
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic activity of transition metal doped Cu(111) surfaces for ethanol synthesis from acetic acid hydrogenation: a DFT study
Minhua Zhang ab,
Rui Yaoab,
Haoxi Jiang*ab,
Guiming Liab and
Yifei Chenab
ab,
Rui Yaoab,
Haoxi Jiang*ab,
Guiming Liab and
Yifei Chenab
aKey Laboratory for Green Chemical Technology of Ministry of Education, R&D Center for Petrochemical Technology, Tianjin University, Tianjin 300072, PR China. E-mail: hxjiang@tju.edu.cn; Fax: +86-22-27406119; Tel: +86-22-27406119
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, PR China
First published on 5th January 2017
Abstract
Transition metal (Co, Ni, Ru, Rh, Pd and Pt) doped Cu(111) models are selected to examine the effects of transition metals on Cu surface for ethanol synthesis from acetic acid hydrogenation using density functional theory (DFT) calculations. On these surfaces, the adsorption of the main intermediates and reaction barriers of key elementary steps are investigated. The calculation results indicate that oxophilic metals are projected to be more active in acetic acid adsorption and acetaldehyde adsorption compared to less-oxophilic metals. Those metals with larger C adsorption energies generally have better C–OH bond cracking activity. Additionally, a good linear Brønsted–Evans–Polanyi (BEP) correlation is established for predicting the preferences of C–OH bond scission of acetic acid on other metals. Finally, O–H bond formation in C2-oxygenates (CH3CO, CH3CHO, CH3CH2O) hydrogenation is examined on all these surfaces. The reactions are more likely to occur on less-oxophilic metal-doped Cu surfaces. Therefore, it appears to involve an intricate balance between C–OH cracking and O–H bond formation reactions. That means those metal-doped Cu-based catalysts that are capable of preferentially activating C–OH bond without considerably inhibiting O–H bond formation of C2-oxygenates are predicted to achieve optimum catalytic activity for ethanol synthesis from acetic acid hydrogenation. The results can provide theoretical guidance for related experiments as well as the designing of Cu-based catalysts for ethanol synthesis.
1. Introduction
Acetic acid, as one of the main short chain fatty acids, can be produced by anaerobic fermentation or pyrolysis of biomass.1 At the same time, the syngas route has also become an alternative route for acetic acid production. In recent years, the overcapacity in the acetic acid industry has brought about sustained decreases in the acetic acid price.2,3 Therefore, it is imperative to purposely synthesize more valuable chemicals using acetic acid as a raw material. The hydrogenation of acetic acid (CH3COOH + 2H2 → CH3CH2OH + H2O) is an alternative route for ethanol production and has not been systematically investigated hitherto. Ethanol has been regarded as a potential carbon-neutral fuel source over the past decades and also can be used for producing many value-added chemicals.4,5 In addition, ethanol has received wide attentions as a substitute for a series of biomass-derived molecules.6 Above all, it is rather meaningful of transferring acetic acid to ethanol through consecutive hydrogenation reactions from an economical and feasible point of view.Currently, the catalysts applied in the process of acetic acid hydrogenation to ethanol are mainly noble metals, including Pt, Pd and Ru, alloyed with a second metal (such as Co, Ni and Cu) as promoter. These noble metal-based catalysts have been proved to show excellent catalytic performance and some have realized industrialization in many countries in recent years. However, the underlying reaction mechanism of the hydrogenation of acetic acid to ethanol has not been investigated profoundly. Only a few researchers have focused on this issue recently. Rachmady et al.7 applied Pt/TiO2 catalyst for acetic acid hydrogenation to ethanol and acquired a selectivity of 70%. They proposed that the reaction take place via adsorbed hydrogen atoms on metal and acetyl groups on TiO2 support rather than acetic acid or acetate groups and the product ethanol mainly forms from acetaldehyde. Alcala et al.8 compared acetic acid conversion over silica-supported Pt and Pt–Sn catalysts. Their results presented that the addition of Sn can help inhibit C–O bond and C–C bond cleavage in oxygenated hydrocarbons. Compared with the main products CO, CH4 and C2H6 over Pt catalyst, the products on Pt–Sn catalyst are mainly ethanol, acetaldehyde and ethyl acetate. Pallassana and co-workers9 postulated the most preferable pathway of acetic acid hydrogenolysis to ethanol on Pd catalyst involves acetic acid dissociation to acetyl, followed by successive hydrogenation to acetaldehyde and ethanol. They suggested that the rate-determining step is C–OH bond activation. For Ru catalyst, Olcay and co-workers10,11 concluded that the high activity is attributed to its strong ability of activating the initial scission of C–OH bond.
Although the noble metal-based catalysts show nice catalytic performance, applications of such noble metal catalysts in industry might be greatly restricted because of their high price and limited resources. Therefore, it is not only economically-valuable, but also resource-saving, if we can exploit a cheaper and more active catalytic system for acetic acid hydrogenation compared to noble metals.
Copper, one of the non-noble metals, has been widely used in many hydrogenation reactions12–19 as an effective catalyst, such as CO or CO2 hydrogenation to methanol or ethanol, acetyl acetate hydrogenation to ethanol, and 2-furfuraldehyde hydrogenation. This means Cu-based catalysts have great potentials to be applied in the process of acetic acid hydrogenation. More importantly, it would significantly reduce the cost if the effective Cu-based catalysts are developed for ethanol synthesis from acetic acid. Onyestyák et al.20–22 investigated the performance of Cu-based for acetic acid hydrogenation, and their experiment results23–25 indicated that the main products are ethanol, acetyl acetate, acetaldehyde and water on pure Cu catalyst. However, the yield of ethanol is relatively low and it can be improved through doping oxide In2O3 due to the alloy phase Cu2In formation.
Although Cu catalyst has been proved to be an effective catalyst in hydrogenation processes, some researchers6,17,26 still believe that C–O bond and C–C bond scission of oxygenates might not occur easily on Cu surface compared to other noble metals. Dumesic et al.26 believed that compared with ethyl acetate dissociation to acetyl and acetate, acetic acid is more difficult to dissociate on Cu catalyst, which leads to lower ethanol yield with the main products ethanol, acetaldehyde and ethyl acetate. Olcay and co-workers10 also suggested that acetic acid conversion must involve C–OH bond scission followed by hydrogenation reactions, and the initial C–OH bond cleavage is the rate-determined step. Therefore, acetic acid dissociation to acetyl and hydroxyl is the indispensable descriptor for acetic acid conversion. This elementary step (CH3COOH → CH3CO + OH) is chosen as one of main concerns for screening alloyed Cu catalysts.
In addition, O–H bond formation for C1-oxygenates and C2-oxygenates was reported to be more difficult than C–H bond formation on Cu catalyst.14,15,17 However, O–H bond formation is an unavoidable step for acetic acid hydrogenation. Therefore, O–H bond formation is considered as another descriptor which could have considerable effects on the selectivity and yield of ethanol.
Alloying second noble metal is a promising way to promote the performance of non-noble metal catalysts.27–41 Various noble metals (Au, Pd, Rh, Pt, Ni) were doped on Cu(111) in the hydrogenation of CO2 to methanol, and Ni/Cu(111) was reported to have the highest methanol yield, followed by Pt, Rh, Pd and Au.18 Spivey et al.27 suggested the ethanol yield can be greatly enhanced on bimetallic catalysts because of synergistic effect. Zhao and co-workers35 reported that Rh-decorated Cu alloys show the lowest reaction barriers of CO insertion and the noticeable increase of the selectivity of C2-oxygenates through DFT calculations.
At present, there are few theoretical and experimental evidences about which metal can enhance the performance of Cu catalyst for acetic acid hydrogenation. So our work mainly focus on the effects of transition metal dopants into Cu surface for acetic acid hydrogenation to ethanol, which is conductive for us to screen potential Cu-based catalysts with excellent catalytic performance. So in this paper, we determined the mechanism of acetic acid hydrogenation to ethanol on the Cu(111) surface doped by other transition metals (Co, Ni, Ru, Rh, Pd and Pt) using DFT calculations. According to the descriptors mentioned above, this work is aimed to provide insights into the effects of doped-metals on the intermediates and transition states of key elementary steps.
2. Computational methods
All our DFT calculations of adsorption energies and reaction barriers were performed using Dmol3 module in Material Studio Package with the generalized gradient approximation (GGA) for the exchange and correlation functional in the form of Perdew–Burke–Ernzerhof (PBE).42,43 DFT semi-core pseudo-potential (DSPP) was set for the core treatment to balance calculation accuracy and computational efficiency and the basic set was expanded in terms of a double numerical plus polarization (DNP). The surface Brillouin zone was sampled by a 3 × 3 × 1 k-point mesh according to Monkhorst–Pack method and a Methfessel–Paxton smearing of 0.005 Ha was conducted to accelerate convergence. The convergence criteria included threshold values of 2 × 10−5 Ha for energy, 0.004 Ha Å−1 for maximum force, and 0.005 Å for maximum displacement, with the self-consistent-field (SCF) density convergence threshold value of 1 × 10−5 Ha. Spin polarization was also involved in our calculations for all metal surfaces.The Cu(111) surface was modelled using a three-layer slab with a (4 × 4) unit cell and only the top layer was allowed to relax while the two bottom layers were fixed in the optimized bulk position as literatures.18,26 A 15 Å of vacuum space between the periodic slabs was utilized to eliminate spurious interactions. Three Cu atoms on the Cu(111) surface were substituted by other transition metals in each unit cell as alloy models, which means the corresponding coverage is 3/16 ML. It has been proved that among all the dopants, Pd, Pt favor to stay in the Cu surface, while Ni, Co, Ru and Rh prefer the bulk.44 However, we believe that the effect of the experimental environment should also be considered. According to the calculations from Fu,45 they concluded that under the hydrogen-rich environments, most of the alloyed metal atoms (except Fe), can stably occupy the surface layer. And also, according to the study by Greeley et al.,46 adsorbed hydrogen on the surface can draw the metal atom to the surface layer of the alloy system under the actual conditions of hydrogenation. Similarly, according to the experiments by Chorkendorff et al.47 for CO hydrogenation to CH3OH, Ni atoms can be pulled out to the surface by the active adsorbates such as CO. Therefore, the surface segregation between doped transition metals and Cu was not included in our present study.
The transition state (TS) searches of elementary reactions were carried out at the same theoretical level as those for the optimization of reactants and products with complete linear synchronous transit/quadratic synchronous transit (LST/QST) method.48,49 Each TS structure was confirmed by the vibrational analysis with only one imaginary frequency along the reaction coordinate. Zero-point energy corrections (ZPE) were considered with harmonic oscillator approach based on the calculations of vibrational frequencies.
The adsorption energies of all species on metal surfaces were calculated according to the following formula:
| Eads = ECu/M + Eadsorbates − Eadsorbates-Cu/M |
The activation barriers and reaction energies are defined as:
| Ea = E(TS) − E(IS) |
| ΔE = E(FS) − E(IS) |
3. Results and discussion
In the present study, DFT calculations were performed to study the transition states (TS) and intermediates of acetic acid hydrogenation to ethanol on pure Cu and Cu–metal alloys for the first time. In order to figure out proper transition metals which can have positive effects on Cu catalysts, six kinds of transition metals (Co, Ni, Ru, Rh, Pd and Pt) are doped into Cu(111) surface as representative models. The two descriptors mentioned above on different metal-doped Cu surfaces are calculated and discussed in detail in the following sections, and those on pure Cu surface are also examined for comparison.3.1 Acetic acid scission
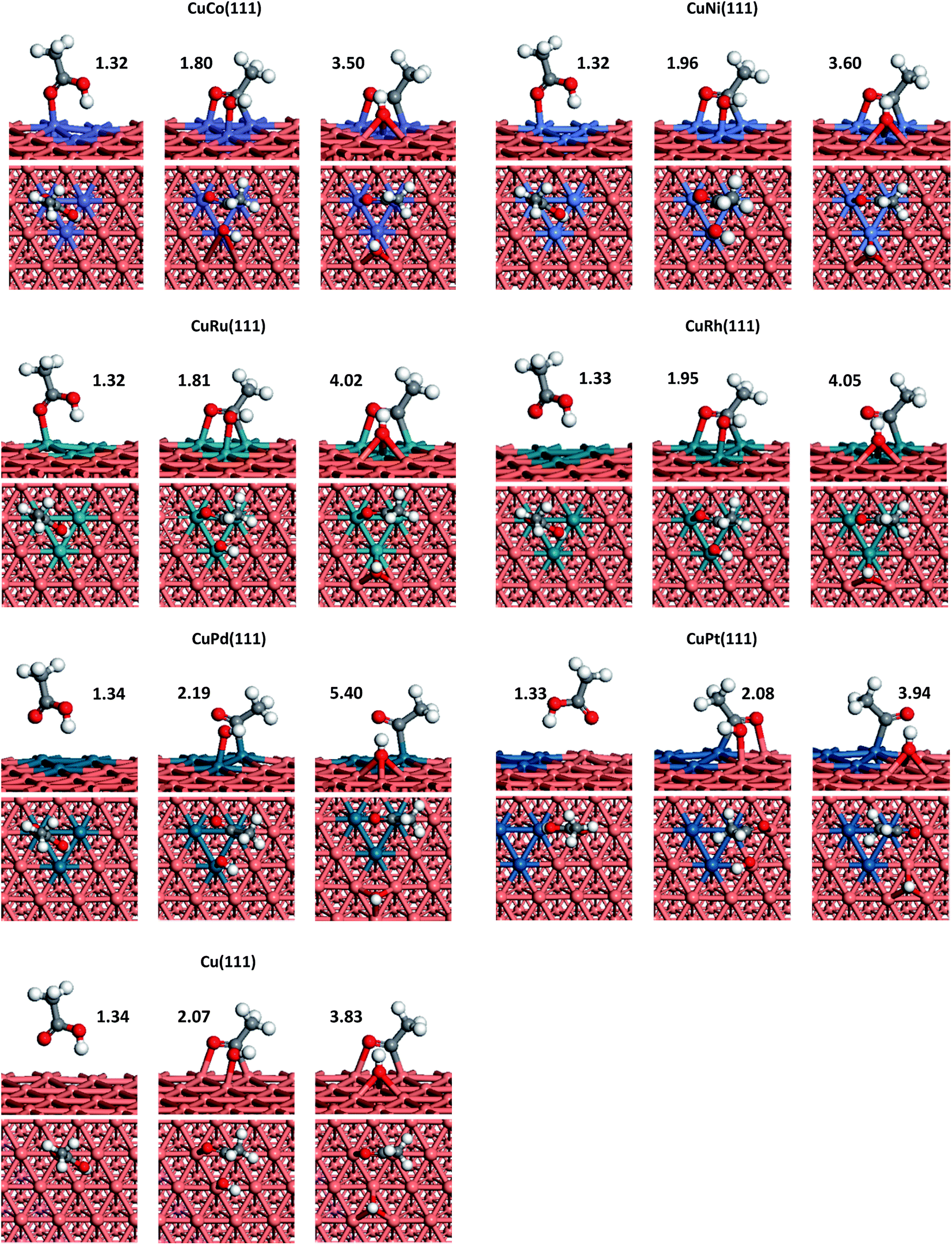 | ||
| Fig. 1 Optimized configurations of the initial states (IS, left), transition states (TS, central) and final states (FS, right) for CH3COOH → CH3CO + OH reaction on Cu(111) and six doped-metal Cu(111) surfaces. C, O, H, and Cu atoms are shown in grey, red, white and orange balls, respectively. The length of C–OH is labelled and given in Å. | ||
| Surface | Adsorption energy | CH3COOH → CH3CO + OH | |
|---|---|---|---|
| (CH3COOH) | Reaction barrier (Ea) | Reaction energy (ΔE) | |
| CuCo(111) | 0.84 | 0.14 | −0.38 |
| CuNi(111) | 0.47 | 0.55 | 0.08 |
| CuRu(111) | 0.75 | 0.66 | −0.09 |
| CuRh(111) | 0.46 | 0.74 | 0.24 |
| CuPd(111) | 0.32 | 1.47 | 0.74 |
| CuPt(111) | 0.37 | 1.23 | 0.59 |
| Cu(111) | 0.30 | 1.53 | 0.92 |
We can see from Fig. 1 that CH3COOH prefer to adsorb on Cu(111), CuCo(111), CuNi(111), CuRu(111), CuRh(111) and CuPd(111) surfaces with one O(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) residing on top of a Cu or doped-metal atom, while on CuPt(111) surface, O atom (CO) is more likely to adsorb at the top of Cu atom which is adjacent to a Pt atom. The adsorption energies of acetic acid on Co, Ni, Ru, Rh, Pd and Pt doped Cu(111) are 0.84, 0.47, 0.75, 0.46, 0.32 and 0.37 eV, respectively. Compared with that on Cu(111) surface, additions of Co and Ru are able to significantly increase acetic acid adsorption energy. Meanwhile, Ni and Rh can also enhance the adsorption energy to a certain extent. However, doping Pt and Pd into Cu(111) is proved to be not effective to enhance the adsorption. Above result means more acetic acid species can be adsorbed on CuCo and CuRu surfaces.
O) residing on top of a Cu or doped-metal atom, while on CuPt(111) surface, O atom (CO) is more likely to adsorb at the top of Cu atom which is adjacent to a Pt atom. The adsorption energies of acetic acid on Co, Ni, Ru, Rh, Pd and Pt doped Cu(111) are 0.84, 0.47, 0.75, 0.46, 0.32 and 0.37 eV, respectively. Compared with that on Cu(111) surface, additions of Co and Ru are able to significantly increase acetic acid adsorption energy. Meanwhile, Ni and Rh can also enhance the adsorption energy to a certain extent. However, doping Pt and Pd into Cu(111) is proved to be not effective to enhance the adsorption. Above result means more acetic acid species can be adsorbed on CuCo and CuRu surfaces.
CH3COOH → CH3CO + OH. CH3CH2OH synthesis from acetic acid and H2 is limited by the higher reaction barrier of CH3COOH dissociation and this reaction is the rate-determining step on Cu catalyst according to some studies mentioned above. In addition, the activation energy of acetic acid dissociation to acetyl on pure Cu surface is 1.53 eV though our calculations, which means this step is more difficult to occur on Cu under normal experimental conditions. Therefore, for the sake of improving the catalytic performance of Cu catalyst, the selected doped metals should have ability to reduce the reaction barrier of CH3COOH scission directly and hence improve CH3COOH conversion rate. Schematic representations of transition states (TSs) corresponding to the elementary reaction of CH3COOH → CH3CO + OH on each doped metal Cu(111) surface are also depicted in Fig. 1 and the reaction barriers and reaction energies on various metal-doped Cu surfaces are listed in Table 1. From Table 1, we can see that the reaction barriers for breaking C–OH bond of acetic acid on six different surfaces vary greatly. Co, Ni, Ru and Rh doped Cu surfaces, are considered to be more catalytically active than pure Cu(111), with reaction barriers 0.14, 0.55, 0.66, and 0.74 eV, while Pt only shows slight improvement for C–OH bond scission (1.23 eV). In addition, on CuCo(111), the reaction energy of this reaction is found to be the most exothermic, which is in consistent with its lowest reaction barrier among all metals. Interestingly, the adsorption energy of acetic acid on CuCo(111) surface is larger than its dissociation barrier, which means acetic acid is more likely to dissociate to acetyl and hydroxyl than to desorb from surface. However, on other metal-doped surfaces and pure Cu surface, acetic acid does not interact strongly enough with metal surfaces and tends to desorb from those into vapour phase.
| Atom | Adsorption energy (eV) | ||||||
|---|---|---|---|---|---|---|---|
| CuCo | CuNi | CuRu | CuRh | CuPd | CuPt | Cu | |
| O | 6.57 | 5.88 | 5.60 | 5.02 | 3.99 | 3.81 | 4.81 |
| C | 7.60 | 6.92 | 7.06 | 6.82 | 5.34 | 5.72 | 4.88 |
As can be seen from Table 2, metals with higher C adsorption energies usually possess higher O adsorption energies in general (except Cu(111) surface). In the meanwhile, we find that the ability of C–OH scission appears to have a strong linear relationship with C adsorption energy (shown in Fig. 2a). However, O adsorption energy seems to be weaker dependent on activation barrier, indicating that the O adsorption energy has a secondary effect (Fig. 2b). Therefore, those metals with higher C (O adsorption energies in most cases) adsorption energies show better activity than metals with weaker ones. The reason is that stronger C adsorption can significantly improve C–metal interaction, which can weaken C–OH bond. Similarly, the metals which have stronger O adsorption energy can also prompt O–metal bond formation to some extent. Besides, as shown in Fig. 1, the products of C–OH bond scission of CH3COOH are CH3CO and OH, and CH3CO prefer to adsorb on metal surfaces via both C and O(CO) atoms, while OH favours to adsorb on metal surfaces with O atom. Therefore, on metals with higher C and even O adsorption energies, the co-adsorption of the terminal CH3CO and OH species is more stable and thus the reverse action is less likely to occur. From another perspective, the strong C–metal interactions on these metal surfaces can withdraw electrons from C–OH bond and result in weakened C–OH bond. Herewith, it is not surprising that Co Ni, Ru, and even Rh with stronger C–metal and also O–metal interactions can enhance C–OH bond scission to varying degrees.
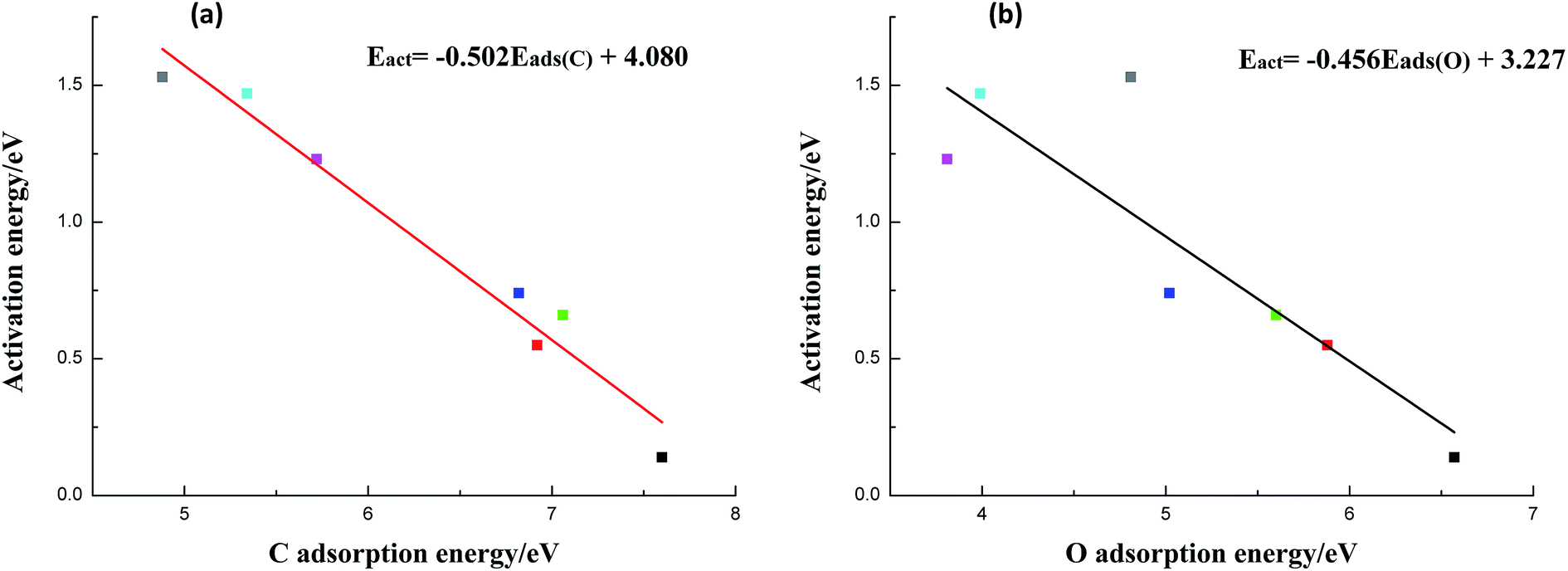 | ||
| Fig. 2 Correlations between reaction barriers for the CH3COOH → CH3CO + OH reaction and: (a) adsorption energies of atomic C, (b) adsorption energies of atomic O on Cu(111) and six metal-doped Cu(111) surfaces (black: CuCo(111), red: CuNi(111), green: CuRu(111), dark blue: CuRh(111), pink: CuPt(111), light blue: CuPd(111) and grey: Cu(111)). | ||
In this section, the species-level BEP correlation is established to describe the relation between reaction energies and activation barriers for C–OH bond scission reactions on pure Cu and transition metal-doped Cu surfaces. As presented in Fig. 3, it is clear that there is a good linear relationship for C–OH bond cracking reactions of acetic acid on pure Cu and six metal-doped Cu surfaces. The correlation line whose fitted parameter is R2 = 0.951 and slope is 1.079, is displayed in Fig. 3. Given that C–OH bond dissociation is an indispensable step for ethanol formation from acetic acid, current BEP correlation can be applied easily and directly to estimate the reaction barriers on other metal-doped Cu surfaces for C–OH bond scission of acetic acid when the adsorption configurations of reactants and products are similar qualitatively. It is so meaningful to examine a wide range of metals by only calculating adsorption energies of CH3COOH and co-adsorption energies of CH3CO and OH without determining transition states.
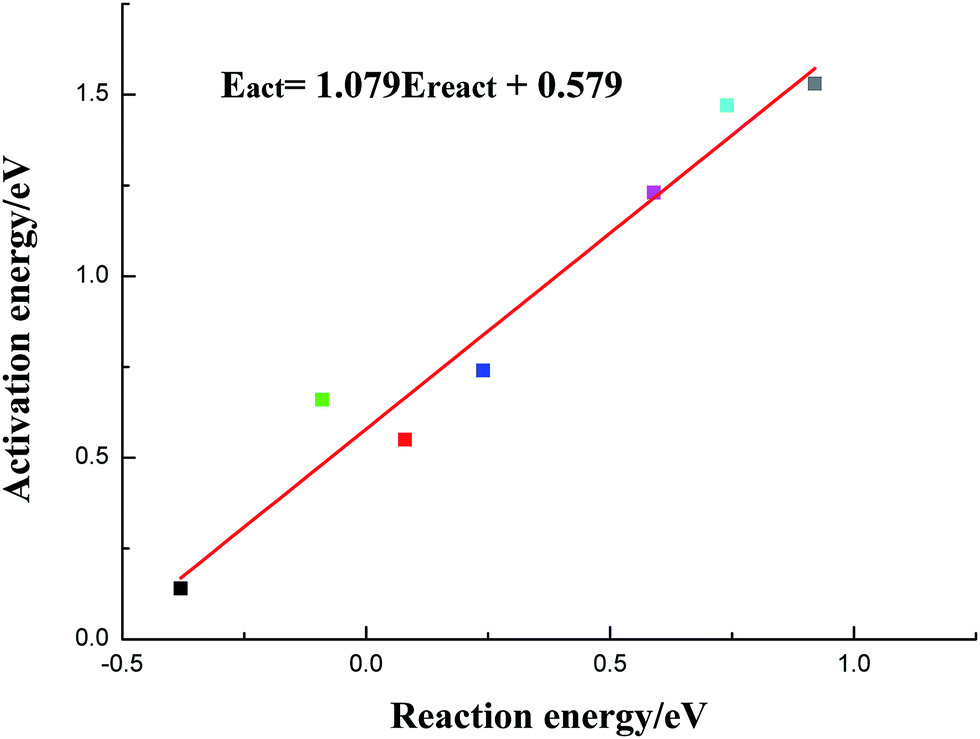 | ||
| Fig. 3 Brønsted–Evans–Polanyi correlation for C–OH scission reaction of acetic acid on Cu(111) and six metal-doped Cu(111) surfaces (black: CuCo(111), red: CuNi(111), green: CuRu(111), dark blue: CuRh(111), pink: CuPt(111), light blue: CuPd(111) and grey: Cu(111)). | ||
3.2 O–H bond formation of C2-oxygenates
The most stable adsorption configurations and adsorption energies of CH3CHO on pure Cu and doped-metal Cu surfaces are shown in Fig. 4 and Table 3. CH3CHO binds to CuNi(111) surface in the bidentate configuration with O atom residing on top of one Ni atom and H atom residing on top site of neighbouring Ni atom. Likewise, the same case could be adopted on other metal-doped surfaces except on CuPt(111). The CH3CHO adsorption energies on CuNi, CuCo, CuRu, CuRh and CuPd surfaces are 0.36, 0.71, 0.58, 0.36 and 0.21 eV, respectively. In contrast, CH3CHO is preferentially adsorb on CuPt(111) via O atom near Cu top site and H atom on the adjacent Pt atom with adsorption energy 0.20 eV.
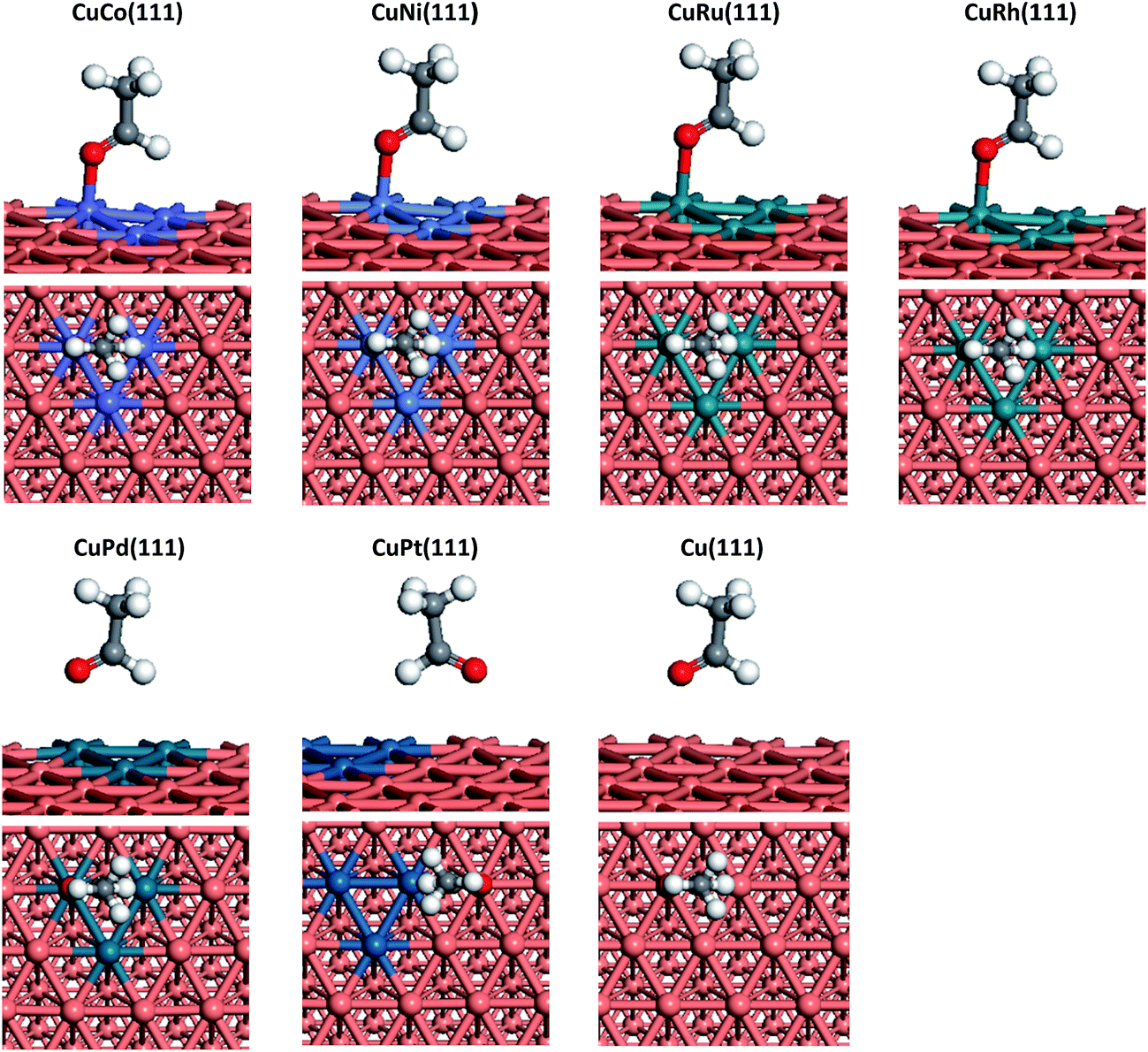 | ||
| Fig. 4 Optimized adsorption configurations of CH3CHO on Cu(111) and six doped-metal Cu(111) surfaces. | ||
| Surface | Adsorption energy (eV) | ||
|---|---|---|---|
| CH3CO | CH3CHO | CH3CH2O | |
| CuCo(111) | 2.92 | 0.71 | 3.37 |
| CuNi(111) | 2.32 | 0.36 | 2.87 |
| CuRu(111) | 2.69 | 0.58 | 2.64 |
| CuRh(111) | 2.37 | 0.36 | 2.27 |
| CuPd(111) | 1.69 | 0.21 | 1.78 |
| CuPt(111) | 1.91 | 0.20 | 1.27 |
| Cu(111) | 1.37 | 0.22 | 2.30 |
Similar to CH3COOH adsorption, compared with the pure Cu(111) surface, doping of Co and Ru could greatly enhance CH3CHO adsorption. Rh, Ni also show a minor contribution, while Pd and Pt have no positive effects on that.
CH3CO + H → CH3COH. As described above, CH3CHO can easily desorb from Cu surface and serves as a by-product. However, compared with CH3COH, CH3CHO is more likely to generate from CH3CO hydrogenation because of its low barrier as identified by others.15 Therefore, if CH3COH species formation from CH3CO can be promoted, ethanol selectivity can be facilitated.
The most stable adsorption on pure Cu and all doped-metal Cu surfaces is tested, and the adsorption energies of CH3CO are listed in Table 3. Similar to Cu(111), CH3CO prefer to adsorb on the CuNi(111), CuCo(111), CuRu(111), CuRh(111), CuPd(111) surfaces via O atom on top site of doped metals and α-C atom at neighbouring top site. While on CuPt(111), O atom more likely to adsorb on top site of Cu atom rather than Pt atom. The reaction barriers for CH3CO hydrogenation to CH3COH on Ni-, Co-, Ru-, Rh-, Pd- and Pt-doped surfaces are 1.09, 1.09, 0.62, 0.76, 0.81 and 0.90 eV, respectively. Nevertheless, comparing with that on pure Cu surface (0.73 eV), only Ru can improve O–H bond formation. Other metals have no promotion effect (Pd and Rh) or even inhibit this elementary step (Pt, Ni and Co).
CH3CHO + H → CH3CHOH. We examined the reaction barriers of CH3CHOH formation from CH3CHO on Cu(111) and six metal-doped Cu(111) surfaces as displayed in Table 4. The reaction barriers are 0.65 eV for CuNi(111), 0.85 eV for CuCo(111), 0.82 eV for CuRu(111), 0.52 eV for CuRh(111), 0.50 eV for CuPd(111) and 0.45 eV for CuPt(111), respectively. From these data, we can conclude the trend that less-oxophilic metals (Pt and Pd) might have better performance on this elementary step.
| Surfaces | CH3CO + H → CH3COH | CH3CHO + H → CH3CHOH | CH3CH2O + H → CH3CH2OH | |||
|---|---|---|---|---|---|---|
| Reaction barrier (Ea) | Reaction energy (ΔE) | Reaction barrier (Ea) | Reaction energy (ΔE) | Reaction barrier (Ea) | Reaction energy (ΔE) | |
| CuCo(111) | 1.09 | 0.79 | 0.85 | 0.19 | 1.69 | 1.21 |
| CuNi(111) | 1.09 | 0.55 | 0.65 | 0.17 | 1.46 | 0.92 |
| CuRu(111) | 0.62 | 0.24 | 0.82 | 0.27 | 1.30 | 0.60 |
| CuRh(111) | 0.76 | 0.19 | 0.52 | 0.03 | 1.10 | 0.22 |
| CuPd(111) | 0.81 | 0.44 | 0.50 | 0.10 | 0.59 | −0.35 |
| CuPt(111) | 0.90 | 0.28 | 0.45 | 0.01 | 0.19 | −0.80 |
| Cu(111) | 0.73 | 0.41 | 0.93 | −0.08 | 1.11 | 0.13 |
CH3CH2O + H → CH3CH2OH. CH3CH2O is an important intermediate on Cu catalyst in acetic acid hydrogenation process since the reaction barrier of CH3CH2O hydrogenation to CH3CH2OH is high (1.11 eV), and the ethoxy adsorption is relatively strong (2.30 eV for adsorption energy), which means CH3CH2O species can be accumulated on Cu surface, the adsorption energies of CH3CH2O and reaction barriers of CH3CH2O hydrogenation are calculated and the results are listed in Tables 3 and 4.
CH3CH2O preferentially adsorbs on metal-doped Cu surfaces through O binding to doped-metals on hollow sites with C–C axis almost parallel to the surface. The only exception is CuPt(111), on which CH3CH2O adsorb on bridge site between two Pt atoms via O atom. The reaction barriers of CH3CH2O hydrogenation to CH3CH2OH are 1.69 eV for CuCo(111), 1.46 eV for CuNi(111), 1.30 eV for CuRu(111), 1.10 eV for CuRh(111), 0.59 eV for CuPd(111) and 0.19 eV for CuPt(111) in such a descending order. Compared with that on pure Cu surface (1.11 eV), Co, Ni, Ru and Rh can lead to CH3CH2O species adsorb more stably on Cu surface via O, resulting in more difficult hydrogenation on O atom of ethoxy species. In contrast, less-oxyphilic metal-doped surfaces, such as Pd and Pt, can greatly enhance the ability of ethoxy hydrogenation (CuPt and CuPd also exhibit high exothermicity).
The key descriptors over transition metal-doped Cu(111) surfaces and Cu(111) surface are summarized in Fig. 5. It can be concluded that weak adsorbates CH3COOH and CH3CHO can be stabilized significantly by oxophilic metals, especially for the case of dopants of Co and Ru. Less-oxyphilic metals (like Pt and Pd) seems to have negative effects on adsorption of CH3COOH and CH3CHO, while Ni and Rh have a certain promoting effect.
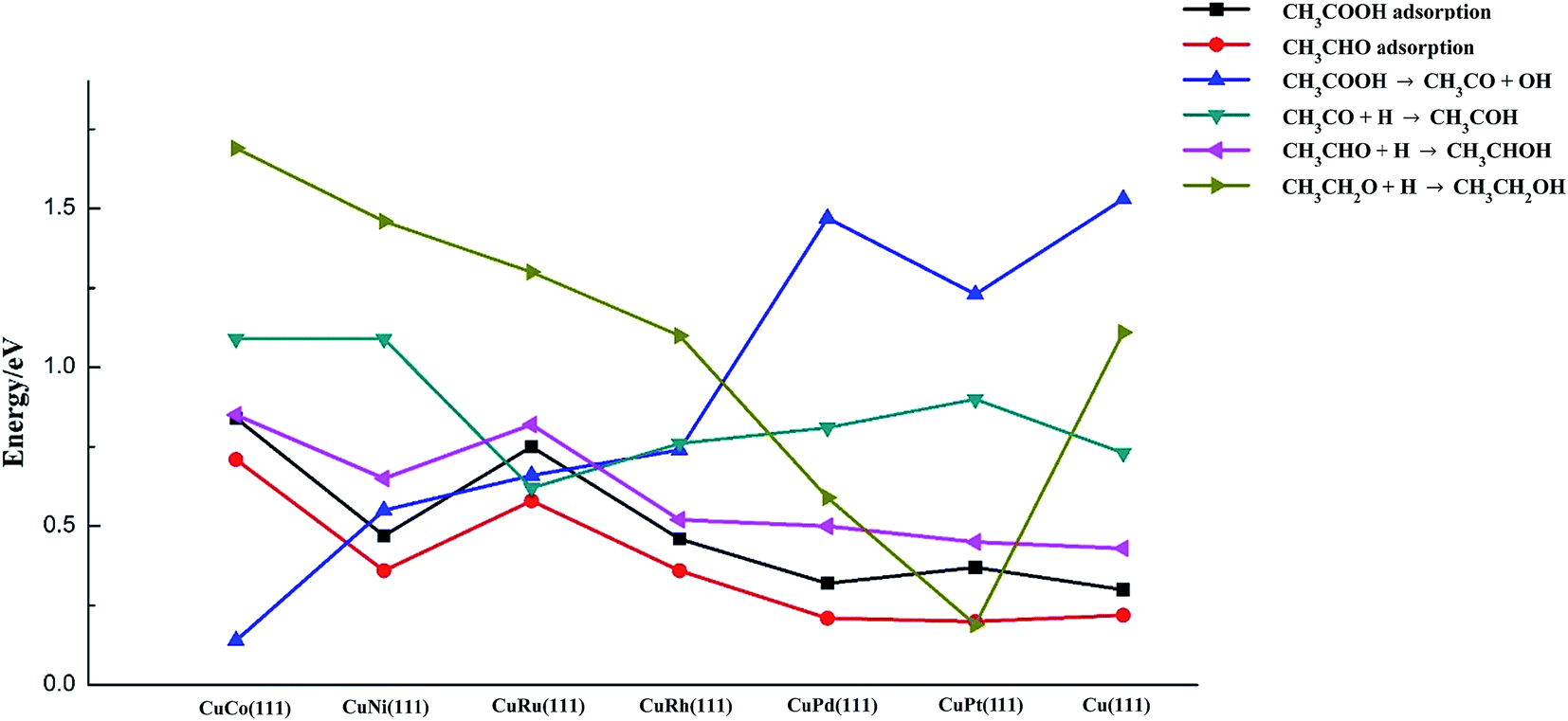 | ||
| Fig. 5 The key descriptors for acetic acid hydrogenation to ethanol on Cu(111) and six metal-doped Cu(111) surfaces. | ||
CH3COOH disassociation to CH3CO and OH is the most indispensable reaction on Cu catalyst since it might considerably influence ethanol productivity. The dopants of Co, Ni, Ru and Rh into Cu surface can activate C–OH bond of acetic acid greatly, and the most oxophilic metal Co shows the highest activity.
As for C2-oxygenates hydrogenation reactions (O–H bond formation), in a general way, those metals with stronger oxophilicity are more likely to strengthen oxygenates via O atom, resulting in more difficult formation of O–H bond for C2-oxygenates. However, the addition of less-oxophilic metals (Pt, Pd and even Rh) might lower the reaction barriers of O–H bond formation.
Therefore, optimum catalytic activity of Cu-based catalysts can be projected to be achieved on those metals which are capable of preferentially activating C–OH bond without considerably inhibiting O–H bond formation of C2-oxygenates. We can see from Fig. 5 that CuCo and CuNi have more activity for C–OH bond cracking, while for O–H bond formation reactions of C2-oxygenates, the barriers on these metals are relatively high, even higher than those on pure Cu surface. In contrast, C2-oxygenates hydrogenation reactions are more likely to occur on CuPd and CuPt, but C–OH cleave ability is so fragile on these surfaces. Therefore, it seems like CuRh and CuRu possess moderate C–OH bond scission barriers and three C2-oxygenates hydrogenation barriers, which means CuRh and CuRu are likely to possess better performance for ethanol synthesis from acetic acid hydrogenation (between these two metals, CuRh has slightly lower barriers for O–H bond formation reactions, while CuRu has stronger adsorption for acetic acid and acetaldehyde as well as lower C–OH bond breaking barrier). Olcay et al.10 reported findings of experimental study on the aqueous-phase hydrogenation of acetic acid catalyzed by transition metals such as Ru, Rh, Pd, Ni, Cu, Ir, and Pt, and the result indicated that Ru and Rh exhibit better performance for acetic acid hydrogenation to ethanol compared to other metals. Their result might be a support for our conclusion in a certain degree.
The species-level BEP correlations for each C2-oxygenate (CH3CO, CH3CHO and CH3CH2OH) hydrogenation reaction are also developed, as presented in Fig. 6 with corresponding parameters listed in Table 5. More than that, all the points for three hydrogenations reactions of C2-oxygenates on Cu(111) and metal-doped Cu(111) surfaces are regressed together to develop an all-metal BEP relationship for O–H bond formation (“universal” BEP correlation for O–H). The slope is 0.749 with intercept 0.644 and R2 0.744, which indicates that O–H formation steps of C2-oxygenates show a tendency to have the mid-to-late transition states.6,53 Similarly, these correlations can also be used to achieve more insights into the chemical trends of these elementary steps on a series of metals, and meanwhile, it can provide a reference for further studies and can be consummated with regressing more calculation data.
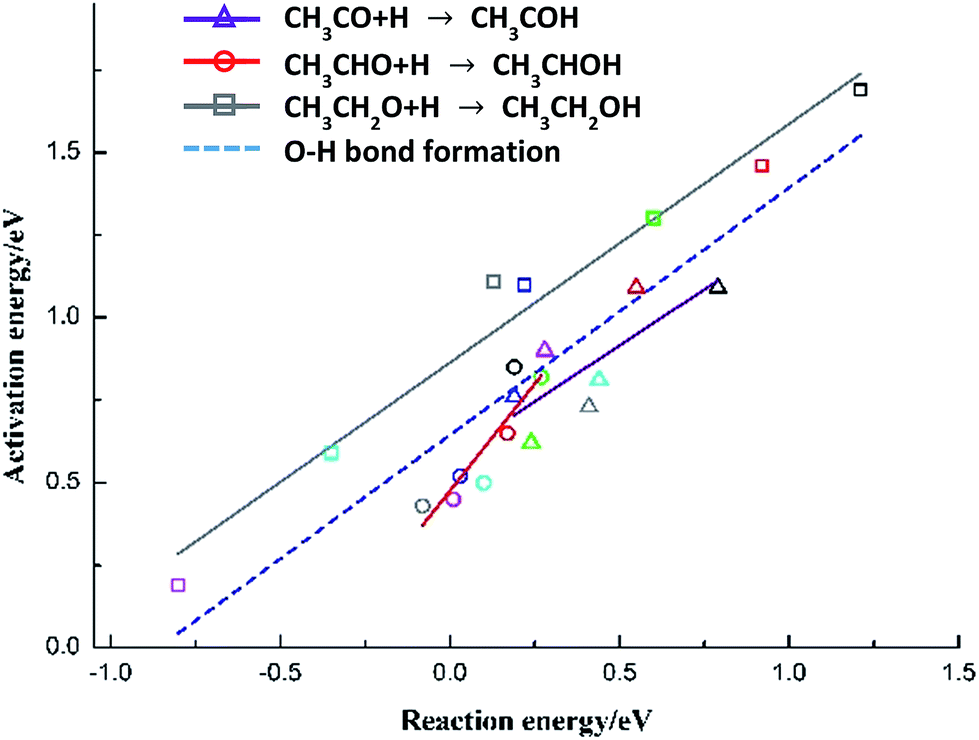 | ||
| Fig. 6 Brønsted–Evans–Polanyi correlations for O–H bond formation reactions of C2-oxygenates on Cu(111) and six metal-doped Cu(111) surfaces (black: CuCo(111), red: CuNi(111), green: CuRu(111), dark blue: CuRh(111), pink: CuPt(111), light blue: CuPd(111) and gray: Cu(111)). | ||
| Reaction | Slope (eV) | Intercept (eV) | R2 |
|---|---|---|---|
| CH3CO + H → CH3COH | 0.678 ± 0.241 | 0.575 ± 0.110 | 0.537 |
| CH3CHO + H → CH3CHOH | 1.296 ± 0.281 | 0.475 ± 0.041 | 0.771 |
| CH3CH2O + H → CH3CH2OH | 0.723 ± 0.056 | 0.864 ± 0.039 | 0.966 |
| O–H formation of C2-oxygenates | 0.749 ± 0.097 | 0.644 ± 0.048 | 0.744 |
4. Conclusions
In the present work, in order to investigate the effects of transition metal dopants on the catalytic activity of Cu(111) surface toward ethanol synthesis via acetic acid hydrogenation, the adsorption of key species and key elementary reactions on pure Cu and six metal-doped Cu surfaces were calculated using density functional theory. The performance of different doped transition metals for C–OH bond scission of acetic acid was systematically investigated. The results indicated that oxophilic metals, such as Co, Ni, Ru, are projected to greatly enhance acetic acid adsorption and C–OH bond scission. The activation energies for C–OH cracking reactions generally decline with increasing C adsorption energies, while O adsorption energy shows a second effect, which means the cleavage of C–OH bond in acetic acid is more facile on oxophilic metal-doped surfaces (especially Co, Ru and Ni). The BEP correlation for C–OH cracking reaction on all metal-doped Cu surfaces showed a good relation between activation energies and reaction energies. It is meaningful for us to test the activity of other Cu–metal surfaces only with adsorption energy calculations of reactants and products. CH3CHO adsorption and the reaction barriers of O–H bond formation of C2-oxygenates are also examined. Metals with more oxophilicity (Co and Ni) could enhance CH3CHO adsorption but inhibit O–H bond formation.In conclusion, those metal-doped Cu-based catalysts which are capable of preferentially activating C–OH bond without considerably inhibiting O–H bond formation of C2-oxygenates, are predicted to achieve optimum catalytic activity for ethanol formation from acetic acid hydrogenation (like Ru and Rh). This work can provide helpful and theoretical guidance for experimental studies and designing of proper Cu-based catalysts for acetic acid hydrogenation to ethanol.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21406159).References
- H. N. Chang, N. Kim, J. Kang and C. M. Jeong, Biotechnol. Bioprocess Eng., 2010, 15, 1–10 CrossRef CAS
.
- W. Chen, Y. Ding, D. Jiang, T. Wang and H. Luo, Catal. Commun., 2006, 7, 559–562 CrossRef CAS
- B. Q. Xu and W. Sachtler, J. Catal., 1998, 180, 194–206 CrossRef CAS
- N. Bion, D. Duprez and F. Epron, ChemSusChem, 2012, 5, 76–84 CrossRef CAS PubMed
- A. Bshish, Z. Yaakob, B. Narayanan, R. Ramakrishnan and A. Ebshish, Chem. Pap., 2011, 65, 251–266 CAS
- J. E. Sutton and D. G. Vlachos, Ind. Eng. Chem. Res., 2015, 54, 4213–4225 CrossRef CAS
- W. Rachmady and M. A. Vannice, J. Catal., 2000, 192, 322–334 CrossRef CAS
- R. Alcala, J. W. Shabaker, G. W. Huber, M. A. Sanchez-Castillo and J. A. Dumesic, J. Phys. Chem. B, 2005, 109, 2074–2085 CrossRef CAS PubMed
- V. Pallassana and M. Neurock, J. Catal., 2002, 209, 289–305 CrossRef CAS
- H. Olcay, L. Xu, Y. Xu and G. W. Huber, ChemCatChem, 2010, 2, 1420–1424 CrossRef CAS
- H. Olcay, Y. Xu and G. W. Huber, Green Chem., 2014, 16, 911–924 RSC
- X. Sun, R. Zhang and B. Wang, Appl. Surf. Sci., 2013, 265, 720–730 CrossRef CAS
- J. Wang, X. Zhang, Q. Sun, S. Chan and H. Su, Catal. Commun., 2015, 61, 57–61 CrossRef CAS
- X. Xu, J. Su, P. Tian, D. Fu, W. Dai, W. Mao, W. Yuan, J. Xu and Y. Han, J. Phys. Chem. C, 2015, 119, 216–227 CAS
- H. Zheng, R. Zhang, Z. Li and B. Wang, J. Mol. Catal. A: Chem., 2015, 404–405, 115–130 CrossRef CAS
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS
- J. Wang, Y. Kawazoe, Q. Sun, S. Chan and H. Su, Surf. Sci., 2016, 645, 30–40 CrossRef CAS
- Y. Yang, M. G. White and P. Liu, J. Phys. Chem. C, 2012, 116, 248–256 CAS
- R. Zhang, G. Wang and B. Wang, J. Catal., 2013, 305, 238–255 CrossRef CAS
- S. Harnos, G. Onyestyák and J. Valyon, Appl. Catal., A, 2012, 439–440, 31–40 CrossRef CAS
- G. Onyestyák, S. Harnos and D. Kalló, Catal. Commun., 2012, 26, 19–24 CrossRef
- S. Harnos, G. Onyestyák and D. Kalló, Microporous Mesoporous Mater., 2013, 167, 109–116 CrossRef CAS
- G. Onyestyák, S. Harnos, M. Štolcová, A. Kaszonyi and D. Kalló, Catal. Commun., 2013, 40, 32–36 CrossRef
- G. Onyestyák, S. Harnos, C. Badari, E. Drotár, S. Klébert and D. Kalló, Open Chem., 2015, 13, 517–527 Search PubMed
- G. Onyestyák, S. Harnos, S. Klébert, M. Štolcová, A. Kaszonyi and D. Kalló, Appl. Catal., A, 2013, 464–465, 313–321 CrossRef
- P. Ferrin, D. Simonetti, S. Kandoi, E. Kunkes, J. A. Dumesic, J. K. Nørskov and M. Mavrikakis, J. Am. Chem. Soc., 2009, 131, 5809–5815 CrossRef CAS PubMed
- N. D. Subramanian, G. Balaji, C. S. S. R. Kumar and J. J. Spivey, Catal. Today, 2009, 147, 100–106 CrossRef CAS
- N. Schumacher, K. Andersson, L. C. Grabow, M. Mavrikakis, J. Nerlov and I. Chorkendorff, Surf. Sci., 2008, 602, 702–711 CrossRef CAS
- G. Prieto, S. Beijer, M. L. Smith, M. He, Y. Au, Z. Wang, D. A. Bruce, K. P. de Jong, J. J. Spivey and P. E. de Jongh, Angew. Chem., Int. Ed., 2014, 53, 6397–6401 CrossRef CAS PubMed
- Y. Zhao, K. Sun, X. Ma, J. Liu, D. Sun, H. Su and W. Li, Angew. Chem., Int. Ed., 2011, 50, 5335–5338 CrossRef CAS PubMed
- M. Behrens, F. Studt, I. Kasatkin, S. Kuehl, M. Haevecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schloegl, Science, 2012, 336, 893–897 CrossRef CAS PubMed
- F. Studt, F. Abild-Pedersen, Q. Wu, A. D. Jensen, B. Temel, J. Grunwaldt and J. K. Nørskov, J. Catal., 2012, 293, 51–60 CrossRef CAS
- J. Liu, H. Su, D. Sun, B. Zhang and W. Li, J. Am. Chem. Soc., 2013, 135, 16284–16287 CrossRef CAS PubMed
- G. Li, Q. Wang, D. Li, X. Lue and J. He, Phys. Lett. A, 2008, 372, 6764–6769 CrossRef CAS
- Y. Zhao, M. Yang, D. Sun, H. Su, K. Sun, X. Ma, X. Bao and W. Li, J. Phys. Chem. C, 2011, 115, 18247–18256 CAS
- V. Glezakou, J. E. Jaffe, R. Rousseau, D. Mei, S. M. Kathmann, K. O. Albrecht, M. J. Gray and M. A. Gerber, Top. Catal., 2012, 55, 595–600 CrossRef CAS
- W. Luo and A. Asthagiri, J. Phys. Chem. C, 2014, 118, 15274–15285 CAS
- R. Zhang, G. Wang, B. Wang and L. Ling, J. Phys. Chem. C, 2014, 118, 5243–5254 CAS
- R. A. van Santen, M. Ghouri and E. M. J. Hensen, Phys. Chem. Chem. Phys., 2014, 16, 10041–10058 RSC
- A. J. Medford, A. C. Lausche, F. Abild-Pedersen, B. Temel, N. C. Schjodt, J. K. Norskov and F. Studt, Top. Catal., 2014, 57, 135–142 CrossRef CAS
- A. N. Pour, Z. Keyvanloo, M. Izadyar and S. M. Modaresi, Int. J. Hydrogen Energy, 2015, 40, 7064–7071 CrossRef
- Y. K. Zhang and W. T. Yang, Phys. Rev. Lett., 1998, 80, 890 CrossRef CAS
- M. Ernzerhof, J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1998, 80, 891 CrossRef
- B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71–129 CAS
- Q. Fu and Y. Luo, J. Phys. Chem. C, 2013, 117, 14618–14624 CAS
- J. Greeley and M. Mavrikakis, Nat. Mater., 2004, 3, 810–815 CrossRef CAS PubMed
- J. Nerlov, S. Sckerl, J. Wambach and I. Chorkendorff, Appl. Catal., A, 2000, 191, 97–109 CrossRef CAS
- B. Delley, J. Phys. Chem. C, 1990, 92, 508 CrossRef CAS
- B. Delley, J. Phys. Chem. C, 2000, 113, 7756 CrossRef CAS
- J. L. C. Fajín, M. N. D. S. Cordeiro, F. Illas and J. R. B. Gomes, J. Catal., 2014, 313, 24–33 CrossRef
- J. E. Sutton and D. G. Vlachos, ACS Catal., 2012, 2, 1624–1634 CrossRef CAS
- S. Wang, V. Vorotnikov, J. E. Sutton and D. G. Vlachos, ACS Catal., 2014, 4, 604–612 CrossRef CAS
- E. D. German and M. Sheintuch, J. Electroanal. Chem., 2011, 660, 261–275 CrossRef CAS
- S. Wang, B. Temel, J. Shen, G. Jones, L. C. Grabow, F. Studt, T. Bligaard, F. Abild-Pedersen, C. H. Christensen and J. K. Nørskov, Catal. Lett., 2011, 141, 370–373 CrossRef CAS
- J. Cheng, P. Hu, P. Ellis, S. French, G. Kelly and C. M. Lok, J. Phys. Chem. C, 2008, 112, 1308–1311 CAS
- R. A. V. Santen, M. Neurock and S. G. Shetty, Chem. Rev., 2010, 110, 2005–2048 CrossRef PubMed
- D. Loffreda, F. Delbecq, F. Vigné and P. Sautet, Angew. Chem., Int. Ed., 2009, 48, 8978–8980 CrossRef CAS PubMed
- T. Bligaard, J. K. Nørskov, S. Dahl, J. Matthiesen, C. H. Christensen and J. Sehested, J. Catal., 2004, 224, 206–217 CrossRef CAS
- M. A. N. Santiago, M. A. Sánchez-Castillo, R. D. Cortright and J. A. Dumesic, J. Catal., 2000, 193, 16–28 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2017 |