
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Utilizing super-atom orbital ideas to understand properties of silver clusters inside ZSM-5 zeolite†
Takashi Yumura *a,
Mitsuhiro Kumondaia,
Yasushige Kurodab,
Takashi Wakasugia and
Hisayoshi Kobayashia
*a,
Mitsuhiro Kumondaia,
Yasushige Kurodab,
Takashi Wakasugia and
Hisayoshi Kobayashia
aDepartment of Chemistry and Materials Technology, Kyoto Institute of Technology, Matsugasaki, Sakyo-ku, Kyoto, 606-8585, Japan. E-mail: yumura@kit.ac.jp
bDepartment of Chemistry, Graduate School of Natural Science and Technology, Okayama University, Tsushima, Kita-ku, Okayama 700-8530, Japan
First published on 17th January 2017
Abstract
The energetic properties of Agn clusters in ZSM-5 zeolite were investigated using density functional theory (DFT) calculations with the B3PW91 functional. Several optimized geometries (Agn–ZSM-5(Alm), 3 ≤ n ≤ 6 and 1 ≤ m ≤ 5) were obtained using this method, where m is the number of Al atoms substituted for Si atoms of a ten-membered ring of ZSM-5. DFT calculations found that an Agn cluster is well stabilized within a ZSM-5(Alm) cavity at n = m + 2. The stabilization conditions can be explained by frontier orbital theory because the HOMO of Agm+2–ZSM-5(Alm) zeolites is composed by totally symmetric 5s-based orbitals on silver atoms. The totally symmetric 5s-based orbital corresponds to a superatom S-orbital in cluster chemistry. Accordingly, the Agm+2–ZSM-5(Alm) zeolites have an S2 electronic configuration, being similar to magic-number silver clusters in the gas phase. Time-dependent DFT calculations found significant oscillator strength at the electronic transition between 5s-based orbitals from the totally symmetric orbital (S-orbital) to that with one node (P-orbital). The S → P electronic transitions in Agm+2–ZSM-5(Alm) follow the selection rule of electronic transitions of bare clusters. Because the excitation energies (λmax) change with an increase in the number of contained silver atoms, the S → P electronic transitions of Agm+2–ZSM-5(Alm) could be used to identify the state of the inner silver atoms.
Introduction
When silver atoms with an electronic configuration of 4d105s1 aggregate to form clusters, the resulting clusters exhibit unique electronic properties dependent on the number of contained atoms and their shapes. In general, their electronic properties are due to frontier orbitals composed of a linear combination of 5s orbitals on silver atoms. These 5s-based orbitals with different numbers of nodes can be categorized by utilizing “superatom orbitals” such as S, P, and D orbitals.1–5 The superatom orbitals, analogous to orbitals in hydrogen-like atoms, have an angular momentum quantum number (L) (L = 0, 1, and 2 for the S, P, and D orbitals, respectively) corresponding to the number of nodes of the 5s-based orbitals. Using superatom orbital ideas, the magic number of bare silver clusters and the selection rule in their electronic transitions (ΔL = ±1) can be determined.1–5To modulate the electronic properties of silver clusters, one needs to control the size of silver clusters, and the use of nanometre-sized cavities of zeolites is promising for the stabilization of relatively small silver clusters.6–47 Zeolite cavities have been shown to encapsulate silver clusters from 2 to 8 atoms in size.6–47 For example, Faujasite and Linde-type A (LTA) zeolites contain Ag3 and Ag6 clusters in their cavities, respectively.47 Previously, we combined UV-vis measurements with density functional theory (DFT) calculations to elucidate the state of silver atoms within ZSM-5 zeolites, whose ten-membered rings cavities are on the nanometre scale.48,49 UV-vis measurements in previous studies found maximum peaks ranging from 300–320 nm.43,49 These peaks can be assigned by electronic transitions of triangle Ag3 and butterfly Ag4 clusters inside ZSM-5, whose structures were optimized by DFT calculations (Fig. S1 (ESI†)).
Furthermore, DFT calculations48,49 revealed that structures of Ag3 or Ag4 clusters within a ten-membered ring of ZSM-5 zeolite are influenced by the degree of substitution of Al atoms for Si atoms in SiO2 frameworks located within the ten-membered ring. The Si → Al substitution in zeolites creates negative charges on framework oxygen atoms and positive charges on silver atoms contained within clusters. The oppositely charged silver clusters and framework oxygen atoms interact through electrostatic attraction, stabilizing the inner cluster. In addition, the Si → Al substitution is directly linked to the number of 5s electrons in silver clusters within a ZSM-5 cavity based on simple electron count arguments.48,49 Accordingly, the Si → Al substitution is the dominant factor in the strength of the orbital interactions between adjacent Ag cations, which is in turn responsible for determining the cluster structures within a zeolite cavity.
Although previous studies provided preliminary insight, knowledge is still lacking on the structural properties of ZSM-5 zeolites containing silver clusters larger than Ag3 and Ag4 clusters, which are experimentally proposed to be responsible for the UV-vis peaks at higher wavelengths. This study extends previous works to obtain atomistic information on the contained silver clusters and to examine the structural features of larger silver clusters inside a ZSM-5 cavity. This extension can allow us to find out conditions of stabilizing silver clusters inside a ZSM-5 cavity with the Si → Al substitution. Moreover, the present paper newly applies superatom orbital ideas1–5 to interpret the properties of silver clusters inside a ZSM-5 cavity with particular focus on the role of the substituted Al atoms in determining stability of inner silver clusters.
Computational methods
The current study employed DFT calculations with the B3PW91 functional50 to elucidate the properties of silver clusters inside a nanometre-sized cavity of ZSM-5 zeolite. Several previous computational studies,51–57 including ours,48,49 indicated that the B3PW91 functional can reproduce results obtained from more reliable calculations using coupled-cluster singlet double (CCSD)58 methods for the stability of neutral Ag3 isomers. Following our previous studies, periodic ZSM-5 structure was modelled using an aluminium-free ZSM-5 cluster (Si92O151H66).59–65 Si atoms within a ten-membered ring of ZSM-5 zeolite were then substituted by Al atoms, where the number of the Al atoms is m. The ZSM-5 cavity containing the substituted Al atoms is denoted as ZSM-5(Alm). Using the finite-size model, the initial structures were constructed by placing an Agn cluster into a ten-membered ring of ZSM-5(Alm) zeolite. Then, we fully optimized initial structures for an Agn cluster contained within a ZSM-5(Alm) cavity, designated by Agn–ZSM-5(Alm). Note that the formal charge of an Agn cluster inside a ZSM-5(Alm) is m, and therefore the inner Agn cluster has n–m 5s electrons. In our calculations, we considered the Agn–ZSM-5(Alm) structure with n–m being even (odd) in the singlet (doublet) state during the B3PW91 optimization.During the optimization, we used Gaussian 09 code66 instead of Gaussian 03 code67 used in the previous studies.48,49 This study re-optimized Ag3–ZSM-5(Alm) and Ag4–ZSM-5(Alm) geometries in Gaussian 09 code, although both codes yielded almost same results in terms of the energetics and geometrical features. The CEP-121G basis set was used for Ag atoms,68,69 the 6-31G* basis set for substituted Al atoms and for the two O atoms bound to a substituted Al atom,70–72 and the 3-21G basis set for all other atoms,73–78 due to computational resource limitations. For example, Ag5–ZSM-5(Al1) and Ag6–ZSM-5(Al1) involve the 2857 and 2888 contracted basis functions, respectively. First, we checked reliability of the cluster-model calculations by comparing them with QM/MM ONIOM calculations79 of a triangle Ag3 cluster inside a ZSM-5(Al1) cavity. As shown in Fig. S2 (ESI†), optimized silver separations of the inner Ag3 cluster in the cluster-model calculations are consistent with those in QM/MM ONIOM calculations, indicating accuracy in our cluster-model calculations. Furthermore, time-dependent DFT calculations were performed to investigate electronic transitions of silver cluster inside a ZSM-5 cavity, where the number of states was considered to be 50.
Results and discussion
Super-atom orbital ideas on bare small silver clusters
Let us first apply super-atom orbital ideas to understand frontier orbitals of a triangle Ag3 cluster and square and tetragonal Ag4 clusters. Fig. 1 displays frontier orbitals of prototype cluster structures: a triangle Ag3 cluster and square and tetragonal Ag4 clusters. Irrespective of the cluster structure, the most stable 5s-based orbital does not possess any nodes (totally symmetric). 5s-based orbitals with nodes have greater energy above the totally symmetric orbital. Based on superatom orbital ideas,1–5 these 5s-based orbitals can be distinguished by the number of nodes; the orbital without any node corresponds to the S-orbital, and those with one and two nodes correspond to the P- and D-orbitals. When two electrons occupy the symmetric 5s-based orbital (S-orbital), their in-phase interaction makes adjacent silver atoms have attractive interactions, stabilizing the cluster structure. In fact, we optimized structures for Ag3+, and Ag42+ clusters, where the S orbital is doubly occupied.48,49 As the number of 5s electrons in a silver cluster increases beyond two, 5s-based orbitals with some nodes are occupied. Because of the presence of some nodes, the electron occupation in the orbitals leads to a partial weakening of Ag–Ag interactions, distorting the symmetric cluster.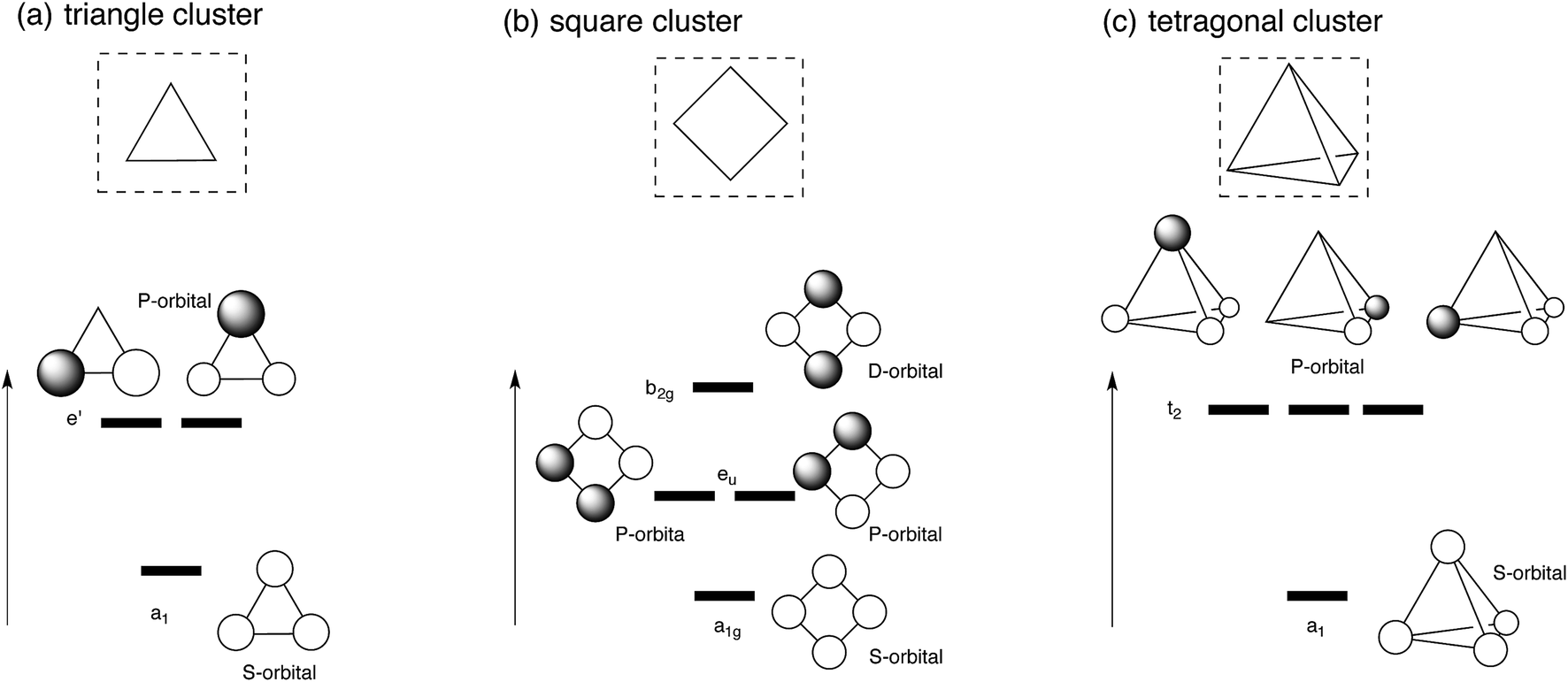 | ||
| Fig. 1 Schematic view of frontier orbitals of prototype silver clusters: (a) triangle Ag3 cluster (b) square Ag4 cluster, and (c) tetragonal Ag4 cluster. | ||
Situations are more complicated in Ag5 and Ag6 clusters. We tried to obtain local minima of bare Ag5m+ and Ag6m+ clusters. As a result of computation, we obtained only Ag5, Ag5+, and Ag52+ clusters as well as Ag6, Ag6+, and Ag62+ clusters (Fig. 2). Optimization of Ag53+ and Ag64+ clusters, which would have doubly occupied S orbital, resulted in decomposition into small fragments such as Ag33+ and Ag42+ clusters plus single Ag+ cations.80 These results indicate that Ag5m+ and Ag6m+ clusters at m being large cannot exist in the gas-phase. Thus, not only attractive interactions from 5s-based orbital interactions, but also repulsive electrostatic interactions between silver cations are important in determining the structure of small silver clusters.
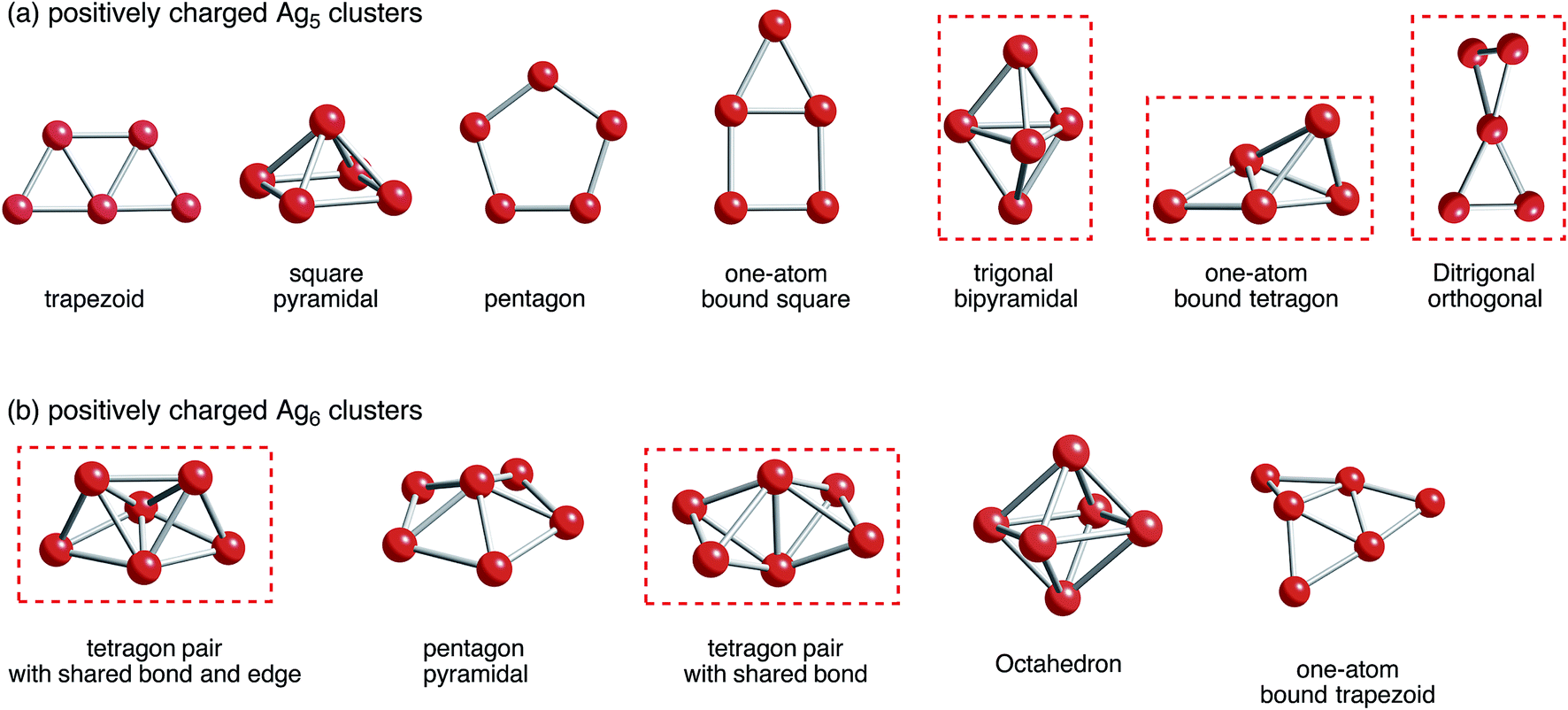 | ||
| Fig. 2 B3PW91 optimized geometries for (a) Ag5 and (b) Ag6 clusters charged positively. The cut-off length used to indicate formation of Ag–Ag bonds is 3 Å. Detailed information can be found in Tables 1 and 2. Clusters that correspond to a ZSM-5 pore and at the same time are energetically stable are marked in red. | ||
With respect to the optimized structures in Fig. 2, we found seven types of Ag5 clusters and five types of Ag6 cluster. Detailed information on geometrical features of silver clusters is listed in Tables 1 and 2. As shown in Fig. 2, Tables 1 and 2, DFT calculations found different optimized geometries for positive charged Ag5 clusters. For Ag5+, a ditrigonal orthogonal cluster is the most energetically stable. The next stable clusters take one-atom bound tetragon and trigonal bipyramidal forms. We obtained three energetically comparable Ag52+ clusters; one-atom bound tetragon, trigonal bipyramidal, and ditrigonal orthogonal forms. Similarly, there are two energetically comparable Ag6+ clusters: a tetragon pair with a shared bond and edge and a pair with shared bonds.
| Spin state | Shapea | Silver separationsb | ΔEc | |
|---|---|---|---|---|
| a Optimized structures can be seen in Fig. 2(a).b Separations between adjacent silver atoms less than 3 Å are listed.c ΔE: relative energies of Ag5m+ are in kcal mol−1. The most stable cluster is given by 0. | ||||
| Ag5 | Doublet | Trapezoid | 2.72, 2.72, 2.74, 2.74, 2.79, 2.79, 2.79 | 0 |
| Doublet | Trigonal bipyramidal | 2.70, 2.70, 2.80, 2.80, 2.85, 2.85, 2.85, 2.85 | 12.4 | |
| Ag5+ | Singlet | Ditrigonal orthogonal | 2.65, 2.65, 2.78, 2.78, 2.78, 2.78 | 0 |
| Singlet | One-atom bound tetragon | 2.63, 2.65, 2.75, 2.75, 2.99, 2.99 | 6.7 | |
| Singlet | Trigonal bipyramidal | 2.73, 2.73, 2.73, 2.86, 2.86, 2.86, 2.86, 2.87, 2.87 | 12.2 | |
| Triplet | Square pyramidal | 2.77, 2.77, 2.77, 2.77, 2.87, 2.87, 2.87, 2.87 | 22.5 | |
| Triplet | Pentagon | 2.73, 2.73, 2.73, 2.73, 2.73 | 38.3 | |
| Ag52+ | Doublet | One-atom bound tetragon | 2.71, 2.76, 2.93, 2.93, 2.94, 2.94, 2.94, 2.94 | 0 |
| Doublet | Trigonal bipyramidal | 2.80, 2.80, 2.80, 2.93, 2.93, 2.94, 2.94, 2.95, 2.95 | 1.7 | |
| Doublet | Ditrigonal orthogonal | 2.81, 2.81, 2.83, 2.83, 2.83, 2.83 | 3.5 | |
| Doublet | One-atom bound square | 2.68, 2.74, 2.88, 2.88, 2.88, 2.88 | 11.6 | |
| Spin state | Shapea | Silver separationsb | ΔEc | |
|---|---|---|---|---|
| a Optimized structures can be seen in Fig. 2(b).b Separations between adjacent silver atoms less than 3 Å are listed.c ΔE: relative energies of Ag6m+ are in kcal mol−1. The most stable cluster is given by 0. | ||||
| Ag6 | Singlet | One-atom bound trapezoid | 2.72, 2.72, 2.72, 2.72, 2.72, 2.73, 2.82, 2.82, 2.82 | 0.0 |
| Singlet | Pentagon pyramidal | 2.72, 2.72, 2.72, 2.72, 2.72, 2.86, 2.86, 2.86, 2.86, 2.87 | 5.3 | |
| Singlet | Tetragon pair with shared bond and edge | 2.66, 2.73, 2.78, 2.78, 2.78, 2.78, 2.91, 2.91, 2.91, 2.91, 2.92, 2.92 | 16.0 | |
| Singlet | Octahedron | 2.69, 2.69, 2.69, 2.69, 2.69, 2.69 | 26.4 | |
| Ag6+ | Doublet | Tetragon pair with shared bond and edge | 2.67, 2.78, 2.78, 2.85, 2.85, 2.85, 2.85, 2.88, 2.90, 2.90, 2.90, 2.90 | 0 |
| Doublet | Tetragon pair with shared bond | 2.72, 2.74, 2.74, 2.75, 2.75, 2.75, 2.75 | 3.7 | |
| Doublet | One-atom bound trapezoid | 2.73, 2.73, 2.73, 2.73, 2.73, 2.84, 2.84, 2.88, 2.88 | 5.6 | |
| Ag62+ | Triplet | Pentagon pyramidal | 2.80, 2.80, 2.80, 2.80, 2.80, 2.95, 2.95, 2.95, 2.95, 2.95 | 0 |
| Triplet | Octahedron | 2.78, 2.78, 2.79, 2.79, 2.79, 2.79 | 8.2 | |
Optimized geometries for Ag5 or Ag6 clusters inside a ZSM-5 cavity
Following the previous studies,48,49 which are summarized in Fig. S1,† Ag5 and Ag6 clusters that are formed from single-atom and double-atom additions to the tetragonal or butterfly Ag4 cluster, respectively (Fig. S3 (ESI†)), can be encapsulated into a ten-membered ring of ZSM-5 zeolites. Using these criteria, we chose energetically stable Ag5 and Ag6 clusters marked by red colour in Fig. 2 as candidates suitable to be encapsulated into a ZSM-5 cavity. To obtain optimized structures for Ag5 or Ag6 clusters inside a ZSM-5 cavity, clusters coloured red in Fig. 2 were put into the cavity to construct initial geometries, and their initial structures were fully optimized. Then, the Al atoms were substituted for Si atoms of zeolite SiO2 frameworks (m) over the range of 1 to 5. Among configurations considered in this study (Fig. S4 and S5, ESI†), the most stable optimized structures for Ag5 and Ag6 clusters inside a ZSM-5 cavity (Ag5–ZSM-5(Alm) and Ag6–ZSM-5(Alm)) are displayed in Fig. 3 and 4, respectively. Their key parameters of inner silver clusters are listed in Tables 3 and 4, respectively.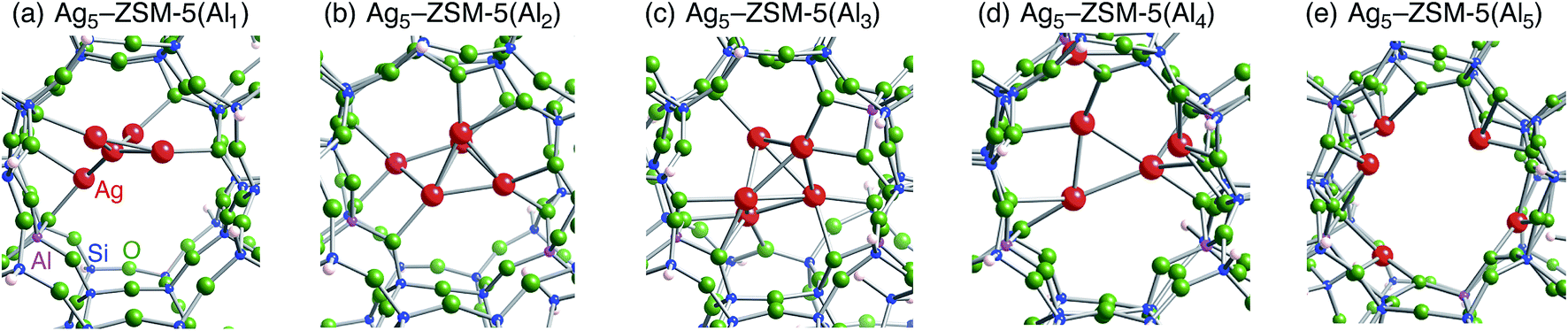 | ||
| Fig. 3 Energetically stable B3PW91 optimized geometries for Ag5–ZSM-5(Alm). (a) m = 1, (b) m = 2, (c) m = 3, (d) m = 4, and (e) m = 5. The cut-off length used to indicate formation of Ag–Ag bonds is 3 Å. Other local minima are displayed in Fig. S4.† Key parameters are listed in Table 3. | ||
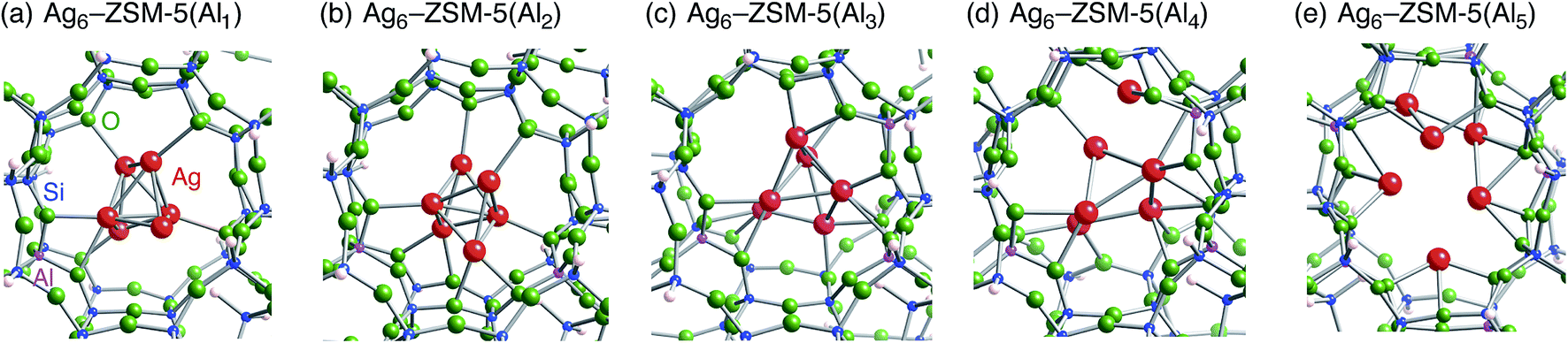 | ||
| Fig. 4 Energetically stable B3PW91 optimized geometries for Ag6–ZSM-5(Alm). (a) m = 1, (b) m = 2, (c) m = 3, (d) m = 4, and (e) m = 5. The cut-off length used to indicate formation of Ag–Ag bonds is 3 Å. Other local minima are displayed in Fig. S5.† Key parameters are listed in Table 4. | ||
| m | Structure labelb | Silver separationsc | ΔEd |
|---|---|---|---|
| a Optimized structures for Ag5–ZSM-(Alm) can be seen in Fig. S4.b Structure labels are displayed in Fig. S4.c Separations between adjacent silver atoms less than 3 Å are listed.d ΔE: relative energies of Ag5–ZSM-(Alm) are in kcal mol−1. The most stable structure is given by 0. | |||
| 1 | a (i) | 2.62, 2.68, 2.73, 2.78, 2.80, 2.80 | 0 |
| 1 | a (ii) | 2.73, 2.76, 2.77, 2.79, 2.82, 2.83, 2.86, 2.88, 2.93 | 1.6 |
| 1 | a (iii) | 2.66, 2.72, 2.72, 2.73, 2.78, 2.83, 2.84, 2.98 | 2.7 |
| 2 | b (i) | 2.74, 2.74, 2.76, 2.80, 2.86, 2.89, 2.91 | 0 |
| 2 | b (ii) | 2.68, 2.80, 2.80, 2.80, 2.82, 2.82, 2.87, 2.96 | 10.0 |
| 2 | b (iii) | 2.78, 2.80, 2.80, 2.86, 2.86, 2.87, 2.90, 2.90, 2.91 | 11.2 |
| 3 | c (i) | 2.79, 2.80, 2.81, 2.85, 2.87, 2.90, 2.92, 2.96 | 0 |
| 3 | c (ii) | 2.71, 2.76, 2.81, 2.82, 2.87, 2.89, 2.96 | 6.4 |
| 3 | c (iii) | 2.73, 2.76, 2.81, 2.84, 2.84, 2.85, 2.86, 2.88, 2.91, 2.92 | 8.2 |
| 4 | d (i) | 2.86, 2.97, 2.98 | 0 |
| 4 | d (ii) | 2.84, 2.87, 2.94 | 5.7 |
| 4 | d (iii) | 2.74, 2.81, 2.82, 2.84, 2.88, 2.90, 2.97 | 34.7 |
| 5 | e | — | — |
| m | Structure labelb | Silver separationsc | ΔEd |
|---|---|---|---|
| a Optimized structures for Ag6–ZSM-(Alm) can be seen in Fig. S5.b Structure labels are displayed in Fig. S5.c Separations between adjacent silver atoms less than 3 Å are listed.d ΔE: relative energies of Ag6–ZSM-(Alm) are in kcal mol−1. The most stable structure is given by 0. | |||
| 1 | a (i) | 2.73, 2.78, 2.78 2.79, 2.79, 2.79, 2.82, 2.85, 2.90, 2.90, 2.91, 2.91 | 0 |
| 1 | a (ii) | 2.70, 2.75, 2.79, 2.81, 2.81, 2.81, 2.84, 2.84, 2.86, 2.87, 2.89, 2.94 | 8.7 |
| 2 | b (i) | 2.71, 2.72, 2.77, 2.83, 2.83, 2.86, 2.87, 2.89, 2.90, 2.91, 2.95 | 0 |
| 2 | b (ii) | 2.70, 2.70, 2.79, 2.79, 2.81, 2.83, 2.84, 2.87, 2.88, 2.88, 2.99 | 10.7 |
| 3 | c (i) | 2.73, 2.76, 2.81, 2.84, 2.84, 2.85, 2.86, 2.88, 2.90, 2.92 | 0 |
| 3 | c (ii) | 2.75, 2.79, 2.81, 2.81, 2.82, 2.83, 2.83, 2.85, 2.90, 2.96, 2.98 | 10.4 |
| 4 | d (i) | 2.70, 2.77, 2.78, 2.81, 2.90, 2.94 | 0 |
| 4 | d (ii) | 2.77, 2.77, 2.80, 2.84, 2.87, 2.87, 2.87, 2.87, 2.90 | 5.3 |
| 5 | e (i) | 2.78, 2.84, 2.91 | 0 |
| 5 | e (ii) | 2.76, 2.78, 2.84, 2.88, 2.91, 2.91, 2.91, 2.92, 2.99 | 22.6 |
Fig. S4 and S5† show that the different structures of Ag5 or Ag6 clusters are dependent on the number of substituted Al atoms. Within ZSM-5(Al1) and ZSM-5(Al2) cavities, the most stable Ag5 clusters take ditrigonal-orthogonal and one-atom bound tetragon forms, respectively. Their structures are similar to those of the stable Ag5+ and Ag52+ clusters without ZSM-5 surroundings in terms of silver separations (Tables 1 and 3), although slight distortions were found. The inner silver clusters are bound to adjacent framework oxygen atoms near the substituted Al atoms, whose separations are listed in Table S1 (ESI†). As shown in Table S1,† the shortest distance between a silver atom and a framework oxygen atom is ∼2.3 Å. Similarly, Ag6 clusters inside ZSM-5(Al1) and ZSM-5(Al2) cavities take a stable shape where the tetragon pair with share a bond and edge. Orientation of the Ag6 cluster with respect to the ZSM-5 cavity is important; the cluster oriented along the straight channel is energetically stable relative to that perpendicular to the straight channel, as shown in Fig. S5.† Between the two structures, different interactions between inner silver cations and framework oxygen atoms are expected from Table S2 (ESI†) to stabilize an inner cluster along the straight channel.
With an increase in m, the clusters are deformed by lengthening the Ag–Ag bond lengths, as shown in Tables 3 and 4. Although the ZSM-5(Al3) cavity still contains a one-atom bound tetragonal Ag5 cluster (Fig. 3(c)) (Ag6 cluster taking a shape with the tetragonal pair with shared bond (Fig. 4(c))), separations between silver atoms increase (Tables 3 and 4). Containing Ag5 cluster inside a restricted cavity of ZSM-5(Al3) is unique, because an Ag53+ cluster, which cannot exist in the gas-phase, can be stabilized by the electrostatic interactions with framework oxygen atoms, as seen from Table S1.† When the number of Al atoms is 4, the number of Ag–Ag bonds less than 3 Å decreases.81 As a result, smaller clusters exist on the inside of ZSM-5(Al4) instead of forming Ag5 or Ag6 clusters. Further increase of the number of Al atom leads silver atoms to exist as isolated cations. These single cations strongly coordinate to three or two oxygen atoms within the framework, which are separated by less than 2.2 Å. See Tables S1 and S2.†
Recently, Cuong et al. reported optical properties of hydrated charged silver tetramer and hexamer clusters inside the sodalite cavity of LTA-type zeolite models ((H2O)4Na4Al8Si16O36H24, (H2O)5Na3Al7Si17O36H24, and (H2O)6Na2Al6Si18O36H24).82,83 Their B3LYP calculations found that butterfly Ag4 or Oh-like Ag6 clusters inside four- and six-membered cavities of the LTA models.82,83 Silver atoms on the inner Ag6 clusters are not directly bound to framework oxygen atoms in LTA zeolite models, whose separations are larger than ∼2.6 Å. Note that these separation ranges are larger than our ZSM-5 cases. Instead, the clusters are bound to OH− groups formed from water molecules near Al atoms of (H2O)5Na3Al7Si17H24O36, and (H2O)6Na2Al6Si18H24O36.82,83 Reflecting from different coordination environment of inner silver atoms between LTA and our ZSM-5 models, the two types of zeolite contain different Ag6 clusters. More interestingly, the structures of Oh-like Ag6 clusters in LTA models remain almost unchanged irrespective of various number of contained Al atoms (m = 6–8). In terms of dependences of m on inner Ag6 structures, their results are different from our results, due to weaker interactions between inner silver atoms and LTA framework oxygen atoms than ZSM-5 cases. These results indicate that zeolite surroundings, including the presence or absence of water molecules, have a strong impact on determining inner cluster structures and their properties.
Stability of Agn–ZSM-5(Alm) dependent on the number of substituted Al atoms
In this section, the stability of Agn–ZSM-5(Alm) using Estabilize values defined as Etotal[Agn–ZSM-5(Alm)] − Etotal[Agn−1–ZSM-5(Alm)] − Etotal[Ag] is discussed. Here, Etotal[Agn–ZSM-5(Alm)], Etotal[Agn−1–ZSM-5(Alm)], and Etotal[Ag] are the total energies of Agn–ZSM-5(Alm), Agn−1–ZSM-5(Alm), and the single silver atom, respectively. Fig. 5 shows the Estabilize values plotted as a function of the number of silver atoms (n). See also Fig. S6 (ESI†), including Tables S3 and S4.† From Fig. 5 and S6,† we found negative Estabilize values for Agn–ZSM-5(Alm). These negative Estabilize values indicate that Agn–ZSM-5(Alm) is energetically stable relative to the dissociation limit towards Agn−1–ZSM-5(Alm) and the single silver atom.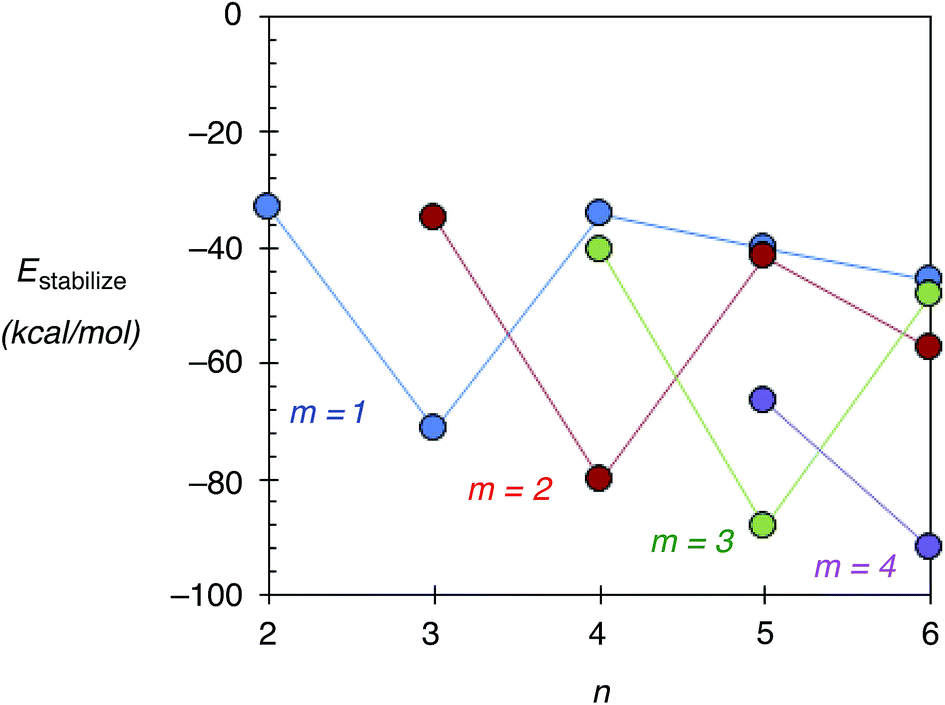 | ||
| Fig. 5 Estabilize values, defined in the main text, as a function of the number of atoms in silver clusters within ZSM-5(Alm): m = 1 (blue), m = 2 (red), m = 3 (green) and m = 4 (purple). Lines are provided for visual assistance. | ||
More interestingly, the Estabilize values depend on the number of substituted Al atoms (m) and silver atoms (n). When m = 1, 2, and 3, the most significant Estabilize values were found at n = 3, 4, and 5. The DFT results indicate that a ZSM-5(Alm) cavity stabilizes an Agm+2 cluster (m ≤ 3). The stabilization conditions (n = m + 2) can be explained by applying the superatom orbital ideas of Agn–ZSM-5(Alm). As previously mentioned, an Agn cluster inside a ZSM-5(Alm) cavity has n–m 5s electrons. Thus, an Agm+2 cluster inside a ZSM-5(Alm) cavity has two 5s electrons. The two electrons are occupied in the totally symmetric 5s-based orbital (S-orbital), indicating that the S-based orbital is the HOMO in Agm+2–ZSM-5(Alm), as confirmed by ref. 48 and 49 and Fig. S7 and S8 (ESI†). Doubly occupation of an S-orbital can be also found in the formation of magic-number silver clusters in the gas phase.1–5
The stabilization conditions can be validated only at m ≤ 3, when the 5s-orbital interactions are mainly responsible for determining the inner cluster structures. When 4 ≤ m, electrostatic interactions between silver cations and negatively charged framework oxygen atoms become more important in determining inner cluster structures, in addition to the 5s-orbital interactions. Although a significant Estabilize value was found at n = 6 and m = 4, the optimized structure does not contain an Ag6 cluster inside a ZSM-5 cavity. Instead, the optimized geometry can be viewed as constructed by the smaller silver clusters in the following manner: an Ag5 cluster near three substituted Al atoms, together with a single silver cation near one Al atom, which separates from the cluster by larger than 3.00 Å. Therefore, our extension study suggests that Ag5 clusters can exist within a ZSM-5(Alm) cavity at m = 3 and 4.
Electronic absorption of stable Agm+2–ZSM-5(Alm) structures
Next, we performed TD-DFT calculations to investigate the electronic transitions of energetically stable Agm+2–ZSM-5(Alm) structures. For TD-DFT calculations, we used smaller Agm+2–ZSM-5(Alm) models constructed by removing atoms far from the ten-membered ring containing the cluster (Fig. S9, ESI†). Note that the small models have frontier 5s-based orbitals whose energies are close to those in the whole Agm+2–ZSM-5(Alm) models, as shown in Fig. S7 and S8.† The use of the smaller models is sufficient to reproduce TD-DFT results of the whole Agm+2–ZSM-5(Alm) model because of the energetic similarity. Fig. 6 shows oscillator strengths (f) of the Ag5–ZSM-5(Al3) and Ag6–ZSM-5(Al4) structures as a function of excitation energy (λ in nm). Fig. 6(a) shows two strong peaks of the oscillator strength at 346 and 263 nm due to the 5s-based orbital excitations from totally symmetric orbital to an orbital with one node. Using the definition of the surperatom orbitals, this electronic excitation occurs from an S-orbital to a P-orbital, where ΔL increases by 1 (ΔL = +1). Similarly, the Ag6–ZSM-5(Al4) structure, composed of Ag5–ZSM-5(Al3) plus Ag1–ZSM-5(Al1), has a maximum oscillator strength value at 374 nm, as shown in Fig. 6(b). The electronic excitation behaviours are different from Oh-like Ag6 clusters in LTA models that have maximum peaks around 530 nm judging from their oscillator strengths, as seen from Table S5 (ESI†).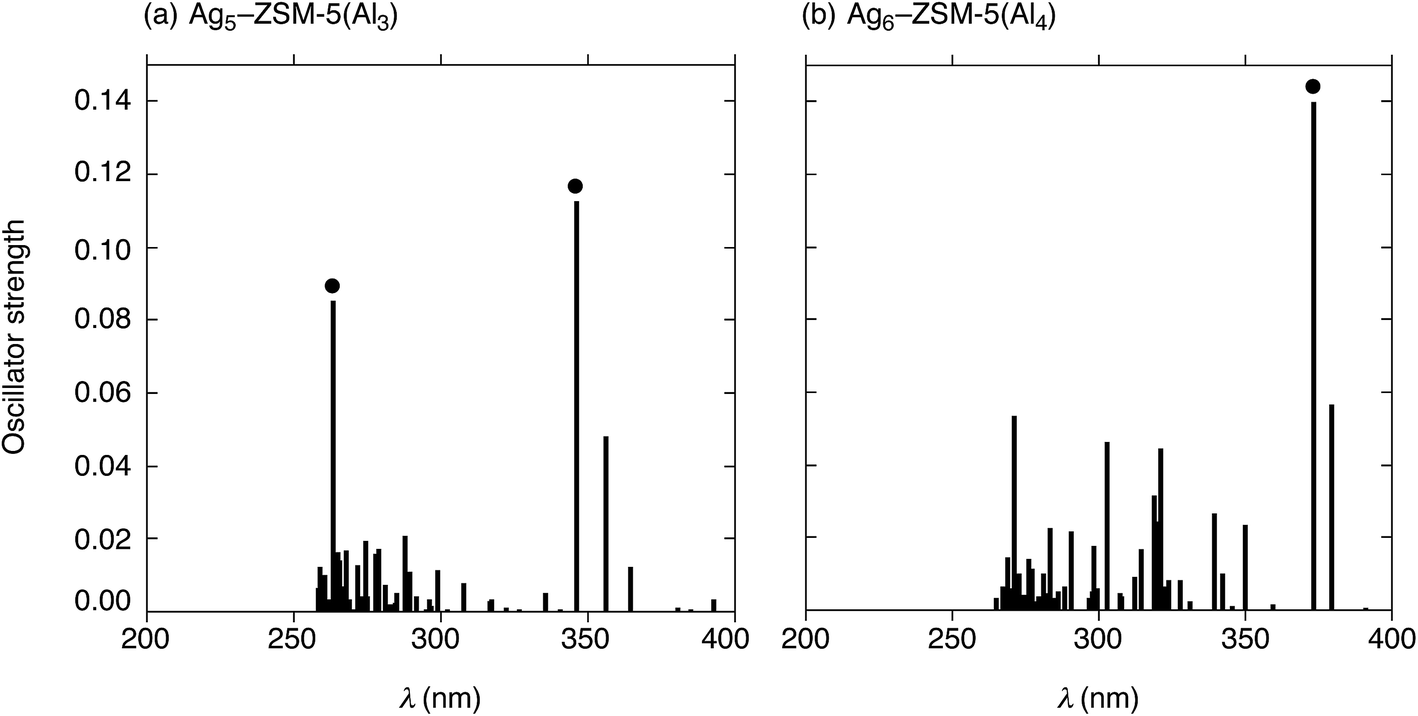 | ||
| Fig. 6 Electronic transitions of truncated Agm+2–ZSM-5(Alm) structures obtained from time-dependent DFT (TD-DFT) calculations with the B3PW91 functional. (a) Ag5–ZSM-5(Al3) and (b) Ag6–ZSM-5(Al4). The oscillatory strengths (f) of Agm+2–ZSM-5(Alm) are plotted as a function of the excitation energies (λ in nm). The black dots indicate fingerprint absorptions originating from electronic transitions between 5s-based orbitals: from the completely symmetric occupied orbital (S-orbital) to an unoccupied orbital with one node (P-orbital). | ||
Combining the current and previous TD-DFT studies, we compared the Agm+2–ZSM-5(Alm) structures in terms of excitation energies at the electronic transition with the strongest oscillator strength (λmax), as listed in Table 5. Independent of m, the maximum absorption in Agm+2–ZSM-5(Alm) comes from the S → P electronic transition, whose selection rule is ΔL = +1. The λmax values increase from 303 nm to 374 nm, when m increases from 1 to 4 in Agm+2–ZSM-5(Alm). Note that the zeolite at m = 4 containing a Ag5 cluster plus a single silver cation has a larger λmax value than that at Ag5–ZSM-5(Al3), due to slight interactions between the cluster and cation at m = 4. The DFT findings suggest that measurement of the absorptions due to the S → P electronic transition is a potential tool for identification of cluster sizes and structures.
Conclusions
Using density functional theory (DFT) calculations with the B3PW91 functional, we investigated the properties of Ag5 and Ag6 clusters inside a ZSM-5 zeolite whose ten-membered ring contains different numbers of Al atoms substituted for Si atoms of the SiO2 framework (m) (Agn–ZSM-5(Alm)). The current study achieved several optimized geometries for Ag5–ZSM-5(Alm) and Ag6–ZSM-5(Alm), and the results were compared with those of Agn–ZSM-5(Alm) where n ≤ 3 in terms of energetics. DFT calculations found that a ZSM-5(Alm) cavity stabilizes an Agm+2 cluster (m ≤ 3). The stabilization conditions (n = m + 2) can be understood through electron counting arguments based on 5s-based frontier orbitals in Agn–ZSM-5(Alm), in which the number of 5s electrons in a cluster is n–m. When an Agm+2 cluster is on the inside of a ZSM-5(Alm) cavity, the cluster has two 5s electrons populating the totally symmetric frontier orbitals, which effectively stabilize the cluster structure. The totally symmetric 5s-based orbital corresponds to a superatom S-orbital. Accordingly, the Agm+2–ZSM-5(Alm) zeolites have an S2 electronic configuration, being similar to magic-numbered silver clusters in the gas phase. Furthermore, time-dependent (TD) DFT calculations were performed to examine the electronic transition of Agm+2–ZSM-5(Alm). Agm+2–ZSM-5(Alm) zeolites were found to exhibit maximum absorption peaks due to electronic transitions based on 5s-based orbitals from the totally symmetric occupied orbital (S-orbital) to an unoccupied orbital with one node (P-orbital). The S → P electronic transitions follow the selection rule in bare cluster cases. In the case of maximum absorption, the excitation energies (λmax) in Agm+2–ZSM-5(Alm) increase from 303 to 374 nm when m increases from 1 to 4. DFT calculations revealed that the S → P electronic transitions of Agm+2–ZSM-5(Alm) are a potential fingerprint for identifying the state of inner silver atoms.Acknowledgements
This work was partly supported by a Grant-in-Aid for Young Scientists (B) (T. Y., No. 26790001) from the Japan Society for the Promotion of Science (JSPS), as well as by a Grant-in-Aid for Scientific Research on the Innovative Area “Stimuli-responsive Chemical Species for the Creation of Fundamental Molecules (No. 2408)” (JSPS KAKENHI Grant Number JP15H00941 for T. Y.).References
- W. D. Knight, K. Clemenger, W. A. de Heer, W. A. Saunders, M. Y. Chou and M. L. Cohen, Electronic shell structure and abundances of sodium clusters, Phys. Rev. Lett., 1984, 52, 2141–2143 CrossRef CAS.
- S. N. Khanna and P. Jena, Assembling crystals from clusters, Phys. Rev. Lett., 1992, 69, 1664–1667 CrossRef CAS PubMed.
- W. A. de Heer, The physics of simple metal clusters: experimental aspects and simple models, Rev. Mod. Phys., 1993, 65, 611–676 CrossRef CAS.
- D. M. P. Mingos, Structural and bonding patterns in gold clusters, Dalton Trans., 2015, 44, 6680–6695 RSC.
- E. B. Guidez and C. M. Aikens, Quantum mechanical origin of the plasmon: from molecular systems to nanoparticles, Nanoscale, 2014, 6, 11512–11527 RSC.
- H. Beyer, P. A. Jacobs and J. B. Uytterhoeven, Redox behaviour of transition metal ions in zeolites. Part 2—Kinetic study of the reduction and reoxidation of silver-Y zeolites, J. Chem. Soc., Faraday Trans. 1, 1976, 72, 674–685 RSC.
- P. A. Jacobs, J. B. Uytterhoeven and K. H. Beyer, redox behaviour of transition metal ions in zeolites. Part 6—Reversibility of the reduction reaction in silver zeolites, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 1755–1762 RSC.
- P. A. Jacobs, J. B. Uytterhoeven and K. H. Beyer, some unusual properties of activated and reduced AgNaA zeolites, J. Chem. Soc., Faraday Trans. 1, 1979, 75, 56–64 RSC.
- Y. Kim and K. Seff, The octahedral hexasilver molecule. Seven crystal structures of variously cacuum-dehydrated fully Ag+-exchanged zeolite A, J. Am. Chem. Soc., 1978, 100, 6989–6997 CrossRef CAS.
- T. Sun and K. Seff, Silver clusters and chemistry in zeolites, Chem. Rev., 1994, 94, 857–870 CrossRef CAS.
- S. Y. Kim, Y. Kim and K. Seff, Two crystal structures of fully dehydrated, fully Ag+-exchanged zeolite X. Dehydration in oxygen prevents Ag+ reduction. Without oxygen, Ag8n+ (Td) and cyclo-Ag4m+ (near S4), form, J. Phys. Chem. B, 2003, 107, 6938–6945 CrossRef CAS.
- L. R. Gellens, W. J. Mortier, R. A. Schoonheydt and J. B. Uytterhoeven, The nature of the charged silver clusters in dehydrated zeolites of type A, J. Phys. Chem., 1981, 85, 2783–2788 CrossRef CAS.
- R. A. Schoonheydt and H. Leeman, Formation of the silver hexameric (Ag6x+) cluster in zeolite A, J. Phys. Chem., 1989, 93, 2048–2053 CrossRef CAS.
- G. A. Ozin and F. Hugues, Selective photoactivation of carbon–hydrogen bonds in paraffinic hydrocarbons. Dimerization of alkanes, J. Phys. Chem., 1982, 86, 5174–5179 CrossRef CAS.
- G. A. Ozin and F. Hugues, Silver atoms and small silver clusters stabilized in zeolite Y: optical spectroscopy, J. Phys. Chem., 1983, 87, 94–97 CrossRef CAS.
- G. A. Ozin, F. Hugues, S. M. Mattar and D. F. McIntosh, Low nuclearity silver clusters in faujasite-type zeolites: optical spectroscopy, photochemistry and relationship to the photodimerization of alkanes, J. Phys. Chem., 1983, 87, 3445–3450 CrossRef CAS.
- M. D. Baker, G. A. Ozin and J. Godber, Direct observation of the reversible redox couple Ag32+ ↔ Ag30 in silver zeolite A by Fourier transform far-infrared spectroscopy, J. Phys. Chem., 1984, 88, 4902–4904 CrossRef CAS.
- M. D. Baker, G. A. Ozin and J. Godber, Far-infrared studies of silver atoms, silver ions, and silver clusters in zeolites A and Y, J. Phys. Chem., 1985, 89, 305–311 CrossRef CAS.
- D. R. Brown and L. Kevan, Comparative electron spin resonance and optical absorption studies of silver-exchanged sodium Y zeolites: silver centers formed on dehydration, oxidation, and subsequent γ-irradiation, J. Phys. Chem., 1986, 90, 1129–1133 CrossRef CAS.
- B. Xu and L. Kevan, Formation of silver ionic clusters and silver metal particles in zeolite rho studied by electron spin resonance and far-infrared spectroscopies, J. Phys. Chem., 1991, 95, 1147–1151 CrossRef CAS.
- J. Texter, R. Kellerman and T. Gonsiorowski, Formation of charged silver clusters and their reversible silver ion desorption in zeolite A, J. Phys. Chem., 1986, 90, 2118–2122 CrossRef CAS.
- E. Gachard, J. Belloni and M. A. Subramanian, Optical and epr spectroscopic studies of silver clusters in Ag, Na-Y zeolite by γ-irradiation, J. Mater. Chem., 1996, 6, 867–870 RSC.
- H. Yahiro, K. Manabe, Y. Itagaki and M. Shiotani, Epr of silver-ammonia adducts in γ-irradiation AgNa-A zeolite with low silver content, J. Chem. Soc., Faraday Trans., 1998, 94, 805–808 RSC.
- J. Sadlo, M. Danilczuk and J. Michalik, Interaction of tetrameric silver with ammonia in AgCs-rho zeolite, Phys. Chem. Chem. Phys., 2001, 3, 1717–1720 RSC.
- J. Sadlo, J. Michalik and L. Kevan, EPR and ESEEM study of silver clusters in ZK-4 molecular sieves, Nukleonika, 2006, 51, S49–S454 CAS.
- J. Michalik, M. Danilczuk, J. Turek and J. Sadlo, Stabilisation of reactive intermediates in molecular sieves, Res. Chem. Intermed., 2007, 33, 793–806 CrossRef CAS.
- W. X. Zhang, H. Yahiro, N. Mizuno, M. Iwamoto and J. Izumi, Silver ion-exchanged zeolites as highly effective adsorbents for removal of NOx by pressure swing adsorption, J. Mater. Sci. Lett., 1993, 12, 1197–1198 CAS.
- M. Anpo, M. Matsuoka and H. Yamashita, In situ investigations of the photocatalytic decomposition of NOx on ion-exchanged silver(I) ZSM-5 catalysts, Catal. Today, 1997, 35, 177–181 CrossRef CAS.
- M. Anpo, S. G. Zhang, H. Mishima, M. Matsuoka and H. Yamashita, Design of photocatalysts encapsulated within the zeolite framework and cavities for the decomposition of NO into N2 and O2 at normal temperature, Catal. Today, 1997, 39, 159–168 CrossRef CAS.
- S. M. Kanan, M. A. Omary, H. H. Patterson, M. Matsuoka and M. Anpo, Characterization of the excited states responsible for the action of silver(I)-Doped ZSM-5 zeolites as photocatalysts for nitric oxide decomposition, J. Phys. Chem. B, 2000, 104, 3507–3517 CrossRef CAS.
- W.-S. Ju, M. Matsuoka, K. Iino, H. Yamashita and M. Anpo, the local structures of silver(I) ion catalysts anchored within zeolite cavities and their photocatalytic reactivities for the elimination of N2O into N2 and O2, J. Phys. Chem. B, 2004, 108, 2128–2133 CrossRef CAS.
- S. M. Kanan, M. C. Kanan and H. H. Patterson, Photophysical properties of Ag(I)-exchanged zeolite A and the photoassisted degradation of malathion, J. Phys. Chem. B, 2001, 105, 7508–7516 CrossRef CAS.
- J. Shibata, Y. Takada, A. Shichi, S. Satokawa, A. Satsuma and T. Hattori, Ag cluster as active species for SCR of NO by propane in the presence of hydrogen over Ag-MFI, J. Catal., 2004, 222, 368–376 CrossRef CAS.
- J. Shibata, K. Shimizu, Y. Takada, A. Shichi, H. Yoshida, S. Satokawa, A. Satsuma and T. Hattori, Structure of active Ag clusters in Ag zeolites for SCR of NO by propane in the presence of hydrogen, J. Catal., 2004, 227, 367–374 CrossRef CAS.
- K. Shimizu, K. Sugino, K. Kato, S. Yokota, K. Okumura and A. Satsuma, Formation and redispersion of silver clusters in Ag-MFI zeolite as investigated by time-resolved QXAFS and UV-vis, J. Phys. Chem. C, 2007, 111, 1683–1688 CAS.
- K. Sawabe, T. Hiro, K. Shimizu and A. Satsuma, Density functional theory calculation on the promotion effect of H2 in the selective catalytic reduction of NOx over Ag–MFI zeolite, Catal. Today, 2010, 153, 90–94 CrossRef CAS.
- K. Shimizu, K. Sawabe and A. Satsuma, Unique catalytic features of Ag nanoclusters for selective NOx reduction and green chemical reactions, Catal. Sci. Technol., 2011, 1, 331–341 CAS.
- T. Baba, N. Komatsu, H. Sawada, Y. Yamaguchi, T. Takahashi, H. Sugisawa and Y. Ono, 1H magic angle spinning NMR evidence for dissociative adsorption of hydrogen on Ag+-exchanged A- and Y-zeolites, Langmuir, 1999, 15, 7894–7896 CrossRef CAS.
- T. Baba and H. Sawada, Conversion of methane into higher hydrocarbons in the presence of ethylene over H-ZSM-5 loaded with silver cations, Phys. Chem. Chem. Phys., 2002, 4, 3919–3923 RSC.
- T. Baba, H. Sawada, T. Takahashi and M. Abe, Chemisorption study of hydrogen and methane by 1H MAS NMR and conversion of methane in the presence of ethylene on Ag-Y zeolite, Appl. Catal., A, 2002, 231, 55–63 CrossRef CAS.
- H. Yoshida, T. Hamajima, Y. Kato, J. Shibata, A. Satsuma and T. Hattori, Active Ag Species in MFI zeolite for direct methane conversion in the light and dark, Res. Chem. Intermed., 2003, 29, 897–910 CrossRef CAS.
- S. Miao, Y. Wang, D. Ma, Q. Zhu, S. Zhou, L. Su, D. Tan and X. Bao, Effect of Ag+ cations on nonoxidative activation of methane to C2-hydrocarbons, J. Phys. Chem. B, 2004, 108, 17866–17871 CrossRef CAS.
- Y. Kuroda, T. Mori, H. Sugiyama, Y. Uozumi, K. Ikeda, A. Itadani and M. Nagao, On the possibility of AgZSM-5 zeolite being a partial oxidation catalyst for methane, J. Colloid Interface Sci., 2009, 333, 294–299 CrossRef CAS PubMed.
- G. D. Cremer, Y. Antoku, M. B. J. Roeffaers, M. Sliwa, J. V. Noyen, S. Smout, J. Hofkens, D. E. D. Vos, B. F. Sels and T. Vosch, Photoactivation of silver-exchanged zeolite A, Angew. Chem., Int. Ed., 2008, 47, 2813–2816 CrossRef PubMed.
- G. D. Cremer, E. Coutino-Gonzalez, M. B. J. Roeffaers, B. Moens, J. Ollevier, M. V. d. Auweraer, R. Schoonheydt, P. A. Jacobs, F. C. D. Schryver and J. Hofkens, et al., Characterization of fluorescence in heat-treated silver-exchanged zeolites, J. Am. Chem. Soc., 2009, 131, 3049–3056 CrossRef PubMed.
- G. D. Cremer, B. F. Sels, J. Hotta, M. B. J. Roeffaers, E. Bartholomeeusen, E. Coutino-Gonzalez, V. Valtchev, D. E. D. Vos, T. Vosch and J. Hofkens, Optical encoding of silver zeolite microcarriers, Adv. Mater., 2010, 22, 957–960 CrossRef PubMed.
- G. De Cremer, E. Coutiño-Gonzalez, M. B. J. Roeffaers, D. E. De Vos, J. Hofkens, T. Vosch and B. F. Sels, In situ Observation of the emission characteristics of zeolite-hosted silver species during heat treatment, ChemPhysChem, 2010, 11, 1627–1631 CrossRef CAS PubMed.
- T. Yumura, T. Nanba, H. Torigoe, Y. Kuroda and H. Kobayashi, Behavior of Ag3 clusters inside a nanometer-sized space of ZSM-5 zeolite, Inorg. Chem., 2011, 50, 6533–6542 CrossRef CAS PubMed.
- T. Yumura, A. Oda, H. Torigoe, A. Itadani, Y. Kuroda, T. Wakasugi and H. Kobayashi, Combined experimental and computational approaches to to elucidate the structures of silver clusters inside the ZSM-5 cavity, J. Phys. Chem. C, 2014, 118, 23874–23877 CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671 CrossRef CAS.
- M. Muniz-Miranda and M. F. Ottaviani, Silver nanoclusters in mesoporous silica, as obtained by visible-laser irradiation, Laser Phys., 2004, 14, 1533–1538 CAS.
- S. Yanagisawa, T. Tsuneda and H. Hirao, An investigation of density functionals: the first-row transition metal dimer calculations, J. Chem. Phys., 2000, 112, 545 CrossRef CAS.
- J. Yoon, K. S. Kim and K. K. Baeck, Ab initio study of the low-lying electronic states of Ag3−, Ag3, and Ag3+: a coupled-cluster approach, J. Chem. Phys., 2000, 112, 9335 CrossRef CAS.
- R. Fournier, Theoretical study of the structure of silver clusters, J. Chem. Phys., 2001, 115, 2165 CrossRef CAS.
- H. M. Lee, M. Ge, B. R. Sahu, P. Tarakeshwar and K. S. Kim, Geometrical and electronic structures of gold, silver, and gold–silver binary clusters: origins of ductility of gold and gold–silver alloy formation, J. Phys. Chem. B, 2003, 107, 9994–10005 CrossRef CAS.
- S. Zhao, Z.-H. Li, W.-N. Wang, Z.-P. Liu, K.-N. Fan, Y. Xie and H. F. Schaefer III, Is the uniform electron gas limit important for small Ag clusters? assessment of different density functionals for Agn (n ≤ 4), J. Chem. Phys., 2006, 124, 184102 CrossRef PubMed.
- G. N. Khairallah and R. A. J. O'Hair, Gas phase synthesis and reactivity of Agn+ and Agn−1H+ clyster cations, Dalton Trans., 2005, 2702–2712 RSC.
- J. Cizek, Correlation problem in atomic and molecular systems. calculation of wave function components in ursell-type expansion using quantum-field theoretical methods, J. Chem. Phys., 1966, 45, 4256 CrossRef CAS.
- A. Itadani, H. Sugiyama, M. Tanaka, T. Ohkubo, T. Yumura, H. Kobayashi and Y. Kuroda, Potential for C–H activation in CH4 utilizing a CuMFI-Type zeolite as a catalyst, J. Phys. Chem. C, 2009, 113, 7213–7222 CAS.
- T. Yumura, H. Yamashita, H. Torigoe, H. Kobayashi and Y. Kuroda, Site-specific Xe additions into Cu–ZSM-5 zeolite, Phys. Chem. Chem. Phys., 2010, 12, 2392–2400 RSC.
- A. Itadani, T. Yumura, T. Ohkubo, H. Kobayashi and Y. Kuroda, Existence of dual species composed of Cu+ in CuMFI being bridged by C2H2, Phys. Chem. Chem. Phys., 2010, 12, 6455–6465 RSC.
- H. Torigoe, T. Mori, K. Fujie, T. Ohkubo, A. Itadani, K. Gotoh, H. Ishida, H. Yamashita, T. Yumura and H. Kobayashi, et al., Direct information on structure and energetic features of Cu+–Xe species formed in MFI-Type zeolite at room temperature, J. Phys. Chem. Lett., 2010, 1, 2642–2650 CrossRef CAS.
- T. Yumura, S. Hasegawa, A. Itadani, H. Kobayashi and Y. Kuroda, The variety of carbon-metal bonds inside Cu-ZSM-5 zeolites: a density functional theory study, Materials, 2010, 3, 2516–2535 CrossRef CAS.
- T. Yumura, M. Takeuchi, H. Kobayashi and Y. Kuroda, Effects of ZSM-5 zeolite confinement on reaction intermediates during dioxygen activation by enclosed dicopper cations, Inorg. Chem., 2009, 48, 508–517 CrossRef CAS PubMed.
- T. Yumura, Y. Hirose, T. Wakasugi, Y. Kuroda and H. Kobayashi, Roles of water molecules in modulating the reactivity of dioxygen-bound Cu-ZSM-5 toward methane: a theoretical prediction, ACS Catal., 2016, 6, 2487–2495 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- M. J. Frisch, et al., Gaussian 03, Gaussian, Inc., Pittsburgh, PA, 2003 Search PubMed.
- W. J. Stevens, H. Basch and M. Krauss, Compact effective potentials and efficient shared-exponent basis sets for the first- and second-row atoms, J. Chem. Phys., 1984, 81, 6026 CrossRef.
- W. J. Stevens, M. Krauss, H. Basch and P. G. Jasien, Relativistic compact effective potentials and efficient, shared-exponent basis sets for the third-, fourth-, and fifth-row atoms, Can. J. Chem., 1992, 70, 612–630 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, Self-consistent molecular orbital methods. xii. Further extensions of gaussian-type basis sets for use in molecular orbital studies of organic molecules, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, The influence of polarization functions on molecular orbital hydrogenation energies, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- J. S. Binkley, J. A. Pople and W. J. Hehre, Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements, J. Am. Chem. Soc., 1980, 102, 939–947 CrossRef CAS.
- M. S. Gordon, J. S. Binkley, J. A. Pople, W. J. Pietro and W. J. Hehre, Self-consistent molecular-orbital methods. 22. Small split-valence basis sets for second-row elements, J. Am. Chem. Soc., 1982, 104, 2797–2803 CrossRef CAS.
- W. J. Pietro, M. M. Francl, W. J. Hehre, D. J. DeFrees, J. A. Pople and J. S. Binkley, Self-consistent molecular orbital methods. 24. Supplemented small split-valence basis sets for second-row elements, J. Am. Chem. Soc., 1982, 104, 5039–5048 CrossRef CAS.
- K. D. Dobbs and W. J. Hehre, Molecular orbital theory of the properties of inorganic and organometallic compounds 4. extended basis sets for third-and fourth-row, main-group elements, J. Comput. Chem., 1986, 7, 359–378 CrossRef CAS.
- K. D. Dobbs and W. J. Hehre, Molecular orbital theory of the properties of inorganic and organometallic compounds 5. Extended basis sets for first-row transition metals, J. Comput. Chem., 1987, 8, 861–879 CrossRef CAS.
- K. D. Dobbs and W. J. Hehre, Molecular orbital theory of the properties of inorganic and organometallic compounds. 6. Extended basis sets for second-row transition metals, J. Comput. Chem., 1987, 8, 880–893 CrossRef CAS.
- S. Dapprich, I. Komáromi, K. S. Byun, K. Morokuma and M. J. Frisch, A new oniom implementation in Gaussian 98. 1. The calculation of energies, gradients and vibrational frequencies and electric field derivatives, J. Mol. Struct.: THEOCHEM, 1999, 462, 1–21 CrossRef.
- To obtain Ag53+ and Ag64+ clusters, which would have the HOMO composed of the S orbital, we tried to optimize their geometries by using initial geometries taken from the optimized geometries for Ag5+ and Ag6+ in Fig. 2. However, the initial geometries for Ag53+ clusters were decomposed into two Ag+ cations and one Ag3+ cluster as well as two Ag+ cations and one Ag42+ cluster during the optimization. The decomposition into two Ag+ cations and one Ag3+ cluster is about 200 kcal mol−1 stable relative to initial Ag53+ clusters, and the decomposition into two Ag+ cations and one Ag42+ cluster is about 160 kcal mol−1 stable. Similarly the optimization of Ag64+ clusters results in the decomposition into one Ag42+ cluster and two Ag+ cations. The decomposed form is about 390 kcal mol−1 stable relative to the initial geometry.
- Lengthening Ag–Ag separations are expected because electrostatic interactions between a silver ion and framework oxygen atoms become stronger than the 5s orbital interactions between silver atoms.
- N. T. Cuong, H. M. T. Nguyen, M. P. Pham-Ho and M. T. Nguyen, Optical properties of the hydrated charged silver tetramer and silver hexamer encapsulated inside the sodalite cavity of an LTA-type zeolite, Phys. Chem. Chem. Phys., 2016, 18, 18128–18136 RSC.
- N. T. Cuong, H. M. T. Nguyen and M. T. Nguyen, Theoretical modeling of optical properties of Ag8 and Ag14 silver clusters embedded in an LTA sodalite zeolite cavity, Phys. Chem. Chem. Phys., 2013, 15, 15404–15415 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized geometries for Ag3–ZSM-5(Alm) and Ag4–ZSM-5(Alm) obtained in ref. 48 and 49 (S1); QM/MM ONIOM calculations of Ag3–ZSM-5(Al1) (S2); selected Ag5 and Ag6 clusters that are could be formed by the single-atom and dual-atom additions into Ag4 clusters (S3); optimized geometries for Ag5–ZSM-5(Alm) obtained in the current study (S4); optimized geometries for Ag6–ZSM-5(Alm) obtained in the current study (S5); Estabilize values obtained from a different basis-set system (S6); frontier orbitals of Ag5–ZSM-5(Al3) and its truncated model (S7); frontier orbitals of Ag6–ZSM-5(Al3) and its truncated model (S8); models for time-dependent DFT calculations (S9); time-dependent B3PW91 DFT calculations of Ag8 clusters inside LTA zeolites, whose geometries were taken from ref. 82 (S10); and full lists in ref. 66 and 67 (S11). See DOI: 10.1039/c6ra26492a |
| This journal is © The Royal Society of Chemistry 2017 |