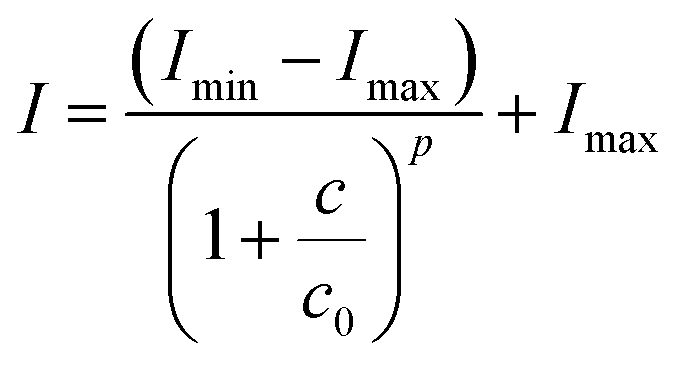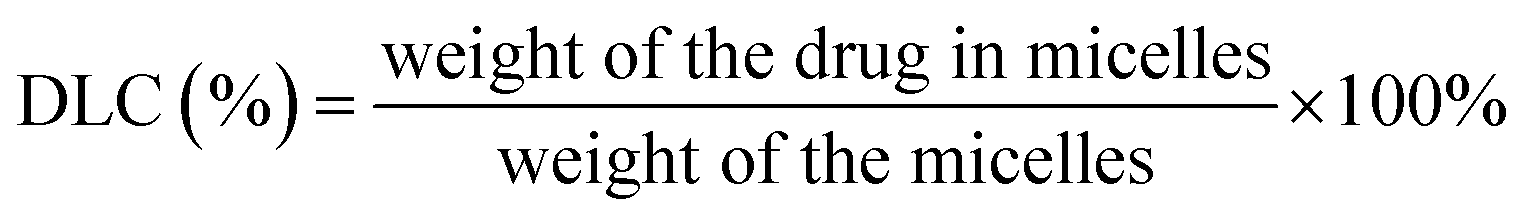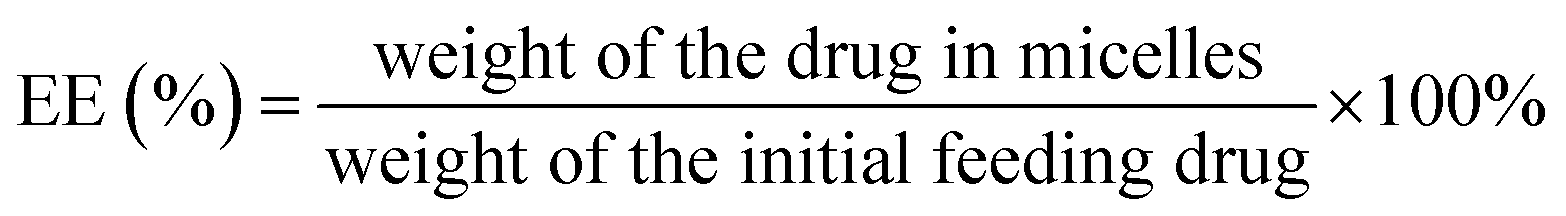DOI:
10.1039/C6RA26669J
(Paper)
RSC Adv., 2017,
7, 11158-11169
Imidazole-modified deferasirox encapsulated polymeric micelles as pH-responsive iron-chelating nanocarrier for cancer chemotherapy†
Received
11th November 2016
, Accepted 4th February 2017
First published on 13th February 2017
Abstract
Deferasirox (Def) is an iron-chelating drug used to reduce iron overload in β-thalassemia patients. After the discovery of its tumor growth inhibition, this drug gained tremendous attention in cancer chemotherapy. Herein, deferasirox and its derivatives including methoxy (mDef) and imidazole-modified (iDef) deferasirox were encapsulated in polymeric micelles. Results showed that the release of deferasirox from polymeric micelles was faster at pH 7.4 (physiological condition) than at pH 4.5 (lysosomal condition). However, the release of mDef was pH-independent where both Def and mDef are not suitable for in vivo applications. Therefore, iDef was synthesized to change the pKa from 3.7 (carbonyl group of Def) to 6.8 (imidazole group of iDef) without interfering the iron-chelating efficacy. Interestingly, the release rate of imidazole-modified deferasirox was conversed from that of deferasirox where it exhibited slow release at physiological condition and faster release at the lysosomal condition. This release profile showed a pH-response and an ON–OFF release behavior where iDef is encapsulated in micelles during systemic circulation and released inside cancer cells after intracellular endosomes/lysosome. Cytotoxicity of deferasirox and modified deferasirox were also investigated, and it was found that the IC50 against PC-3 and HepG2 cell lines were in the range of micromolar to submicromolar. Flow cytometry analysis confirmed a decrease in the amount of iron inside lysosome when cells were treated by iDef-loaded micelles. Therefore, iDef-loaded micelles have potential application in cancer treatment as a pH-responsive iron-chelating nanocarrier.
1. Introduction
Iron is essential for many proteins in the body to maintain normal cellular function. It can also act as a cofactor in many biological pathways such as synthesis of ATP, DNA, and neurotransmitters. However, an excess iron level in cells can cause toxicity since it can generate highly reactive oxygen species (ROS), hydroxyl radical (HO˙) or ferryl [Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O]2+ by Fenton's reaction which can damage cells.1 Normally, cells maintain iron homeostasis by regulating iron absorption, transportation, efflux and storage. Iron is stored inside cells in two major forms, i.e., ferritin and hemosiderin. Studies have been reported that cancer cells express higher iron level due to reprogrammed iron metabolism. This results in an increase in transferrin receptor (influx iron) and a decrease in ferroportin (efflux iron) on cancer cell membrane.2 Therefore, inhibiting iron metabolism in cancer cells is a novel strategy for the treatment of cancer.3 The antiproliferation along with antineoplasticity of cancer cells were found when the iron level in cancer cells depleted after treatment with iron chelating drugs such as deferiprone, desferrioxamine and deferasirox.4,5 Hence, new iron-chelating compounds were designed and synthesized as chemotherapeutic agent including Dp44mT for breast and melanoma cancer,6 triapine and series of pyridoxal isonicotinoyl hydrazone for leukemia.7
O]2+ by Fenton's reaction which can damage cells.1 Normally, cells maintain iron homeostasis by regulating iron absorption, transportation, efflux and storage. Iron is stored inside cells in two major forms, i.e., ferritin and hemosiderin. Studies have been reported that cancer cells express higher iron level due to reprogrammed iron metabolism. This results in an increase in transferrin receptor (influx iron) and a decrease in ferroportin (efflux iron) on cancer cell membrane.2 Therefore, inhibiting iron metabolism in cancer cells is a novel strategy for the treatment of cancer.3 The antiproliferation along with antineoplasticity of cancer cells were found when the iron level in cancer cells depleted after treatment with iron chelating drugs such as deferiprone, desferrioxamine and deferasirox.4,5 Hence, new iron-chelating compounds were designed and synthesized as chemotherapeutic agent including Dp44mT for breast and melanoma cancer,6 triapine and series of pyridoxal isonicotinoyl hydrazone for leukemia.7
Recently, deferasirox has been tested as an anticancer drug for many cancer cell lines. For example, IC50 against liver cancer cell line (HepaRG) was 6.5 μM8 which is similar to that of doxorubicin (IC50 = 5.5 μM).9 In addition, it was reported that cytotoxicity of deferasirox in normal liver cell line was less than that of liver cancer cell line.10 The cytotoxicity of deferasirox in various types of cancer cell lines have been reported.11–15 Deferasirox also shows synergistic in vitro anticancer effects with many well-known drugs such as docetaxel,16 sorafenib17 and bevacizumab.18 Ohara T. et al. studied the pathology of cancer cells in iron-deficient mouse model treated with bevacizumab. They reported that the pathology was similar to cancer cells treated with bevacizumab and deferasirox in the in vitro studies. However, the in vivo study did not indicated the synergistic effect between the two drugs because iron depletion from deferasirox was not specific at the tumor site.12 Currently, few studies have been reported where deferasirox has been used to treat cancer in human. Results revealed a complete remission in a 73 year-old patient who had chemotherapy-resistance acute monocytic leukemia after 12 month daily dose of deferasirox.19 The efficiency of cancer treatment can be elevated by deferasirox-substituted calix[4]arene that was synthesized and showed the improvement in an antiproliferation and antineoplasticity.20 However, the applications of this iron-chelating compound against cancers are limited to only small molecules for both in vitro and in vivo. Herein, it is the first time that pH-responsive iron-chelator-loaded micelles are fabricated to be used as a cancer chemotherapy. Although oral administration of deferasirox is likely to make patients compliant, its bioavailability (the percentage of the drug absorbed compared to its initial dosage) is highly variable including the low tissue distribution (Vss is 14.37 L) results from high plasma protein binding of deferasirox (>98%).21 This may be the reason why using deferasirox to treat hepatocellular carcinoma in human has not been achieved.22 To overcome those problems, the nanotechnology was applied to change the route of administration of deferasirox form oral to intravenous injection. Furthermore, nanoparticle therapeutic systems have several advantages over small molecule-based chemotherapies including enhanced bioavailability, longer plasma retention time and increased accumulation of a drug to tumor tissues.23 The targeting ligand can be attached to nanoparticles in order to increase the selective uptake resulting in enhancement of drug delivery in tumors and reduce side effects.24
Polymeric micelles are self-assembly nanocarrier made from block copolymers which comprises of hydrophobic core and hydrophilic shell. The core can encapsulate the hydrophobic drug, which increases its solubility and stability.25 The shell can prolong the circulation half-life of the drug delivery system improving its pharmacokinetics. Poly(ethylene glycol) (PEG) is widely used as the shell due to its stealth property and hydrophilicity which allows micelles to avoid mononuclear phagocyte system (MPS).26 Additionally, nanoscale carriers including micelles with a diameter between 10 to 200 nm can be accumulated within solid tumor by enhanced permeability and retention (EPR) effect.27 Micelles can be internalized into cancer cells via endocytosis pathway where cells produce endocytic vesicles that are transformed to early endosomes, late endosomes and finally to lysosomes. Intracellular pH gradients were reported along this path, i.e., early endosome (pH 5.9–6.2), late endosome (pH 5.0–5.5) and lysosome (pH 4.5–5.0).28 This provides the ideal condition for the controlled release of drugs only under the acidic condition. Therefore, most of the drugs will be released inside the cells and the release rate will be very slow during blood circulation (pH 7.4). Currently, two common strategies are being used to develop the pH-responsive of polymeric nanocarriers. One is the conjugation of drugs to polymers by pH-responsive linker.29 The other strategy is the conjugation of pH-responsive moieties to polymers backbone.30 In this present work, unlike strategies mentioned above, the modification of deferasirox by conjugation of pH-sensitive moiety was prepared to change pKa while maintaining its iron binding efficiency. These modified drugs were encapsulated into PEG-b-PCL micelles without any further modification. The properties of micelles were determined and in vitro release profiles were investigated for their performance as a pH-responsive nanocarrier including fluorescence microscope and flow cytometry. IC50 of all formulations were also determined to evaluate their potential use for cancer chemotherapy.
2. Results and discussion
2.1 Synthesis and characterization of methoxy and imidazole-modified deferasirox (mDef and iDef)
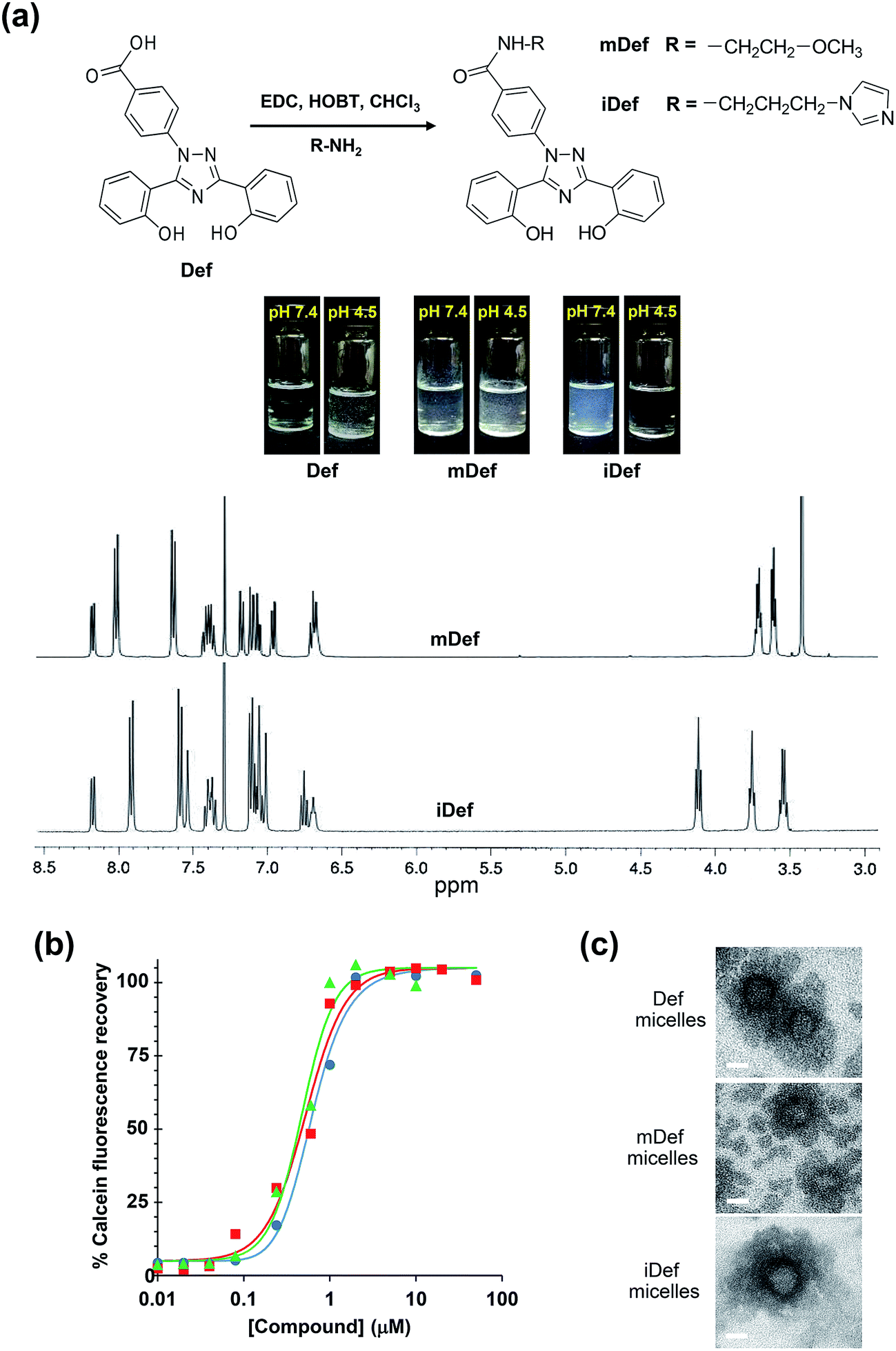
The synthesis was carried out using a modified method according to a literature report31 and the schematic diagram of the synthetic method was shown in Fig. 1a. Methoxy-modified deferasirox was synthesized by EDC/HOBT coupling between the primary amine group of 2-methoxyethylamine and the carboxyl group of deferasirox. The successful conjugation was confirmed by proton nuclear magnetic resonance (1H-NMR) and mass spectroscopy. The chemical shifts of mDef (Fig. 1a) were assigned as follows 1H-NMR (CDCl3) δ: 3.42 (s, 3H), 3.61 (t, J = 4.8 Hz, 2H), 3.71 (q, J = 5.0 Hz, 2H), 6.65–6.69 (m, 2H), 6.94 (dd, J = 8.0, 1.2 Hz, 1H), 7.03–7.09 (m, 2H), 7.15 (d, J = 8.4 Hz, 1H), 7.33–7.41 (m, 2H), 7.60 (d, J = 8.4 Hz, 2H), 7.98 (d, J = 8.4 Hz, 2H) 8.14 (dd, J = 7.8, 1.4 Hz, 1H). NMR spectrum showed the signals of ethylene protons at 3.61 and 3.71 and methoxy protons at 3.42 ppm at the ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3, respectively. The conjugation was also confirmed by mass spectroscopy. The molecular weight (MW) of the original compound (Def) [M + Na]+ was 374 Da.32 The mass spectrum indicated that the MW of mDef [M + Na]+ was 453.15 Da, whereas the calculated MW [M + H]+ was 431.16 Da based on its molecular structure (C24H22N4O4) (ESI, Fig. S1†). Synthesis of imidazole-modified deferasirox was similar to that of methoxy-modified deferasirox but here 1-(3-aminopropyl) imidazole was used instead. The chemical shifts of iDef (Fig. 1a) were assigned as follows 1H-NMR (CDCl3) δ: 3.53 (dd, J = 12, 8 Hz, 2H), 3.74 (t, J = 8 Hz, 2H), 4.10 (t, J = 8 Hz, 2H), 6.67 (bs, 1H), 6.73 (t, J = 8 Hz, 1H), 6.98–7.09 (m, 6H), 7.32–7.39 (m, 2H), 7.50 (s, 1H), 7.55 (d, J = 8 Hz, 2H), 7.88 (d, J = 8 Hz, 2H), 8.14 (dd, J = 12 Hz, 1H). NMR spectrum showed the signal of imidazole protons at 6.67 and 7.50. The mass spectrum indicated that the molecular weight of iDef [M + H]+ was 481.20 Da whereas the calculated MW [M + H]+ was 481.19 Da based on its molecular structure (C27H24N6O3) (ESI, Fig. S1†). Both conjugation introduced methoxy and imidazole to the carboxyl group of deferasirox resulting in the change in pKa as shown in Table 1. The pKa of both derivatives was estimated by MarvinSketch software. The pKa of mDef was switched to 8.9 whereas the pKa of iDef was 6.8. The solubility of all compounds was compared by visual observation as shown in Fig. 1 and Table 1. Each compound was dissolved in different aqueous environment including PBS pH at 7.4 and PBS pH at 4.5 at the same concentration of 100 μg mL−1. Acidic compounds containing proton-donating group such as Def and mDef are soluble if the pH of the solution is considerably higher than pKa. For basic compound containing proton-accepting group, like iDef, then the compound is soluble (if pH of the solution is considerably lower than pKa). The results displayed that mDef was insoluble in all conditions while Def was insoluble in PBS pH 4.5. Interestingly, iDef was insoluble in PBS pH at 7.4 but dissolved very well in acidic condition pH at 4.5. Solubility of Def and its derivatives depends on the pKa of each compound.
:2:3, respectively. The conjugation was also confirmed by mass spectroscopy. The molecular weight (MW) of the original compound (Def) [M + Na]+ was 374 Da.32 The mass spectrum indicated that the MW of mDef [M + Na]+ was 453.15 Da, whereas the calculated MW [M + H]+ was 431.16 Da based on its molecular structure (C24H22N4O4) (ESI, Fig. S1†). Synthesis of imidazole-modified deferasirox was similar to that of methoxy-modified deferasirox but here 1-(3-aminopropyl) imidazole was used instead. The chemical shifts of iDef (Fig. 1a) were assigned as follows 1H-NMR (CDCl3) δ: 3.53 (dd, J = 12, 8 Hz, 2H), 3.74 (t, J = 8 Hz, 2H), 4.10 (t, J = 8 Hz, 2H), 6.67 (bs, 1H), 6.73 (t, J = 8 Hz, 1H), 6.98–7.09 (m, 6H), 7.32–7.39 (m, 2H), 7.50 (s, 1H), 7.55 (d, J = 8 Hz, 2H), 7.88 (d, J = 8 Hz, 2H), 8.14 (dd, J = 12 Hz, 1H). NMR spectrum showed the signal of imidazole protons at 6.67 and 7.50. The mass spectrum indicated that the molecular weight of iDef [M + H]+ was 481.20 Da whereas the calculated MW [M + H]+ was 481.19 Da based on its molecular structure (C27H24N6O3) (ESI, Fig. S1†). Both conjugation introduced methoxy and imidazole to the carboxyl group of deferasirox resulting in the change in pKa as shown in Table 1. The pKa of both derivatives was estimated by MarvinSketch software. The pKa of mDef was switched to 8.9 whereas the pKa of iDef was 6.8. The solubility of all compounds was compared by visual observation as shown in Fig. 1 and Table 1. Each compound was dissolved in different aqueous environment including PBS pH at 7.4 and PBS pH at 4.5 at the same concentration of 100 μg mL−1. Acidic compounds containing proton-donating group such as Def and mDef are soluble if the pH of the solution is considerably higher than pKa. For basic compound containing proton-accepting group, like iDef, then the compound is soluble (if pH of the solution is considerably lower than pKa). The results displayed that mDef was insoluble in all conditions while Def was insoluble in PBS pH 4.5. Interestingly, iDef was insoluble in PBS pH at 7.4 but dissolved very well in acidic condition pH at 4.5. Solubility of Def and its derivatives depends on the pKa of each compound.
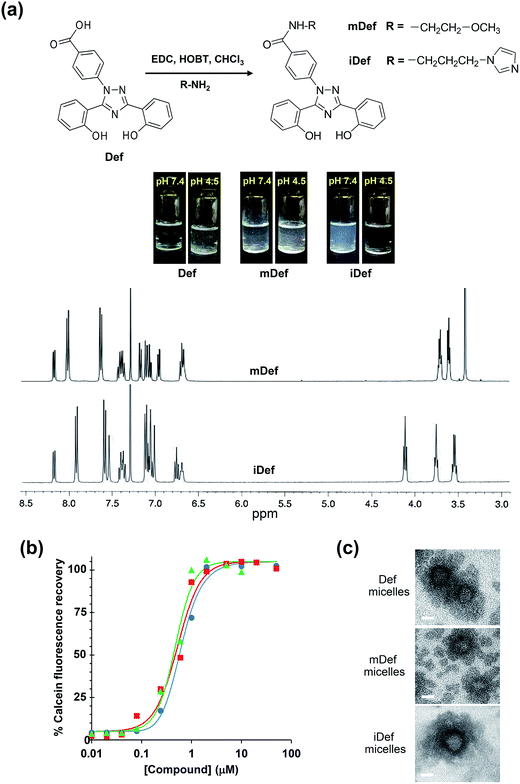 |
| | Fig. 1 (a) Schematic diagram of the synthesis and chemical structure of deferasirox (Def), methoxy-modified deferasirox (mDef) and imidazole-modified deferasirox (iDef). Solubility of Def, mDef and iDef at 100 μg mL−1 in PBS at pH 7.4 and PBS at pH 4.5, respectively. 1H-NMR spectrum of mDef and iDef. (b) The iron-chelating efficiency of deferasirox and its derivatives determined by calcein fluorescence measurement. Calcein fluorescence was quenched by adding iron(II) and subsequently recovered after adding deferasirox (Def) (●), methoxy-modified (mDef) (■) or imidazole-modified deferasirox (iDef) (▲). (c) TEM images of Def-loaded micelles, mDef-loaded micelles and iDef-loaded micelles. Scale bars were 20 nm. | |
Table 1 Properties of free and micelle form of deferasirox (Def), methoxy-modified deferasirox (mDef) and imidazole-modified deferasirox (iDef)
| Compound name |
Compound properties |
Micellar properties |
| m/z [M + H]+ |
pKa |
Solubilityc at 100 μg mL−1 |
Chelator affinity (CR50) |
DLC (%) |
EE (%) |
Sized (nm)/DPI |
Zeta potentiale (mV) |
Time to 50% release (day) |
| pH 4.5 |
pH 7.4 |
pH 7.4 |
pH 4.5 |
| [M + Na]+. Calculated pKa using MarvinSketch (ChemAxon Ltd.). Fig. 1a. Average particle size. Fig. S3. |
| Def |
374 |
3.7 |
Precipitate |
Soluble |
0.62 |
4.8 ± 0.6 |
93.2 ± 1.8 |
21.8/0.248 |
−11.5 ± 0.7 |
0.5 |
>7.0 |
| mDef |
453a |
8.9b |
Precipitate |
Precipitate |
0.53 |
4.6 ± 0.3 |
93.3 ± 6.7 |
25.4/0.104 |
−0.9 ± 0.2 |
4.0 |
4.0 |
| iDef |
481 |
6.8b |
Soluble |
Precipitate |
0.47 |
4.8 ± 0.5 |
95.4 ± 1.0 |
23.2/0.238 |
−1.7 ± 0.7 |
>7.0 |
0.9 |
Fluorescence quenching of calcein by iron was used to determine the chelating efficiency of deferasirox and its derivatives by competition method where the fluorescence intensity of calcein is decreased or quenched by iron.8 When the iron-chelator (deferasirox) is added, it competitively binds to iron resulting in the recovery of calcein fluorescence. Chelator affinity (RC50) is defined as the concentration of chelator required to achieve half-maximal fluorescence recovery. The 4-parameter logistic (4PL) nonlinear regression model was used for curve-fitting to determine RC50 value as well as dose–response curves for the determination of IC50 according to Rodbard.33
I is the fluorescence intensity,
Imin and
Imax are minimal and maximal fluorescence intensity, respectively,
c is the drug concentration in logarithmic scale,
c0 is the inflection point which is RC
50 or IC
50 in logarithmic scale and
p is the slope at the inflection point of the RC
50 values where the chelator efficiency is inversely proportional to the RC
50. RC
50 values of all compounds were not significantly different (
Fig. 1b) and they were in the same order of magnitude as shown in
Table 1. This was due to the iron complexation which occurred at the two phenolate groups and one of the nitrogen atoms of the heterocycle.
34 Therefore, the modification of deferasirox through the carboxyl group did not affect the binding efficiency. This result is similar to the previous reports on deferasirox conjugation.
35,36
2.2 Preparation and characterization of iron-chelator loaded polymeric micelles
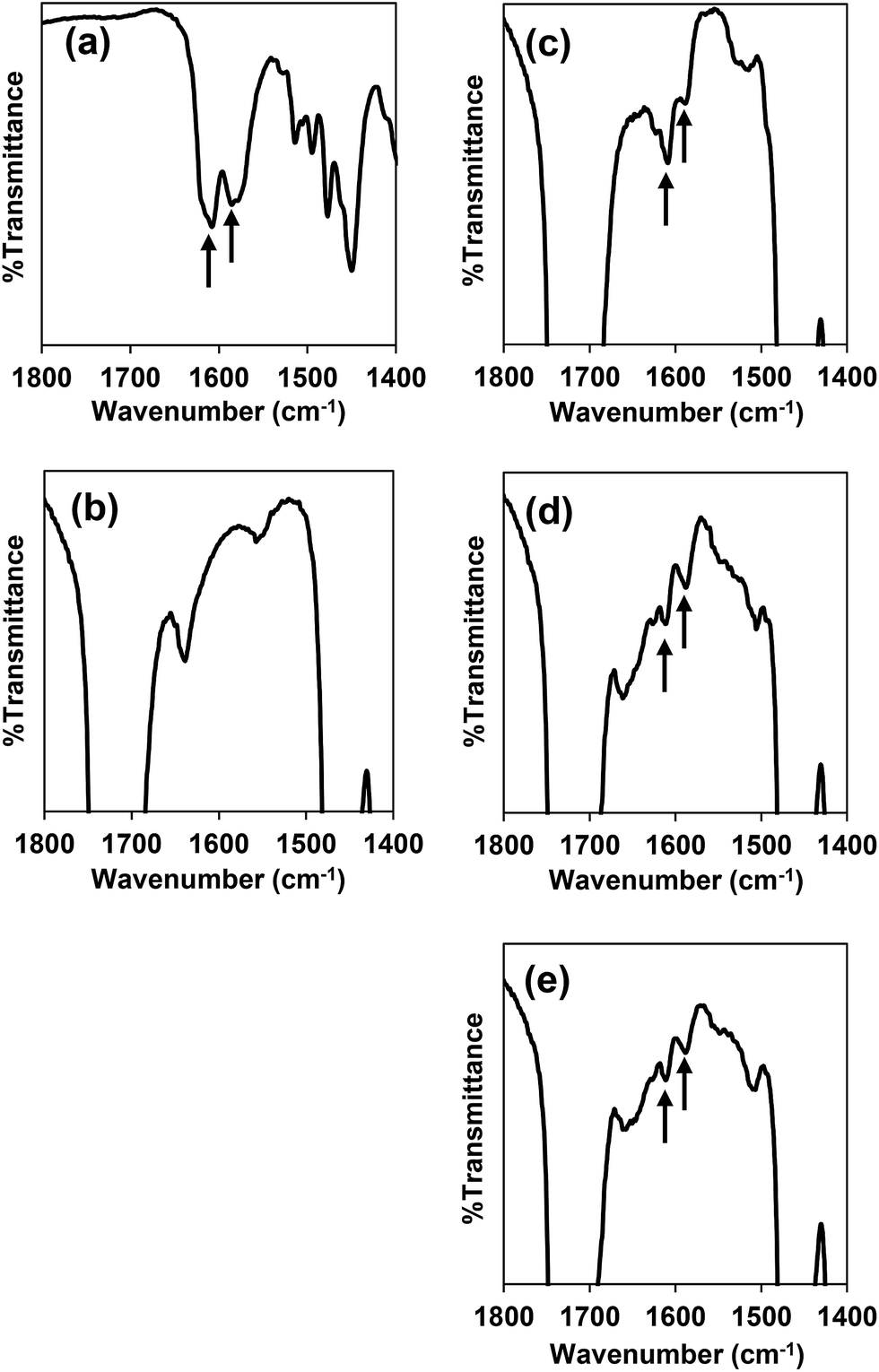
PEG-b-PCL was synthesized through ring-opening polymerization of ε-caprolactone using stannous(II) octanoate as the catalyst. The molecular weight of PCL was calculated by integral intensity of 1H-NMR spectrum to compare methylene proton of PEG at 3.63 ppm and methylene proton of PCL at 4.04 ppm. The molecular weight of the PCL segment is 5.07 kDa and the PDI from GPC is 1.19 (ESI, Fig. S2†). Deferasirox is the only iron-chelating drug that has a hydrophobic property with aqueous solubility of 0.4 mg mL−1 at pH 7.4 (ref. 34) thus it has the potential to be loaded into polymeric micelles. Encapsulation of deferasirox and its derivative in PEG-b-PCL micelles was successfully prepared by solvent evaporation method. The FT-IR spectrum of deferasirox and bare micelles were shown in Fig. 2a and b, respectively. The arrows indicated peaks of deferasirox and its derivative at 1585 cm−1 (aromatic, CC stretching) and 1607 cm−1 (CN stretching). The FT-IR spectrum of deferasirox-loaded micelles (Fig. 2c), mDef-loaded micelles (Fig. 2d) and iDef-loaded micelles (Fig. 2e) showed two new peaks which are characteristic of deferasirox and its derivative. The observed peaks confirmed that deferasirox and its derivatives were successfully encapsulated into micelles. Table 1 shows properties such as drug loading content (DLC) and drug loading efficiency (EE) of these compounds inside micelles. High drug loading efficiency (EE) (>93%) was found for all compounds. The average particle size of different types of micelles was not significantly different (ESI, Fig. S3†). Moreover, all formulation showed narrow size distribution with the PDI less than 0.25. The zeta potential of mDef- and iDef-loaded micelles was found to be neutral (Table 1 and ESI, Fig. S3†). It was also found that the zeta potential of the deferasirox-loaded micelles was slightly lower than those of mDef- and iDef-loaded micelles as a result of the presence of deferasirox on the micelle surface. TEM images confirmed Def, mDef and iDef loaded micelles had the spherical shape (Fig. 1c). It should be noted that DLC, EE and the size of Def, mDef and iDef were not significantly different.
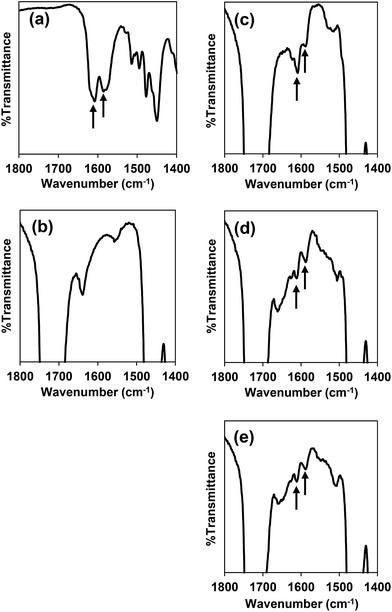 |
| | Fig. 2 FT-IR spectra of (a) deferasirox, (b) bare micelles, (c) deferasirox-loaded micelles, (d) mDef-loaded micelles, (e) iDef-loaded micelles. Arrows indicate characteristic peaks of deferasirox and its derivatives. | |
2.3 pH-dependent release of deferasirox and derivatives from polymeric micelles
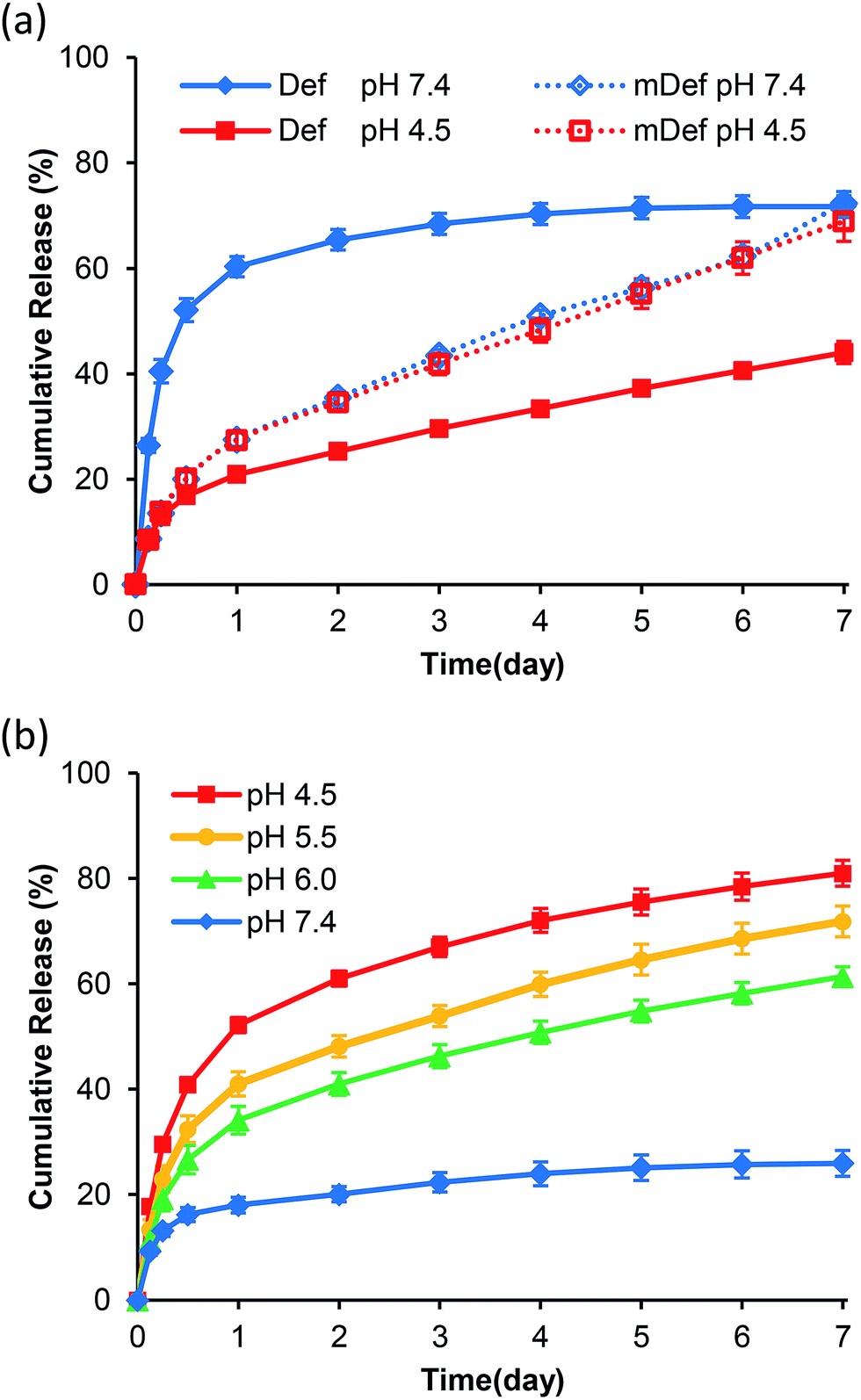
Release studies were carried out in PBS pH 7.4 (physiological condition) and pH 4.5 (lysosomal condition). Results showed that the release rate of deferasirox from micelles at pH 7.4 was faster than that of pH 4.5 as shown in Fig. 3a. Time to reach 50% release was increased from 12 hours at pH 7.4 to be more than 7 days at pH 4.5. For mDef-loaded micelles, the time to reach 50% release was unchanged by the environmental pH as shown in Fig. 3a. To clarify this phenomenon, the Henderson–Hasselbalch equation was used to determine the ratio of ionized and non-ionized form of these compounds in different pH environments. It should be noted that the ionized form of deferasirox derivative is water-soluble, which quickly diffuses out of micelles. Instead, the non-ionized form shows much slower release because it is uncharged and hydrophobic. Considering that the pKa of deferasirox is approximately 3.7 (ref. 37) so its ionized form was almost 100% at pH 7.4 while the ionized form was reduced to 86% at pH 4.5 resulting in a slower release rate. The ionized form of methoxy-modified deferasirox (pKa = 8.9) was only 3% at pH 7.4. When the pH was lowered to 4.5, the ionized form was reduced to approximately 0%. Thus, the release profile of methoxy-modified deferasirox between pH 7.4 and 4.5 are not different.
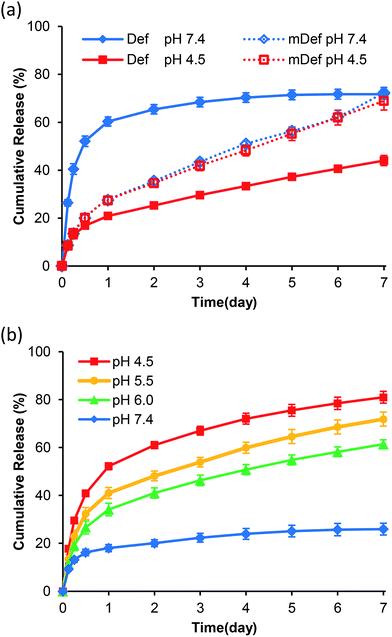 |
| | Fig. 3 (a) Release profiles at different pH conditions (PBS pH 7.4 and 4.5) of deferasirox- and mDef-loaded micelles. (b) iDef-loaded micelles release profiles at different pH conditions (PBS pH 7.4, 6.0, 5.5 and 4.5). | |
The conceptual release profile of deferasirox from micelles should have minimal release in circulation system, but the release should be faster after endocytosis to avoid systemic toxicity. Therefore, the appropriate pKa of deferasirox should be between 7.4 and 4.5. This will be an alternative strategy for pH-responsive release where the release rate at pH 7.4 is slower than that of pH 4.5. To prove this concept, the imidazole-modified deferasirox (iDef) that had pKa around 6.8 was synthesized. In theory, an ionized form of iDef at pH 7.4 is 20%. When the pH was decreased to 4.5, the percentage of an ionized form is increased up to 100%. The release profile of iDef was shown in Fig. 3b. Interestingly, the faster release rate was found at pH 4.5 which was converted from that of deferasirox. The time for 50% release of all compounds was shown in Table 1. For iDef-loaded micelles, the time to reach 50% release was decreased from more than 7 days at pH 7.4 to 22 hours (0.9 day) at pH 4.5. To confirm the pH-responsive properties of iDef-loaded micelles, release studies were tested under simulated lysosomal condition at pH 6.0 and 5.5 representing different stages of endocytosis from early endosome, respectively. Results showed that a small change in pH environment has a significant effect on the release rate as shown in Fig. 3b. The higher acidic environment provided the faster release rate where the time to reach 50% release at the pH value of 5.5 and 6.0 was 2.3 and 3.8 days, respectively. These results indicated that imidazole-modified deferasirox-loaded polymeric micelles possess a pH-responsive characteristic. Moreover, these micelles have potential for intravenous administration because iDef showed much slower release rate at pH 7.4 and higher release rate after cellular uptake.
Next, in-depth analysis of pH-responsive property of iDef-loaded micelles was carried out by switching the pH of the release buffer between pH 7.4 and 4.5 (Fig. 4a). At day 1, micelles were in pH 7.4 and the burst release was observed at the rate of 6.8 μg per hour which was then decreased to 2.7 μg per hour. It should be noted that the burst release is common for all polymeric drug delivery systems.38 After 24 hours, the buffer solution was switched to pH 4.5 and the rate was increased abruptly to 26.6 μg per hour (approximately 10-fold) which was much faster than that of pH 7.4. The release rate was successfully controlled as ON–OFF characteristic as demonstrated by the reversible switching between high and low release rate. It was found that the average particle size and PDI of micelles did not change significantly during the pH-switching between 7.4 and 4.5 as shown in Fig. 4b and S4 (ESI†). These results indicated that acid–base switching could change the ionized form of iDef inside the micelle core without affecting the size of polymeric micelles. For general pH-responsive polymeric micelles, drug release can be triggered by the protonation of pH-responsive moieties inside the hydrophobic core under acidic condition. The electrostatic repulsion among polymer chains and loss of hydrophobic interaction lead to the destabilization and disruption of polymeric micelles.39 In opposite, PEG-b-PCL in this work is the neutral polymer and cannot be protonated at the acidic condition resulting in stable micelles. There are two possible acid–base mechanisms which support these results. First, protons or hydroxyls in an environment diffuse into micelles resulting in a pH gradient in the core of micelles leading to the formation of ionized form of the encapsulated compound. This result is similar to the previous reports in which protons diffuse through the membrane of vesicles and polymersomes.40,41 For the other mechanism, proton exchanging between encapsulated compound and acidic environment may occur at the core–shell interface of polymeric micelles.
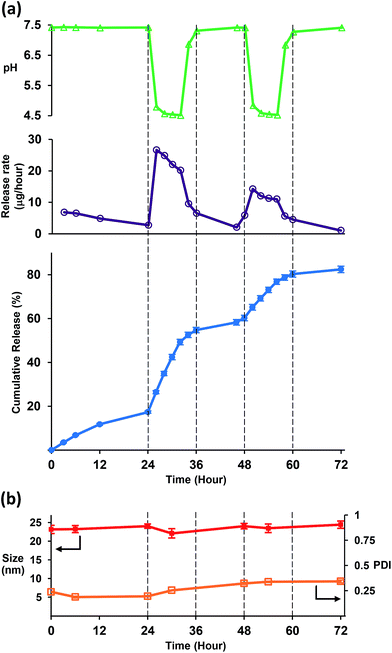 |
| | Fig. 4 (a) ON–OFF release behavior of iDef-loaded micelles (●), release rate (μg per hour) (○), measured pH value (▲). (b) Average particles size (■) and PDI (□). | |
iDef-loaded micelles is a good platform for the new alternative strategy for pH-responsive nanocarrier. In summary, this system consists of two main characteristics: (i) the encapsulated compound should be conjugated with pH-responsive molecules with pKa around 6.5–7.0 such as imidazole or tertiary amine (pentamethyleneimine hexamethyleneimine, dimethylamine, diethylamine, diisopropylamine, dibutylamino and morpholinopropyl group); and (ii) the linkage should be amide or ester because it can be degraded in physiological environment or by protease. The acidic cleavable linkage such as hydrazone and imine can also be used. Results mentioned above demonstrate the strategy for pH-stimuli controlled release which consists of several advantages: (i) this system possesses reversible process of fast and slow release depending on environmental pH which can be called “pH-stimuli controlled drug release”. (ii) It uses common biodegradable amphiphilic block copolymer such as PEG-b-PCL, PEG-b-PLA, PEG-b-PLGA and poloxamer without any further modifications of these polymers; (iii) this strategy does not only have quick response but the micelle size is also not significantly changed at different pH; and (iv) this system can maintain the controlled-release property at endosome/lysosome target site while other pH-responsive polymeric micelles were disrupted when the pH was decreased resulting in the loss of controlled-release function.42,43
2.4 Cytotoxicity
The antiproliferative property of compounds were determined by utilizing MTT assay after 72 hours of incubation. The dose response curves of free compounds and micelle formulation of Def, mDef and iDef against human prostate cancer cell line (PC-3), human hepatocellular carcinoma cell line (HepG2) and mouse fibroblast L929 cell (normal cell line) were shown in Fig. 5. IC50 values were determined by curve fitting as shown in Table 2. It was found that deferasirox derivatives exhibited higher cytotoxicity when compared to deferasirox. IC50 of iDef against PC-3 is in the sub-micro molar level and this is applied to both free iDef and iDef-loaded micelles. Considering the binding effect, the modification of deferasirox at the carboxyl position had no effect on antiproliferative efficiency because the carboxylic groups were not involved in the iron chelation.36 The increase in cytotoxicity of deferasirox derivatives is primarily due to the increase in the lipophilicity of derivatives compared to the original form. It should be noted that small molecules penetrate through cellular membranes by passive diffusion based on concentration gradient.28 Thus methoxy and imidazole-modified deferasirox that were highly lipophilic at pH 7.4 (Fig. 1) could cross the cell membranes easier than deferasirox (lower lipophilicity).44 The internalization route of nanoparticles and small molecules were totally different. Nanoparticles could be taken up by tumor cells via endocytosis. For drug-loaded micelles, IC50 was also correlated to the release rate of the drug in acidic condition and was slightly higher than that of a free compound. In general, encapsulation of anticancer drug in the micelle core showed slightly higher IC50 than that of free drugs. This is due to the prolonged release of drugs from micelles.38 For anticancer drug such as doxorubicin, the target organelle is the nuclei so doxorubicin has to be liberated from micelles and move out of endocytosis-related organelles to the nuclei.45,46 while free doxorubicin can cross the cell membrane and diffuses directly to nuclei. In the case of an iron-chelating compound and iron-chelating compound loaded micelles, the target organelle is endosome and lysosome.3 Therefore, there is not much difference in the IC50 of free compounds and micelle formulation. To evaluate safety and potential application in the field of medicine, the cytotoxicity of all micelle formulations were compared to normal cell line, i.e., L929. Def- and mDef-loaded micelles did not exhibit significant difference in cytotoxicity compared to cancer cell line and normal cell line (Fig. 5 and Table 2). Interestingly, IC50 of iDef-micelles against the normal cell was 4.7-time and 2.2-time higher than that of PC-3 and HepG2, respectively. These IC50 values indicated that iDef-loaded micelles have promising antiproliferative activity with minimal cytotoxicity to the normal cell lines.
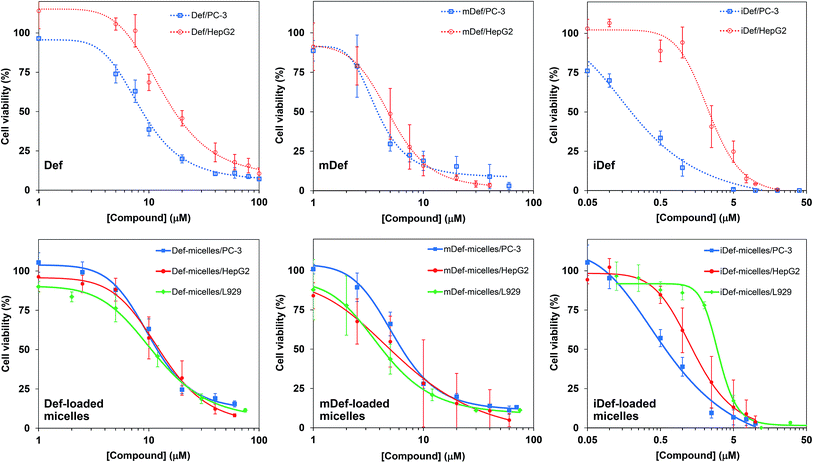 |
| | Fig. 5 Inhibition of cell growth of Def, mDef and iDef in the free form (dotted line) and micelles (solid line) against PC-3 (blue line), HepG2 (red line) and L929 (green line) cell lines. All data points were means ± standard deviation. Graphs were obtained by curve fitting of each data set. | |
Table 2 IC50 of deferasirox and its derivatives in the free form and micelle formulation
| Compound |
IC50 values (μM) |
| PC-3 |
HepG2 |
L929 |
| Free drug |
Micelles |
Free drug |
Micelles |
Micelles |
| Def |
8.44 ± 0.48 |
10.23 ± 1.12 |
13.56 ± 2.80 |
12.67 ± 3.92 |
11.18 ± 1.11 |
| mDef |
3.77 ± 1.08 |
5.50 ± 0.63 |
5.02 ± 1.59 |
6.46 ± 1.50 |
4.14 ± 1.07 |
| iDef |
0.27 ± 0.03 |
0.66 ± 0.12 |
2.32 ± 0.59 |
1.43 ± 0.69 |
3.13 ± 1.09 |
2.5 Iron binding of iDef-loaded polymeric micelles in lysosome
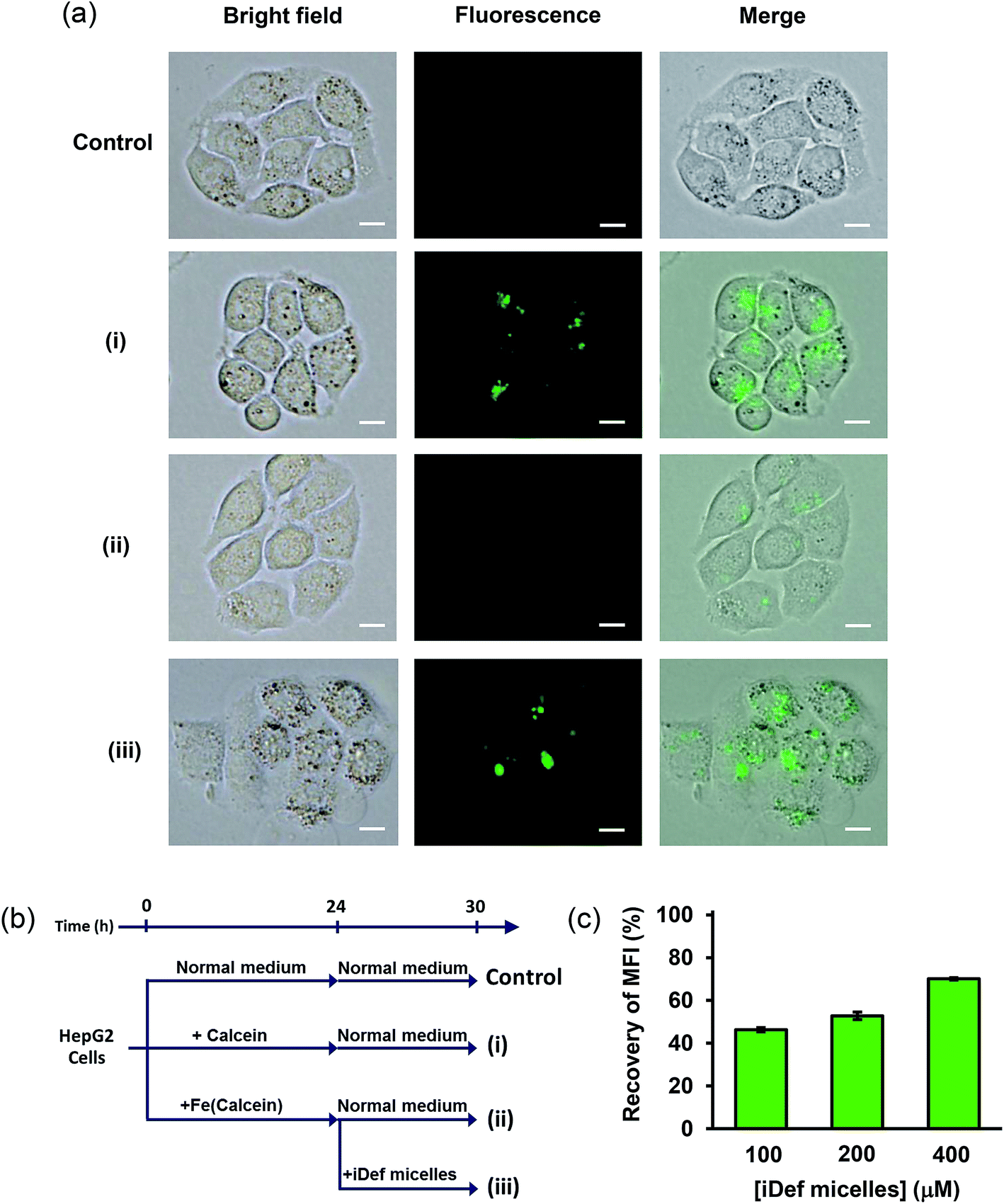
The decrease of iron in lysosome as a result of iDef-loaded micelles was confirmed by calcein fluorescence microscopy and flow cytometry. Calcein is an iron chelator with strong green fluorescence property. As aforementioned, it was successfully used as iron-chelating efficiency assay where the fluorescence can be quenched when it binds to the iron atom. Since, calcein is not able to penetrate cell membrane thus it is barely used as a biomarker. The derivative of calcein, calcein AM, was developed to increase lipophilic property and ability to permeate cell membrane and is widely used for labile iron pool assay.47 The uptake of calcein and calcein AM in human cell line was intensely investigated by Tenopoulou M. et al.48 It was reported that high concentration and long incubation time was required for calcein internalization and accumulation into endosome/lysosome by endocytosis. It should be noted that calcein was not found in other cytoplasmic organelles. Therefore, calcein may be used as an iron probe in the lysosome. The limitation of calcein is the sensitivity which has been lowered about 10-time in lysosome due to acidic environment. To implement calcein as iron probe in lysosome, very high concentration and long incubation time were applied to detect its fluorescence signal. Therefore, we evaluated the cytotoxicity of calcein (ESI, Fig. S5†). Results demonstrated no cytotoxic effect of calcein up to 200 μM after incubation for 24 and 72 hours. Thus, 50 μM concentration of calcein with the incubation time of 24 hours was selected for calcein uptake study by fluorescence microscopy and flow cytometry. It was found that calcein fluorescence in lysosome of HepG2 cells was still detected after 6 hours as shown in Fig. 6a(i). Next, iron in the form of FAS was added to calcein-containing medium and allowed for complete iron-calcein complexation. HepG2 cells were then treated with this complex (Fe(calcein)) as shown in Fig. 6a(ii). The recovery of calcein fluorescence in lysosome was found after cells were incubated with 200 μM of iDef (micelle form) for 6 hours as shown in Fig. 6a(iii). These results suggested that iron in the form of iron-calcein complexation in lysosome were competitively bound by iDef from micelles. This resulted in the liberation and dequenching of calcein. The experimental design of each condition (control, (i), (ii) and (iii)) was represented by schematic diagram in Fig. 6b. Flow cytometry was also used to evaluate fluorescence intensity of calcein in the lysosome. Mean fluorescence intensity (MFI) was measured as shown in Fig. 6c. Condition (i) was defined as the 100% fluorescence intensity. MFI was quenched and dropped to 28% in the presence of iron (condition (ii)). After adding iDef-micelle, the recovery of MFI was increased with the rising concentration of iDef-micelles which confirmed the reduction of iron in lysosome. MFI was recovered to approximately 70% when using 400 μM of iDef in micelles.
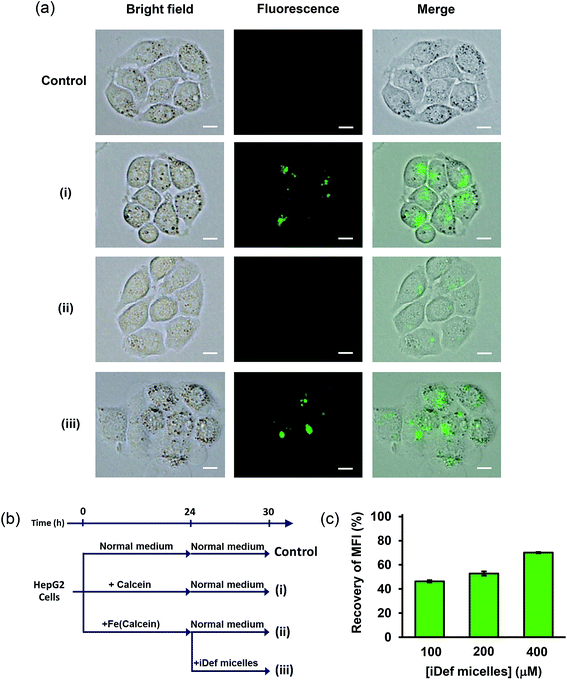 |
| | Fig. 6 (a) Images of HepG2 cells after incubation by different conditions for 24 hour and then incubated with normal medium for 6 hours: (i) 50 μM of calcein, (ii) 50 μM of Fe(calcein), (iii) 50 μM of Fe(calcein) then dequenching by the addition of 200 μM of iDef-loaded micelles for 6 hours. The control was HepG2 cells that were incubated with normal medium for 30 hours without any treatments. Scale bars were 10 μm. (b) Experimental design of cell uptake and competitive binding. (c) Recovery of mean fluorescence intensity (%) of calcein calculated from flow cytometry after treated with various iDef-loaded micelles concentration (100, 200 and 400 μM) for 6 hours. | |
3. Experimental
3.1 Materials
Methoxy-terminated poly(ethylene glycol) with the molecular weight of 5000 Dalton (MeO-PEG-OH) was purchased from Fluka and purified by recrystallization with ethyl acetate. ε-Caprolactone was purchased from Sigma-Aldrich and purified by vacuum distillation over calcium hydride. Stannous(II) octoate (Sn(Oct)2) was purchased from Aldrich. Deferasirox (Def) was purchased from Shanghai Renyoung Pharmaceutical Co., Ltd. N-Ethyl-N′-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), 1-hydroxybenzotrizole hydrate (HOBT) and 1-(3-aminopropyl)imidazole were purchased from Aldrich. 2-Methoxyethylamine was purchased from Acros. Potassium bisulfate (KHSO4), sodium bicarbonate (NaHCO3) and ammonium iron(II) sulfate hexahydrate (FAS) were purchased from Ajax finechem. HEPES and calcein were purchased from Sigma-aldrich. Lysosomal buffer preparation was modified from the protocol of Arbab et al.49 Phosphate buffered saline (PBS) pH 4.5 was prepared as same as PBS pH 7.4 but added with 20 mM sodium citrate and adjusted to pH 4.5 with 1 M hydrochloric acid. All organic solvents were of analytical grade. Toluene was refluxed with sodium-benzophenone.
3.2 Synthesis of methoxy-modified deferasirox (mDef)
Deferasirox (180 mg, 0.48 mmol) was suspended in 12 mL of CHCl3. Triethylamine (0.3 mL, 2.10 mmol) was added to dissolve deferasirox. 1-Hydroxybenzotriazole (174 mg, 1.14 mmol) was added and the mixture was cooled to 0 °C, then EDC (240 mg, 1.25 mmol) was added. After 10 minutes, 2-methoxyethylamine (0.122 mL, 1.4 mmol) was added. The reaction was stirred at 0 °C for 1 hour in ice bath at room temperature for 12 hours, followed by addition of 100 mL of CHCl3. The mixture was extracted with 5% KHSO4 solution (3 × 100 mL) followed by 5% NaHCO3 solution (3 × 100 mL) and finally water (3 × 100 mL). Each extraction step was repeated for 2 times. The organic phase was dried by NaSO4 and the solvent was removed by rotary evaporation.
3.3 Synthesis of imidazole-modified deferasirox (iDef)
The synthesis was similar to the synthesis of mDef. Deferasirox (180 mg, 0.48 mmol) was suspended in 6 mL of CHCl3 and triethylamine (0.3 mL, 2.1 mmol) was added to dissolve deferasirox. 1-Hydroxybenzotriazole (174 mg, 1.14 mmol) was added, and then the reaction mixture was allowed to cool to 0 °C followed by addition of EDC (240 mg, 1.25 mmol). After 10 minutes, the solution of 1-(3-aminopropyl) imidazole (0.183 mL, 1.4 mmol) in 3 mL of MeOH was added. The reaction mixture was stirred at 0 °C for 1 hour in ice bath at room temperature for 12 hours. The crude material was purified by silica column chromatography (CH2Cl2/MeOH as the mobile phase).
3.4 Synthesis of poly(ethylene glycol)-block-poly(ε-caprolactone) (PEG-b-PCL)
PEG-b-PCL was synthesized by ring-opening polymerization of ε-caprolactone as reported earlier.50 MeO-PEG-OH (2.0 g, 0.4 mmol) was vacuum-dried overnight at 65 °C then 20 mL of anhydrous toluene was added to dissolve polymer under dry argon atmosphere. Next, ε-caprolactone (2.0 g, 17.5 mmol) was added into polymer solution and the reaction was heated to 140 °C. Then, a few drops of Sn(Oct)2 was added into reaction flask. The polymerization proceeded for 18 hours under magnetic stirring and argon atmosphere at 140 °C. The product was collected by precipitation in diethyl ether then polymer was purified by dissolving in THF and precipitated in diethyl ether.
3.5 Preparation of drug-loaded polymeric micelles
Drug-loaded polymeric micelles were obtained by a solvent evaporation method.50 Briefly, PEG-b-PCL (120 mg) and dried powder of iron-chelating compound (6.3 mg) were mixed and dissolved in 2 mL of THF then the mixture was added drop-wise into the distilled water (20 mL) under sonication (SONIC, Model VCX 130) with output power of 80%. THF was removed by slow evaporation overnight at room temperature. Micelles were filtered by using syringe filter with the pore size of 0.45 μm to remove precipitation of drug and polymer, then micelles were subjected to Vivaspin centrifugal filter (Mw cut-off: 50 kDa) to separate unencapsulated drugs and to increase micelle concentration.
3.6 Determination of drug loading content (DLC) and drug loading efficiency (EE)
First, micelles were lyophilized to determine the weight of micelles. Next, freeze-dried micelles were dissolved in THF then the amount of iron-chelating compound in micelles was determined by the absorbance at the wavelength of 305 nm (Thermo Scientific, Model G10S UV-Vis). Finally, the calibration curve was prepared by measuring the absorbance of the various known concentrations of iron-chelating compound. The DLC and EE were calculated following below equations.
3.7 Particle size and morphology determination
Micelles were filtered through a 0.45 μm syringe filter and then the solution was diluted to 2 mg mL−1. Average particle size, polydispersity index (PDI) and zeta potential were measured by dynamic light scattering (Malvern Instruments Ltd., Zetasizer Nano-ZS). The reflective index (RI) and absorption value of micelles for zetasizer are 1.40 and 0.00, respectively. The morphology of micelles was carried out by transmission electron microscopy (TEM) a Philips TECNAI 20 at the acceleration voltage of 80 kV. The specimens were prepared by dropping the micelle solution at the concentration of 1 mg L−1 into a copper grid, followed by negative staining by adding another drop of 2 wt% phosphotungstic acid.
3.8 In vitro release studies
Deferasirox micelle (2 mL) was transferred into a dialysis bag (Mw cut-off: 50 kDa) and immersed either in PBS pH 7.4 or buffer pH 4.5 plus 20 mM sodium citrate. They were kept at 37 °C in an incubator shaker. At selected time intervals, buffer solution outside the bag was collected and replaced with the same volume of fresh buffer solution. The amount of releasing deferasirox was calculated based on the absorbance at 290 nm. The release studies of deferasirox derivatives were performed by the same method as for the deferasirox micelle. Next, iDef-loaded micelle was prepared then transferred into a dialysis bag (Mw cut-off: 50 kDa). At the first period, the dialysis bag was placed into 20 mL PBS pH 7.4, collected and measured release at predetermined time point during the 24 hours. After collecting buffer solution, fresh buffer pH 4.5 was replaced at the same amount. The release was monitored, the buffer was collected then the pH of the buffer was switched back to PBS pH 7. This process was repeated twice. The amount of imidazole-modified deferasirox release was calculated based on the absorbance at 290 nm. The pH of buffer solution was measured at every point of replacement. The particle size was also measured during experiment by zetasizer.
3.9 Cytotoxicity
Human prostate carcinoma (PC-3) cell line was purchased from ATCC (Manassas, VA, USA). Human hepatocellular carcinoma (HepG2) and mouse fibroblast (L929) cell lines were purchased from Japanese Collection of Research Bioresources Cell Bank (Tokyo, Japan). Cells in logarithmic growth phase were used in this experiment. Cell lines were seeded on a 96-well plate at the cell density of 3 × 103 cells per well in 100 μL EMEM medium for HepG2 (using RPIM medium for PC-3) and grew for 72 hours in a humidified atmosphere with 5% CO2. The stock solution of deferasirox or its derivatives including micelle formulation was sterilized by filtration through 0.22 mm syringe filter. Then cells were incubated for 72 hours in the media containing deferasirox or its derivatives at the concentration of 0.01 to 100 μM. For MTT assay, the media was removed, and cells were washed twice with PBS. 3-(4,5-Dimethyl-thiazol-yl)-2,5-diphenyltetrazolium bromide (MTT) was dissolved in PBS at the concentration of 5 mg mL−1 and RPMI phenol red free medium was mixed at the ratio of 1:10. The cancer cells were incubated in a growth medium containing MTT agent for an additional 4 h at 37 °C. After that, the solution was removed and DMSO (100 μL) was added to each well to ensure solubilization of formazan crystals. Finally, the optical density was performed using microplate reader at the wavelength of 550 nm. The 4PL nonlinear regression curve fitting was used to determine the half maximal inhibitory concentration (IC50).
3.10 Cell uptake and competitive binding
HepG2 cells were cultured for 24 h in 50 μM of calcein-containing medium or 50 μM of Fe(calcein)-containing medium (50 μM calcein and 100 μM FAS was mixed 30 minutes before added in culture medium). Next, all cultured cells were washed with PBS. Then, cells were treated by calcein was incubated with EMEM for 6 hours as shown in Fig. 6b(i). Cells treated by Fe(calcein) were divided into two subgroups. First, cells were incubated with EMEM for 6 hours as shown in Fig. 6b(ii). Second, cells were incubated with 200 μM of iDef-loaded micelles for 6 hours as shown in Fig. 6b(iii). For control group, HepG2 cells were cultured for 24 h in EMEM and washed with PBS then cultured for 6 hours in EMEM. The calcein fluorescence in HepG2 cells was monitored using a flow cytometry and fluorescence microscope.
4. Conclusions
Deferasirox derivatives were successfully synthesized and displayed good chelating efficiency and cytotoxicity towards cancer cells. Deferasirox and its derivatives were encapsulated in micelles via solvent evaporation method with high drug loading content. All of these micelles demonstrated anti-proliferative properties suggesting that they have potential applications for cancer treatment, especially micelles loaded with imidazole-modified deferasirox. These micelles showed cytotoxic effect against PC-3 cell line in submicromolar level. Flow cytometry showed a decrease in the amount of iron in lysosome after cells were incubated with micelles loaded with imidazole-modified deferasirox. Furthermore, this type of micelles performed pH-responsive and ON–OFF release behavior by the modification of pKa of encapsulated compounds. This report demonstrates an alternative strategy to develop pH-responsive nanoparticles for anticancer chemotherapy for which further in vivo experiments are needed in the future to evaluate antitumor efficiency.
Acknowledgements
This research project was supported by Mahidol University. The authors thank Dr S. Charoonniyomporn for assistance in interpreting 1H-NMR spectrum. Man Theerasilp was partially supported by the Center for Innovation in Chemistry (PERCH-CIC), Commission on Higher Education, Ministry of Education is gratefully acknowledged.
Notes and references
- S. J. Stohs and D. Bagchi, Free Radical Biol. Med., 1995, 18, 321–336 CrossRef CAS PubMed.
- J. Boult, K. Roberts, M. J. Brookes, S. Hughes, J. P. Bury, S. S. Cross, G. J. Anderson, R. Spychal, T. Iqbal and C. Tselepis, Clin. Cancer Res., 2008, 14, 379–387 CrossRef CAS PubMed.
- S. V. Torti and F. M. Torti, Nat. Rev. Cancer, 2013, 13, 342–355 CrossRef CAS PubMed.
- D. S. Kalinowski and D. R. Richardson, Pharmacol. Rev., 2005, 57, 547–583 CrossRef CAS PubMed.
- M. R. Bedford, S. J. Ford, R. D. Horniblow, T. H. Iqbal and C. Tselepis, J. Clin. Pharmacol., 2013, 53, 885–891 CrossRef CAS PubMed.
- M. Whitnall, J. Howard, P. Ponka and D. R. Richardson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14901–14906 CrossRef CAS PubMed.
- R. A. Finch, M. C. Liu, S. P. Grill, W. C. Rose, R. Loomis, K. M. Vasquez, Y. C. Cheng and A. C. Sartorelli, Biochem. Pharmacol., 2000, 59, 983–991 CrossRef CAS PubMed.
- F. Gaboriau, A. M. Leray, M. Ropert, L. Gouffier, I. Cannie, M. B. Troadec, O. Loreal, P. Brissot and G. Lescoat, BioMetals, 2010, 23, 231–245 CrossRef CAS PubMed.
- L. Barraud, P. Merle, E. Soma, L. Lefrançois, S. Guerret, M. Chevallier, C. Dubernet, P. Couvreur, C. Trépo and L. Vitvitski, J. Hepatol., 2005, 42, 736–743 CrossRef CAS PubMed.
- K. Chantrel-Groussard, F. Gaboriau, N. Pasdeloup, R. Havouis, H. Nick, J. L. Pierre, P. Brissot and G. Lescoat, Eur. J. Pharmacol., 2006, 541, 129–137 CrossRef CAS PubMed.
- G. Y. L. Lui, P. Obeidy, S. J. Ford, C. Tselepis, D. M. Sharp, P. J. Jansson, D. S. Kalinowski, Z. Kovacevic, D. B. Lovejoy and D. R. Richardson, Mol. Pharmacol., 2013, 83, 179–190 CrossRef CAS PubMed.
- T. Ohara, K. Noma, S. Urano, S. Watanabe, S. Nishitani, Y. Tomono, F. Kimura, S. Kagawa, Y. Shirakawa and T. Fujiwara, Int. J. Cancer, 2013, 132, 2705–2713 CrossRef CAS PubMed.
- J. H. Ohyashiki, C. Kobayashi, R. Hamamura, S. Okabe, T. Tauchi and K. Ohyashiki, Cancer Sci., 2009, 100, 970–977 CrossRef CAS PubMed.
- L. Vazana-Barad, G. Granot, R. Mor-Tzuntz, I. Levi, M. Dreyling, I. Nathan and O. Shpilberg, Leuk. Lymphoma, 2013, 54, 851–859 CrossRef CAS PubMed.
- S. J. Ford, P. Obeidy, D. B. Lovejoy, M. Bedford, L. Nichols, C. Chadwick, O. Tucker, G. Y. L. Lui, D. S. Kalinowski, P. J. Jansson, T. H. Iqbal, D. Alderson, D. R. Richardson and C. Tselepis, Br. J. Pharmacol., 2013, 168, 1316–1328 CrossRef CAS PubMed.
- A. Lipton, L. Witters and H. Nick, Cancer Res., 2009, 69, 5075 Search PubMed.
- S. Urano, T. Ohara, S. Watanabe, K. Noma, Y. Tomono, H. Tazawa, M. Kataoka, N. Kazuhiro, Y. Shirakawa and T. Fujiwara, Proceedings of the 104th Annual Meeting of the American Association for Cancer Research, 2013, vol. 69, p. 5608 Search PubMed.
- T. Ohara, K. Noma, S. Watanabe, T. Ohnishi, S. Nishitani, Y. Tomono, Y. Hashimoto, H. Tazawa and T. Fujiwara, Proceedings of the 102nd Annual Meeting of the American Association for Cancer Research, 2011, vol. 71, p. 4247 Search PubMed.
- T. Fukushima, H. Kawabata, T. Nakamura, H. Iwao, A. Nakajima, M. Miki, T. Sakai, T. Sawaki, Y. Fujita, M. Tanaka, Y. Masaki, Y. Hirose and H. Umehara, Anticancer Res., 2011, 31, 1741–1744 Search PubMed.
- P. Rouge, A. Dassonville-Klimpt, C. Cezard, S. Boudesocque, R. Ourouda, C. Amant, F. Gaboriau, I. Forfar, J. Guillon, E. Guillon, E. Vanquelef, P. Cieplak, F. Y. Dupradeau, L. Dupont and P. Sonnet, ChemPlusChem, 2012, 77, 1001–1016 CrossRef CAS PubMed.
- R. Séchaud, A. Robeva, R. Belleli and S. Balez, J. Clin. Pharmacol., 2008, 48, 919–925 CrossRef PubMed.
- I. Saeki, N. Yamamoto, T. Yamasaki, T. Takami, M. Maeda, K. Fujisawa, T. Iwamoto, T. Matsumoto, I. Hidaka, T. Ishikawa, K. Uchida, K. Tani and I. Sakaida, World J. Gastroenterol., 2016, 22, 8967–8977 CrossRef PubMed.
- S. Movassaghian, O. M. Merkel and V. P. Torchilin, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 691–707 CrossRef CAS PubMed.
- E. Blanco, C. W. Kessinger, B. D. Sumer and J. Gao, Exp. Biol. Med., 2009, 234, 123–131 CrossRef CAS PubMed.
- S. Puntawee, M. Theerasilp, S. Reabroi, R. Saeeng, P. Piyachaturawat, A. Chairoungdua and N. Nasongkla, Pharm. Dev. Technol., 2016, 21, 437–444 CAS.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS PubMed.
- H. Maeda, G. Y. Bharate and J. Daruwalla, Eur. J. Pharm. Biopharm., 2009, 71, 409–419 CrossRef CAS PubMed.
- I. Canton and G. Battaglia, Chem. Soc. Rev., 2012, 41, 2718–2739 RSC.
- J. Li, L. Zhang, Y. Lin, H. Xiao, M. Zuo, D. Cheng and X. Shuai, RSC Adv., 2016, 6, 9160–9163 RSC.
- W. Hao, D. Liu, Y. Shang, J. Zhang, S. Xu and H. Liu, RSC Adv., 2016, 6, 29149–29158 RSC.
- M. Arduini, F. Felluga, F. Mancin, P. Rossi, P. Tecilla, U. Tonellato and N. Valentinuzzi, Chem. Commun., 2003, 9, 1606–1607 RSC.
- G. J. Bruin, T. Faller, H. Wiegand, A. Schweitzer, H. Nick, J. Schneider, K. O. Boernsen and F. Waldmeier, Drug Metab. Dispos., 2008, 36, 2523–2538 CrossRef CAS PubMed.
- D. Rodbard and S. W. McClean, Clin. Chem., 1977, 23, 112–115 CAS.
- U. Heinz, K. Hegetschweiler, P. Acklin, B. Faller, R. Lattmann and H. P. Schnebli, Angew. Chem., Int. Ed., 1999, 38, 2568–2570 CrossRef CAS PubMed.
- J. Liu, D. Obando, L. G. Schipanski, L. K. Groebler, P. K. Witting, D. S. Kalinowski, D. R. Richardson and R. Codd, J. Med. Chem., 2010, 53, 1370–1382 CrossRef CAS PubMed.
- P. Rouge, M. Becuwe, A. Dassonville-Klimpt, S. D. Nascimento, J. F. Raimbert, D. Cailleu, E. Baudrin and P. Sonnet, Tetrahedron, 2011, 67, 2916–2924 CrossRef CAS.
- S. Steinhauser, U. Heinz, M. Bartholomä, T. Weyliermüller, H. Nick and K. Hegetschweiler, Eur. J. Inorg. Chem., 2004, 4177–4192 CrossRef CAS.
- X. Shuai, H. Ai, N. Nasongkla, S. Kim and J. Gao, J. Controlled Release, 2004, 98, 415–426 CrossRef CAS PubMed.
- C. J. F. Rijcken, O. Soga, W. E. Hennink and C. F. V. Nostrum, J. Controlled Release, 2007, 120, 131–148 CrossRef CAS PubMed.
- J. Wu and A. Eisenberg, J. Am. Chem. Soc., 2006, 128, 2880–2884 CrossRef CAS PubMed.
- Y. Yu, X. Jiang, S. Gong, L. Feng, Y. Zhong and Z. Pang, Nanoscale, 2014, 6, 3250–3258 RSC.
- Y. Cao, J. Zhao, Y. Zhang, J. Liu, J. Liu, A. Dong and L. Deng, RSC Adv., 2015, 5, 28060–28069 RSC.
- L. Fu, L. Liu, Z. Ruan, H. Zhang and L. Yan, RSC Adv., 2016, 6, 40312–40322 RSC.
- Q. Al-Awqati, Nat. Cell Biol., 1999, 1, E201–E202 CrossRef CAS PubMed.
- N. Nasongkla, E. Bey, J. Ren, H. Ai, C. Khemtong, J. S. Guthi, S. F. Chin, A. D. Sherry, D. A. Boothman and J. Gao, Nano Lett., 2006, 6, 2427–2430 CrossRef CAS PubMed.
- N. Nasongkla, X. Shuai, H. Ai, B. D. Weinberg, J. Pink, D. A. Boothman and J. Gao, Angew. Chem., Int. Ed., 2004, 43, 6323–6327 CrossRef CAS PubMed.
- Y. Ma, V. Abbate and R. C. Hider, Metallomics, 2015, 7, 212–222 RSC.
- M. Tenopoulou, T. Kurz, P. T. Doulias, D. Galaris and U. T. Brunk, Biochem. J., 2007, 403, 261–266 CrossRef CAS PubMed.
- A. S. Arbab, L. B. Wilson, P. Ashari, E. K. Jordan, B. K. Lewis and J. A. Frank, NMR Biomed., 2005, 18, 383–389 CrossRef CAS PubMed.
- M. Theerasilp and N. Nasongkla, J. Microencapsulation, 2013, 30, 390–397 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26669j |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Punlop Chalermpanapun,
Kanyawan Ponlamuangdee,
Dusita Sukvanitvichai and
Norased Nasongkla
,
Punlop Chalermpanapun,
Kanyawan Ponlamuangdee,
Dusita Sukvanitvichai and
Norased Nasongkla