
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOH- and O3-initiated atmospheric degradation of camphene: temperature dependent rate coefficients, product yields and mechanisms†
Elizabeth Gaona-Colmána,
María B. Blancoa,
Ian Barnesb,
Peter Wiesenb and
Mariano A. Teruel *a
*a
aInstituto de Investigaciones en Fisicoquímica de Córdoba (I.N.F.I.Q.C.), Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Ciudad Universitaria, 5000 Córdoba, Argentina. E-mail: mteruel@fcq.unc.edu.ar
bUniversity of Wuppertal, School of Mathematics and Natural Science, Institute of Atmospheric and Environmental Research, Gauss Strasse 20, 42119 Wuppertal, Germany
First published on 12th January 2017
Abstract
Gas-phase rate coefficients for the reactions of OH and O3 with camphene have been measured over the temperature range 288–311 K using the relative rate method. The experiments were carried out in an environmental chamber using long-path FTIR spectroscopy to monitor the reactants. Room temperature rate coefficients (in cm3 per molecule per s) of k(camphene+OH) = (5.1 ± 1.1) × 10−11 and k(camphene+O3) = (5.1 ± 1.1) × 10−19 were obtained for the OH and O3 reactions, respectively. The temperature dependence of the reactions are best fit by the Arrhenius expressions (in cm3 per molecule per s) k(camphene+OH) = (4.1 ± 1.2) × 10−12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp[(754 ± 44)/T] for the OH reaction and k(O3+camphene) = (7.6 ± 1.2) × 10−18exp[−(805 ± 51)/T] for the O3 reaction. To the best of our knowledge, this is the first report of the temperature dependencies for the reactions of OH and O3 with camphene. In addition, product studies have been performed at (298 ± 2) K and 760 Torr of synthetic air for the reaction of OH + camphene in the absence and presence of NOx, and for O3 molecules + camphene at (298 ± 2) K and 750 Torr of synthetic air. For the OH reaction the following molar product yields were obtained: acetone (10 ± 2)% and (33 ± 6)%, and formaldehyde (3.6 ± 0.7)% and (10 ± 2)% in the absence and presence of NOx, respectively. Formaldehyde with a molar yield of (29 ± 6)% was the only product uniquely identified and quantified for the O3 reaction.
exp[(754 ± 44)/T] for the OH reaction and k(O3+camphene) = (7.6 ± 1.2) × 10−18exp[−(805 ± 51)/T] for the O3 reaction. To the best of our knowledge, this is the first report of the temperature dependencies for the reactions of OH and O3 with camphene. In addition, product studies have been performed at (298 ± 2) K and 760 Torr of synthetic air for the reaction of OH + camphene in the absence and presence of NOx, and for O3 molecules + camphene at (298 ± 2) K and 750 Torr of synthetic air. For the OH reaction the following molar product yields were obtained: acetone (10 ± 2)% and (33 ± 6)%, and formaldehyde (3.6 ± 0.7)% and (10 ± 2)% in the absence and presence of NOx, respectively. Formaldehyde with a molar yield of (29 ± 6)% was the only product uniquely identified and quantified for the O3 reaction.
Introduction
Camphene (2,2-dimethyl-3-methylenebicyclo[2.2.1]heptane) is a bicyclic monoterpene, which is emitted to the atmosphere by vegetation. It is one of the most abundant monoterpenes emitted from plants such as Pinus sylvestris, Abies alba and Echinacea species.1–3 The release of biogenic volatile organic compounds (BVOCs) from vegetation can have several functions such as the attraction of pollinators, serving in signaling between plants or as a defense mechanism against herbivores.4–10 Apart from being a well-known biogenic compound, it also has been employed in diverse industrial activities, such as in the production of isobornyl acetate and isoborneol, components of perfumes, in the production of isobornyl thiocyano acetate that is a component in insecticides and in the manufacture of the chlorinated camphene (toxaphene), which is a pesticide.11–13 Once in the atmosphere, biogenic volatile organic compounds react with trace oxidants such as OH and NO3 radicals, and O3 molecules leading to the formation of a wide variety of oxygenated compounds and secondary organic aerosol (SOA).14,15There have been previous room temperature studies on the kinetics of the reactions of camphene with trace oxidants. Atkinson et al.16 have determined rate coefficients for the reactions of OH radicals, NO3 radicals and O3 molecules with camphene at 296 K and 1 atm. The experiments were performed in a Teflon chamber, whereby relative rate methods were used to determine the rate coefficients for the reactions of OH and NO3 radicals with camphene utilizing Gas Chromatography/Flame Ionization Detector CG/FID for the analysis. An absolute rate coefficient for the ozonolysis reaction was determined using a chemiluminescence analyzer. Johnson et al.17 have determined the rate coefficient for the reaction of O3 with camphene, using a relative rate method at 298 K and 760 Torr. The experiments were performed in a static reaction chamber in combination with analysis using GC/FID. There have also been computational studies on the kinetics of the ozonolysis of camphene.18,19 In addition, Peeters et al.20 have estimated the rate coefficient for the OH–camphene reaction using a Structure-Activity Relationship (SAR).
A number of studies have been published in the literature for the gas-phase products formed in the reactions of OH and O3 with camphene. Jay and Stieglitz21 have studied the products from the ozonolysis of camphene in the absence and in the presence of SO2. The experiments were carried out at 40 °C in a glass flask using Gas Chromatography with Mass Spectrometry (GC/MS) for the detection of products. Hakola et al.22 have studied the product formation from the reaction of OH radicals (in the presence of NO) and O3 with camphene at 297 K and 740 Torr. The experiments were performed in a Teflon chamber using GC/MS and Gas Chromatography/Fourier Transform Infrared spectroscopy (GC/FTIR) for product identification and GC/FID for the quantification. Reissell et al.23 have measured the acetone yield from the reaction of OH with camphene at 298 K and 740 Torr in a Teflon chamber using GC/FID and GC/MS. Finally, Oliveira and Bauerfeldt18 in a computational study present a chemical model for the ozonolysis of camphene leading to carbonyl final products.
In this work we present, in addition to room temperature determinations of the rate coefficients for the reactions of OH and O3 with camphene, a first-time determination of their temperature dependencies over the range 288–311 K. An investigation of the products formed in the reactions of OH and O3 with camphene is also presented whereby the OH reaction was investigated in the presence and absence of NOx.
Since the monoterpenes together with isoprene are the main biogenic compounds emitted to the atmosphere,24,25 detailed experimental studies of their atmospheric reaction pathways are fundamental for the provision of data for atmospheric models which are used to assess their contribution to the oxidative capacity of the atmosphere and secondary aerosol formation (SOA).
Experimental section
A quartz chamber of 1080 L capacity was employed for the kinetic and product studies. The reactor consists of two cylindrical quartz glass vessels, each 3 m in length and 45 cm inner diameter, joined in the middle with both open ends closed by aluminium flanges. The reactants and bath gas were introduced through ports located on the end flanges. Three fans with Teflon blades are mounted inside the chamber and ensure homogeneous mixing of the reactants. The reactor can be evacuated to 10−3 Torr by a pumping system consisting of a turbomolecular pump backed by a double stage rotary fore pump. The reactor can be temperature regulated over the range 284–313 K with a precision of ±2 K. The chamber is equipped with 32 low-pressure mercury lamps (Philips TUV40W, λmax = 254 nm), which are spaced evenly around the reaction vessel. A white-type mirror system is mounted inside the chamber and is coupled to a FTIR spectrometer (Thermo Nicolet Nexus) equipped with a liquid nitrogen cooled mercury–cadmium–telluride (MCT) detector. The setup facilitates long path (484.7 m) “in situ” monitoring of reactants and products in the infrared range 4000–700 cm−1. Infrared spectra were collected with a spectral resolution of 1 cm−1. The chamber is described in greater details in Barnes et al.26,27The kinetic experiments on the reaction of OH with camphene were studied over the temperature range 288–311 K in 760 Torr of nitrogen whereas the kinetic experiments on the reaction of O3 with camphene were studied over the temperature range 288–311 K in 750 Torr of nitrogen. The relative rate technique was used to determine the rate coefficients for the reactions of OH radicals and O3 molecules with camphene, i.e. the decay rate of camphene with OH or O3 as oxidant was compared with that of the corresponding decay of reference compounds:
| Oxidant + camphene → products, kcamphene | (1) |
| Oxidant + reference → products, kreference | (2) |
If camphene and the reference compound are lost only by reactions (1) and (2) it can be shown that:
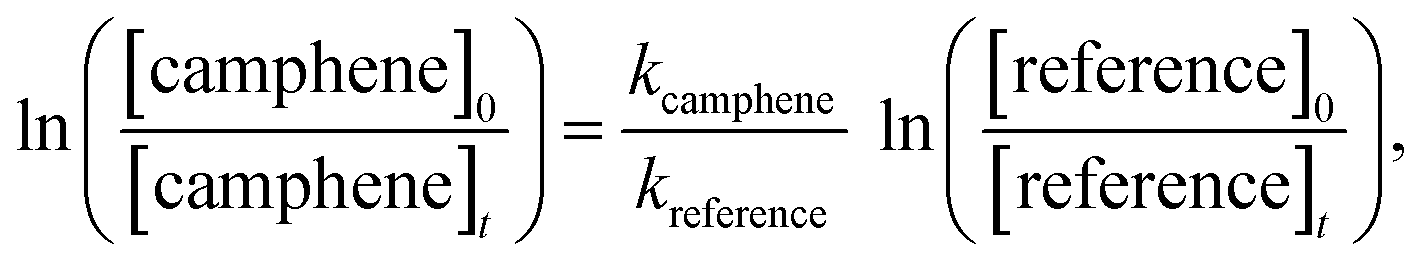 | (3) |
The 254 nm photolysis of hydrogen peroxide (H2O2) was employed as the source of hydroxyl radicals. Monitoring of camphene and the reference compounds in the dark in the reactor showed that wall loss of the compounds was negligible in all cases. Monitoring of camphene/reference compound/H2O2/nitrogen mixtures before irradiation showed there was no reaction of H2O2 with either camphene or the reference compounds. In a typical experiment 60 interferograms were co-added per spectrum and 15 such spectra were collected whereby the first five were without irradiation. The reference compounds and associated rate coefficients employed for the OH kinetic experiments were: 2,3-dimethyl-2-butene kOH(298 K) = (1.09 ± 0.04) × 10−10 cm3 per molecule per s28 and isobutene using the following Arrhenius equation kOH = 9.47 × 10−12 cm3 per molecule per s exp(504/T).29 Both 2,3-dimethyl-2-butene and isobutene were employed to determinate the rate coefficient for the reaction of OH with camphene at 298 K, while isobutene was employed for the determination of the rate coefficients of OH with camphene over the temperature range 288–311 K.
For the ozonolysis experiments, ozone was produced by flowing O2 through an electrical discharge and was added stepwise to ppm mixtures of camphene and the reference compound in 750 Torr of nitrogen. Prior to the addition of O3, the concentrations of camphene and the reference compounds were monitored to check for possible wall loss; wall loss was negligible in all of the experiments. In a typical experiment 30 interferograms were co-added per spectrum and 16 such spectra were recorded. The kinetic experiments were performed over the temperature range 288–311 K. The reference compounds and associated O3 rate coefficients used for the ozonolysis experiments were: ethene kO3 = (5.1 ± 1.0) × 10−15 cm3 per molecule per s exp[−(2446 ± 179)/T]30 and o-cresol kO3(298 K) = (2.55 ± 0.39) × 10−19 cm3 per molecule per s.31
The product studies were performed in synthetic air at 298 K and 760 Torr for the reaction of OH radicals with camphene and at 298 K and 750 Torr for the reaction of O3 with camphene. To identify and quantify products formed in the reaction of OH with camphene mixtures of H2O2/camphene/air (with and without NOx) were irradiated. Typically 60 interferograms were co-added per spectrum over a period of approximately 1 min and 18 such spectra were collected. In the product study on the ozonolysis of camphene, ozone was continuously added to a mixture of camphene/air while simultaneously recording infrared spectra with the FTIR spectrometer. Typically 30 interferograms were co-added per spectrum and 18 such spectra were collected.
Reactants and products were quantified by comparison with calibrated reference spectra contained in the IR spectral databases of the laboratory in Wuppertal. The reactants were monitored at the following infrared absorption frequencies (in cm−1): camphene 882; 2,3-dimethyl-2-butene 2800–3050; isobutene 890; o-cresol 700–800 and ethene 950. The reactions products were monitored at the following infrared absorption frequencies (in cm−1): acetone 1217 and formaldehyde 2766.
The initial concentrations of the organic compounds (1 ppmv = 2.46 × 1013 molecule per cm3 at 298 K) were: 0.97–1.5 ppmv for camphene; 2–2.3 ppmv for 2,3-dimethyl-2-butene; 1.6–1.9 ppmv for isobutene; 2.5–3.0 ppmv for o-cresol; 2–2.8 ppmv for ethene; 7–7.5 ppmv for H2O2; 2.3–3 ppmv for NO. The chemicals used in the experiments had the following purities as given by the manufacturer and were used as supplied: nitrogen (Air Liquide, 99.999%), synthetic air (Air Liquide, 99.999%), H2O2 (Interox, 85%), camphene (Aldrich, 95%), 2,3-dimethyl-2-butene (Aldrich, 98%), nitrogen oxide (Messer Griesheim, 99%), isobutene (Messer Griesheim, 99%), o-cresol (Aldrich, 99+%) and ethene (Messer Griesheim, 99%).
Results and discussion
OH reaction
Fig. 1 shows the kinetic data, plotted according to eqn (3), obtained from experiments on the reaction of OH with camphene at 298 K measured relative to the reference compounds 2,3-dimethyl-2-butene and isobutene. Each plot represents three experiments for each reference compound. As seen from Fig. 1 linear relationships were obtained in all experiments. The rate coefficient ratios kcamphene/kreference obtained from the individual plots for each experiment are listed in Table 1 together with the absolute values of the rate coefficients for the reactions of camphene with OH radicals at 298 K calculated using the appropriate room temperature rate coefficient value for the reference reaction. Since the values of the rate coefficients obtained in the individual measurements with each reference compound, and the values obtained with both reference compounds, are in excellent agreement we prefer to report a final rate coefficient for the reaction of OH radicals with camphene at 298 K, which is the average of all the determinations:| k(camphene+OH) = (5.1 ± 1.1) × 10−11 cm3 per molecule per s |
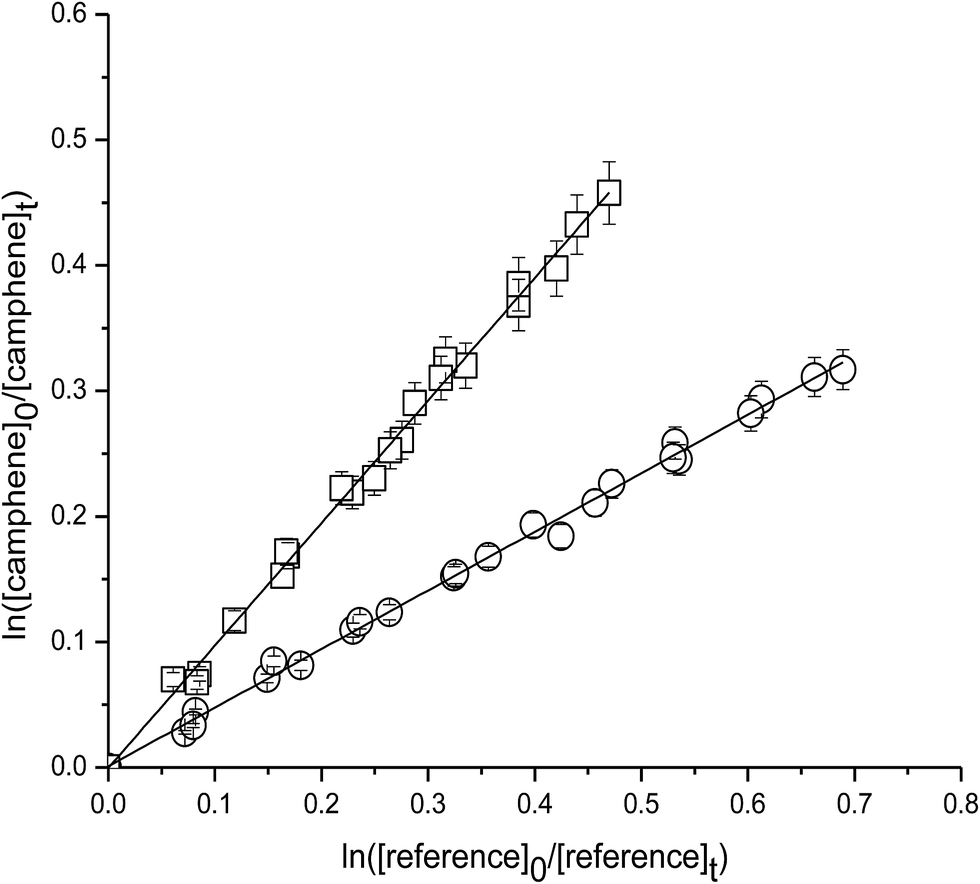 | ||
| Fig. 1 Plot of the kinetic data for the reaction of OH radicals with camphene measured relative to (○) 2,3-dimethyl-2-butene and (□) isobutene at (298 ± 2) K and (760 ± 10) Torr total pressure. | ||
| Reaction | Reference | kcamphene/kreference | kcamphene × 1011 (cm3 per molecule per s) |
|---|---|---|---|
| Camphene + OH | 2,3-Dimethyl-2-butene | 0.46 ± 0.01 | 5.0 ± 0.3 |
| 2,3-Dimethyl-2-butene | 0.47 ± 0.01 | 5.1 ± 0.3 | |
| 2,3-Dimethyl-2-butene | 0.47 ± 0.01 | 5.1 ± 0.2 | |
| Isobutene | 0.97 ± 0.01 | 5.0 ± 1.0 | |
| Isobutene | 0.99 ± 0.02 | 5.1 ± 1.1 | |
| Isobutene | 1.01 ± 0.03 | 5.2 ± 1.1 | |
| Average | 5.1 ± 1.1 |
| Reaction | Reference | kcamphene/kreference | kcamphene × 1019 (cm3 per molecule per s) |
|---|---|---|---|
| Camphene + O3 | Ethane | 0.38 ± 0.01 | 5.2 ± 1.1 |
| Ethane | 0.38 ± 0.01 | 5.2 ± 1.1 | |
| Ethane | 0.37 ± 0.01 | 5.1 ± 1.1 | |
| o-Cresol | 1.97 ± 0.04 | 5.0 ± 0.9 | |
| o-Cresol | 1.94 ± 0.04 | 4.9 ± 0.9 | |
| o-Cresol | 1.96 ± 0.03 | 5.0 ± 0.8 | |
| Average | 5.1 ± 1.1 |
The errors quoted are twice the standard deviation obtained from the least-squares fits of the straight lines plus a contribution to take into account uncertainties in the reference rate coefficients.
The room temperature rate coefficient for the reaction of OH radicals with camphene obtained in this work can be compared with previous kinetic data. Atkinson et al.16 have reported a value of (5.33 ± 0.20) × 10−11 cm3 per molecule per s for the reaction at 296 ± 2 K and 735 Torr total pressure of synthetic air measured relative to the reaction of OH with isoprene. Their study was performed in a 6400 L Teflon chamber using CG/FID for the analysis and methyl nitrite (CH3ONO) as the OH radical precursor. On the other hand, Peeters et al.20 have estimated a value of 5.95 × 10−11 cm3 per molecule per s for the rate coefficient of the camphene–OH reaction at 298 K using SAR prediction. The value of the rate coefficient obtained in this work for the reaction at 298 K of (5.1 ± 1.1) × 10−11 cm3 per molecule per s is in excellent agreement within experimental errors with the value reported by Atkinson et al.16 and with the estimated value of Peeters et al.20
The temperature dependence of the rate coefficient for the reaction of OH with camphene was measured over the temperature range 288–311 K using isobutene as the reference compound. Fig. S1 (see ESI†) shows plots of the kinetic data according to eqn (3) for two experiments performed at temperatures of 288, 293, 303 and 311 K. Good linear correlations were obtained at all temperatures. The rate coefficients obtained in the two experiments at each temperature are very similar and give the averaged rate coefficients listed in Table 2. The errors for the rate coefficients are twice the standard deviation resulting from the straight line least-squares fits of the data plus an additional 20% to account for the uncertainty in the rate coefficient for isobutene. The rate coefficients are plotted in Arrhenius form in Fig. 2, the following Arrhenius expression has been derived from a linear least-squares treatment of the plot:
| k(OH+camphene) = (4.1 ± 1.2) × 10−12 exp[(754 ± 44)/T] |
| Reaction | Temperature (K) | kcamphene × 1011 (cm3 per molecule per s) |
|---|---|---|
| Camphene + OH | 288 | 5.6 ± 1.1 |
| 293 | 5.4 ± 1.1 | |
| 298 | 5.1 ± 1.1 | |
| 303 | 4.9 ± 1.0 | |
| 311 | 4.6 ± 1.0 |
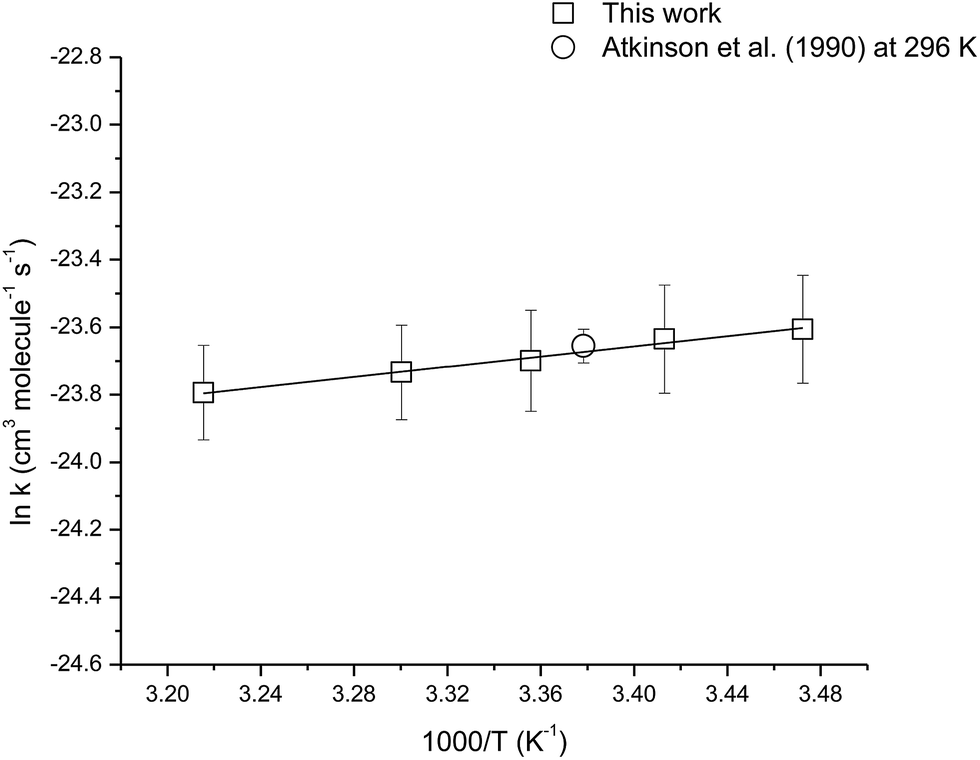 | ||
| Fig. 2 Arrhenius plot of the kinetic data for the reaction of OH radicals with camphene over the temperature range 288–311 K. A comparison with a previous literature value is also shown. | ||
The rate coefficients for the reaction of OH with camphene were found to decrease with increasing temperature over the temperature range 288–311 K, which is indicative of an addition mechanism. There are no previous temperature dependence studies for this reaction with which a comparison can be made. However, it is interesting to compare the results obtained here for the temperature dependence of the reaction of OH with camphene with that of OH with β-pinene, which is also a bicyclic monoterpene with an exocyclic ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 group. The temperature dependence of the reaction of OH with β-pinene has been studied by several authors who all found that the rate coefficient for this reaction decreases with increasing temperature and report values of Ea/R that vary from −358 to −610 K. Kleindienst et al.32 report an Ea/R value of (−358 ± 57) K determined using an absolute method (FP-RF) over the temperature range 297–424 K in argon at a total pressure of 50 Torr. Chuong et al.33 have reported a value of Ea/R of (−610 ± 50) K determined over the temperature range 300–435 K in helium at 5 Torr using discharge-flow systems coupled with resonance fluorescence and laser-induced fluorescence detection of OH. Gill and Hites34 obtained a value of Ea/R of (−467 ± 50) K determined over the temperature range 298–362 K at a total pressure 760 Torr using a relative kinetic method and on-line mass spectrometry for the detection of reactants. Finally, Montenegro et al.35 have reported a value of Ea/R of (−470 ± 17) K measured over the temperature range 240–340 at a total pressure of 1–8 Torr using a relative rate/discharge flow/mass spectrometry method. The value of Ea/R for the reaction of OH with camphene measured in this study is 20% higher than the highest value of Ea/R reported in the literature for the reaction of OH with β-pinene, however, it is the range generally reported for both exo- and endocylic double bonds in bicyclic monoterpenes.36,37
CH2 group. The temperature dependence of the reaction of OH with β-pinene has been studied by several authors who all found that the rate coefficient for this reaction decreases with increasing temperature and report values of Ea/R that vary from −358 to −610 K. Kleindienst et al.32 report an Ea/R value of (−358 ± 57) K determined using an absolute method (FP-RF) over the temperature range 297–424 K in argon at a total pressure of 50 Torr. Chuong et al.33 have reported a value of Ea/R of (−610 ± 50) K determined over the temperature range 300–435 K in helium at 5 Torr using discharge-flow systems coupled with resonance fluorescence and laser-induced fluorescence detection of OH. Gill and Hites34 obtained a value of Ea/R of (−467 ± 50) K determined over the temperature range 298–362 K at a total pressure 760 Torr using a relative kinetic method and on-line mass spectrometry for the detection of reactants. Finally, Montenegro et al.35 have reported a value of Ea/R of (−470 ± 17) K measured over the temperature range 240–340 at a total pressure of 1–8 Torr using a relative rate/discharge flow/mass spectrometry method. The value of Ea/R for the reaction of OH with camphene measured in this study is 20% higher than the highest value of Ea/R reported in the literature for the reaction of OH with β-pinene, however, it is the range generally reported for both exo- and endocylic double bonds in bicyclic monoterpenes.36,37
O3 reaction
Fig. 3 shows plots of the kinetic data obtained from experiments on the reaction of camphene with O3 molecule measured relative to the reference compounds ethene and o-cresol at 298 K. Each plot represents three experiments for each reference compound. As seen from Fig. 3 linear relationships were obtained in all the experiments. The rate coefficient ratios kcamphene/kreference obtained from the individual plots from each experiment are listed in Table 1 together with the absolute values of the rate coefficients for the reactions of camphene with O3 molecule at 298 K. The rate coefficients obtained in the individual measurements with each reference compound are in good agreement as are the values obtained with both reference compounds. We, therefore, prefer to quote a final rate coefficient for the reaction of camphene with O3 radicals at 298 K which is the average of all the determinations:| k(camphene+O3) = (5.1 ± 1.1) × 10−19 cm3 per molecule per s |
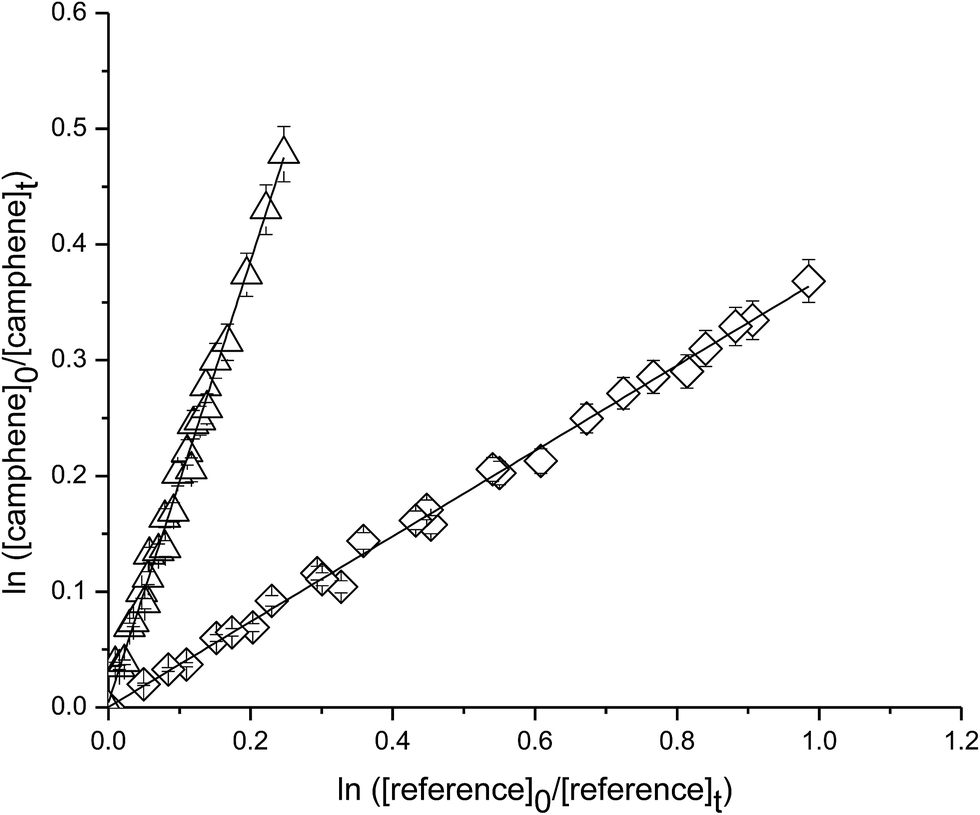 | ||
| Fig. 3 Plot of the kinetic data for the reaction O3 molecules with camphene measured relative to (◊) ethene and (△) o-cresol at (298 ± 2) K and (750 ± 10) Torr total pressure. | ||
The errors are twice the standard deviation arising from the least-squares fit of the straight lines plus a contribution to take into account the uncertainties in the reference rate coefficients.
There are two previous experimental studies of the rate coefficient at room temperature for the reaction of O3 molecules with camphene. Atkinson et al.16 have obtained a rate coefficient for the reaction of (9.0 ± 1.7) × 10−19 cm3 per molecule per s at 296 ± 2 K and atmospheric pressure. The experiments were performed in a 160 L reaction chamber using an absolute method, the ozone concentration was monitored by chemiluminescence and the camphene concentration by GC-FID. Johnson et al.17 have obtained a rate coefficient of (4.5 ± 0.2) × 10−19 cm3 per molecule per s for the reaction at (298 ± 2) K and (760 ± 10) Torr using the relative rate method. They performed the experiments in a static reaction chamber in the presence of an excess of cyclohexane to scavenge any OH radicals produced in the reaction and used GC-FID for the reactant analysis.
The rate coefficient obtained in this work for the reaction of O3 with camphene is much lower than that obtained by Atkinson et al.16 but is, within the experimental errors, in excellent agreement with that obtained by Johnson et al.17 In their density functional theory computational study of the ozonolysis of camphene Oliveira and Bauerfeldt18 report a rate coefficient of 1.12 × 10−18 cm3 per molecule per s for the reaction which is more in line with the value reported by Atkinson et al.16 However, in a more recent study Oliveira and Bauerfeld19 have obtained a rate coefficient of 4.61 × 10−19 cm3 per molecule per s at 298 K for the ozonolysis of camphene in a variational transition state investigation based on the microcanonical variational method. This more recent computed rate coefficient value is in very good agreement with the values obtained by Johnson et al.17 and in this study.
The present experiments were performed in the absence of an OH radical scavenger since the large concentrations of a scavenger, such as CO or cyclohexane, that would have been necessary to effectively scavenge any OH formed in the system would have rendered monitoring of the reactants in the infrared impossible. The experiments, however, have been performed in nitrogen in an attempt to reduce inference from OH radical production. The OH radical production in the ozonolysis of both camphene and ethene have been reported to be relatively low.37 In addition, since the reference compounds have rate coefficients for reaction with O3 which differ by a factor of ∼2 and rate coefficients with OH radicals that differ by a factor of ∼6 the excellent agreement between the values of the rate coefficient for the reaction of O3 with camphene obtained with ethene and o-cresol supports that interference in the experiments by OH radical production has not been occurring at a significant level. The reason for the much higher rate coefficient of Atkinson et al.16 is not clear. Their sample of camphene had a stated purity of 85%, however, the authors corrected their reported rate coefficient for the impurity and it is unlikely that this is the reason for the large difference. Ozonolysis reactions are generally slow and difficult to measure. Thus it is not unusual to find quite large differences in the reported literature values.38
It is interesting to compare the rate coefficients for the ozonolysis of camphene with those for the ozonolysis of two bicyclic monoterpenes with exocyclic double bonds, i.e. β-pinene and sabinene. The room temperature rate coefficient for the ozonolysis of camphene is two orders of magnitude lower than those for the ozonolysis of β-pinene and sabinene.
In the literature, the reported room temperature rate coefficients for the reaction of O3 with β-pinene are in the range (1.2–2.35) × 10−17 cm3 per molecule per s16,17,38–41 and for the reaction of O3 with sabinene a room temperature rate coefficient of 8.07 × 10−17 cm3 per molecule per s has been reported.16 Both Atkinson et al.16 and Johnson et al.17 ascribe the slowness of the ozonolysis of camphene compared to the other monoterpenes to steric hindrance of the approach of O3 to the double bond in camphene by the two adjacent methyl (CH3–) groups and the methylene (–CH2–) bridge group.
The temperature dependence of the reaction of camphene with O3 molecules was studied over the temperature range 288–311 K using ethene as the reference compound. Fig. S2† shows the kinetic data obtained from the experiments performed at 288, 293, 303 and 311 K plotted according to eqn (3). Good linear correlations were obtained at all temperatures. The average rate coefficients obtained from two experiments at each temperature are listed in Table 3. The errors are twice the standard deviation from straight line least-squares fits of the data, plus an additional 20% to account for the uncertainty in the rate coefficient for ethene. An Arrhenius plot of the kinetic data is shown in Fig. 4 and the following Arrhenius expression has been derived from a linear least-squares treatment of the plot:
| k(O3+camphene) = (7.6 ± 1.2) × 10−18 exp[−(805 ± 51)/T] |
| Reaction | Temperature (K) | kcamphene × 1019 (cm3 per molecule per s) |
|---|---|---|
| Camphene + O3 | 288 | 4.7 ± 1.3 |
| 293 | 4.9 ± 1.1 | |
| 298 | 5.1 ± 1.1 | |
| 303 | 5.3 ± 1.1 | |
| 311 | 5.8 ± 1.2 |
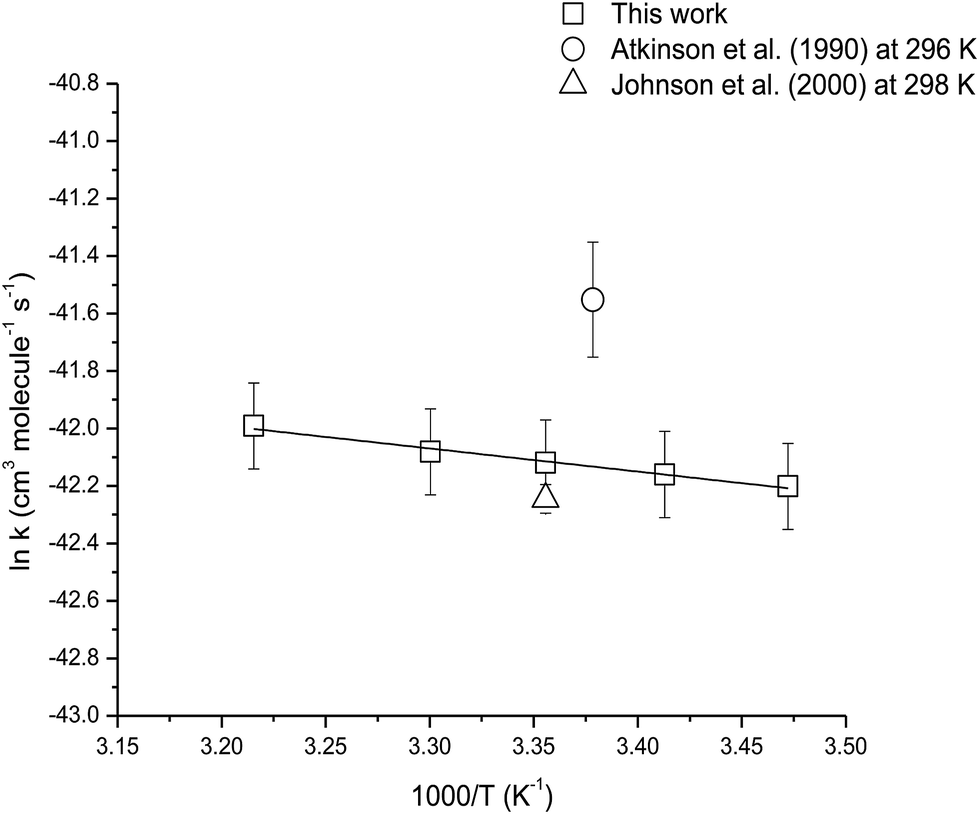 | ||
| Fig. 4 Arrhenius plot of the kinetic data for the reaction of O3 molecules with camphene over the temperature range 288–311 K. A comparison with previous literature values is also shown. | ||
The reaction of O3 molecule with camphene was found to exhibit slight positive temperature dependence over the temperature range 288–311 K. To our knowledge, this is the first kinetic temperature dependence study for the reaction of O3 with camphene. However, Khamaganov and Hites41 have investigated the temperature dependence for the reaction of O3 with β-pinene over the temperature range 242–363 K using the relative rate technique and GC/MS for the analysis. They obtained an Ea/R value of (1297 ± 75) K for the reaction which is somewhat higher than the Ea/R value of (805 ± 51) K obtained in this work for the reaction of O3 with camphene.
Product studies
OH reaction
The considerable slope in the product spectrum above ∼2700 cm−1, which is particularly evident in Fig. S4,† is indicative for the formation of significant levels of aerosol in the reaction system. Formation of acetone, formaldehyde and CO were observed in the experiments performed in the absence and in the presence of NOx. Carbon monoxide was quite evidently a secondary reaction product and is not considered further in the discussion. Concentration–time profiles of camphene and the identified products in the absence and in the presence of NO are presented in Fig. S5 and S6,† respectively. The concentration–time profiles support that these products are to a large extend primary products under our experimental conditions. Plots of the concentrations of the identified products against the amount of camphene obtained from studies on the reaction of OH with camphene performed in the absence and in the presence of NOx are shown in Fig. S7 and S8,† respectively. The plots of the concentrations of acetone and formaldehyde as a function of consumed camphene show reasonable linearity. Formaldehyde can be consumed in a secondary reaction with OH in the reaction system, therefore, to estimate the extent of this secondary consumption, the formation yield correction method outlined in Tuazón et al.42 was used. The IUPAC43 recommended rate coefficient for the reaction of OH with formaldehyde at 298 K is 8.5 × 10−12 cm3 per molecule per s. The calculated secondary consumption in the reaction system for this carbonyl compounds was <1% in all cases, therefore, no correction has been applied to the HCHO formation yields. The formation yields of the oxidation products identified in the OH-radical initiated oxidation of camphene in the presence and absence of NOx are listed in Table 4.
| Reaction | Identified products | Yield (%) in absence of NOx | Yield (%) in presence of NOx |
|---|---|---|---|
| Camphene + OH | Acetone | 10 ± 2 | 33 ± 6 |
| Formaldehyde | 3.6 ± 0.7 | 10 ± 2 | |
| Camphene + O3 | Formaldehyde | 29 ± 6 | — |
The formation yield of acetone obtained in this work due to the reaction of OH radical with camphene in the absence of NOx was (10 ± 2)%, while in the presence of NOx it was (33 ± 6)%, i.e. an enhancement by a factor of 3 over the NOx-free system. Formation of acetone in the photooxidation is expected since the camphene structure contains the C(CH3)2 entity. Reissell et al.23 in their study on the reaction of OH with camphene, performed in a Teflon chamber at 298 ± 2 K and 740 Torr in the presence of NO using GC/FID and GC/MS for the product analysis, obtained a formation yield for acetone of (39 ± 5)%. Their yield of acetone is in very good agreement, within the experimental errors, with that obtained in this study in the presence of NOx. Hakola et al.22 have performed experiments on the reaction of OH with camphene in an attempt to identify and quantify possible high molecular weight carbonyl products. The experiments were performed in a Teflon chamber at 297 ± 2 K and 740 Torr total pressure of air in the presence of NOx, whereby, the products were identified by GC/MS and GC/FTIR and quantified by GC/FID. They established that the yields of camphenilone (3,3-dimethylbicyclo[2.2.1]heptan-2-one) and 6,6-dimethyl-ε-caprolactone-2,5-methylene (4,4-dimethyl-3-oxabicyclo[3.2.1]octan-2-one) in the reaction are both <0.02, i.e. negligible. The formaldehyde yields obtained in this work were (3.6 ± 0.7) and (10 ± 2)% in the absence and presence of NOx, respectively. To our knowledge, formaldehyde has not been previously reported as a product of the reaction of OH with camphene. However, it is worth noting that the yields of formaldehyde and acetone only account for a small fraction of the reacted carbon.
Camphene has a bicyclic molecular structure with an exocyclic double bond of the type CCH2. The reaction of OH radicals with camphene will proceed mainly by addition of OH to the exocyclic double bond to form hydroxyalkyl radicals. The addition of the OH radical can occur at either of the carbon atoms on the double bond, however, according to Reissell et al.23 and Orlando et al.44 OH radical addition at the terminal carbon atom of the double bond is favored. Under our experimental conditions the hydroxyalkyl radicals will rapidly add molecular oxygen to form hydroxyalkylperoxy radicals. When the reaction is performed in the absence of NOx, the hydroxyalkylperoxy radicals undergo self-reaction and reaction with other peroxy radicals to form mainly hydroxyalkoxy radicals, although molecular channels are also possible.45,46 Conversion of the hydroxyalkylperoxy radicals to hydroxyalkoxy radicals is overwhelmingly favored in the presence of NO over peroxy–peroxy reactions. In the presence of NO formation of hydroxyalkylnitrates is also a possibility. The hydroxyalkoxy radicals can either decompose or react with O2 leading to the formation of multifunctional products.
A simplified mechanism for the possible reactions following OH addition to the terminal carbon atom of the double bond in camphene, indicated as C8, is shown in Fig. 5. As outlined above addition generates an alkyl radical, which adds O2 molecules forming a hydroxyalkylperoxy radical. The hydroxyalkylperoxy radical reacts with other peroxy radicals or with NO forming mainly hydroxyalkoxy radicals. The hydroxyalkoxy radical can undergo decomposition by scission of the C2–C3 and C3–C4 and C3–C8 bonds.
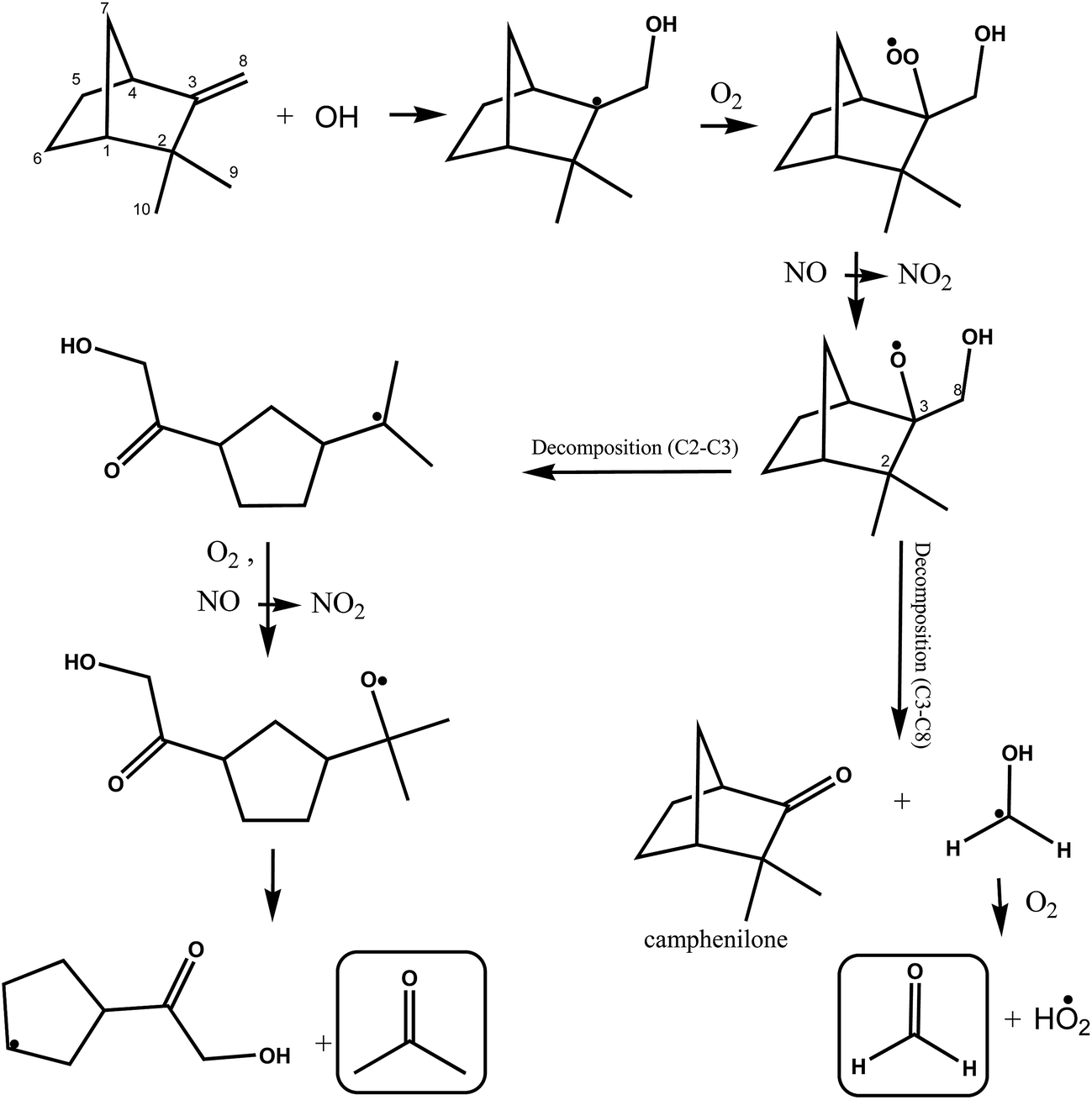 | ||
| Fig. 5 Simplified mechanism for the OH-radical initiated oxidation of camphene via addition of OH to the C8 carbon of the double bond. | ||
The decomposition of the hydroxyalkoxy radical by scission of the C3–C8 bond will result in the formation of camphenilone and the H2C˙(OH) radical which will react with molecular O2 to form formaldehyde and a HO2˙ radical. The relatively low yield of HCHO observed in the system immediately leads to the conclusion that scission of the C3–C8 is minor compared to possible scission of the C2–C3 and C3–C4 bonds. This also implies that the formation yield of camphenilone should be low which is in agreement with the experimental findings of Hakola et al.22
Reissell et al.23 note in their study on the reaction of OH with camphene that C2–C3 bond scission is thermochemically favored and Orlando et al.44 in their study on the reaction of OH with β-pinene have suggested that ring-strain energy release of the bicyclo structure though breaking C–C bond is a favorable process. Decomposition via scission of the C2–C3 bond opens the bicyclic structure forming a five-membered ring with a lateral alkyl radical chain, where the radical center is located on the tertiary carbon atom of the [–C˙(CH3)2] group. Addition of O2 to this alkyl radical followed by reaction with NO or other peroxy radicals will form an alkoxy radical, which can decompose with the formation of acetone and a radical co-product containing a five-membered ring. The alkoxy radical could alternatively eject a methyl group and form a carbonyl compound (not shown), however, further reactions of the methyl radical would form HCHO and since the yield of HCHO is low in the system this reaction channel can be considered relatively unimportant.
A simplified mechanism for the possible reactions following OH addition to the C3 on the double bond of camphene is shown in Fig. 6. The initially formed alkyl radical will undergo the same cascade of reactions described above to form a hydroxyalkoxy radical. This hydroxyalkoxy radical can react with O2 to form a 1,2-hydroxycarbonyl compound and a HO2 radical (not shown) or decompose by a C3–C8 bond scission to form HCHO and a hydroxylated cyclic alkyl radical as shown in Fig. 6. Acetone can be formed in further consecutive reactions of the radical with O2 and NO. However, here again the relatively low yields of formaldehyde obtained in this work support that addition of the OH radical to the C3 atom on the double bond in camphene is a fairly minor reaction pathway.
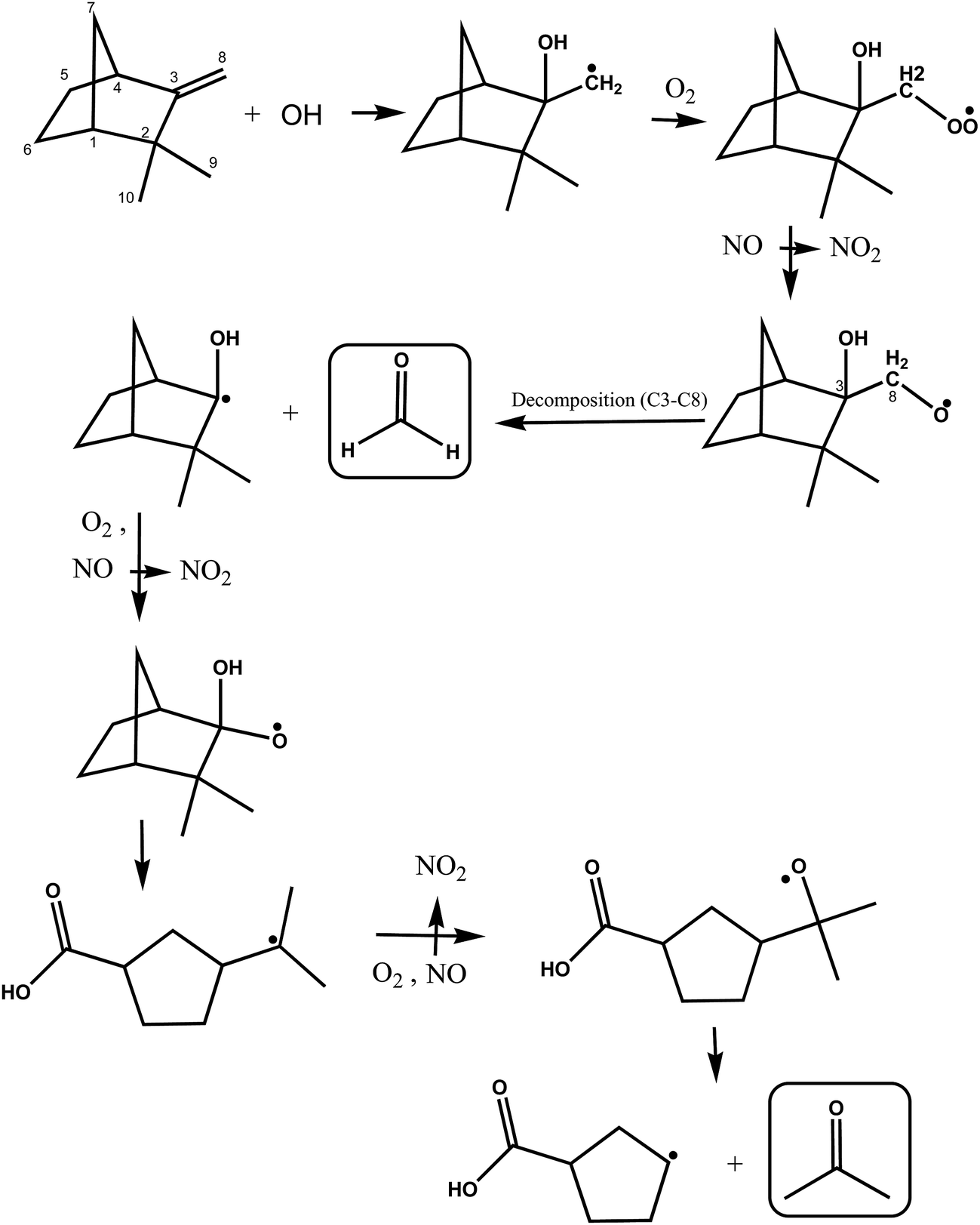 | ||
| Fig. 6 Simplified mechanism for the OH-radical initiated oxidation of camphene via addition of OH to the C3 carbon of the double bond. | ||
In summary, the reaction of the OH radical with camphene proceeds mainly by addition of OH to the double bond terminal carbon atom (C8). Decomposition of the intermediate hydroalkoxy radical via scission of the C2–C3 bond is much more favored over scission of the C3–C8 bond. However, the magnitude of the product yields for acetone and HCHO obtained in the presence of NO would suggest that a considerable fraction of the reaction will proceed via scission of the C3–C4 bond which like C2–C3 scission opens the bicyclic ring structure. The further reaction of the radical results from C3–C4 bond scission is expected to result in the formation of multifunctional products.
O3 reaction
To the best of our knowledge there are no reports in the literature on the yields of formaldehyde from the ozonolysis of mono- or bicyclic compounds containing an exocyclic double bond. There is information, however, on the HCHO yield from the ozonolysis of acyclic alkenes with structure R1R2CCH2 (R = alkyl group). For the few acyclic alkenes that have been investigated the fragmentation of the ozonide to form the most substituted Criegee Intermediate (CI) is preferred, i.e. fragmentation to form R1R2C˙OO˙ and HCHO, with yields of ∼70%.33 This is completely opposite to what we have observed for the ozonolysis of camphene where fragmentation to form the least substituted CI (˙CH2OO˙) seems to be preferred.
In previous camphene ozonolysis studies, camphenilone, 6,6-dimethyl-ε-caprolactone-2,5-methylene and cis- and trans-camphenylanaldehyde have been identified as reaction products.21,22,47 Jay and Stieglitz21 have studied the reaction in the absence and presence of SO2 at 40 °C in a glass flask using GC/MS for the product analysis. They found that camphenilone and 6,6-dimethyl-ε-caprolactone-2,5-methylene were formed in 1:1 ratio in the absence of SO2 but on adding 0.4 mg of SO2 the ratio changed to 8:1 which they interpreted as demonstrating that the Criegee biradical participated in the oxidation of SO2. On the other hand, Hakola et al.22 have reported fractional yields of (36 ± 6)% for camphenilone and (20 ± 3)% for 6,6-dimethyl-ε-caprolactone-2,5-methylene. The authors studied the ozonolysis at 297 K and 740 Torr total pressure in a Teflon chamber using GC/MS and GC/FTIR for the product identification and GC/FID for the quantification. The computational study of the ozonolysis of camphene by Oliveira and Bauerfeldt18 using a chemical model predicts 58 and 42% for the formation of camphenilone and 6,6-dimethyl-ε-caprolactone-2,5-methylene, respectively.
Ozone molecules react with unsaturated compounds through a concerted 1,3-dipolar cycloaddition to the double bond to form a thermally unstable 1,2,3-trioxolane adduct also known as a primary ozonide. This initially formed adduct fragments by rupture of one of the O–O–O bonds and the C–C bond. Depending on which of the O–O bonds ruptures two pairs of products result each pair consisting of a carbonyl compound and an energy rich biradical known as a Criegee biradical (CI). Subsequence reactions of the CI lead to the formation of numerous products.
A simplified mechanism for the reaction of ozone with camphene is shown in Fig. 7. The addition of O3 to camphene double bond forms a primary ozonide that can decompose through two pathways, A and B. Decomposition of the ozonide through pathway A will lead to the formation of camphenilone and the ˙CH2OO˙ biradical, while decomposition of the ozonide via pathway B forms formaldehyde along with a bicyclic Criegee biradical. Jay and Stieglitz.21,47 have proposed that this bicyclic Criegge biradical can form a dioxirane, the rearrangement of which can form 6,6-dimethyl-ε-caprolactone-2,5-methylene as shown in Fig. 7, pathway B.
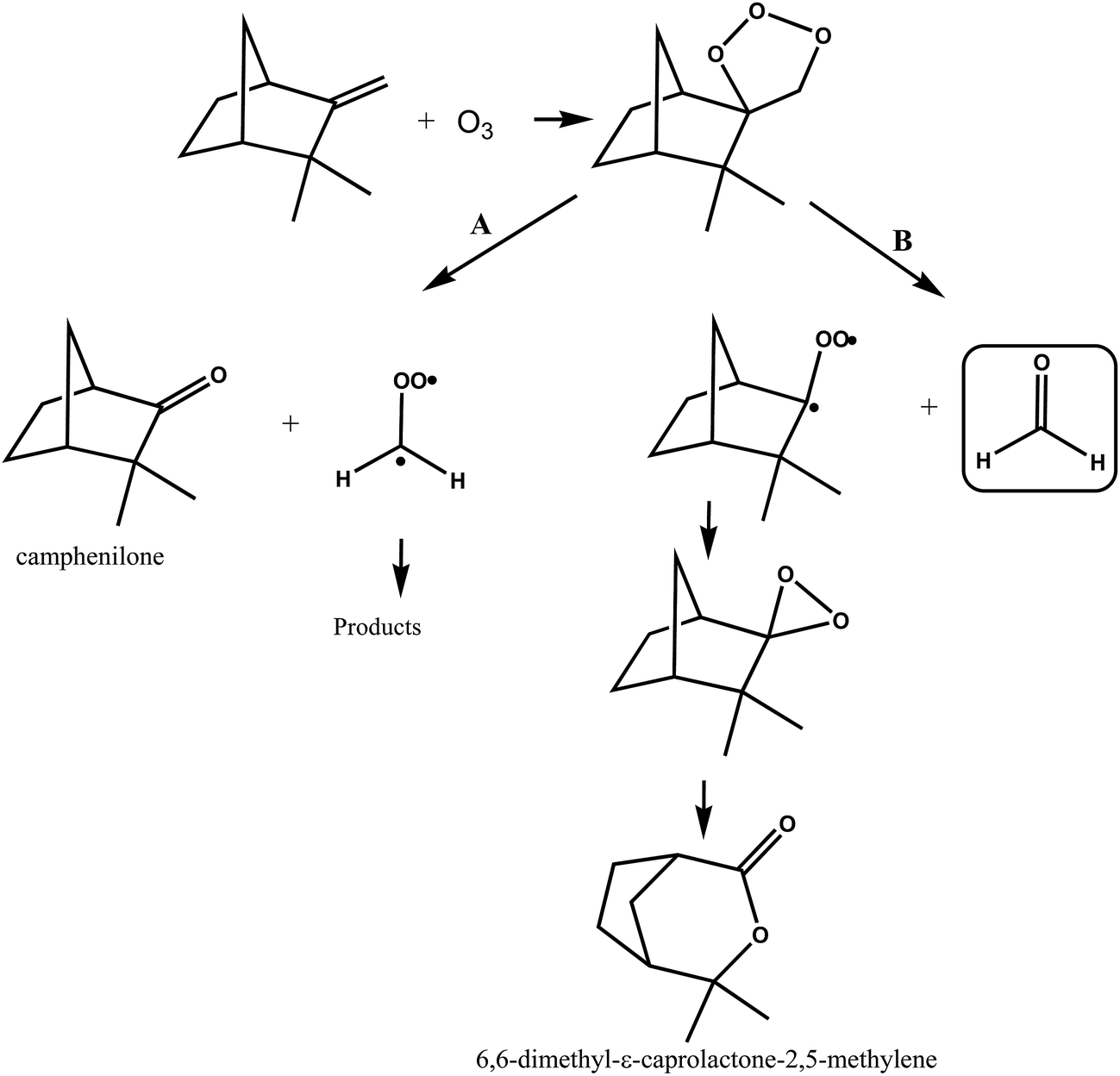 | ||
| Fig. 7 Simplified mechanism for the O3-molecule initiated oxidation of camphene via addition of O3 to the double bond. | ||
Working on the premise that the sum of the yields of the primary carbonyls formed from the two possible splitting routes of the primary ozonide must be unity the following observations can be made. If we assume that the HCHO formation observed in the ozonolysis of camphene stems overwhelmingly from the breakdown of the primary ozonide then the preferred reaction channel of the primary ozonide is decomposition to form the less substituted Criegee intermediate ˙CH2OO˙ by ∼70%, i.e. pathway A in Fig. 7. Assuming that the decomposition of the primary ozonide is the main source of HCHO in the reaction system is not unreasonable since although the ozonolysis of camphene will produce OH radicals the yield is low37 and the work here has shown that the yield of HCHO from the reaction of OH with camphene in the absence of NO is also very low. Irrespective of the source of HCHO measured in the present work its yield represents in the worst case an upper limit for the fraction of the ozonolysis proceeding by pathway B in Fig. 7. The 20% yield of 6,6-dimethyl-ε-caprolactone-2,5-methylene measured by Hakola et al.22 is also an indication that the pathway producing the bicyclic Criegee biradical (Fig. 7, pathway B) may not be the dominant O–O bond rupture channel for the primary ozonide.
The yield of camphenilone of (36 ± 6)% measured by Hakola et al.22 is much less than the ∼70% predicted from this work for rupture of the ozonide O–O bond via pathway A (Fig. 7). Indeed combining this yield with the yield for HCHO determined in this study only gives a yield of ∼66% for the primary carbonyls resulting from the primary ozonide fragmentation. Assuming that the splitting ratio for the fragmentation of the primary ozonide determined here is correct, then either the yield of camphenilone has been underestimated in the work of Hakola et al.22 or there is another possible fragmentation channel in addition to that forming camphenilone.
Atmospheric implications
The tropospheric lifetimes for the reaction of camphene with OH radicals, O3 molecules and NO3 radicals are given in Table 5. These lifetimes were calculated using the expression: τx = 1/kx[X] whereby X = OH, O3 or NO3, kx is the rate coefficient for the reaction of the oxidant X with camphene and [X] is the tropospheric concentration of the oxidant. The following typical oxidant concentrations have been used in the calculations: [OH] = 2 × 106 radical cm3,48 [O3] = 7 × 1011 molecule cm3,49 and [NO3] = 5 × 108 radical cm3.50 Photolytic loss of camphene will be of negligible importance in the troposphere since it does not have a functional group that absorbs significantly in the atmospheric actinic region. The calculated lifetimes for camphene indicate that the main degradation pathways are reaction with OH radicals during the daytime and NO3 radicals during the nighttime. The lifetime of camphene due to reaction of O3 is several days. However, the O3 concentration used in the calculations was representative of the typical concentration in remote areas, therefore in urban environments and under smog conditions were O3 levels could be much higher, loss of camphene due to ozonolysis may play a contributing role. The calculated lifetimes of camphene indicate that this compound is degraded close to its emission source.The work has validated that the reaction of OH with camphene is a relevant source of acetone, particularly in polluted environments, however, the majority of the other oxygenated products still remain to be identified. The yield of formaldehyde from the ozonolysis of camphene has been determined for the first time and with the yields of the oxygenated compounds determined previously by Jay and Stieglitz21,47 and Hakola et al.22 helps in assessing the relative contributions of the branching ratios for the various reaction channels. Acetone and formaldehyde are sources of HOx radicals and acetone is also a source of peroxy acetyl nitrate (PAN)51–54 and the present work will help to better assess the contribution of camphene to the atmospheric global acetone source strength and the HOx radical pool.
Monoterpenes are known to impact on SOA formation55–57 and recent studies have demonstrated that infected tree pines induced the release of monoterpenes, which include camphene, leading to the formation of SOA.58,59 Camphene is also one of the monoterpenes emitted from soil and leaf litter.60 Although SOA was not a topic of this investigation the work showed that SOA formation was prevalent in the OH-radical mediated oxidation of camphene, particularly in the presence of NO.
Acknowledgements
We gratefully acknowledge the financial support for this research provided by the EU project EUROCHAMP2 (E2-2013-02-27-0086), CONICET, PIP-GI 2010-2012-cod: 11220090100623 (Argentina), SECyT-UNC 14306/24. (Argentina) and FONCyT Préstamo BID 1201/OC-AR (Argentina). E. G.-C. wish to thank CONICET for a PhD fellowship. M. B. Blanco wishes to acknowledge the Alexander von Humboldt Foundation (Germany) for support.References
- M. Komenda and R. Koppmann, J. Geophys. Res., 2002, 107(D13), 4161, DOI:10.1029/2001JD000691.
- S. Moukhtar, C. Couret, L. Rouil and V. Simon, Sci. Total Environ., 2006, 354, 232–245 CrossRef CAS PubMed.
- G. Mazza and T. Cottrell, J. Agric. Food Chem., 1999, 47, 3081–3085 CrossRef CAS PubMed.
- J. Kesselmeier and M. Staudt, J. Atmos. Chem., 1999, 33, 23–88 CrossRef CAS.
- J. Kesselmeier, U. Kuhn, A. Wolf, M. O. Andreae, P. Cicciolo, E. Brancaleoni, M. Frattoni, A. Guenther, J. Greenberg, P. De Castro Vaconcellos, P. Telles de Oliva, T. Tavares and P. Artaxo, Atmos. Environ., 2000, 34, 4063–4072 CrossRef CAS.
- I. T. Baldwin, R. Halitschke, A. Paschold, C. C. von Dahl and C. A. Preston, Science, 2006, 311, 812–815 CrossRef CAS PubMed.
- K. L. Metlen, E. T. Aschehoug and R. M. Callaway, Plant, Cell Environ., 2009, 32, 641–653 CrossRef CAS PubMed.
- A. Novoplansky, Plant, Cell Environ., 2009, 32, 726–741 CrossRef PubMed.
- J. K. Holopainen and J. Gershenzon, Trends Plant Sci., 2010, 15, 176–184 CrossRef CAS PubMed.
- A. R. War, M. G. Paulraj, T. Ahmad, A. A. Buhroo, B. Hussain, S. Ignacimuthu and H. C. Sharma, Plant Signaling Behav., 2012, 7, 1306–1320 CrossRef PubMed.
- H. Panda, Aromatic plants cultivation, processing and uses, Asia Pacific Business Press Inc., 2005 Search PubMed.
- P. F. Landrum, G. A. Pollock and J. N. Seiber, Chemosphere, 1976, 5, 63–69 CrossRef CAS.
- A. Trukhina, F. Kruchkova, L. K. Hansenb, R. Kallenbornc, A. Kiprianovaa and V. Nikiforov, Chemosphere, 2007, 67, 1695–1700 CrossRef PubMed.
- A. Calogirou, B. R. Larsen and D. Kotzias, Atmos. Environ., 1999, 33, 1423–1439 CrossRef CAS.
- R. Atkinson and J. Arey, Atmos. Environ., 2003, 37, S197–S219 CrossRef CAS.
- R. Atkinson, S. M. Aschmann and J. Arey, Atmos. Environ., Part A, 1990, 24, 2647–2654 CrossRef.
- D. Johnson, A. R. Rickard, C. D. McGill and G. Marston, Phys. Chem. Chem. Phys., 2000, 2, 323–328 RSC.
- R. C. d. M. Oliveira and G. F. Bauerfeldt, J. Chem. Phys., 2012, 137, 134306 CrossRef PubMed.
- R. C. d. M. Oliveira and G. F. Bauerfeldt, J. Phys. Chem. A, 2015, 119, 2802–2812 CrossRef CAS PubMed.
- J. Peeters, W. Boullart, V. Pultau, S. Vandenberk and L. Vereecken, J. Phys. Chem. A, 2007, 111, 1618–1631 CrossRef CAS PubMed.
- K. Jay and L. Stieglitz, Atmos. Environ., 1989, 23(6), 1219–1221 CrossRef CAS.
- H. Hakola, J. Arey, S. M. Aschmann and R. Atkinson, J. Atmos. Chem., 1994, 18, 75–102 CrossRef CAS.
- A. Reissell, C. Harry, S. M. Aschmann, R. Atkinson and J. Arey, J. Geophys. Res., 1999, 104, 13869–13879 CrossRef CAS.
- A. Guenther, C. Hewitt, D. Erickson, R. Fall, C. Geron, T. Graedel, P. Harley, L. Klinger, M. Lerdau, W. McKay, T. Pierce, B. Scholes, R. Steinbrecher, R. Tallamraju, J. Taylor and P. Zimmerman, J. Geophys. Res., 1995, 100, 8873–8892 CrossRef CAS.
- W. Zhihui, B. Yuhua and Z. Shuyu, Atmos. Environ., 2003, 37, 3771–3782 CrossRef.
- I. Barnes, K. H. Becker and T. Zhou, J. Atmos. Chem., 1993, 17, 353–373 CrossRef CAS.
- I. Barnes, K. H. Becker and N. Mihalopoulos, J. Atmos. Chem., 1994, 18, 267–289 CrossRef CAS.
- R. Atkinson, S. M. Aschmann and W. P. Carter, Int. J. Chem. Kinet., 1983, 15, 1161–1177 CrossRef CAS.
- R. Atkinson, J. Phys. Chem. Ref. Data, 1997, 26, 215–290 CrossRef CAS.
- J. Treacy, M. El Hag and D. O'Farrel, Ber. Bunsen-Ges. Phys. Chem., 1992, 3, 422–427 CrossRef.
- R. Atkinson, S. M. Aschmann, D. R. Fitz, A. M. Winer and J. N. Pitts Jr, Int. J. Chem. Kinet., 1982, 14, 13–18 CrossRef CAS.
- T. E. Kleindienst, G. W. Harris and J. R. Pitts, Environ. Sci. Technol., 1982, 16, 844–846 CrossRef CAS PubMed.
- B. Chuong, M. Davis, M. Edwards and P. S. Stevens, Int. J. Chem. Kinet., 2002, 34, 300–308 CrossRef CAS.
- K. J. Gill and R. A. Hites, J. Phys. Chem. A, 2002, 106, 2538–2544 CrossRef CAS.
- A. Montenegro, J. S. A. Ishibashi, P. Lam and Z. Li, J. Phys. Chem. A, 2012, 116, 12096–12103 CrossRef CAS PubMed.
- J. G. Calvert, R. Atkinson, J. A. Kerr, A. Madronich, G. K. Moorgat, T. J. Wallington and G. Yarwood, The mechanisms of atmospheric oxidation of alkenes, Oxford University Press, Oxford, 2000 Search PubMed.
- J. G. Calvert, J. J. Orlando, W. R. Stockwell and T. J. Wallington, The mechanisms of reactions influencing atmospheric ozone, Oxford University Press, Oxford, 2015 Search PubMed.
- R. Atkinson, A. M. Winer and J. N. Pitts Jr, Atmos. Environ., 1982, 16, 1017–1020 CrossRef CAS.
- F. Nolting, W. Behnke and C. A. Zetzsch, J. Atmos. Chem., 1988, 6, 47–59 CrossRef CAS.
- D. Grosjean, E. L. Williams II, E. Grosjean, J. M. Andino and J. H. Seinfeld, Environ. Sci. Technol., 1993, 27(13), 2754–2758 CrossRef CAS.
- V. G. Khamaganov and R. Hites, J. Phys. Chem., 2001, 105, 815–822 CrossRef CAS.
- E. C. Tuazón, H. Mac Leod, R. Atkinson and W. P. L. Carter, Environ. Sci. Technol., 1986, 20, 383–387 CrossRef PubMed.
- IUPAC Task Group on Atmospheric Chemical Kinetic Data Evaluation, http://iupac.pole-ether.fr, 2007.
- J. J. Orlando, B. Noziére, G. S. Tyndall, G. E. Orzechowska, S. E. Paulson and Y. Rudich, J. Geophys. Res., 2000, 105(9), 11561–11572 CrossRef CAS.
- P. D. Lightfoot, R. A. Cox, J. N. Crowley, M. Destriau, G. D. Hayman, M. E. Jenkin, G. K. Moortgat and F. Zabel, Atmos. Environ., Part A, 1992, 26, 1805–1961 CrossRef.
- J. J. Orlando and G. S. Tyndall, Chem. Soc. Rev., 2012, 41, 6294–6317 RSC.
- K. Jay and L. Stieglitz, Proceedings of an International Symposium, France, 18–22 May 1987, pp. 542–547 Search PubMed.
- R. Hein, P. J. Crutzen and M. Heimann, Global Biogeochem. Cycles, 1997, 11, 43–76 CrossRef CAS.
- J. A. Logan, J. Geophys. Res., 1985, 90(D6), 10463–10482 CrossRef.
- Y. Shu and R. Atkinson, J. Geophys. Res., 1995, 100(D4), 7275–7281 CrossRef CAS.
- S. A. Mc Keen, G. Mount, F. Eisele, E. Williams, J. Harder, P. Goldan, W. Kuster, S. C. Liu, K. Baumann, D. Tanner, A. Fried, S. Sewell, C. Cantrell and R. Shetter, J. Geophys. Res., 1997, 102, 6467–6493 CrossRef CAS.
- J. F. Müller and G. Brasseur, J. Geophys. Res., 1999, 104, 1705–1715 CrossRef.
- A. H. Goldstein and G. W. Schade, Atmos. Environ., 2000, 34, 4997–5006 CrossRef CAS.
- E. V. Fischer, D. J. Jacob, R. M. Yantosca, M. P. Sulprizio, D. B. Millet, J. Mao, F. Paulot, H. B. Singh, A. Roiger, L. Ries, R. W. Taulbot, K. Dzepina and S. Pandey Deolal, Atmos. Chem. Phys., 2014, 14, 2679–2698 Search PubMed.
- M. Kanakidou, J. H. Seinfeld, S. N. Pandis, I. Barnes, F. J. Dentener, M. C. Facchini, R. van Dingenen, B. Ervens, A. Nenes, C. J. Nielsen, E. Swietlicki, J. P. Putaud, Y. Balkanski, J. Fuzzi, J. S. Horth, G. K. Moortgat, R. Winterhalter, C. E. L. Myhre, K. Tsigaridis, E. Vignati, E. G. Stephanou and J. Wilson, Atmos. Chem. Phys., 2005, 5, 1053–1123 CrossRef CAS.
- L. M. Russell, R. Bahadur and P. Ziemann, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3516–3521 CrossRef CAS PubMed.
- A. Kahnt, Y. Linuma, A. Mutzel, O. Böge, M. Claeys and H. Herrmann, Atmos. Chem. Phys., 2014, 14, 719–736 Search PubMed.
- H. Amin, P. T. Atkins, R. S. Russo, A. W. Brown, B. Sive, A. G. Hallar and K. E. Huff Hartz, Environ. Sci. Technol., 2012, 46, 5696–5703 CrossRef CAS PubMed.
- R. P. Ghimire, J. M. Markkanen, M. Kivimäenpää, P. Lyytikäinen-Saarenmaa and J. K. Holopainen, Environ. Sci. Technol., 2013, 47, 4325–4332 CrossRef CAS PubMed.
- C. L. Faiola, G. S. VanderSchelden, M. Wen, F. C. Elloy, D. R. Cobos, R. J. Watts, B. T. Jobson and T. M. VanReken, Environ. Sci. Technol., 2014, 48, 938–946 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Extensive supporting information, mainly in the form of diagrams as indicated in the text. See DOI: 10.1039/c6ra26656h |
| This journal is © The Royal Society of Chemistry 2017 |