
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile hydrothermal synthesis of BaZrxTi1−xO3 nanoparticles and their application to a lead-free nanocomposite generator†
Changyeon Baek‡
a,
Ji Eun Wang‡ a,
Soojy Ryua,
Joo-Hyung Kima,
Chang Kyu Jeongab,
Kwi-Il Park*c and
Do Kyung Kim*a
a,
Soojy Ryua,
Joo-Hyung Kima,
Chang Kyu Jeongab,
Kwi-Il Park*c and
Do Kyung Kim*a
aDepartment of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon, 34141, Republic of Korea. E-mail: kipark@gntech.ac.kr; Fax: +82-55-751-3889; Tel: +82-55-751-3884
bKAIST Institute for the NanoCentury (KINC), 291 Daehak-ro, Yuseong-gu, Daejeon, 34141, Republic of Korea
cDepartment of Energy Engineering, Gyeongnam National University of Science and Technology (GNTECH), 33 Dongjin-ro, Jinju-si, Gyeongsangnam-do 52725, Republic of Korea. E-mail: dkkim@kaist.ac.kr; Fax: +82-42-350-3310; Tel: +82-42-350-4118
First published on 12th January 2017
Abstract
A facile form of hydrothermal synthesis was utilized to obtain BaZrxTi1−xO3 (BZT) nanoparticles (NPs) with a wide range of Zr concentrations. The amorphous B-site precursor prepared by the hydrolysis of alkoxide using an ammonia solution, efficiently promotes the reaction with the A-site precursor solution. Subsequently, the BZT NPs were adopted for a flexible piezoelectric energy harvester (PEH) by employing a simple, low-cost, and scalable spin-casting method. The output performance achieved by the fabricated flexible PEH depends on the Zr/Ti ratio inside a piezoelectric nanocomposite. The BZT NP-embedded PEH with Zr concentration of 32 mol% generated a stable output voltage of ∼20 V and a current signal of 400 nA without performance decay.
Introduction
An increasing interest in energy issues has led many researchers to develop various devices for harvesting energy from electrostatic, thermal, and mechanical sources that dissipate (waste) energy in real-life. Among many kinds of energy conversion technologies, the energy harvesters based on piezoelectricity have been considered one of the most versatile tools to glean energy from various mechanical sources including hydrokinetic flows, vibration, pressing, and innumerable movements of organisms that are easily available anywhere.1–9For these reasons, many types of energy harvesting devices have been introduced to more effectively generate electrical output from even trivial mechanical motion. This nanocomposite generator (NCG) technology has been considered one of the more promising candidates owing to the simple, scalable processes used for fabrication of such devices, at relatively low cost.10–17 The first demonstration of a composite-based piezoelectric energy harvester (PEH) with embedded BaTiO3 nanoparticles (NPs) was provided by Park et al. in 2012.16 Since then, numerous studies have been performed to improve the output performance of PEH devices by replacing lead-based piezoelectric NPs such as PbZrxTi1−xO3 (PZT)15 and (1 − x)Pb(Mg1/3Nb2/3)O3–xPbTiO3 (PMN–PT).18,19 Recently, in response to consideration of potential applications of energy harvesters for real-life or in vivo uses, composite-based PEHs made of less toxic and more eco-friendly materials were demonstrated by a number of researchers. These materials included such as 0.942(K0.480Na0.535)NbO3–0.058LiNbO3 (KNLN),10 0.5(Ba0.7Ca0.3)TiO3–0.5Ba(Zr0.2Ti0.8)O3 (BCTZ),20–22 and alkaline metal-niobate (MNbO3) (M = Li, Na, K).11,12,23
As one of the lead-free materials, BaZrxTi1−xO3 (BZT) solid solutions have been studied to investigate their tuneable properties, which depend on the degree of substitution at B-sites with Zr ions.24–28 Unique variations in dielectric behaviour, including ferroelectric (x ≤ 0.15), diffuse phase transition (0.15 ≤ x ≤ 0.30), and relaxor behaviour (x ≥ 0.30), were achieved by adjusting the Zr-to-Ti molar ratio. This made the corresponding compounds promising candidates for various applications such as piezoelectric transducers, dynamic random access memory, and tuneable capacitors owing to their low leakage current and high breakdown field strength.24,28–35 For the synthesis of BZT particles, a solid-state reaction at very high temperature (∼1200 °C) is needed due to the low reactivity of Zr ions. It is still difficult to obtain a homogeneous solid solution, especially in higher Zr concentrations.30,35 Few studies have been made to synthesize BZT NPs via wet chemical routes.35–38 Recent studies demonstrated an interesting approach to synthesis of BZT NPs with a broad region of Zr concentration (0 ≤ x ≤ 1) under supercritical fluid conditions (∼400 °C, 23 MPa).37,38
Herein, we report a facile hydrothermal reaction to synthesize BZT NPs having Zr concentration from x = 0 to 0.32. The synthesis route is scalable and easy to control the Zr/Ti molar ratio. The synthesized BZT NPs were carefully characterized via scanning electron microscopy (SEM), X-ray diffraction (XRD), and Raman spectroscopy. The BZT particles were applied to the piezoelectric nanocomposite, which was sandwiched between two indium tin oxide (ITO)-coated flexible substrates to prepare the flexible PEH. The output performance of the energy generation devices, which corresponded to the Zr concentration, was monitored and its potential for operating small electronic devices such as LCD and LEDs, was confirmed.
Experimental procedure
Hydrothermal synthesis and characterization of BZT NPs
Alkoxide precursors which contain B-site ions were used to obtain the designed molar ratio of ZrxTi1−xO2 homogeneous precipitates. The stoichiometric amount of titanium butoxide (Ti[O(CH2)3CH3]4, reagent grade 97%, Sigma-Aldrich) and zirconium butoxide (Zr[O(CH2)3CH3]4, 80 wt% in 1-butanol, Sigma-Aldrich) precursors were mixed in ethanol (10 mL) and heated to 80 °C with magnetic stirring in a water bath. The amorphous ZrxTi1−xO2 particles were precipitated by the addition of 4 mL of ammonia solution (25% NH3 in H2O, Sigma-Aldrich) to the mixture. Barium hydroxide was dissolved in 10 mL of preheated deionized water in parallel (barium and titanium precursors in a 1.5 = Ba/(Zr + Ti) molar ratio). The barium hydroxide aqueous solution was added to the ZrxTi1−xO2 precipitates, and the resultant suspension was stirred for an hour. The mixed solution was transferred to a 100 mL Teflon container with a fill factor of 70%, and then the container was sealed in a stainless-steel autoclave. A hydrothermal reaction was conducted at 200 °C for 24 h under autogenous pressure. After the reaction, the autoclave was cooled to room temperature. The as-synthesized BZT particle was washed repeatedly using a mixture of high-purity ethanol and deionized water, and subsequently dried at 80 °C for 24 h in air.Changes in morphology of the synthesized BZT particles occurred in accordance with the Zr molar ratio, and were observed via field-emission scanning electron microscope. The particle sizes were measured using the Image J program, and the average particle sizes and size distributions were calculated based on the measured values. The crystal structures of the obtained BZT particles were investigated via X-ray diffraction (XRD) (Rigaku D/Max-RB (12 KW), Tokyo, Japan) with Cu Kα radiation (λ = 1.5406 Å) operating at 40 kV and 300 mA with a step size of 0.01° at 2° min−1 in a 2θ range from 20° to 80°. The lattice parameter change with respect to the Zr molar ratio, was simply calculated from the average d-spacing values for four diffraction peaks (100, 110, 111, and 200) based on Bragg's law (nλ = 2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ). The vibration modes in the synthesized BZT particles at room temperature were confirmed from the Raman spectra range from 100 to 1000 cm−1 using a high-resolution dispersive Raman microscope (514.5 nm line of Ar+ laser, LabRAM HR UV/Vis/NIR, Horiba Jobin Yvon, Longjumeau, France).
sinθ). The vibration modes in the synthesized BZT particles at room temperature were confirmed from the Raman spectra range from 100 to 1000 cm−1 using a high-resolution dispersive Raman microscope (514.5 nm line of Ar+ laser, LabRAM HR UV/Vis/NIR, Horiba Jobin Yvon, Longjumeau, France).
Fabrication and evaluation of BZT NPs-based flexible PEHs
To make the BZT–CNT NPs, synthesized BZT NPs were mixed with carbon nanotubes (average diameter of 10–15 nm and length of 20–30 μm, CM-95, Injin Nanotech Co.) by magnetic stirring with ethanol in the ratio of 10 wt% BZT to 1 wt% CNT. The well mixed nanomaterials were dried sufficiently and the mixture sieved using a testing sieve (mesh #200) to get granulated fine particles. The 11 wt% of mixed BZT–CNT powder was poured into a PDMS matrix (Sylgard 184, Dow Corning, containing curing agent in ratio of 0.1) to produce the BZT–CNT nanocomposite (BZT–CNT NC). A PDMS elastomer was spin-coated at 1500 rpm onto an ITO-coated plastic substrate; then, BZT–CNT NC and PDMS layers were coated sequentially. Between each coating step, 10 min of curing at 80 °C in a dry oven was done to prevent undesirable intermixing of layers. Cured PDMS/BZT–CNT NC/PDMS layers were diced into pieces 3.0 × 3.0 cm2. Each diced layer was peeled and sandwiched between two ITO-coated PET films. A device with a sandwiched structure was fully hardened for a day and the Cu wires were connected to the conductive ITO with conductive epoxy (Chemtronics). Finally, the fabricated device was polarized to improve piezoelectric performance by applying 1 kV of electric field for 12 h at 120 °C.Results and discussion
Simple and versatile synthesis of NPs with the designed stoichiometry has always been favoured for minimizing unnecessary expense while getting reproducible products. A homogenous solid solution of BZT NPs was simply achieved via a facile hydrothermal reaction starting from a mixture of metal alkoxide precursors without any complicated experimental set-up. The Fig. 1 shows the SEM images of the synthesized BZT NPs and their size and distribution changes with respect to the zirconium (Zr) concentration.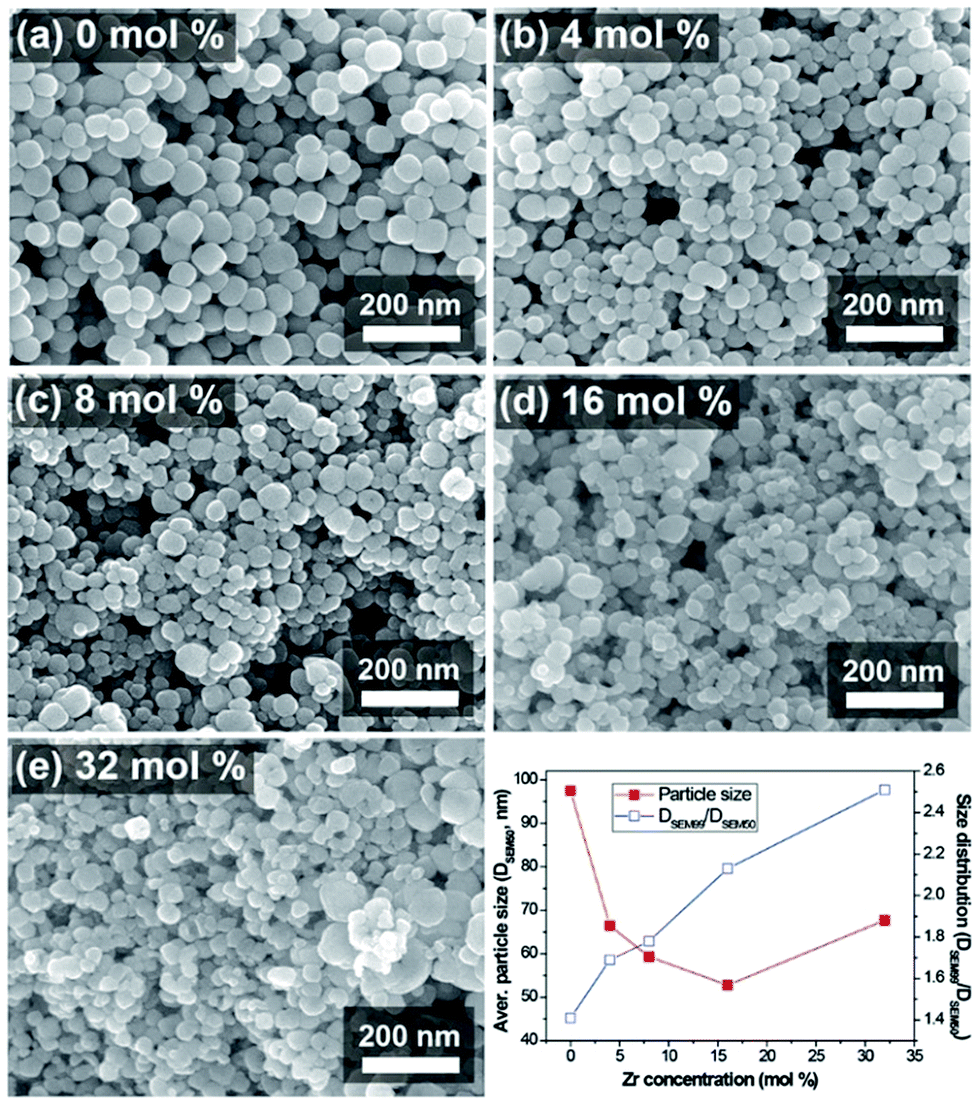 | ||
| Fig. 1 SEM images of the synthesized BZT NPs with different Zr molar concentrations (a) 0 mol%, (b) 4 mol%, (c) 8 mol%, (d) 16 mol%, and (e) 32 mol%. The graph shows the apparent size and size distribution change measured from SEM images. | ||
The size of the obtained BZT NPs was varied and expected to be reduced with the increasing Zr molar ratio because the added amount of the organic solvents (1-butanol) contained in the precursor solution (zirconium butoxide), was subordinately increased with the Zr concentration. It is commonly accepted that the presence of organic solvent in the hydrothermal condition affects the growth rate of particles due to change in the nucleation rate and polarity change associated with the lower dielectric constant of the organic solvents.39–41 The n-butanol solvent is not miscible with water, which is the hydrothermal reaction solvent. The immiscible nature of the n-butanol solution with water may cause irregular reactions and morphologies in the hydrothermal reaction mixture.42 The increase in particle size above a certain Zr molar ratio was caused by the agglomeration of particles due to the van der Waals force originating from the large surface area forming rather larger secondary particles. The deterioration in particle size distribution might be attributable to the reasons above.
The crystal structures of the synthesized BZT NPs are shown in Fig. 2a. The particles obtained were a single phase solid solution with cubic Pm3m structure; without any distinguished impurity peaks such as barium carbonate or other undesirable phases (or phase segregation) originating from the imperfection of the solid solution. As the concentration of zirconium increased, the diffraction patterns shifted toward lower angles that represented the expansion of the lattice structure. This change in lattice spacing showed reverse trends in relation to our previous study of Ba1−xSrxTiO3 NP synthesis, and can be attributed to the size effect that occurred due to the ionic repulsion when the size of the particles became smaller than a few tens of nanometers.43 However, the lattice parameter changes that could originate from the size effect were less distinguishable than was the interplanar spacing that would be caused by the dopant concentration.41,44 Because the ionic radius of the Zr4+ ions is larger than that of the Ti4+ ions, it is expected that the interplanar spacing would also be wider in the portion of Zr4+ ions.37,38 Although synchrotron X-ray analysis and the Rietveld refinement process would be needed for exact analysis of the crystal structures of the BZT NPs; here, we simply calculated the lattice parameters (d-spacing) from normal X-ray diffraction patterns and the results are summarized in Table 1. The changes in the lattice spacing, with respect to the Zr concentration, correspond to the reference patterns. Consistent with our previous research, the amorphous phase of hydrolysed Zr–Ti precursor was formed by the addition of ammonia solution, as shown in Fig. S1.† The highly reactive and well homogenized amorphous B-site precursor (ZrxTi1−xO2) promoted the solid solution.33,43,44 The reaction between the amorphous B-site precursor and A-site ions occurred instantaneously to form the initial BZT NPs, and this rapid reaction was confirmed via X-ray diffraction patterns (Fig. S2†) of the well-crystallized particles at the initial stage of the reaction (5 min). This implies that the composition of the NPs could easily be tuned via this facile hydrothermal route. The Raman spectrum of the synthesized BZT NPs is shown in Fig. 2b and the typical aspect of the Raman spectrum was observed for all NPs. Among all Raman modes, the Raman band appeared at around 307 cm−1, which is known to be strongly related to the presence of tetragonal distortion in the materials.25,28,30,35,37 This tetragonal phase related peak got weaker and flatter as the Zr concentration increased, and the spectra became comparable with the typical aspects of relaxor perovskite materials.
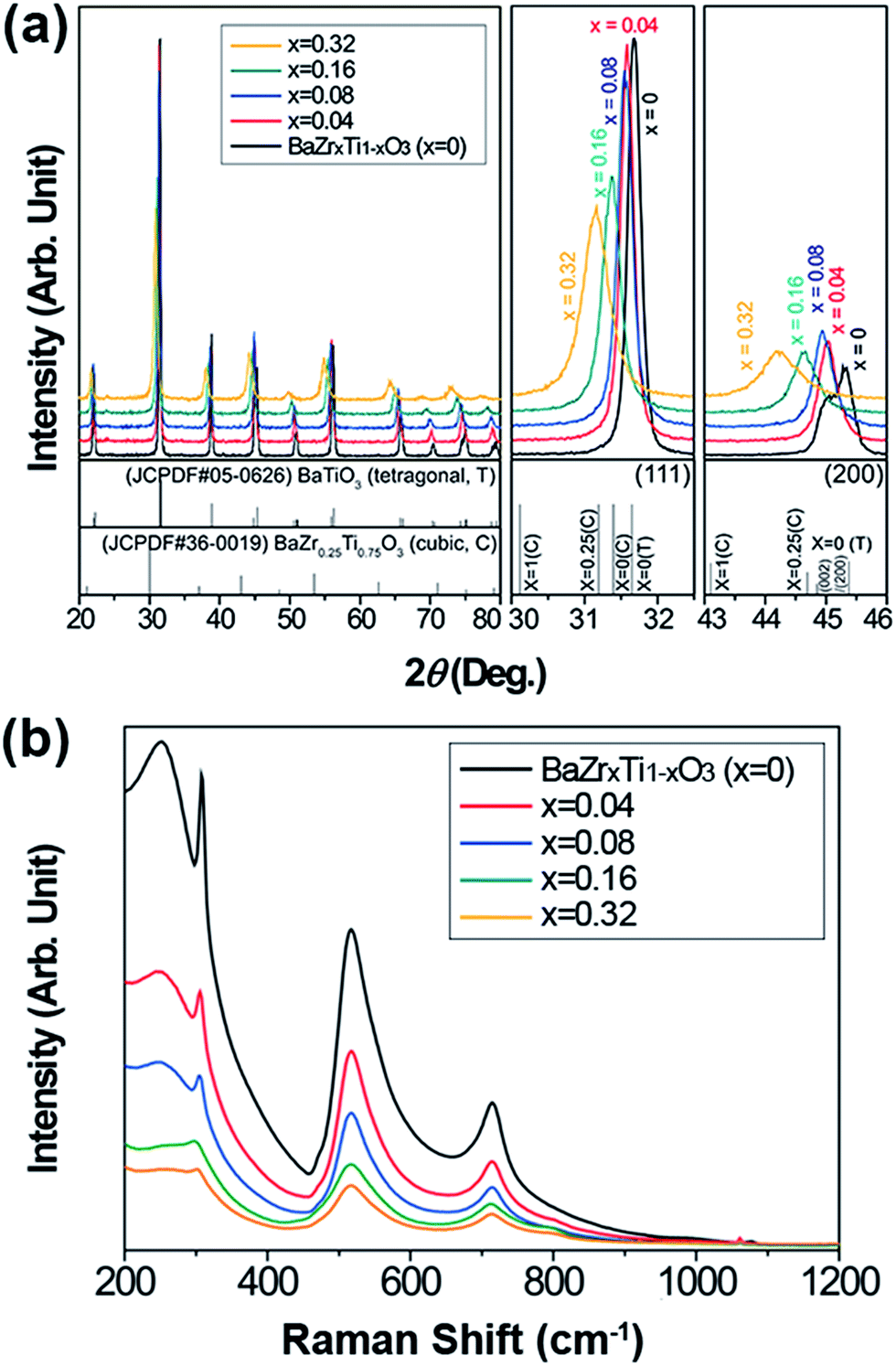 | ||
| Fig. 2 (a) X-ray diffraction patterns of the synthesized BZT NPs with enlarged diffraction angles (111 peak: 30–32.5°, 200 peak: 43–46°). (b) Raman spectrum of the BZT NPs. | ||
| Zr conc. | Lattice parameter (Å) | ||||
|---|---|---|---|---|---|
| (100) | (110) | (111) | (200) | Average (±standard deviation) | |
| x = 0 | 4.0100 | 4.0108 | 4.0108 | 4.0039 | 4.0089 (0.0037) |
| x = 4 | 4.0315 | 4.0258 | 4.0257 | 4.0215 | 4.0261 (0.0017) |
| x = 8 | 4.0334 | 4.0308 | 4.0317 | 4.0309 | 4.0317 (0.0012) |
| x = 16 | 4.0607 | 4.0585 | 4.0570 | 4.0600 | 4.0591 (0.0041) |
| x = 32 | 4.0865 | 4.0893 | 4.0929 | 4.0949 | 4.0909 (0.0034) |
Fig. 3a delineates the schematic illustration of the lamellar structure of the BZT NP-polymeric composite-based PEH. The p-NC layer adhered well to any surface and was thin enough to distinguish objects placed behind the layer. The mixture of BZT NPs and CNTs was well distributed throughout the PDMS matrix without any regions of segregation.
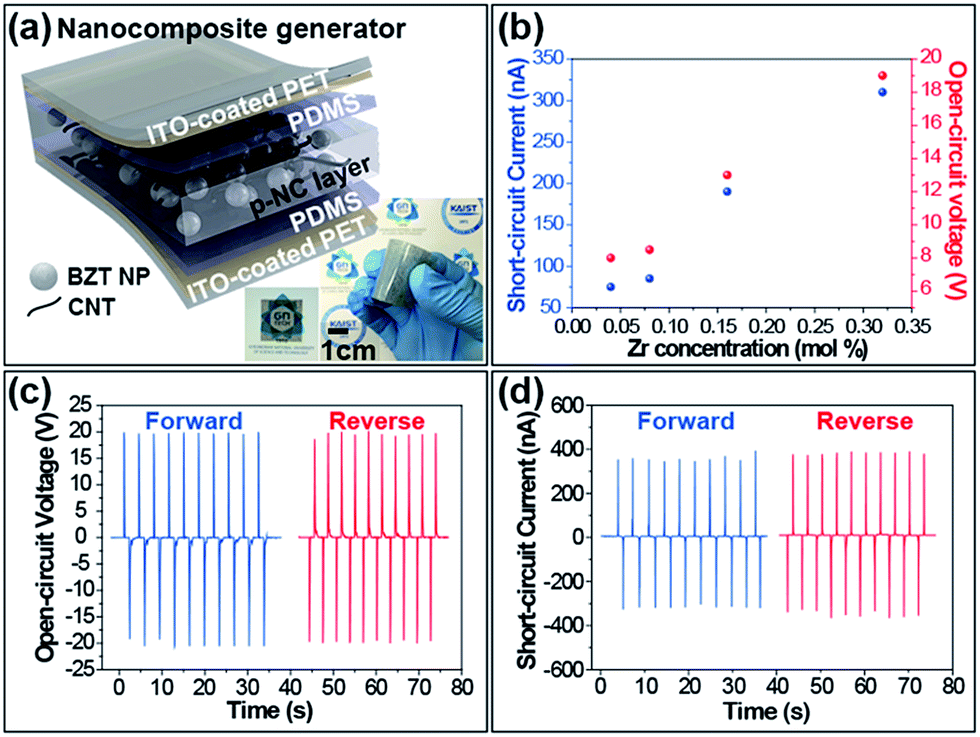 | ||
| Fig. 3 (a) Illustration of the fabricated BZT NPs-embedded composite-based PEH with photographs of the p-NC layer and the NCG device. (b) Output performance of BZT NPs-flexible PEH corresponding to Zr molar concentration. The generated open-circuit voltage (c) and short-circuit current (d) of flexible PEH when forward and reverse connected with a measurement kit. | ||
The flexibility of the p-NC is attributed to the mechanical support of the CNT-supplemented PMDS matrix, and the sandwiched structure enabled the device to harvest electrical signals under various modes of mechanical deformation. The electrons generated when the device was mechanically deformed, flowed along the external circuit because the thin layer of PDMS acts as a passivation barrier. This stream of electrons towards the external circuit generates the electric signals. The averaged short-circuit currents (Isc) and the open-circuit voltages (Voc) obtained from NCG devices fabricated with an effective area of 3 × 3 cm2 are shown in Fig. 3b during bending and unbending motions. The performance varied with the different molar concentration of Zr in the embedded BZT NPs. The enhancement of the output performance was closely related to the piezoelectric property of the embedded BZT NPs. In recent research, G. Philippot et al.,38 reported that the microstrain of the lattice structure of the BZT NP, varied with the dopant (Zr ion) concentration, and was maximal at the Zr composition of 60 mol%. The increase of the intra-structural microstrain in the ranges we synthesized could provide a plausible reason for the improvement of the device performance. The electrical energy output from the BaZr0.32Ti0.68O3 NPs embedded in the NCG device under mechanical deformation, are shown in Fig. 3c and d. The open-circuit voltage and short-circuit current reached ∼20 V and ∼400 nA, respectively, when the device was repeatedly deformed with a planar displacement of 5 mm, at a rate of 0.2 m s−1. The switching polarity test is crucial to confirm whether the measured electric signals were generated from the piezoelectric potential built into both sides of the p-NC active layer. From coherency in output signals extracted from the forward and reverse connections, and the corresponding positive-to-negative changes, we verified well the harvested output signals from a PEH.
To evaluate the instantaneous power, the load voltage (VL) and current (IL) signals were measured with a wide range of external resistors from 10 kΩ to 1 GΩ, as shown in Fig. 4a and b. The load voltage increased when the resistance increased, and showed an inflection point near a few mega-ohm range and reached the saturated load voltage. The load current followed the inverse trends of load voltage with respect to the external resistance. The multiple of these two output load signals is the instantaneous power; therefore, it represented the definite peak, yielding the maximum power of ∼6 μW at a resistance of around 100 MΩ where the IL–VL curves were crossed. Considering the device for actual applications, its durability should also be determined for persistent use. The BZT NP-based NCG was confirmed to maintain its output performance up to ∼5000 bending cycles, while retaining the initial output voltage without decay.
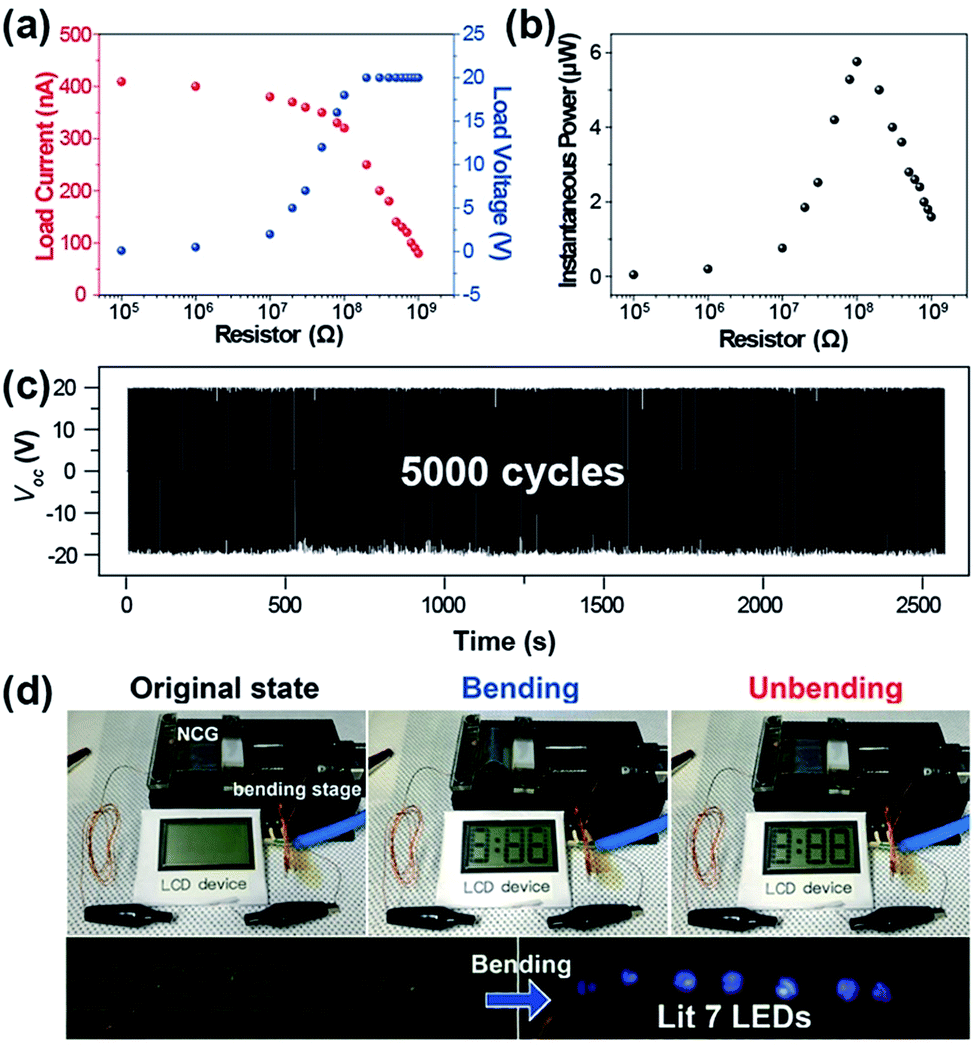 | ||
| Fig. 4 The measured load voltage and current (a) and the calculated instantaneous power (b) with the various external resistors from 100 kΩ to 1 GΩ. (c) The mechanical durability test results of a fabricated flexible PEH under periodically deformed conditions up to 5000 cycles. (d) Operation of the commercial LCD screen and LEDs using the electrical energy generated from a BZT NP-flexible PEH. | ||
Demonstrating the correspondence of the device performance to real-life applications is helpful for estimating its more practical uses. As shown in Fig. 4d, commercial electronic devices (LCD and LEDs) were properly operated by the electricity generated from the BZT NP-based NCG. An LCD window was activated during both the bending and the releasing motions of the NCG device due to its nonpolar nature (it contained no permanent dipoles). The bottom image of Fig. 4d shows the prompt responses of seven blue LEDs that lit up during bending motions with forward external connection. This corresponds to an output voltage of ∼20 V.
Conclusions
A facile hydrothermal method was utilized to synthesize BZT NPs with highly controllable zirconium molar concentration. The amorphous B-site precursor prepared by the hydrolysis of alkoxide using ammonia solution, reacted efficiently with the A-site precursor solution. The resulting initial BZT NPs were well crystallized and homogenized without any carbonate impurities or segregated phases. The particle size can be controlled by varying the temperature and reaction time of the hydrothermal conditions. The synthesized BZT NPs were applied to the composite-based PEH. The output performance of the PEH depended on the Zr/Ti molar ratio of the embedded BZT NPs. The maximum output voltage and current signals reached 20 V and 400 nA, respectively. These generated power sources from a lead-free BZT NPS-PEH were successfully used to drive the small commercial electronic devices.Acknowledgements
This work was supported by the National Research Foundation (NRF) of Korea grant funded by the Korea government (MSIP) (No. 2016R1C1B1006456). This work was also supported by the Future Planning and by the Climate Change Research Hub of KAIST (Grant No. N11160019).Notes and references
- C. R. Bowen, H. A. Kim, P. M. Weaver and S. Dunn, Energy Environ. Sci., 2014, 7, 25–44 CAS.
- J. Briscoe and S. Dunn, Nano Energy, 2015, 14, 15–29 CrossRef CAS.
- G. T. Hwang, Y. Kim, J. H. Lee, S. Oh, C. K. Jeong, D. Y. Park, J. Ryu, H. Kwon, S. G. Lee, B. Joung, D. Kim and K. J. Lee, Energy Environ. Sci., 2015, 8, 2677–2684 CAS.
- G. T. Hwang, H. Park, J. H. Lee, S. Oh, K. I. Park, M. Byun, H. Park, G. Ahn, C. K. Jeong, K. No, H. Kwon, S. G. Lee, B. Joung and K. J. Lee, Adv. Mater., 2014, 26, 4880–4887 CrossRef CAS PubMed.
- S. H. Lee, C. K. Jeong, G. T. Hwang and K. J. Lee, Nano Energy, 2015, 14, 111–125 CrossRef CAS.
- X. D. Wang, Nano Energy, 2012, 1, 13–24 CrossRef CAS.
- Z. L. Wang, J. Phys. Chem. Lett., 2010, 1, 1388–1393 CrossRef CAS.
- Z. L. Wang and W. Z. Wu, Angew. Chem., Int. Ed., 2012, 51, 11700–11721 CrossRef CAS PubMed.
- R. Yang, Y. Qin, C. Li, G. Zhu and Z. L. Wang, Nano Lett., 2009, 9, 1201–1205 CrossRef CAS PubMed.
- C. K. Jeong, K. I. Park, J. Ryu, G. T. Hwang and K. J. Lee, Adv. Funct. Mater., 2014, 24, 2620–2629 CrossRef CAS.
- J. H. Jung, C. Y. Chen, B. K. Yun, N. Lee, Y. S. Zhou, W. Jo, L. J. Ho and Z. L. Wang, Nanotechnology, 2012, 23, 375401 CrossRef PubMed.
- J. H. Jung, M. Lee, J. I. Hong, Y. Ding, C. Y. Chen, L. J. Chou and Z. L. Wang, ACS Nano, 2011, 5, 10041–10046 CrossRef CAS PubMed.
- Z. H. Lin, Y. Yang, J. M. Wu, Y. Liu, F. Zhang and Z. L. Wang, J. Phys. Chem. Lett., 2012, 3, 3599–3604 CrossRef CAS PubMed.
- K. I. Park, S. B. Bae, S. H. Yang, H. I. Lee, K. Lee and S. J. Lee, Nanoscale, 2014, 6, 8962–8968 RSC.
- K. I. Park, C. K. Jeong, J. Ryu, G. T. Hwang and K. J. Lee, Adv. Energy Mater., 2013, 3, 1539–1544 CrossRef CAS.
- K. I. Park, M. Lee, Y. Liu, S. Moon, G. T. Hwang, G. Zhu, J. E. Kim, S. O. Kim, D. K. Kim, Z. L. Wang and K. J. Lee, Adv. Mater., 2012, 24, 2999–3004 CrossRef CAS PubMed.
- H. Sun, H. Tian, Y. Yang, D. Xie, Y. C. Zhang, X. Liu, S. Ma, H. M. Zhao and T. L. Ren, Nanoscale, 2013, 5, 6117–6123 RSC.
- C. K. Jeong, J. Lee, S. Han, J. Ryu, G. T. Hwang, D. Y. Park, J. H. Park, S. S. Lee, M. Byun, S. H. Ko and K. J. Lee, Adv. Mater., 2015, 27, 2866–2875 CrossRef CAS PubMed.
- S. Y. Xu, Y. W. Yeh, G. Poirier, M. C. McAlpine, R. A. Register and N. Yao, Nano Lett., 2013, 13, 2393–2398 CrossRef CAS PubMed.
- M. M. Yuan, L. Cheng, Q. Xu, W. W. Wu, S. Bai, L. Gu, Z. Wang, J. Lu, H. P. Li, Y. Qin, T. Jing and Z. L. Wang, Adv. Mater., 2014, 26, 7432–7437 CrossRef CAS PubMed.
- Z. Zhou, C. C. Bowland, M. H. Malakooti, H. X. Tang and H. A. Sodano, Nanoscale, 2016, 8, 5098–5105 RSC.
- C. Baek, J. H. Yun, J. E. Wang, C. K. Jeong, K. J. Lee, K. I. Park and D. K. Kim, Nanoscale, 2016, 8, 17632–17638 RSC.
- B. K. Yun, Y. K. Park, M. Lee, N. Lee, W. Jo, S. Lee and J. H. Jung, Nanoscale Res. Lett., 2014, 9, 4 CrossRef PubMed.
- V. Buscaglia, S. Tripathi, V. Petkov, M. Dapiaggi, M. Deluca, A. Gajovic and Y. Ren, J. Phys.: Condens. Matter, 2014, 26, 065901 CrossRef PubMed.
- A. Dixit, S. B. Majumder, P. S. Dobal, R. S. Katiyar and A. S. Bhalla, Thin Solid Films, 2004, 447, 284–288 CrossRef.
- D. Hennings, A. Schnell and G. Simon, J. Am. Ceram. Soc., 1982, 65, 539–544 CrossRef CAS.
- D. Y. Liang, X. H. Zhu, J. L. Zhu, J. G. Zhu and D. Q. Xiao, Ceram. Int., 2014, 40, 2585–2592 CrossRef CAS.
- T. Maiti, R. Guo and A. S. Bhalla, J. Am. Ceram. Soc., 2008, 91, 1769–1780 CrossRef CAS.
- J. Bera and S. K. Rout, Mater. Lett., 2005, 59, 135–138 CrossRef CAS.
- T. Maiti, R. Guo and A. S. Bhalla, J. Appl. Phys., 2006, 100, 114109 CrossRef.
- S. Miao, J. Pokorny, U. M. Pasha, O. P. Thakur, D. C. Sinclair and I. M. Reaney, J. Appl. Phys., 2009, 106, 114111 CrossRef.
- I. K. Jeong, C. Y. Park, J. S. Ahn, S. Park and D. J. Kim, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 214119 CrossRef.
- C. Kajtoch, J. Mater. Sci., 2011, 46, 1469–1473 CrossRef CAS.
- T. Maiti, R. Guo and A. S. Bhalla, Ferroelectrics, 2011, 425, 4–26 CrossRef CAS.
- F. A. Rabuffetti and R. L. Brutchey, Chem. Commun., 2012, 48, 1437–1439 RSC.
- J. Y. Choi, C. H. Kim and D. K. Kim, J. Am. Ceram. Soc., 1998, 81, 1353–1356 CrossRef CAS.
- G. Philippot, M. Albino, U. C. Chung, M. Josse, C. Elissalde, M. Maglione and C. Aymonier, Mater. Des., 2015, 86, 354–360 CrossRef CAS.
- G. Philippot, E. D. Boejesen, C. Elissalde, M. Maglione, C. Aymonier and B. B. Iversen, Chem. Mater., 2016, 28, 3391–3400 CrossRef CAS.
- S. G. Kwon, B. H. Park, K. Choi, E. S. Choi, S. Nam, J. W. Kim and J. H. Kim, J. Eur. Ceram. Soc., 2006, 26, 1401–1404 CrossRef CAS.
- H. W. Lee, S. Moon, C. H. Choi and D. K. Kim, J. Am. Ceram. Soc., 2012, 95, 2429–2434 CrossRef CAS.
- M. B. Smith, K. Page, T. Siegrist, P. L. Redmond, E. C. Walter, R. Seshadri, L. E. Brus and M. L. Steigerwald, J. Am. Chem. Soc., 2008, 130, 6955–6963 CrossRef CAS PubMed.
- H. K. Park, D. K. Kim and C. H. Kim, J. Am. Ceram. Soc., 1997, 80, 743–749 CrossRef CAS.
- C. Baek, J. E. Wang, D. S. Kim and D. K. Kim, J. Nanosci. Nanotechnol., 2016, 16, 11652–11657 CrossRef.
- C. Baek, J. E. Wang, S. Moon, C. H. Choi and D. K. Kim, J. Am. Ceram. Soc., 2016, 99, 3802–3808 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26285f |
| ‡ Equal contributors. |
| This journal is © The Royal Society of Chemistry 2017 |