
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of composition and structure on the performance of tin/graphene-containing carbon nanofibers for Li-ion anodes†
Martin K. Dufficy ,
Sheng-Yang Huang,
Saad A. Khan and
Peter S. Fedkiw*
,
Sheng-Yang Huang,
Saad A. Khan and
Peter S. Fedkiw*
Department of Chemical and Biomolecular Engineering, North Carolina State University, Raleigh, NC, USA 27695. E-mail: Fedkiw@ncsu.edu
First published on 8th March 2017
Abstract
Tin is a promising replacement for graphite anodes in Li-ion batteries (994 mA h g−1 for Sn vs. 372 mA h g−1 for graphite), but suffers from particle pulverization upon lithiation that causes capacity fade. Herein, thermally reduced graphene oxide-containing carbon nanofibers (TRGO/CNFs) are used as scaffolds to house Sn/SnO2 particles, enhance anode capacity beyond that of graphite, and prolong cycle life of Sn-based electrodes. This study attempts to elucidate structure–composition relationships of tin-TRGO/CNF electrodes that lead to increased capacity retention. The composition and morphology of tin-TRGO/CNFs are assessed as a function of heat-treatment temperature and Sn loading as a means to understand and correlate electrochemical performance with physical features. We find: (1) the oxidation state of tin in TRGO/CNFs is in part determined by temperature-dependent, thermal-decomposition products of polyacrylonitrile-derived CNFs, and (2) precursor Sn(IV) loadings ≤10 wt% in the tin-TRGO/CNFs lead to Sn(0) or SnO2 particles embedded within the fiber + TRGO matrix. Electrodes with precursor Sn(IV) loading ≤10 wt% have smaller tin particles than electrodes with Sn(IV) loadings >10 wt%, and have longer cycle-lives; reversible capacities of ∼600 mA h g−1 are observed at 0.2C rates, while capacities of ∼400 mA h g−1 are observed after hundreds of cycles at 2C rates. The durable graphene-containing nanofiber matrix, coupled with the high-capacity of tin, provides a promising anode material for Li-ion cells.
1. Introduction
Advancements in mobile technology require electrochemical energy storage devices with an energy and power density beyond that of current Li-ion batteries. Enhanced Li-ion battery performance may be achieved with new active materials that exhibit charge capacities beyond carbon.1–3 Tin is an attractive anode material with a large theoretical charge capacity (994 mA h g−1, 1990 mA h cm−3), but suffers from particle pulverization due to catastrophic mechanical strain at full lithiation.4 Pulverization results in electronic isolation—caused by recurrent solid-electrolyte interphase (SEI) formation—and a short cycle life. One proposed method to cope with the strain of Li-insertion is to form composite tin materials, such as tin alloys or Sn/C materials. A battery using tin alloy anodes, introduced by Sony under the name Nexelion,5,6 saw limited success due to capacity fade and short cycle life. Another common method to cope with the frail Sn–Li complex is to use the oxide of tin. Fuji introduced batteries with tin-composite oxide anodes under the name Stalion,7 which never reached commercialization due to its large capacity fade. Thus, new methods to extend the cycle-life of tin-based anodes are a worthy research thrust. Herein, we report upon tin/carbon composites as a means to enhance Sn-based anode cycle life.Carbon composites have long been a common method to enhance electrode cycle life.8 Moreover, carbon nanofibers (CNFs) provide a favorably-sized support for composite electrodes for fast Li-diffusion and high-rate capabilities.9 CNFs are also durable materials, and thus composite materials (e.g., Sn particles) may impart strains to the CNF matrix during volume expansions to inhibit pulverization; volume expansions in the LiCx complex are small (∼10% graphene layer expansion) relative to the LixSn complex (∼300% volume expansion).10 The CNF electrode is well characterized,11,12 and long cycle lives have been reported13 with use of CNF anodes produced via electrospinning. The nonwoven carbon architecture from electrospinning negates the need for binder and conductive additive,14,15 which enhances electrode gravimetric energy density.
We recently demonstrated that thermally reduced graphene oxide-containing carbon nanofibers (TRGO/CNFs) increase charge capacity and electronic conductivity compared to CNFs.16 As a continuation of our investigation of TRGO/CNFs anodes for LIBs, we study composite CNFs as a matrix for embedding Sn or SnO2 particles to produce Sn- or SnO2-containing TRGO/CNF electrodes. While Sn-containing CNFs have been applied to LIB anodes,12,14–16 such studies frequently highlight the electrochemical cycling behavior and minimize importance of structure evolution. Studies on production of Sn or SnO2/C composites from precursors such as SnCl4 and polyacrylonitrile (PAN, a common CNF precursor) do not discuss simultaneous Sn(II) or (IV) reduction and the carbonization process. Additionally, to our knowledge, the influence of tin loading on fiber morphology has not been reported. Herein, we report the formation and evolution of Sn-TRGO/CNFs electrodes; we explore the influence of heat-treatment temperature (HTT) on Sn oxidation state, and the influence of Sn-precursor concentration on particle size after heat treatment. We relate physical parameters favorable for LIB electrodes (composition, morphology, Sn-particle size, etc.) to electrochemical half-cell cycling performance. The SnO2-based electrodes produced in this work revealed capacities ∼400 mA h g−1 after 900 cycles at 2C rates, and Sn-based electrodes revealed similar capacities ∼400 mA h g−1 after 500 cycles at 2C rates. Long-cycle lives are ascribed to preservation of TRGO/CNF structure, which withstands structural rearrangement of Sn during Li-insertion. This study shows Sn and SnO2-TRGO/CNF is promising electrode material for Li-ion cells.
2. Experimental
2.1 Preparation of electrospinning solution
Appropriate amounts of SnCl4 (Sigma Aldrich, 99.995%) were added to anhydrous dimethylformamide (DMF, Sigma Aldrich, 99.8%) in an argon-filled glove box, such that Sn(IV) content in dry polymer fibers comprised 0, 5, 10, 15, or 30 wt%. Anhydrous ethylene glycol (Sigma Aldrich, 99.8%) was added to the SnCl4/DMF mixture in a ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol ethylene glycol:SnCl4. Graphite oxide16 was then added to the solution, resulting in a loading of 5 wt% relative to dry polymer–fiber content. The mixture was sealed, taken out of the glove box, and sonicated for 1 hour to exfoliate graphite oxide into single-to-multilayer graphene oxide (GO) sheets. After sonication, polyacrylonitrile (PAN, Scientific Polymer Products, 150 kDa) was added to the dispersion in ambient conditions, resulting in an 8 wt% polymer solution, and stirred overnight at 60 °C.
:1 mol ethylene glycol:SnCl4. Graphite oxide16 was then added to the solution, resulting in a loading of 5 wt% relative to dry polymer–fiber content. The mixture was sealed, taken out of the glove box, and sonicated for 1 hour to exfoliate graphite oxide into single-to-multilayer graphene oxide (GO) sheets. After sonication, polyacrylonitrile (PAN, Scientific Polymer Products, 150 kDa) was added to the dispersion in ambient conditions, resulting in an 8 wt% polymer solution, and stirred overnight at 60 °C.
2.2 Preparation of Sn-containing fibers
The electrospinning precursor solution was cooled to room temperature and then electrospun using a needle-and-plate collector system, as described elsewhere.17 Typical process parameters include flow rate, voltage, tip-to-collector plate distance, and relative humidity of 1 mL h−1, 5–7 kV, 17 cm, and 50–60%, respectively. Composite polymer fibers amassed in a nonwoven mat on an Al-foil collector to create Sn-GO/PAN fibers. The mats were thermally treated in a quartz tube furnace using a two-step process. The first thermal treatment, stabilization, was conducted under dry air at 275 °C for 2 hours, using a heating ramp rate of 5 °C min−1 followed by uncontrolled cooling to room temperature. The second heat treatment, carbonization, was conducted under nitrogen at 650 or 850 °C using a heating rate of 2 °C min−1, a dwell time of 1 hour at maximum temperature, and uncontrolled cooling to room temperature (typically 3 hours). The quartz furnace was purged with nitrogen for 3 hours prior to carbonization. The notation used to represent the electrodes produced in this study denotes precursor Sn(IV) wt% in the polymer fibers, followed by the carbonization temperature (in °C). For example, a tin-GO/PAN solution electrospun with 10 wt% Sn(IV) and carbonized at 650 °C is denoted 10Sn650.2.3 Characterization of Sn-containing fibers
Solution rheology was measured using an AR2000 (TA Instruments) stress-controlled rheometer with 40 mm diameter, parallel-plate geometry and a gap size of 100 μm. Ionic conductivity of the electrospinning precursor solution was measured using a Mettler Toledo S47-K conductivity meter. Scanning electron microscope (SEM) images were captured on a FEI Verios 460L XHR equipped with an energy-dispersive X-ray spectroscopy (EDS) detector. Polymer fibers were sputter coated with 4 nm of Au/Pd alloy to increase conductivity before imaging. Fiber-diameter distributions were obtained by measuring diameters from SEM images of 100 fibers from three locations on a nonwoven mat. Transmission electron microscope (TEM) images were taken on a JEOL 2010F, which was also equipped with an EDS detector. To prepare samples on TEM grids, we first dispersed fiber mats in acetone followed by sonication for 1 hour. Thermogravimetric analysis (TGA) of composite-polymer fibers was conducted on a Discovery Series thermogravimetric analyzer (TA Instruments). Bulk structure analysis was measured on a Rigaku SmartLab X-ray diffractometer (Cu Kα, λ = 1.5406 Å). Surface analysis of fibers was conducted on a SPECS X-ray photoelectron spectroscopy (XPS) unit equipped with a dual Al/Mg anode monochromator source. The carbon transition (C 1s, 285 eV) was used to normalize the spectra. Samples were dried in a vacuum oven overnight at 100 °C prior to XPS analysis.2.4 Electrochemical characterization
Carbonized mat working electrodes were punched into 1.11 cm diameter discs from the electrospun mat and dried in a vacuum oven overnight at 100 °C. The discs were transferred to an argon-filled glove box. Carbonized mats (areal loading: 1–1.3 mg cm−2) were placed on a Cu foil current collector and cycled against metallic Li (Sigma Aldrich, 99%) in 2032 half-cells. The electrolyte, 1 M LiPF6 (>99% purity, BASF) dissolved in ethylene carbonate (EC, 99% purity, Sigma Aldrich) and ethyl methyl carbonate (EMC, 99% purity, Sigma Aldrich) comprising a 1:1 wt% EC:EMC solution, was added by pipette onto the working electrode (∼50 μL) and Celgard separator (∼50 μL). Prior to use, the electrolyte was dried in 4 Å molecular sieves (Sigma Aldrich) to a water content <10 ppm. All half-cells underwent a formation cycle at C/24 prior to further cycling. Charge/discharge cycling and cyclic voltammetry (CV) was conducted on a Bio-Logic VMP3 between 2.5 and 0.01 V vs. Li/Li+. Electrochemical measurements were conducted on at least 3 half-cells to assure reproducibility, typically within 10%.
3. Results and discussion
3.1 Characterization of Sn-fibers
Prior to thermal treatment, tin–polymer fibers were analyzed via SEM to observe fiber morphology (Fig. 1a–e). Images in Fig. 1 show (1) a continuous network of fibers with fiber beading at Sn(IV) concentrations >5 wt%, and (2) fiber diameter increases with Sn-loading. As noted in our previous study,16 the appearance of beads may be caused by encapsulated GO in the mats, which was added at 5 wt% to the fibers to increase the charge capacity and conductivity of CNFs. Additional electron microscope images, including TRGO/CNFs in the absence of tin, are available in our previous study.16 However, the beading observed at >5 wt% Sn(IV) loading (Fig. 1c–e) is not due to GO sheets, as evidenced by a different morphology than reported in our previous study on TRGO/CNFs in the absence of tin,16 and may be attributed to decreased “effective” polymer concentration as filler content increased. We measured electrospinning precursor solution properties that govern the electrospinning process, such as solution rheology and ionic conductivity, in attempt to correlate fiber diameter and beading in the fibers with Sn(IV) loading. Steady-shear rheology (Fig. S1†) of the electrospinning solution revealed viscosity (1) to be independent of SnCl4 concentration at high-shear stresses (or shear rates), and (2) to marginally increase with EG addition (added 4:1 mol ratio EG:SnCl4). However, the increased viscosity we measured from EG addition (<1 Pa s−1 at high-shear stresses) is not significant to increase the fiber diameter. Interestingly, increasing SnCl4 concentration, and thus ionic conductivity (Fig. 1f), did not lead to smaller fiber diameters; it is generally accepted that increasing solution conductivity increases whipping instabilities of the polymer jet during electrospinning that reduces fiber diameter.18 Thus, we attribute the increase in fiber diameter to EG swelling in PAN; a larger EG content is associated with higher SnCl4 concentration. Insets of Fig. 1a–e show high-resolution images of a typical fiber extruded during the electrospinning process. The fibers produced are porous, which may be the result of volatilized solvent.
 | ||
| Fig. 1 SEM images of nonwoven GO/PAN mats with (a) 0, (b) 5, (c) 10, (d) 15, and (e) 30 wt% Sn(IV). The scale bars represent 20 μm. The insets show high-resolution SEM images of a typical fiber. Scale bars of insets represent 500 nm; (f) fiber diameter plotted against ionic conductivity of electrospinning solution, with Sn(IV) loading [wt%] labeled on the plot. | ||
Thermally treating (stabilization and carbonization) tin-GO/PAN fibers (to produce Sn- or SnO2-TRGO/CNFs) decreased diameters to values independent of Sn(IV) loading (Fig. S2†); the average fiber diameter ranged from 340 to 470 nm at 650 °C and 280 to 330 at 850 °C, but fiber diameter did not systematically increase with Sn(IV) loading. The diameter distributions (Fig. S2†) suggest EG swelled PAN to increase fiber size, as unevaporated EG generated larger resistances to whipping instabilities during electrospinning. Heat treatments also collapsed large pores (Fig. 1a–e inset), although it is possible that heat-treated fibers retain a porous structure too small to discern in the SEM images.
As apparent by SEM, tin-TRGO/CNFs carbonized at 650 °C produced similar morphologies, irrespective of Sn(IV) loading (Fig. S3†); we did not observe sintering or agglomeration of tin despite carbonization temperatures above the melting point of Sn (232 °C). Increasing carbonization temperature from 650 to 850 °C and Sn(IV) loadings >5 wt% produced Sn particles on the fiber surface (Fig. 2). Loadings of 5 wt% Sn(IV) (Fig. 2a and b) produced fibers with a morphology similar to fibers in the absence of tin (Fig. S4†), suggesting that small Sn particles, not discernable at the magnifications employed, are present. An increased loading of 10 wt% Sn(IV) effected sparsely-decorated Sn particles on fibers (Fig. 2c and d), however the majority of Sn appeared to be within the TRGO/CNF scaffolding. Sn(IV) loadings >10 wt% produced fibers with abundant, large particles on the fiber surface. Mean Sn particle diameters of 830 and 330 nm were observed with 15 (Fig. 2e and f) and 30 wt% Sn(IV) (Fig. 2g and h), respectively. Similar Sn-aggregation has recently been reported with Sn(IV) heat treated at 700 °C.19
 | ||
| Fig. 2 SEM images of TRGO/CNFs carbonized at 850 °C with (a, b) 5, (c, d) 10, (e, f) 15, and (g, h) 30 wt% Sn(IV). Scale bars represent (a, c, e, and g) 10 μm and (b, d, f, and h) 500 nm. | ||
Heat treatments of tin-GO/PAN fibers likely produced temperature-dependent gaseous atmospheres, leading to unique fiber morphologies (previously seen in SEM images) and tin oxidation states. Carbonization of PAN releases gases such as H2, HCN, NH3, and CH4,20,21 and evolution of particular gases during carbonization is temperature dependent.21 It has been proposed that H2 off-gasses (producing a reducing atmosphere) at ∼900 °C21 as a product of reactions between hydrogenated carbons and nitrogen molecules in cyclic compounds, whereas N2 off-gasses at 600 °C as a product of reactions between cyclic nitrogen compounds. We observed evidence of these two reactions (releasing H2 and N2) via derivative thermogravimetric analysis (DTG) of PAN fibers (Fig. S5†). DTG plots produced a broad peak at ∼650 °C with a shoulder appearing at 850 °C, which likely corresponded to thermal decomposition of PAN releasing N2 and H2, respectively. We propose that H2 is a major PAN-decomposition product at 850 °C, facilitating the carbothermic reduction of Sn(IV) to Sn(0) and agglomeration of metallic Sn (as seen in Fig. 2c and d). Such a mechanism is likely, as researchers22 have shown that SnO2 reduces to Sn(0) at temperatures as low as 450 °C in a H2 atmosphere. In contrast, N2 comprised the majority of decomposition gases released at 650 °C, resulting in SnO2 particles in TRGO/CNFs. Sn–O bonds form via condensation reaction between SnCl4 and EG, releasing HCl.23 Thermal treatment then decomposed Sn–EG entities to SnO2.
We employed XPS on tin-GO/PAN fibers and tin-TRGO/CNFs carbonized at 650 and 850 °C in an attempt to detect the presence of contaminants such as Cl, observe polymer degradation through the nitrogen signal, and identify tin oxidation state. Fibers with 10 wt% Sn(IV) loading were analyzed. Chlorine (4:1 mol Cl:Sn in precursor) was not detected on survey scans (Fig. 3a), and produced weak signals on the Cl 2p transition (Fig. S6a†). Absence of Cl from the survey of as-spun fibers suggests a surface (<10 nm) relatively free of Cl or HCl (product from condensation of SnCl4 and EG). The XPS survey also shows decreasing N-content in the fibers, from 22 to 15 to 8 wt% N in as-spun fibers, and fibers heat-treated at 650 and 850 °C, respectively, as PAN thermally degrades into a carbon-rich structure. We observed slightly lower binding energies than expected in the N 1s transition (Fig. S6b†) of tin-TRGO/CNFs, which may be due to metal–nitride (Sn–N) formation, as reported elsewhere.24 Nonetheless, nitrogen-containing compounds in large quantities are undesired,25 as they may result in the irreversible formation of LiN during LIB charge cycles and decrease capacity retention. Interestingly, Sn 3d5/2 (Fig. 3b) peaks in as-spun mats shifted positive ∼1 eV upon carbonization, which suggests formation of the oxide (SnO2 ∼ 486.5–486.7 eV). As previously stated, Sn–O bonds initially formed in as-spun mats via a reaction between SnCl4 and EG. Lower binding energies in as-spun fibers may be due, in part, to remaining Sn–Cl linkages. Broad Sn 3d peaks from fibers carbonized at 650 and 850 °C are similar, and we were unable to distinguish if the open-air environment prior to XPS analysis resulted in (1) formation of a native-surface oxide layer on large Sn(0) particles, or (2) complete oxidation of small Sn(0) particles near the fiber surface to create SnO2 particles. We employed XRD in attempt to make this distinction.
 | ||
| Fig. 3 Surface and bulk characterization of Sn-containing fibers; XPS data of as-spun GO/PAN fibers containing 10 wt% Sn(IV), along with the ensuing spectra of fibers carbonized at 650 and 850 °C showing (a) the survey and (b) high-resolution Sn 3d transition. XRD profiles for tin-TRGO/CNFs carbonized at (c) 650 °C and (d) 850 °C with Sn loadings labeled on the plot. If present, the location of the strongest peaks associated with SnO2 are labeled in (c). | ||
Composite tin fibers carbonized at 650 °C produced X-ray diffraction patterns independent of tin loading without discernible diffraction signatures from Sn/SnO2 (Fig. 3c), presumably due to the small size or amorphous nature of tin. We note that crystalline SnO2 may be present in the form of small particles that do not generate diffraction patterns. Likewise, TRGO/CNFs carbonized at 850 °C with ≤10 wt% Sn(IV) did not produce diffraction patterns (Fig. 3d). Although large (and sparsely dispersed) particulates were observed in SEM images of 10Sn850 (Fig. 2d), the sensitivity of XRD was insufficient to register a diffraction signal. The diffractions pattern of fibers with loadings ≤10 wt% Sn(IV) displayed the amorphous (002) phase of TRGO/CNFs (2θ ≈ 25°, not shown), as we described elsewhere.16 Greater than 10 wt% Sn(IV) loading formed crystalline structures of metallic Sn (Fig. 3d); the peaks are associated with the large Sn particles that were observed via SEM. Thus, it is reasonable to infer that H2 off-gassing occurred during the carbonization process to reduce Sn(IV) to Sn(0).
In an attempt to detect Sn(0) or SnO2 particles that were not found with XRD (e.g., fibers with low, ≤10 wt%, Sn(IV) loading), we examined heat-treated fibers under high-resolution TEM. As expected, tin-TRGO/CNFs heat-treated at 650 °C did not contain crystalline particles; we merely observed the amorphous Sn/C structure (Fig. 4a). On the other hand, tin-TRGO/CNFs carbonized at 850 °C contained crystalline particles of SnO2 that were visible near the fiber surface (Fig. 4b). Particulates near the fiber surface may originate from the mobility of low-molecular weight Sn–EG entities, which migrate to the air-fiber interface during heat treatments. Particles encapsulated within a fiber matrix are favorable for LIB cycling, as they may impart stresses onto the TRGO/CNF scaffolding during volume expansions of lithiation to minimize cracking. Measurements of lattice fringes confirmed presence of SnO2 (JCPDS 41-1445) within TRGO/CNFs carbonized at 850 °C, with a particle size <15 nm. Particles of SnO2 also appeared on TRGO that was protruding through the fiber surface (Fig. 4c). In addition to the roles of TRGO in composite CNF synthesis described in our previous study,16 here we note the presence of tin particles on TRGO (or within few layers) may originate from hydrogen bonding interactions between terminal hydroxyl groups of EG–Sn entities and oxygen functional groups of GO in the electrospinning solution. Appearance of SnO2 on TRGO/CNFs with 10 wt% Sn(IV) and carbonized at 850 °C was not expected since we detected Sn(0) on TRGO/CNFs with >10 wt% Sn(IV) and carbonized at 850 °C using XRD. However, we note that TEM sample preparation (exposure to air and sonication in acetone, which heats the sample to ∼45 °C) likely oxidized small Sn(0) particles on or near the TRGO/CNF surface to SnO2.
 | ||
| Fig. 4 TEM images of (a) 10Sn650 (b, c) 10Sn850 (d) STEM image and corresponding EDS maps of 10Sn650. The element pertaining to each map is labeled in the lower left. Scale bars for EDS represent 1 μm. | ||
Well-dispersed tin-particulates in the anode presumably leads to uniform stress during lithiation and hinders regions of disproportionate stress that may lead to electrode cracking. Thus, a uniform Sn-particle dispersion is necessary to extend the cycle life of high-capacity anodes. We used EDS to determine the dispersion of tin within tin-TRGO/CNF fibers. Fig. 4d shows typical EDS maps (acquired via STEM imaging) for 10Sn650. Regions of Sn appear dispersed throughout many fibers on the EDS image, however Sn-rich regions throughout all fibers could not be resolved (in a manner similar to element maps of carbon and oxygen) due to curvature of the fibers, shadowing effects, and focal plane of the image. It should be noted that a small amount of Cl was observed via EDS, which produced a negligible peak ∼2.6 keV in the EDS spectrum (Fig. S7b†). Coupling EDS and XPS data suggest a Cl content in tin-TRGO/CNFs <0.2 wt%. Typical EDS maps of tin-TRGO/CNFs at 850 °C are shown in Fig. S7 and S8.† Tin also appeared well-dispersed in TRGO/CNFs carbonized at 850 °C and at low Sn(IV) loadings (≤10 wt%), while larger tin loadings produced EDS maps with tin appearing both on large aggregated particles and dispersed throughout the fiber—not all tin aggregated into large Sn(0) particles. Interestingly, we found that EDS maps of Sn and N overlapped (Fig. S7a†). Overlapping Sn and N EDS maps, coupled with XPS data (Fig. S6b†) that show N 1s transitions at more negative binding energies than expected for Sn/C composites, may indicate formation of tin nitride upon thermal treatments. Tin nitride is produced via thermal treatment of Sn(IV) with NH3 and N2 gases26,27 and may exist in small quantities in the fibers since NH3 and N2 are products of PAN carbonization. The lack of SnxNy peaks on XRD suggests that small, amorphous regions of SnxNy may exist. While reports suggest that nitrogen-doped carbons28,29 (and perhaps doping Sn in the process) leads to enhanced cycling, to our knowledge the electrochemical performance of tin nitride has not been assessed.
3.2 Electrochemical characterization of Sn-containing fibers
Since we were unable to conclusively deduce the oxidation state of tin within fibers heat-treated at 650 °C and Sn(IV) loadings <15 wt% in fibers heat-treated at 850 °C using physical characterization methods, we employed CV as an electrochemical means to study composition of tin in the fibers. Working electrodes consisted of 10Sn650 and 10Sn850, identical to samples analyzed via XPS and TEM. Voltammograms of tin-TRGO/CNFs carbonized at 650 °C indicate presence of SnO2 on the first cycle. The first cathodic polarization (Fig. 5a) scan shows two peaks: (1) irreversible reduction of SnO2 (Reaction (1)) and electrolyte reduction causing SEI formation at ∼0.7 V vs. Li/Li+, and (2) three overlapping peaks from SnO reduction (Reaction (2)),30 Sn–Li complexation (Reaction (3)), and Li-intercalation into carbon (until 0.01 mV). The irreversible nature of SnO2 reduction is exemplified via CV, as the high-voltage reduction peak does not appear on the second cathodic cycle.| SnO2 + 2Li+ + 2e− → SnO + Li2O | (1) |
| SnO + 2Li+ + 2e− → Sn + Li2O | (2) |
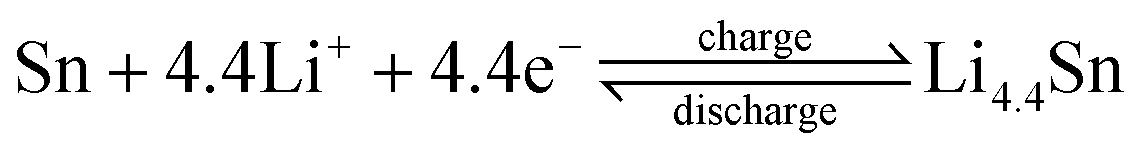 | (3) |
 | ||
| Fig. 5 Typical CV profiles using scan rates of 0.1 mV s−1 for (a) 10Sn650 and (b) 10Sn850 with phase transitions labeled.31 | ||
Unlike the voltammogram of 10Sn650, the first-cycle cathodic polarization of the 10Sn850 sample (Fig. 5b) did not produce a broad peak at 0.7 V vs. Li/Li+ related to SnO2 reduction, which suggests the presence of Sn(0). Oxidized tin in TRGO/CNFs carbonized at 850 °C (native surface oxide layers or SnO2 particles near the electrode surface) did not contribute to appreciable reduction activity in the voltammograms. Thus, we infer the majority of tin in the fibers carbonized at 650 °C and 850 °C resides in the oxide and metallic state, respectively. Distinct phase transitions31 in CV scans (labeled on Fig. 5b) are present over 10 cycles. Reoccurrence of redox peaks over cycling suggest that the TRGO/CNF support provides stability for small Sn particles, and prevents catastrophic cracking of the frail Li–Sn complex. Broad peaks at ∼1.2 V vs. Li/Li+ during anodic polarization of Sn- and SnO2-based electrodes have been attributed to partial oxidation of Sn(0) particles with diameters on the order of 1 nm.32,33 For comparison, voltammograms of TRGO/CNFs in the absence of tin produced similar peaks at 1.2 V vs. Li/Li+ with reduced magnitude (Fig. S9†). Thus, we attribute minimal activity to the oxidation of small Sn(0) particles. The influence of tin oxidation state in TRGO/CNFs and fiber morphology on Li-ion half-cell cycling was subsequently evaluated.
Prior to galvanostatic cycling, we approximated the specific theoretical (calculated) capacities. Theoretical capacities were approximated using Sn composition from EDS analysis (such that specific capacities accounted for entire mat weight) with the following equation:
| Capacitymax ≈ (1000 mA h g−1)(x wt% Sn) + (375 mA h g−1)(1 − x wt% Sn) |
Theoretical capacities, Sn concentration from EDS, and experimentally found, first-cycle lithiation capacities (using symmetrical constant current cycling at 0.05C rates) are listed in Table 1. As a reminder, tin loadings in sample nomenclature are expressed in wt% of Sn(IV) in GO/PAN polymer fibers. Actual Sn content in heat treated fibers approximately doubles. For example, 10Sn650 and 10Sn850 contain ∼20 and 21 wt% Sn, respectively.
| [Sn(VI)] wt% | [Sn]650 (wt%) | Actual650 [mA h g−1] (CE%) | Calculated650 [mA h g−1] | [Sn]850 (wt%) | Actual850 [mA h g−1] (CE%) | Calculated850 [mA h g−1] |
|---|---|---|---|---|---|---|
| 0 | 0 | 1040 (75) | 375 | 0 | 690 (79) | 375 |
| 5 | 11 | 1465 (73) | 445 | 15 | 780 (79) | 473 |
| 10 | 20 | 1320 (72) | 550 | 21 | 700 (80) | 508 |
| 15 | 27 | 1200 (50) | 542 | 23 | 643 (78) | 523 |
| 30 | 42 | 1260 (24) | 636 | 41 | 644 (76) | 632 |
On the first lithiation cycle at 0.05C, working electrodes carbonized at 650 °C exhibited capacities larger than calculated. For example, 5Sn650 electrodes displayed a capacity of 1465 mA h g−1, whereas 445 mA h g−1 was calculated. The discrepancy is largely due to irreversible reduction of oxygen and nitrogen-containing entities (forming Li3N and Li2O) such as the electrochemical reduction of SnO2, as well as SEI formation. Reaction (1)–(3) produce a theoretical charge capacity of 1494 mA h g−1, which is larger than the capacity from Reaction (3) (∼1000 mA h g−1) that we used to determine theoretical capacities for SnO2-TRGO/CNFs. In contrast, Sn-TRGO/CNF electrodes heat treated at 850 °C exhibited first-cycle capacities closer to, but still greater than, calculated values (largely due to SEI formation). The thermal reduction of SnO2 to Sn during carbonization of PAN at 850 °C nullifies much of the irreversible capacity from Reaction (1) and (2) on the first cycle. Increasing the carbonization temperature from 650 to 850 °C produced higher coulombic efficiencies (Table 1), which we attribute to a more reduced electrode with less noncarbonaceous content; weight ratios of active (C + Sn) to inactive (N + O) material increased with carbonization temperature (Fig. S10†), which decreased irreversible capacity on the first cycle. Higher CEs observed in tin-TRGO/CNFs carbonized at 850 °C, compared to those carbonized at 650 °C, may also be due to less SEI formation, as specific surface area of carbonized PAN decreases with carbonization temperature.34
Rate capabilities of the carbonized electrodes (Fig. 6a and b) were assessed using currents from the calculated capacities presented in Table 1. Discharge capacities from the fifth cycle (example of technique in Fig. S11a†) were used to represent the capacity at the respective rates. Rate studies on fibers carbonized at 650 °C revealed high capacities at slower rates, which we attribute to incomplete reduction of non-carbonaceous groups on the formation cycle. For example, TRGO/CNFs with 5 and 10 wt% Sn(IV) both displayed capacities of ∼600 mA h g−1 at 0.2C rates. While PAN carbonized at low temperatures led to a disordered carbon structure and tortuous Li-transport pathways, the absence of mass/electron transport limitations at low currents enabled Li to diffuse through the TRGO/CNF structure, into encapsulated Sn particles, and produce large capacities. Capacities decreased as C-rates increased, in part due to ohmic and Li transport limitations. Electrodes comprising >10 wt% Sn(IV) and carbonized at 650 °C revealed large irreversible capacities (Fig. S11b† and Table 1) and typically failed after a few cycles. Large irreversible capacity loss was likely due to formation of Li2O during irreversible reduction of SnO2 to Sn. Thus, their cycling performance beyond the first cycle was not pursued in this study.
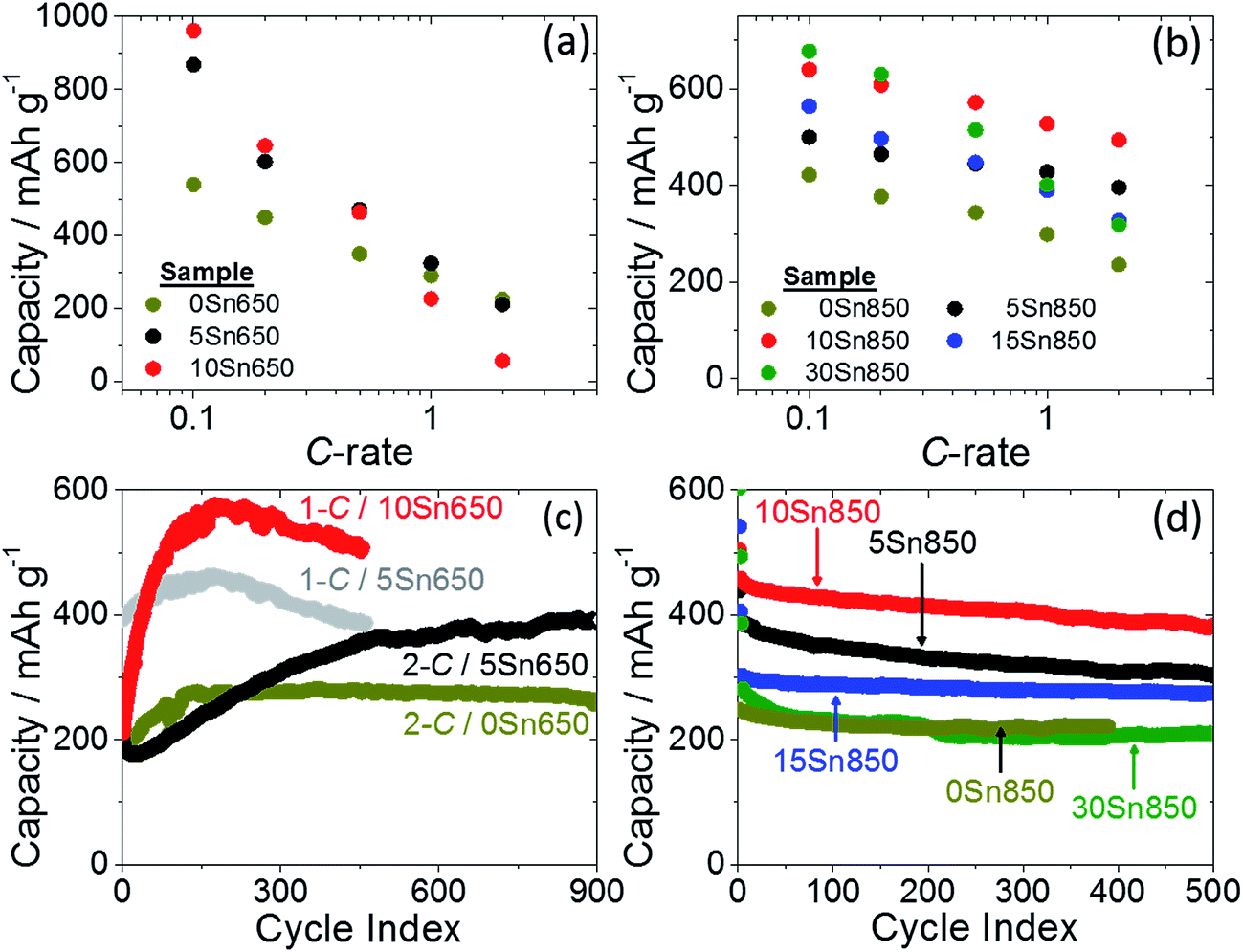 | ||
| Fig. 6 Rate capability measurements of tin-TRGO/CNFs carbonized at (a) 650 °C and (b) 850 °C; symmetrical Li-extraction cycling data of tin-TRGO/CNFs carbonized at (c) 650 °C and (d) 850 °C cycled at 2C rates. | ||
Increasing HTT to 850 °C produced fibers with larger capacities at fast rates when compared to electrodes produced at 650 °C (Fig. 6b), which we attribute to a more ordered carbon scaffolding for Sn particles. For example, at 2C rates, electrodes comprising 10 wt% Sn(IV) and heat-treated at 650 °C produced capacities of ∼50 mA h g−1, whereas electrodes heat-treated at 850 °C produced capacities of ∼500 mA h g−1. A more ordered carbon structure facilitates Li and electron transport in electrodes. The ID/IG ratios (a measure of defects in carbons) calculated from Raman spectroscopy decrease with HTT, which indicates larger graphitic domains and enhanced sp2 ordering (Fig. S12a†). The enhanced sp2 ordering corresponds to an increased electronic conductivity in the fibers (Fig. S12a†) and decreased internal cell resistances. Increasing HTT also created smaller fibers, which reduced Li diffusion pathway length. Nevertheless, electrodes with Sn(IV) loadings >10 wt% did not produce large capacities at fast rates; large Sn particles on the fiber surface of electrodes carbonized at 850 °C initially added capacity at cycles with slow C-rates, but cracked/deactivated after successive cycling with faster C-rates (Fig. 6b). We subsequently investigated electrode cycle life over hundreds of cycles to evaluate the robustness of the tin-containing electrodes. Long cycle lives are required for electrodes to be considered for commercial applications.
Extended cycling was executed in half-cells via symmetrical cycling at fast (≥1C) rates. As reported in our previous study,16 capacities of TRGO/CNFs carbonized at 650 °C increase with cycle number, which suggests incomplete reduction of noncarbonaceous species or restructuring of Li-active material. We observed similar results in this study; after capacity increase and stabilization, TRGO/CNF electrodes registered capacities ∼270 mA h g−1, while 5Sn650 electrodes registered capacities ∼400 mA h g−1 after 900 cycles at 2C rates—the added capacity is attributed to tin (Fig. 6c). High capacities were also observed at 1C rates, however capacities faded after 200 cycles. We attribute capacity fade to pulverization and deactivation of Sn within the TRGO/CNF matrix; volume expansions (during lithiation) of encapsulated particles produced strains too great for the fiber matrix to endure.
Contrary to the capacity growth exhibited by fibers carbonized at 650 °C at 2C rates, fibers carbonized at 850 °C exhibited monotonic capacity fade (Fig. 6d). Fibers with Sn(IV) loadings >10 wt% displayed the largest amount of capacity fade; after 500 cycles at 2C rates, these electrodes revealed capacities similar to TRGO/CNFs in the absence of Sn. The similarity in capacities indicates that the Sn on the surface of the fibers (as seen in Fig. 2e and g) cracked and inactivated. The cracked and inactive Sn particles may be seen in SEM images of 30Sn850 electrodes after 500 cycles in Fig. S13.† Compared to the aforementioned highly loaded Sn electrodes, electrodes with Sn(IV) loadings ≤10 wt% exhibited greater capacity retention over 500 cycles at 2C rates; the 10Sn850 electrodes displayed capacities ∼400 mA h g−1 (>75% capacity retention), nearly doubling the capacity of TRGO/CNFs in the absence of Sn.
Electrodes comprising ≤10 wt% Sn(IV) displayed the largest capacities and capacity retention. We attribute these performance enhancements to the favorable electrode morphology that was produced at low Sn(IV) loadings, which resulted in a mechanically durable electrode and limited catastrophic structure rearrangements during lithiation. Favorable electrode morphologies include: (1) encapsulated particles, yet wetted by electrolyte, in the carbon fiber matrix, which were able to impart stresses to the robust scaffolding; (2) small Li-host particle sizes (≤15 nm), which resulted in small absolute volume expansions upon Li-insertion; (3) uniform dispersions of tin in the fibers, which limited regions of disproportionate stress and reduced fiber cracking. However, capacity fade exhibited by tin-containing electrodes at capacities greater than 400 mA h g−1, warrants the necessity for a tailored fiber morphology.
Researchers have proposed that SnO2 electrodes display longer cycle lives and better capacity retention than Sn electrodes.35 It is thought that the initial reduction of SnO2 (Reaction (1) and (2)) generates lithia-rich regions that hinder Sn aggregation to retain a small particle size during cycling. However, we did not observe better cycling performances in the SnO2-based electrodes produced at 650 °C, compared to Sn-based electrodes produced at 850 °C. Rather, we attribute differences in cycling performance to the TRGO/CNF scaffolding. After hundreds of cycles at 2C rates, SnO2-TRGO/CNF electrodes produced at 650 °C and Sn-TRGO/CNF electrodes produced at 850 °C exhibited similar capacities. Additionally, electrode microstructure of both Sn and SnO2-TRGO/CNFs using Sn(IV) loadings of 5 wt% appeared similar after 500 cycles. SEM images of 5Sn650 (washed in EMC and dried in the glovebox) after 900 cycles at 2C (Fig. 7a) and 500 cycles at 2C in 5Sn850 (Fig. 7b) revealed a nonwoven structure similar to pre-cycled electrodes (picture in Fig. S3† and 2).
 | ||
| Fig. 7 Post cycle SEM images of (a) 5Sn650 after 900 cycles at 2C, (b) 5Sn850 after 500 cycles at 2C, and (c) 15Sn850 after 500 cycles at 2C with EDS mapping of the individual elements. The scale bars represent 1 μm. | ||
The promising capacity retention of tin-TRGO/CNF electrodes is attributed, in part, to high CEs; fibers carbonized at 650 °C displayed average CEs ≥99.8% for 1C rates and ≥99.9 for 2C rates (Table S1†). Average CEs decreased to ≥99.2% for fibers carbonized at 850 °C (Table S2†), which may be associated with capacity fade. The presence of TRGO in the CNFs may have increased electrode tensile properties, such as interfacial shear strength,36 which enabled TRGO/CNFs with small, encapsulated tin particles to cycle without recurrent cracking/SEI formation. Additionally, we did not observe Sn particle agglomeration upon cycling (other than previously observed upon thermal treatments), suggesting that loadings <15 wt% Sn(IV) produced particle sizes within TRGO/CNF favorable for LIB anodes. In contrast to the uncycled electrode particles (Fig. 2f), the Sn particles of 15Sn850 after 500 cycles at 2C rate (Fig. 7c) show a highly roughened surface, indicating significant fracturing. EDS map of the fractured Sn particle reveal SEI formation; concentrated regions of electrolyte salt decomposition products (fluorine and phosphorous maps) and electrolyte solvent reduction and polymerization (oxygen map) appear on the fractured Sn particle. It is likely that formation of the SEI on the Sn particles ohmically limited the electrodes, which decreased capacity at fast rates (as seen in Fig. 6b). As previously mentioned, N-rich regions appear on the EDS maps of pulverized Sn-particle, which is not a decomposition product of the electrolyte. Introduction of nitrogen most likely occurred during electrode processing, and we are currently investigating the electrochemical performance of SnxNy-based electrodes.
4. Summary
In this study, we used thermally reduced graphene oxide-containing carbon nanofibers (TRGO/CNFs) as a matrix for tin particles to produce high-capacity and high-capacity retention anodes for Li-ion battery applications. The Sn-precursor, SnCl4, was electrospun with ethylene glycol, polyacrylonitrile (PAN), and exfoliated graphene oxide, followed by heat treatments to produce Sn or SnO2-TRGO/CNFs. The effects of heat-treatment temperature and Sn(IV) loading on fiber structure and morphology were assessed. Oxidation state of Sn within the fiber mat, inferred from cyclic voltammetry, varied with carbonization temperature; SnO2 and Sn(0) were produced at 650 and 850 °C, respectively. Thermal decomposition of PAN released H2 at 850 °C, facilitating the high-temperature carbothermal reduction of Sn(IV) to Sn(0). Increased HTT also volatilized Sn–EG entities, which brought Sn to the TRGO/CNF surface. Low Sn(IV) loadings, ≤10 wt%, produced small tin particles encapsulated within the TRGO/CNF matrix, whereas high Sn(IV) produced large tin particulates on the fibers. The ensuing fiber morphology was critical to electrochemical performance.Fibers with ≤10 wt% Sn(IV) produced the smallest size particles (<15 nm diameter), which reduced absolute volume expansions and led to higher capacities over hundreds of cycles than fibers with larger tin loadings. For example, TRGO/CNFs with 10 wt% Sn(IV) loading produced capacities ∼400 mA h g−1 after 900 cycles. Fibers with ≤10 wt% Sn(IV) also produced fibers with encapsulated particles within the TRGO/CNF matrix that were able to impart their stresses onto the carbon substrate to withstand the mechanical strains of lithiation. The aforementioned particle size and location within the fibers led to a longer cycling life for Sn and SnO2-TRGO/CNFs. We also attribute the long cycle life at low-Sn(IV) loadings to uniform dispersion of Sn(IV) throughout fiber mats, which created a uniform stress profile across the electrode during lithiation to reduce fiber fragmentation. On the other hand, Sn(IV) loadings >15 wt% produced fibers that were not conducive for LIB anodes; large irreversible capacities in SnO2-based fibers and large particles on the TRGO/CNF surface led to large initial capacities, but a short cycle life. Additional research is needed to take full advantage of the large-charge capacity inherent to Sn/SnO2 anodes. However, this work provides a critical step in producing tin-based anodes that operate at fast rates with high-capacities that endure lithiation over hundreds of cycles.
References
- N. Nitta and G. Yushin, High-Capacity Anode Materials for Lithium-Ion Batteries: Choice of Elements and Structures for Active Particles, Part. Part. Syst. Charact., 2014, 31, 317–336 CrossRef CAS.
- H. Li, Z. Wang, L. Chen and X. Huang, Research on Advanced Materials for Li-Ion Batteries, Adv. Mater., 2009, 21, 4593 CrossRef CAS.
- C.-M. Park, J.-H. Kim, H. Kim and H.-J. Sohn, Li-Alloy Based Anode Materials for Li Secondary Batteries, Chem. Soc. Rev., 2010, 39, 3115–3141 RSC.
- M. Winter and J. O. Besenhard, Electrochemical Lithiation of Tin and Tin-Based Intermetallics and Composites, Electrochim. Acta, 1999, 45, 31–50 CrossRef CAS.
- H. Inoue, in International meeting on lithium batteries abstract #228, Biarritz, France, 2006 Search PubMed.
- J. Wolfenstine, J. L. Allen, J. Read and D. Foster, Chemistry and Structure of Sony's Nexelion Li-Ion Electrode Materials, DTIC Document, 2006 Search PubMed.
- Y. Idota, T. Kubota, A. Matsufuji, Y. Maekawa and T. Miyasaka, Tin-Based Amorphous Oxide: A High-Capacity Lithium-Ion-Storage Material, Science, 1997, 276, 1395–1397 CrossRef CAS.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Challenges in the Development of Advanced Li-Ion Batteries: A Review, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- C. Kim, et al., Fabrication of Electrospinning-Derived Carbon Nanofiber Webs for the Anode Material of Lithium-Ion Secondary Batteries, Adv. Funct. Mater., 2006, 16, 2393–2397 CrossRef CAS.
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novak, Insertion Electrode Materials for Rechargeable Lithium Batteries, Adv. Mater., 1998, 10, 725–763 CrossRef CAS.
- M. Inagaki, Y. Yang and F. Kang, Carbon Nanofibers Prepared Via Electrospinning, Adv. Mater., 2012, 24, 2547–2566 CrossRef CAS PubMed.
- B. Zhang, F. Kang, J.-M. Tarascon and J.-K. Kim, Recent Advances in Electrospun Carbon Nanofibers and Their Application in Electrochemical Energy Storage, Prog. Mater. Sci., 2016, 76, 319–380 CrossRef CAS.
- Y. Wu, M. Reddy, B. Chowdari and S. Ramakrishna, Long-Term Cycling Studies on Electrospun Carbon Nanofibers as Anode Material for Lithium Ion Batteries, ACS Appl. Mater. Interfaces, 2013, 5, 12175–12184 CAS.
- L. Ji and X. Zhang, Electrospun Carbon Nanofibers Containing Silicon Particles as an Energy-Storage Medium, Carbon, 2009, 47, 3219–3226 CrossRef CAS.
- C. Bonino, et al., Electrospun Carbon–Tin Oxide Composite Nanofibers for Use as Lithium Ion Battery Anodes, ACS Appl. Mater. Interfaces, 2011, 3, 2534–2542 CAS.
- M. K. Dufficy, S. A. Khan and P. S. Fedkiw, Hierarchical Graphene-Containing Carbon Nanofibers for Lithium-Ion Battery Anodes, ACS Appl. Mater. Interfaces, 2016, 8, 1327–1336 CAS.
- C. Tang, A. E. Ozcam, B. Stout and S. A. Khan, Effect of Ph on Protein Distribution in Electrospun Pva/Bsa Composite Nanofibers, Biomacromolecules, 2012, 13, 1269–1278 CrossRef CAS PubMed.
- H. Fong, I. Chun and D. Reneker, Beaded Nanofibers Formed During Electrospinning, Polymer, 1999, 40, 4585–4592 CrossRef CAS.
- J. Wang, W.-L. Song, Z. Wang, L.-Z. Fan and Y. Zhang, Facile Fabrication of Binder-Free Metallic Tin Nanoparticle/Carbon Nanofiber Hybrid Electrodes for Lithium-Ion Batteries, Electrochim. Acta, 2015, 153, 468–475 CrossRef CAS.
- M. Rahaman, A. F. Ismail and A. Mustafa, A Review of Heat Treatment on Polyacrylonitrile Fiber, Polym. Degrad. Stab., 2007, 92, 1421–1432 CrossRef CAS.
- B. Saha and G. C. Schatz, Carbonization in Polyacrylonitrile (Pan) Based Carbon Fibers Studied by Reaxff Molecular Dynamics Simulations, J. Phys. Chem. B, 2012, 116, 4684–4692 CrossRef CAS PubMed.
- S. Yang, H. Song and X. Chen, Nanosized Tin and Tin Oxides Loaded Expanded Mesocarbon Microbeads as Negative Electrode Material for Lithium-Ion Batteries, J. Power Sources, 2007, 173, 487–494 CrossRef CAS.
- G. Zhang and M. Liu, Preparation of Nanostructured Tin Oxide Using a Sol–Gel Process Based on Tin Tetrachloride and Ethylene Glycol, J. Mater. Sci., 1999, 34, 3213–3219 CrossRef CAS.
- S. Ye and A. K. Vijh, A New Fuel Cell Electrocatalyst Based on Carbonized Polyacrylonitrile Foam the Nature of Platinum-Support Interactions, J. Electrochem. Soc., 1997, 144, 90–95 CrossRef CAS.
- L. G. Bulusheva, et al., Electrochemical Properties of Nitrogen-Doped Carbon Nanotube Anode in Li-Ion Batteries, Carbon, 2011, 49, 4013–4023 CrossRef CAS.
- S. V. Nand, K. Ankur, K. Brijesh and M. B. Raj, Synthesis of Single Phase Cubic Tin Nitride Nanoparticles by Atmospheric Pressure–Halide Vapor Phase Epitaxy, Solid State Sci., 2008, 10, 569–572 CrossRef CAS.
- A. Othonos and M. Zervos, Carrier Relaxation Dynamics in Snxny Nanowires Grown by Chemical Vapor Deposition, J. Appl. Phys., 2009, 106, 114303 CrossRef.
- X. Zhou, L. J. Wan and Y. G. Guo, Binding SnO2 Nanocrystals in Nitrogen-Doped Graphene Sheets as Anode Materials for Lithium-Ion Batteries, Adv. Mater., 2013, 25, 2152–2157 CrossRef CAS PubMed.
- Z. Zhu, et al., Ultrasmall Sn Nanoparticles Embedded in Nitrogen-Doped Porous Carbon as High-Performance Anode for Lithium-Ion Batteries, Nano Lett., 2013, 14, 153–157 CrossRef PubMed.
- Y.-J. Kim, H. Lee and H.-J. Sohn, Lithia Formation Mechanism in Tin Oxide Anodes for Lithium-Ion Rechargeable Batteries, Electrochem. Commun., 2009, 11, 2125–2128 CrossRef CAS.
- I. Courtney, J. Tse, O. Mao, J. Hafner and J. Dahn, Ab Initio Calculation of the Lithium–Tin Voltage Profile, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 15583 CrossRef CAS.
- Z. Chen, et al., In Situ Generation of Few-Layer Graphene Coatings on SnO2-Sic Core–Shell Nanoparticles for High-Performance Lithium-Ion Storage, Adv. Energy Mater., 2012, 2, 95–102 CrossRef CAS.
- Y. Chen, B. Song, R. M. Chen, L. Lu and J. Xue, A Study of the Superior Electrochemical Performance of 3 Nm SnO2 Nanoparticles Supported by Graphene, J. Mater. Chem. A, 2014, 2, 5688–5695 CAS.
- D.-K. Kim, et al., Electrospun Polyacrylonitrile-Based Carbon Nanofibers and Their Hydrogen Storages, Macromol. Res., 2005, 13, 521–528 CrossRef CAS.
- I. A. Courtney and J. Dahn, Electrochemical and in Situ X-Ray Diffraction Studies of the Reaction of Lithium with Tin Oxide Composites, J. Electrochem. Soc., 1997, 144, 2045–2052 CrossRef CAS.
- X. Zhang, X. Fan, C. Yan, H. Li, Y. Zhu, X. Li and L. Yu, Interfacial microstructure and properties of carbon fiber composites modified with graphene oxide, ACS Appl. Mater. Interfaces, 2012, 4, 1543–1552 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26371b |
| This journal is © The Royal Society of Chemistry 2017 |