical applications. We report a systematic theoretical study of the successive oxidation reactions of the Mo27SxOy nanoparticles and the reaction network, along with the electronic structure changes caused by the oxygen substitution. The replacement of surface sulfur by oxygen atoms is thermodynamically favorable. Our results indicate that the oxidation on the S edge with 100% and 50% coverage is more favored than on the Mo edge. Meanwhile, it is found that the oxidation on the S edge with 100% coverage has similar replacement ability by oxygen as on the S edge with 50% coverage. Thus, sulfur coverage does not play an important role in the oxidation on the S edge. Further comparison shows that the oxidation on corner sites is more favorable than on edge sites. In addition, the replacement of the bulky sulfur on the Mo edge is equally as favored as those of sulfur on the S edge. This work provides important information on the thermodynamics of the Mo27SxOy nanoparticles, and gives new insights into the mechanism of the oxidation of MoS2 and the sulfidation of MoO3.
Introduction
With stringent environmental regulations and the declining quality of fossil energy resources, the sulfur level in fuels has renewed the interests in the understanding of the hydrodesulfurization (HDS) mechanism.1 Molybdenum sulfide catalysts (MoSx) have been widely used as HDS catalysts for more than 80 years. To understand the HDS mechanism and improve deep HDS catalysts it is necessary to have full insight into the structure and composition of the MoSx catalysts.It has been proposed since the 1990s that oxygen plays an important role in the MoS2 catalyzed HDS reactions. Chary et al.2 found a linear correlation between oxygen chemisorption on MoS2 and the HDS activity. They thus propose that oxygen chemisorption on MoS2 and HDS behaves in a similar manner, and that both happen on the coordinatively unsaturated sites (CUS).
The importance of molybdenum oxysulfide clusters is accentuated by the MoS2 preparation process. It is well known that HDS catalysts are initially prepared from molybdenum oxides by sulfidation. The sulfidation process, which is important for understanding the catalysts structure and HDS mechanism, also involves intermediate state like MoSxOy. In 1996, Weber et al.3 studied the sulfidation of MoO3, and found that the early stage of MoO3 sulfidation is the transformation to oxysulfides (MoOS2).
In addition, similar to graphene, MoS2 is known as an important two-dimensional (2D) material of stacks of layers with unique mechanical,4 electric,5–7 and optical properties,8–10 and has become a very promising functional material in nanoelectronics11 and optoelectronics.12 As atomic defects such as the oxidation of MoS2 may be responsible for the variation of its mechanical, electric, optical and catalytic properties,13 understanding the oxidation process is crucial in improving its performance in all of these aspects. For example, controllable bandgap widening from 1.8 to 2.6 eV was recently achieved through oxidized MoS2 sheets that are composed of quilted phases of various MoSxOy flakes.14 Oxygen-incorporated MoS2 nanosheets can be used as catalysts for efficient electrochemical hydrogen evolution15,16 and oxygen reduction reaction17 by regulating the electronic structure of MoS2 with oxygen.
There have been several studies, earlier on mostly experimental and more recently mostly theoretical, on the oxidation of the MoS2. The oxidation characteristics of MoS2 was first reported by Lavik et al.18 with thermogravimetric method and later by Sohn et al.19 with kinetic method. Lince20,21 used the MoS2−xOx model with substitutional oxygen and explained the oxidation of MoS2. In 1999, Fleischauer and Lince22 compared the oxidation and oxygen substitution in MoS2 solid film lubricants. They suggested that oxygen atoms are incorporated into the MoS2 structure. In 2001, Miller et al.23 reported the adsorption of O2 on MoS2 catalysts by EXAFS analysis. They found that sulfur atoms are partially substituted by oxygen in MoS2, and proposed that much of the MoS2 structure remains unchanged. As they suggested, the bond length of Mo–O is from 1.69 to 1.73 Å. Santosh K. C. et al. studied the surface oxidation of monolayer MoS2 with DFT, and found that the dissociative adsorption of molecular oxygen on the MoS2 surface is kinetically limited due to the large energy barrier at low temperature.24 Sen et al. also found by DFT that oxygen molecule only weakly interacts with the surface, and the penetration of oxygen atoms and molecules through a defect-free MoS2 monolayer is prevented by a very high diffusion barrier.25 Both of the studies imply that the oxidation of MoS2 happens on the corner or edge atoms with lower coordination number, rather than on the surface (or bulk). The energetics of oxidation of triangular MoS2 nanoparticles with different S coverages was first reported by Liang et al.26 Using a periodic model, a wide range in the oxidation energy (−0.9 to −2.4 eV) was observed for thee first oxidation step.
Despite these reported works, a systematic study of the thermodynamics of the various oxidation paths and networks are lacking, and the structure of the hexagonal MoSxOy nanocluster is still unclear. Previously, we have carried out systematic DFT investigations into the carburization processes of MoSx clusters to understand their surface structures.27 In this work, on the basis of the same idea, we report systematically the oxidation of Mo27Sx cluster. The MoSxOy clusters are treated with the DFT method. The oxidation processes are performed on the Mo edge (33% sulfur coverage), the S edge (100%, 67% and 50% sulfur coverage) and the bulk Mo27S54 cluster, respectively. The oxidation structure and its formation energies are discussed to understand the oxidation processes of Mo27Sx cluster.
The stoichiometric (Mo27S54) and non-stoichiometric (Mo27Sx) clusters were used to model the MoS2 surfaces. The structures of Mo27S54 (I) with 0% sulfur coverage on Mo edge and 100% sulfur coverage on S edge, Mo27S56 (II) with two bridge S atoms on Mo edge (33% sulfur coverage on Mo edge), Mo27S51 (III) with three bridge S atoms on S edge (50% sulfur coverage on S edge) are shown in Fig. 1. The transformation between various molybdenum sulfide clusters and the corresponding reaction energies are summarized in eqn (1) and (2). In our previous study,28 we reported various Mo27Sx clusters. On the basis of eqn (1) and (2), the formation of Mo27S56 (II) and Mo27S51 (III) is exothermic by 0.19 and 4.61 eV, respectively.
| MonSm + xH2S = MonSm+x + xH2 (adding sulfur to Mo edge), ΔE0(Mo) = [E(MonSm+x) + E(xH2)] − [E(MonSm) + E(xH2S)] | (1) |
| MonSm + xH2 = MonSm−x + xH2S (deleting sulfur from S edge), ΔE0(S) = [E(MonSm−x) + E(xH2S)] − [E(MonSm) + E(xH2)] | (2) |
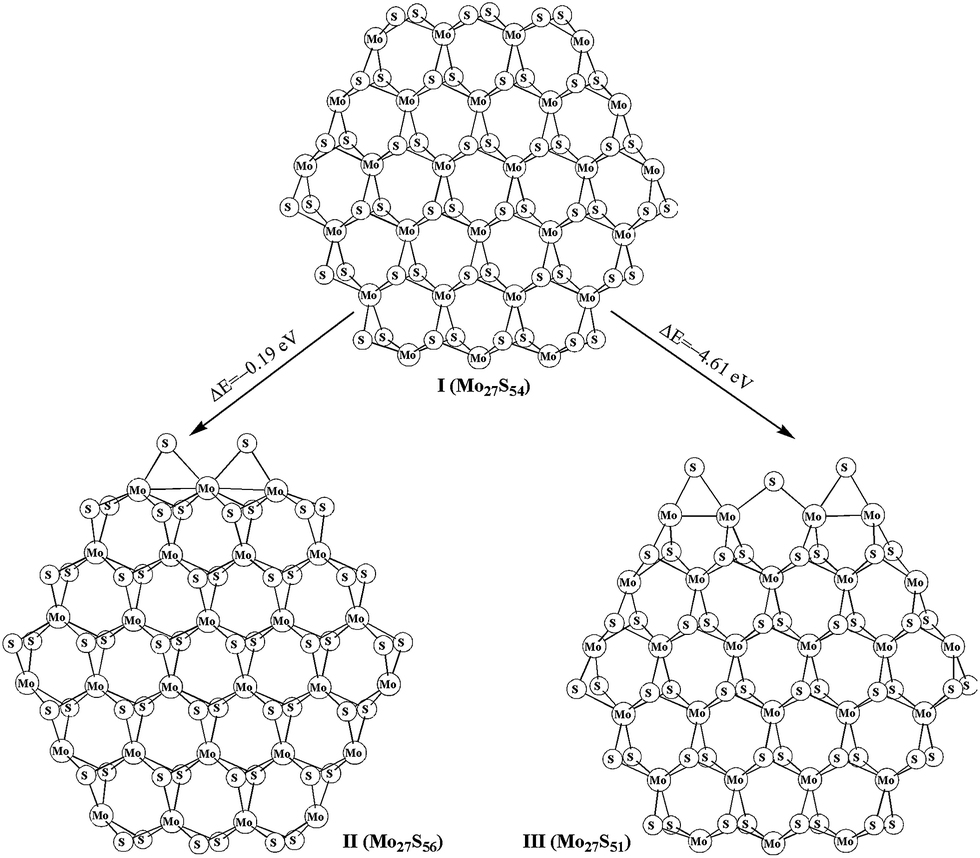 | ||
| Fig. 1 Optimized different Mo27Sx cluster models. | ||
In this work, we used the atom formation energy in eqn (3) to investigate the oxidation process, in which we have used atomic oxygen and sulfur as references. On the basis of eqn (3), the more negative the atom formation energies, the more favorable the process. To address the possibility of other oxidation species instead of atomic oxygen, we also used the following equation for calculating the formation energy: Mo27Sx + nO2 = Mo27Sx−nOn + ½nSO2 + ½nS. Compared to eqn (3), there is only a quantitative difference by −1.71 eV per oxygen (½nS + nO2 = nO + ½SO2), which does not change our qualitative conclusion that the oxygen process is exothermic. In additon, it is convenient for comparing with carburization process with using the atomic formation energy.
| Mo27Sx + nO = Mo27Sx−nOn + nS, ΔEf = [E(Mo27Sx−nOn) + nE(S)] − [E(Mo27Sx) + nE(O)] | (3) |
Computational section
All calculations are performed with the program package DMol29 in the Materials Studio of Accelrys Inc. In DMol29 the physical wave functions are expanded in terms of accurate numerical basis sets.29–31 We have used the doubled numerical basis set with polarization functions for other elements (DNP), while effective core potential (ECP)32 is used for molybdenum. The generalized gradient corrected functional by Perdew and Wang (GGA-PW91) is used33 and the medium quality mesh size is used for the numerical integration. The tolerances of energy, gradient and displacement convergence are 2 × 10−5 au, 4 × 10−3 au Å−1, 5 × 10−3 Å, respectively. The real space cutoff of atomic orbitals is set at 5.5 Å, and a Fermi smearing of 0.0005 was used to count the orbital occupancy. All structures are fully optimized without any constrain. Vibrational frequency calculations were used to confirm the local minima. The atomic position of Mo27Sx cluster and corresponding inputs used are shown in ESI.†Results and discussion
1. Oxidation on Mo edge
The oxidation processes and structures are shown in Fig. 2. The formation energy for Mo27S56 (II) of −13.04 eV is obtained by using S atom as the reactant (Mo27S54 + 2S = Mo27S56). The first replacement step from Mo27S56 (II) to form Mo27S55O (1) is −1.93 eV, and the second step from 1 to Mo27S54O2 (2) is also −1.93 eV. The two steps have the same formation energies, and are both exothermic processes, indicating that the sulfur and oxygen atoms on the Mo edge are roughly independent. A direct oxidation on the Mo edge of Mo27S54 (I) is also favored thermodynamically by −16.91 eV. In addition, it is obvious that oxidation is more favored than sulfidation on the Mo edge with 0% sulfur coverage. In other words, the O atom is more favored than S atom to be bonded with Mo. Other than that, by comparing oxidation with the carburization process of MoS2 in the literature,5 it is clear that the oxidation is also more favorable than carburization on the Mo edge.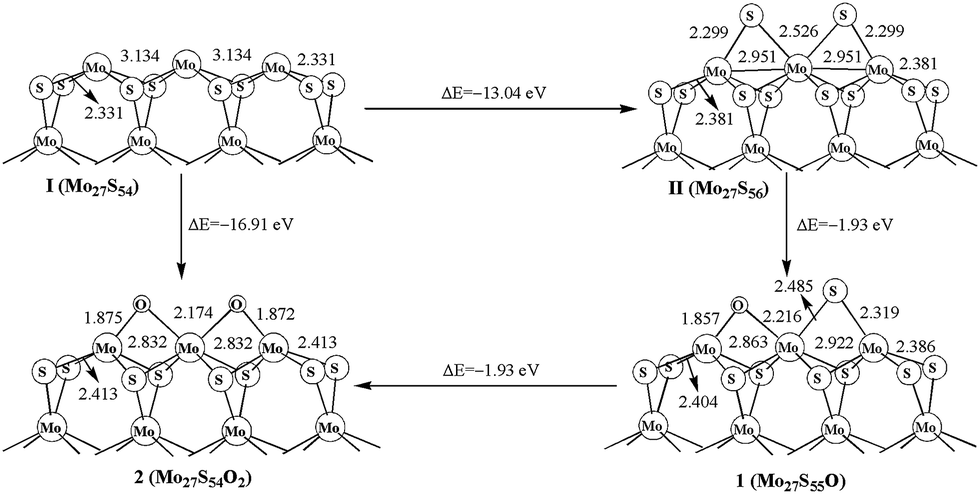 | ||
| Fig. 2 Oxidation on Mo edge. | ||
Liang et al.26 also studied the oxidation of MoS2 edges with DFT, and is worth comparison in the discussion. To begin with, they studied the edge oxidation of triangular MoS2 clusters, while our works studies the edge oxidation of hexagonal MoS2 clusters. Experimentally,34,35 both triangle-shaped and hexagonal MoS2 nanoclusters were observed as stable forms of existence. Because of the difference in topology, coordination environment, and the electronic structures from quantum size effect, the oxidations of the edges of different shaped MoS2 clusters are different. Indeed, their structures of Mo edge are covered with 0%, 50%, and 100% sulfur, while our Mo edge is covered with 33% sulfur.
Second, their work only studied the first oxidation step, equivalent to the step of II (Mo27S56) to 1 (Mo27S55O) in our studies in Fig. 2. Interestingly, comparing the oxidation energies on Mo edge, our value of −1.93 eV at 33% coverage is well within the case of −1.5 eV, −1.6 eV and −1.8 eV at 0% coverage and the case of −2.1 eV and −2.4 eV at 50% coverage in Liang's work.
We have tried to explain this energetic difference among oxidation, carburization and sulfidation by using the bonding energies, but the related experimental data for Mo–S bond(s) is not available. However, on the basis of the Pauling's rule for estimating bond energies,36 the estimated Mo–S bond energy is around 433 kJ mol−1. The Mo–O and Mo–C bond energy is 560.2 ± 20.9 and 481 ± 15.9 kJ mol−1 from the available experimental data,37 respectively. Thus the estimated Mo–S bond energy is much lower than the Mo–O and Mo–C values, and Mo–O has the highest bond energy. This gives a qualitative conclusion that Mo–O bond is stronger than Mo–S or Mo–C bond, and therefore the formation of Mo–O bonds is energetically more favored than Mo–S and Mo–C bonds. This is in agreement with the local electronic density of states (EDOS) study in the literature,26 which states that the oxidation of MoS2 is the result of local competition of binding energy of the covalent bonds.
The changes of the oxidation surface structure (Mo–Mo distance and Mo–S distance) are shown in Fig. 2. The Mo–Mo bond length is 3.134 and 2.951 Å in Mo27S54 (I) and Mo27S56 (II), respectively, while it is shorter in 2 (2.832 Å). It is worth noting that the Mo–Mo distance in Mo27S54 (I) matches very well with the experimental value of 3.15 ± 0.1 Å of the same hexagonal single-layer MoS2 nanocluster from STM.36 The Mo–C distances are 1.875 and 2.174 Å. Meanwhile, the distance (2.413 Å) of corner Mo atom to corner S atom is longer than that of in Mo27S54 cluster (2.331 Å), as shown in Fig. 2. Substitution of the S atoms on the edge with O atoms drew the Mo atoms that they are connecting with closer by about 0.09 Å, making the structure more “compact” on the edge. However, the remaining Mo–S bonds in the bulk are slightly elongated, because O has more electron negativity than S and the Mo–O bonds are stronger than the Mo–S bonds. Similar structural distortions are also found in the oxidation of other edges in the following sections.
2. Oxidation on 100% covered S edge
For replacing one S atom in Mo27S54 (I) by oxygen, there are two possible structures. Structure 3 and 4 with one corner S atom and one edge S atom replaced are shown in Fig. 3, respectively. From formation energy, both replacements are exothermic. The replacement of the corner S atom (3, −2.46 eV) is more favored than the edge S atom (4, −2.35 eV). Both of the oxidation are more easier than that of the S edge with 100% sulfur coverage (−1.3 eV for corner, and −1.9 eV for edge) on the triangular MoS2 cluster reported by Liang et al.26 This can be ascribed to the different chemical environment of the oxidation sites on the hexagonal and triangle-shaped clusters. From bond length parameters in Fig. 3, one can see that the Mo(c)–O and Mo(o)–O bond lengths are 1.875 and 2.044 Å in 3, respectively. In 4, the Mo(o)–O bond lengths are 1.948 and 1.977 Å. In carburization processes, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S species are formed. However, there are not OS species in oxidation processes. That is, the distance of the O atom and the neighbor S atom is 2.804 Å in 3 and 2.776 Å in 4, and is longer than CS (from 1.790 to 1.826 Å) in carburization structures. This shows the difference in structures between carburization and oxidation.
S species are formed. However, there are not OS species in oxidation processes. That is, the distance of the O atom and the neighbor S atom is 2.804 Å in 3 and 2.776 Å in 4, and is longer than CS (from 1.790 to 1.826 Å) in carburization structures. This shows the difference in structures between carburization and oxidation.
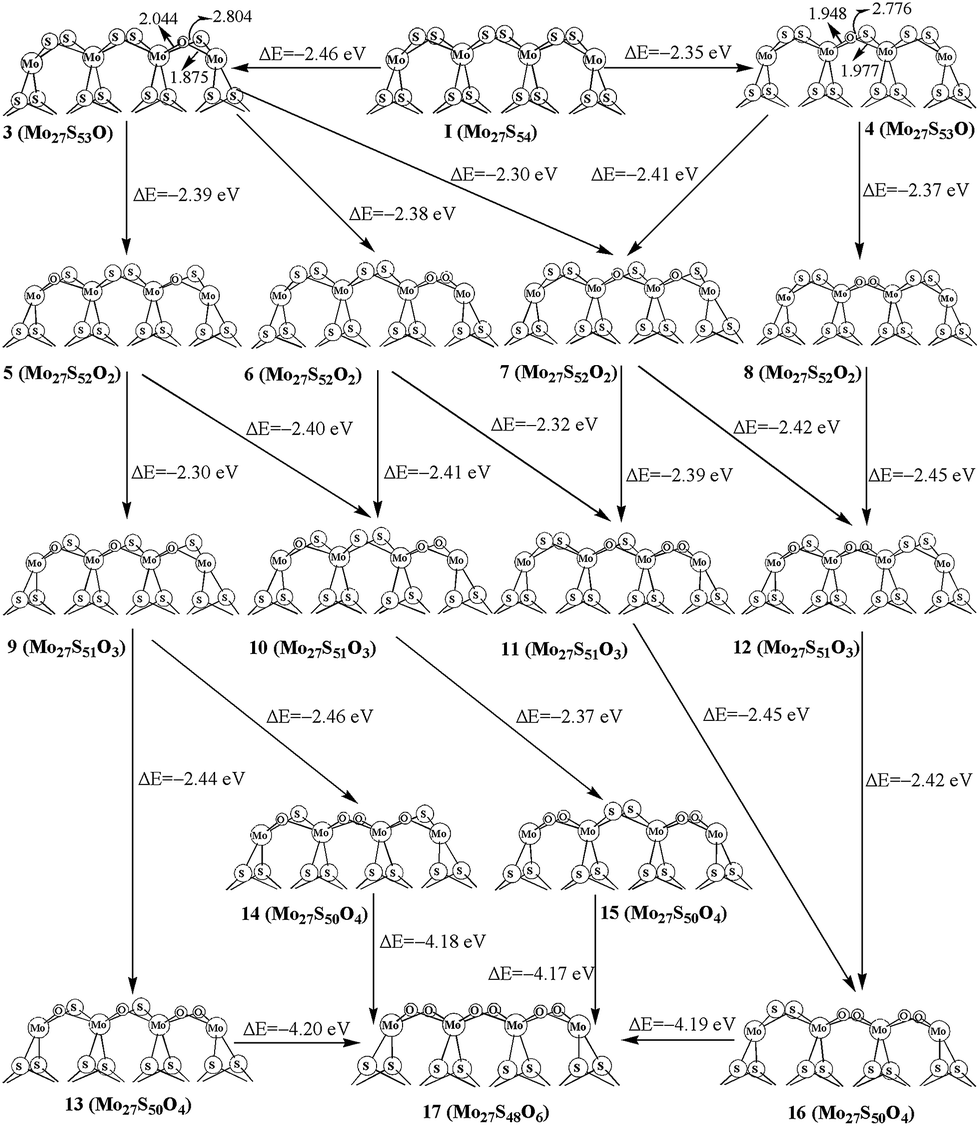 | ||
| Fig. 3 Oxidation on S edge with 100% sulfur coverage. | ||
For replacing two sulfur atoms on 100% covered S edge, there are four structures (5, 6, 7 and 8). As shown in Fig. 3, 5 has two corner S atoms replaced, while 7 has one corner and one edge S atoms replaced. 6 and 8 have two oxygen atoms replacement at the same corner and edge, respectively. The formation energy shows that corner site replacement at the different corner (5, −2.39 eV) and at the same corner (6, −2.38 eV) is more favored than those at edge (7, −2.30 eV). Also, stating from 4, the corner site replacement (7, −2.41 eV) is more favorable than the edge site replacement (8, −2.38 eV). It indicates that the oxidation at corner site on the surface is more favored.
As shown in Fig. 3, there are four structures for replacing three sulfur atoms by carbons atoms (9, 10, 11 and 12). In 10 and 11, there are two corner oxygen atoms and one single oxygen atom on the corner or edge. Structure 12 has two edge oxygen atoms and one single corner oxygen atom. However, 9 has three single oxygen atoms on the same side of the molecule plane. From the calculated formation energy, one can see that the replacement at corner sites is generally more favored than at the edge. In addition, we have also computed the four-oxygen (13, 14, 15 and 16) and full oxygen (17) replacement. All these processes are favored thermodynamically.
3. Oxidation on 50% covered S edge
The oxidation processes on 50% covered S edge in Mo27S51 (III) is illustrated in Fig. 4. For one oxygen replacement, there are two possible structures (18 and 19): one with a corner S atom replaced (18) and one with an edge S atom replaced (19), as shown in Fig. 4. The formation energy difference shows that corner oxygen (18, −2.42 eV) can be more easily formed than the edge one that bridges two Mo atoms (19, −2.27 eV). Similar preference for corner oxidation is observed at the additional S replacement in 20 (−2.43 eV) and 21 (−2.26 eV) from structure 18. This is different from the case in triangle-shaped MoS2,26 where the oxidation corner oxidation (−2.3 eV) is easier than the edge oxidation (−2.1 eV). In addition, the formation of 22 with full replacement (−2.40 eV) from structure 21 is easier than that of from 20 (−2.23 eV). By comparing with carburization on same edge, the carburization at the edge site is more favorable than at the corner, and is different with oxidation processes. This indicates the difference between carburization and oxidation processes.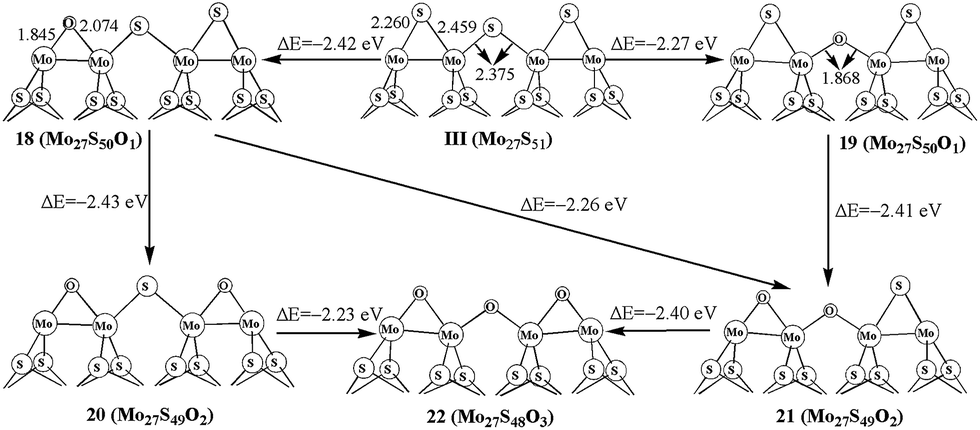 | ||
| Fig. 4 Oxidation on S edge with 50% sulfur coverage. | ||
4. Oxidation of the bulky S in Mo27S54
As shown in the above studies, the oxidation of surface sulfur on Mo edge and S edge has been discussed. It is worth noting of the possibility of replacing the bulky sulfur. As shown in Fig. 5, there are four types of bulky sulfur atoms in Mo27S54 (I). The first bulky sulfur is S1 to the Mo edge, and the second type is S2 close to the S edge. The third type is S3, which is one line deeper than S1, and the fourth type is S4, which is one line deeper than S2. These symbolic bulky S atoms are also used in carburization processes.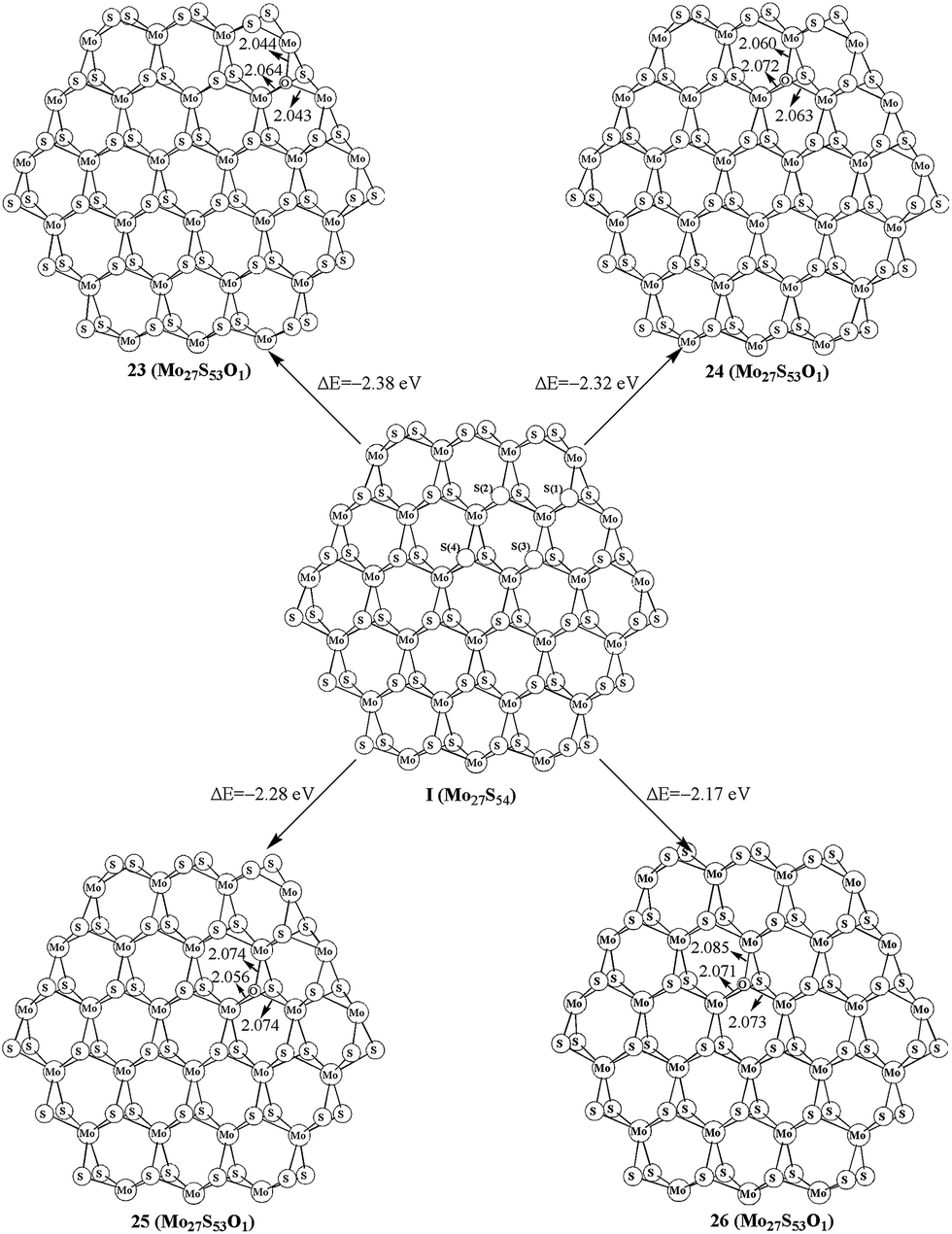 | ||
| Fig. 5 Oxidation on the bulk of Mo27S54. | ||
The replacement of S1 is easier than that of S2, as indicated by the formation energy of 23 (−2.38 eV) and 24 (−2.32 eV). Comparably the replacement of S3 and S4 is very difficult (25, −2.28 eV, and 26, −2.17 eV, respectively). Therefore, S1 is the type of bulky sulfur, which can be replaced most easily. Like previous work on carburization, our results also indicate that the carburization of bulky sulfur on the Mo edge is as equally favored as those of sulfur on the S edge.
We further report the replacements of multiple S1 atoms. Structure 27 and 28 with two S1 atoms replaced on different planes and on the same plane are shown in Fig. 6, respectively. The formation energy shows that replacing two S1 atoms at the same plane (28, −2.42 eV) is more favored than that at different planes (27, −2.31 eV). The same result is also shown in 29. As shown in Fig. 6, the formation of 29 from structure 27 is more easily than that of from 28. In addition, the replacement of four S1 atoms (30, −2.27 eV) is also exothermic. As comparison, the oxidation of the bulky S in triangle-shaped MoS2 cluster is exothermic by only −1.4 eV to −1.7 eV, except for the one (−2.3 eV) that is close to the corner of cluster with 100% sulfur coverage.26
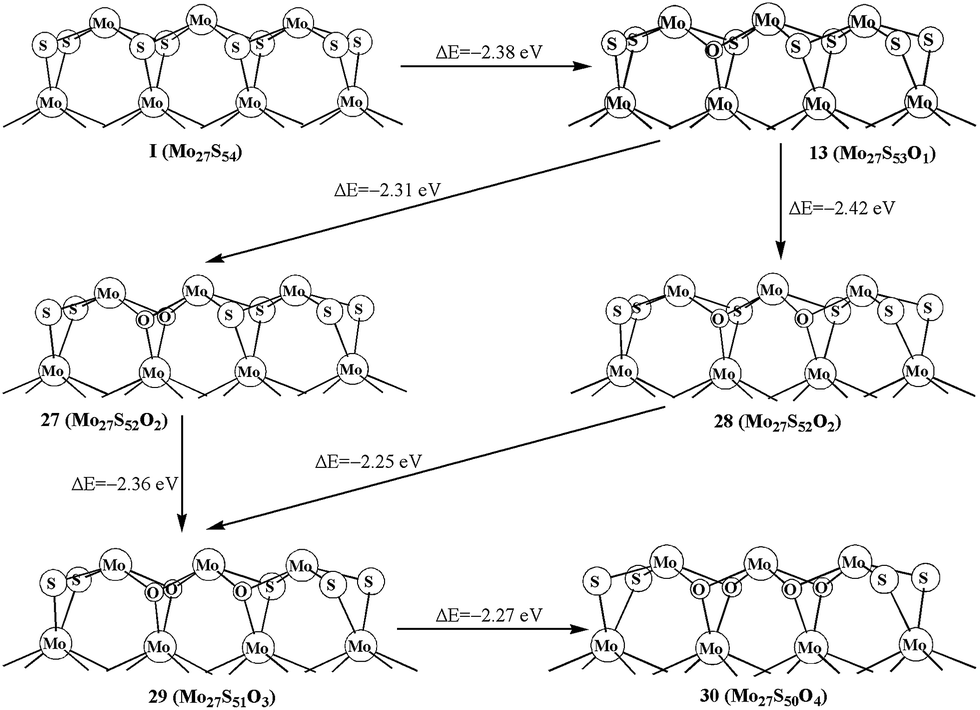 | ||
| Fig. 6 Oxidation on the bulky S1 atoms of Mo27S54. | ||
5. The effect of oxidation on the density of states of Mo27S54
To provide a better understanding of the oxidation process, the electronic structure analysis of the Mo27S54−xOx clusters are presented here. To start with, the density of states (DOS) of the whole clusters is calculated (Fig. 7, left). There are three major peaks in the DOS of the Mo27S54−xOx clusters. The DOS at the energy level of −8.4 to 3.3 eV is mainly filled with the 4d electrons of Mo, 3p electrons of S, and 2p electrons of O. The peak at about −12 eV is identified with the 3s electrons of S and the 5s electrons of Mo, and the peak at about −19 eV corresponds to the 2s electrons of O.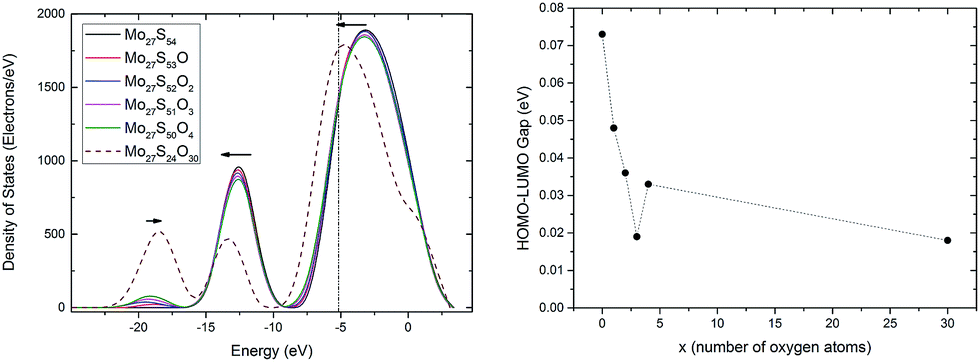 | ||
| Fig. 7 (Left) Density of states of the Mo27S54−xOx clusters; the vertical line represents the HOMO orbital energy of the clusters; (right) the HOMO–LUMO gap of the Mo27S54−xOx cluster subjected to oxidation. | ||
With the substitution, the O (2s) peak at −19 eV intensifies gradually, and a small blue shift is observed. The S (3s) and Mo (5s) peak at −12 eV weakens and shifts to lower energy. The large peak from −8.4 eV to 3.3 eV has a shift in energy to lower values. However, the intensity and area of the peak does not change significantly. This shows that the during oxidation, the overlap of the Mo 4d electrons with the S 3p electrons can be roughly replaced by the overlap of the Mo 4d electrons with the O 2p electrons, and that the total number of electrons involved in these two types of bonding remain unchanged.
It is known that MoO3 is a semiconductor with band gap of around 3.1 eV,38 while MoS2 has a band gap of 1.9 eV.38 By intuition, we would think that the oxidation of Mo27S54 will lead to the increase of the HOMO–LUMO gap. However, interestingly, this is not the case as shown in Fig. 7 (right). On the contrary, we observe a general decrease of the HOMO–LUMO gap of the Mo27S54−xOx clusters with oxidation. This difference in the trend can be attributed to the quantum confinement effect of the nanoscale clusters. In fact, all the Mo27S54−xOx clusters studied show strong metallic nature with very small HOMO–LUMO gaps. Besides, oxidation increases the metallic nature of the Mo27S54 cluster. The HOMO orbital of all clusters has energy of about −5.2 eV. Therefore, the valence band regions of the clusters are from −8.4 eV to −5.2 eV. The width of the valence band is about 3.2 eV, and is significantly smaller than that of either bulk MoO3 or MoS2 (about 6 eV).
To further illuminate the trend of changes of the electronic structures of the local sites, the local electronic density of states (LDOS) of an edge Mo atom and the 4 neighboring S atoms before and after substitution are plotted in Fig. 8, with each graph representing a different oxidation state. Before substitution, the four S atoms (labeled as a–f) of interest in the Mo27S54 cluster are indistinguishable with exactly the same LDOS (Fig. 8a). After substitution, the peak at −19 eV appears clearly for O and Mo and slightly for the other three S atoms (Fig. 8b). The latter fact suggests that after oxidation, there is considerable overlap between O 2s and Mo orbitals, but only a small amount of overlap between O 2s and S orbitals. Another proof of the latter statement is that, after oxidation, the O atom includes some electrons from S, as shown at the peak of −12 eV. This is different from the case in the oxidation of the edge of triangular MoS2 with 100% S coverage reported in literature,26 where no overlap was observed at all between O 2s and the S orbitals. The further substitution of S by O has almost no effect on the bonding of the previously added O, as Fig. 8c to e shows. Comparing Fig. 8e to f, the oxidation of the rest of all the edge S atoms only affected the valence band region (−8.4 eV to −5.2 eV), mainly related to the Mo 4d electrons and the conduction band (−5.2 eV to 3.3 eV). The core level regions are almost not affected.
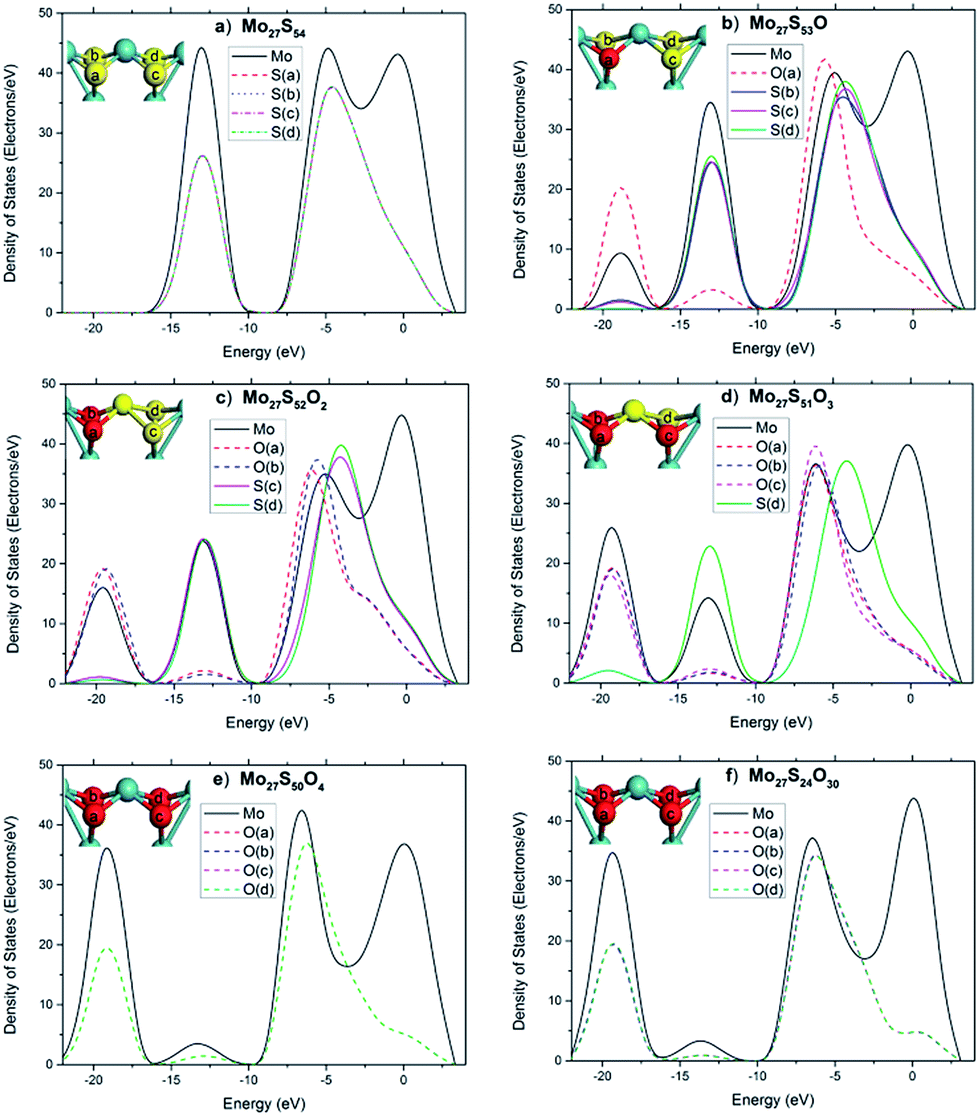 | ||
| Fig. 8 LDOS of the tetra-coordinated Mo atom on the edge of the Mo27S54−xOx cluster. | ||
6. Vibrational frequencies
MoS2 is a semiconductor and have the absorption edge in near-IR region. Previous study39 have shown that the absorption spectra depend strongly on the size of the system. However, the IR spectra of MoS2 nanoparticles with size similar to the clusters used in this work are not available in the literature.The IR spectra of all the clusters before and after oxidation are very similar, in agreement with the finding that it’s absorption is only slightly sensitive to oxidation suggested in the literature.39 Because of the broken symmetry of the clusters compared to the bulk, concerted vibrational modes of atoms in the cluster may not match very well with the experimental IR with large single layers of MoS2. However, some agreements were identified.
Taking structure 27 in Fig. 6 as an example, the calculated IR spectra show a broad region between 420 cm−1 and 520 cm−1, which correspond to the Mo–S vibrations of MoS2 (480 cm−1) reported in experiment.40 For oxygen in the bulk, another broad peak appears at about 600 cm−1 to 650 cm−1, with Mo–O symmetric stretching contained in the vibrational mode with frequency of 609 cm−1. The asymmetric stretching of Mo–O appears at 726 cm−1.
For terminal MoO vibrations, IR calculation on structure 10, for example, show MoO asymmetric stretching at 909 cm−1 to 914 cm−1 and symmetric stretching at 691 cm−1, in agreement with the experimental value41 of 941 cm−1 and 693 cm−1, respectively.
Conclusions
We have carried out systematic investigations into the replacement of the surface sulfur on Mo edge and S edge as well as bulky sulfur to understand the oxidation processes of MoSx. On these surfaces, the replacement of surface sulfur by the oxygen atoms is thermodynamically favorable.22,24,26 By comparing the oxidation processes on the surfaces of different sulfur coverage, our results indicate that the oxidation on the S edge with 100% and 50% coverage is more favored than on the Mo edge. Meanwhile, it is found that the oxidation on the S edge with 100% coverage have similar replacement ability by oxygen as on the S edge with 50% coverage. Thus, sulfur coverage does not play important role in the oxidation on the S edge. Further comparison shows that the oxidation on corner sites is more favorable than on edge sites. In addition, the replacement of the bulky sulfur on the Mo edge is equally as favored as those of sulfur on the S edge. If we consider only the replacement of the S atoms by O, instead of a large variation (−0.9 to −2.4 eV) in the oxidation energy reported by Liang26 with plane-wave basis, we find that the range of the oxidation energy is rather narrow (−1.9 eV to −2.4 eV) for MoSx. This is probably due to the weakness of the plane-wave model in treating finite clusters. The Mo27SxOy nanoclusters have unique electronic properties compared to the bulk. The width of its valence band is about 3.2 eV, and is much smaller than that of either bulk MoO3 or MoS2. The electronic structure of the Mo27SxOy nanoclusters can be easily tuned by the substitution of S atoms with O atoms.By comparing with carburization processes on these surfaces, we find that for Mo edge the oxidation is much easier than carburization from their atomic formation energy. There is a large difference of formation energy (−1.93 vs. −0.77 eV) between oxidation and carburization. By comparing with carburization and sulfidation, it is shown that oxidation is more favored than carburization and sulfidation on Mo edge. However, on the S edge with 100% coverage, there are only small differences in oxidation and carburization ability, although they have different surface structures. As one can see, there are CS and CC species in carburization processes, while OS and OO species are not formed in oxidation processes. This is reflected by the difference between carburization and oxidation in experiments. Further, we find that the oxidation on the 100% and 50% covered S edge have similar reaction energy, while the carburization on the 100% covered S edge is more favored than on the 50% covered S edge. This shows that the carburization depends on the effect of coverage, but oxidation does not. For bulky S, the oxidation and carburization of the bulky sulfur on the Mo edge is equally favored as those of sulfur on S edge. This suggests that the bulky sulfur probably plays important roles in oxidation. From these calculations, we can get the following information:
(1) The S atoms on S edge and in the bulk can be oxidized first. Also, the oxidation on S edge does not depend on its coverage. Following this, the S atoms on Mo edge will be oxidized next. Since the frictional resistance between the MoS2 layers is determined by the van de Waals forces of the S atoms, the substitution of the S atoms by O would inevitably affect its performance as lubricant. Our results suggests that the capability of MoS2 lubricants has link with not only the oxidation of the S edge but also bulky at certain degree.
(2) From these results, we can know about some sulfidation process of MoO3 (precursor). As the opposite processes to oxidation, it is very difficult to sulfurize the O atoms on S edge and bulky, due to the high cost in energy. That is to say, the sulfidation of MoO3 is probably determined by the sulfidation on S edge and the bulk. Although these studies provides important information on the thermodynamic aspects of the oxidation of MoS2, further experimental and theoretical studies are needed to understand structure and other property of the oxidized material.
Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (No. 21473229, No. 91545121, No. 21603252, and No. 21473231), No. 201601D021048 from the Shanxi Province Science Foundation for Youth, and from Synfuels China, Co. Ltd. We also acknowledge the innovation foundation of Institute of Coal Chemistry, Chinese Academy of Sciences, National Thousand Young Talents Program of China, Hundred-Talent Program of Chinese Academy of Sciences and Shanxi Hundred-Talent Program.Notes and references
- H. H. Gary, G. E. Handwerk and M. J. Kaiser, Petroleum Refining: Technology and Economics, CRC Press, 5th edn, 2007 Search PubMed
.
- K. V. R. Chary, H. Ramakrishna, K. S. Rama Rao, G. Murali Dhar and P. Kanta Rao, Catal. Lett., 1991, 10, 27–33 CrossRef CAS
- T. Weber, J. C. Muijsers, J. H. M. C. van Wolput, C. P. J. Verhagen and J. W. Niemantsverdriet, J. Phys. Chem., 1996, 100, 14144–14150 CrossRef CAS
- F. J. Clauss, Solid Lubricants and Self-Lubricating Solids, Academic Press, New York, 1972 Search PubMed
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147–150 CrossRef CAS PubMed
- M. M. Perera, M.-W. Lin, H.-J. Chuang, B. P. Chamlagain, C. Wang, X. Tan, M. M.-C. Cheng, D. Tománek and Z. Zhou, ACS Nano, 2013, 7, 4449–4458 CrossRef CAS PubMed
- S. Kim, A. Konar, W.-S. Hwang, J. H. Lee, J. Lee, J. Yang, C. Jung, H. Kim, J.-B. Yoo, J.-Y. Choi, Y. W. Jin, S. Y. Lee, D. Jena, W. Choi and K. Kim, Nat. Commun., 2012, 3, 1011 CrossRef PubMed
- K. F. Mak, K. He, J. Shan and T. F. Heinz, Nat. Nanotechnol., 2012, 7, 494–498 CrossRef CAS PubMed
- H. Zeng, J. Dai, W. Yao, D. Xiao and X. Cui, Nat. Nanotechnol., 2012, 7, 490–493 CrossRef CAS PubMed
- T. Cao, G. Wang, W. Han, H. Ye, C. Zhu, J. Shi, Q. Niu, P. Tan, E. Wang, B. Liu and J. Feng, Nat. Commun., 2012, 3, 887 CrossRef PubMed
- J. Andrés, S. Berski, M. Feliz, R. Llusar, F. Sensato and B. Silvi, C. R. Chim., 2005, 8, 1400–1412 CrossRef
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed
- J. Hong, Z. Hu, M. Probert, K. Li, D. Lv, X. Yang, L. Gu, N. Mao, Q. Feng, L. Xie, J. Zhang, D. Wu, Z. Zhang, C. Jin, W. Ji, X. Zhang, J. Yuan and Z. Zhang, Nat. Commun., 2015, 6, 6293 CrossRef CAS PubMed
- S. H. Song, B. H. Kim, D.-H. Choe, J. Kim, D. C. Kim, D. J. Lee, J. M. Kim, K. J. Chang and S. Jeon, Adv. Mater., 2015, 27, 3152–3158 CrossRef CAS PubMed
- J. Guo, F. Li, Y. Sun, X. Zhang and L. Tang, J. Power Sources, 2015, 291, 195–200 CrossRef CAS
- J. Xie, J. Zhang, S. Li, F. Grote, X. Zhang, H. Zhang, R. Wang, Y. Lei, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 17881–17888 CrossRef CAS PubMed
- H. Huang, X. Feng, C. Du, S. Wu and W. Song, J. Mater. Chem. A, 2015, 3, 16050–16056 CAS
- M. T. Lavik, T. M. Medved and G. D. Moore, ASLE Trans., 1968, 11, 44–55 CrossRef CAS
- H. Y. Sohn and D. Kim, Metall. Trans. B, 1988, 19, 973–975 CrossRef
- J. R. Lince, J. Mater. Res., 1990, 5, 218–222 CrossRef CAS
- J. R. Lince, M. R. Hilton and A. S. Bommannavar, Surf. Coat. Technol., 1990, 43–44(2), 640–651 CrossRef CAS
- P. D. Fleischauer and J. R. Lince, Tribol. Int., 1999, 32, 627–636 CrossRef CAS
- J. T. Miller, C. L. Marshall and A. J. Kropf, J. Catal., 2001, 202, 89–99 CrossRef CAS
- K. C. Santosh, R. C. Longo, R. M. Wallace and K. Cho, J. Appl. Phys., 2015, 117, 135301 CrossRef
- H. S. Sen, H. Sahin, F. M. Peeters and E. Durgun, J. Appl. Phys., 2014, 116, 083508 CrossRef
- T. Liang, W. G. Sawyer, S. S. Perry, S. B. Sinnott and S. R. Phillpot, J. Phys. Chem. C, 2011, 115, 10606–10616 CAS
- X.-D. Wen, Z. Cao, Y.-W. Li, J. Wang and H. Jiao, J. Phys. Chem. B, 2006, 110, 23860–23869 CrossRef CAS PubMed
- X.-D. Wen, T. Zeng, Y.-W. Li, J. Wang and H. Jiao, J. Phys. Chem. B, 2005, 109, 18491–18499 CrossRef CAS PubMed
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS
- B. Delley, J. Phys. Chem., 1996, 100, 6107–6110 CrossRef CAS
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef
- J. V. Lauritsen, J. Kibsgaard, S. Helveg, H. Topsoe, B. S. Clausen, E. Laegsgaard and F. Besenbacher, Nat. Nanotechnol., 2007, 2, 53–58 CrossRef CAS PubMed
- J. V. Lauritsen, M. V. Bollinger, E. Laegsgaard, K. W. Jacobsen, J. K. Norskov, B. S. Clausen, H. Topsoe and F. Besenbacher, J. Catal., 2004, 221, 510–522 CrossRef CAS
- L. Pauling, The Nature of the Chemical Bond and the Structure of Molecules and Crystals, Cornell University Press, New York, 1960 Search PubMed
- D. R. Lide, Handbook of Chemistry and Physics, CRC Press, 84th edn, 2004 Search PubMed
- Gmelin Handbook of inorganic and organometallic chemistry. [System number 45]. Ge–Organogermanium compounds, Springer-Verlag, Berlin, New York, 8th edn, 1993 Search PubMed
- C. B. Roxlo, M. Daage, A. F. Ruppert and R. R. Chianelli, J. Catal., 1986, 100, 176–184 CrossRef CAS
- S. Liu, X. Zhang, H. Shao, J. Xu, F. Chen and Y. Feng, Mater. Lett., 2012, 73, 223–225 CrossRef CAS
- G. Nagaraju, C. Tharamani, G. Chandrappa and J. Livage, Nanoscale Res. Lett., 2007, 2, 461 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26264c |
| This journal is © The Royal Society of Chemistry 2017 |