
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sm3+ and Eu3+ codoped SrBi2B2O7: a red-emitting phosphor with improved thermal stability†
Liwei Wua,
Yuxing Baia,
Li Wu*a,
Huan Yia,
Yongfa Konga,
Yi Zhang *b and
Jingjun Xua
*b and
Jingjun Xua
aKey Laboratory of Weak-Light Nonlinear Photonics, Ministry of Education, School of Physics, Nankai University, Tianjin 300071, China. E-mail: lwu@nankai.edu.cn; Fax: +86-22-23505409; Tel: +86-22-23506257
bCollege of Electronic Information and Optical Engineering, Tianjin Key Laboratory of Photo-electronic Thin Film Devices and Technology, Nankai University, Tianjin 300071, China. E-mail: yizhang@nankai.edu.cn; Fax: +86-22-23508912; Tel: +86-22-23508572 extn 8018
First published on 20th December 2016
Abstract
The thermal stability of luminescence is very critical for white light-emitting diodes. However, it is a continuous challenge to improve the thermal stability of red phosphors. In this study, Sm3+ and Eu3+ codoped SrBi2B2O7 was synthesized by a high temperature solid state reaction method. It was found that the thermal stability of the synthesized phosphors was improved as Sm3+ was used as the sensitizer for Eu3+ doped into SrBi2B2O7. Combined with a local crystal environment study and the first-principles calculations, the origin of the improvement of this thermal stability was studied in detail. The doped Sm3+ and Eu3+ ions were inclined to occupy Bi(1) (6c) and Bi(2) (6c) sites simultaneously and the crystal structure of the SrBi2B2O7:Sm3+, Eu3+ was more compact at high temperature than that at room temperature. Thus, the defect formation energy was very low in the Sm3+ and Eu3+ codoped SrBi2B2O7 phosphor, which is the main reason to improve the thermal stability with Sm3+ and Eu3+ codoped into SrBi2B2O7. This study provides a new idea for developing a new method to improve the thermal stability of red-emitting phosphors.
1. Introduction
In high-power w-LEDs, when they are working the temperature of the layer deposited on the chip can rise to more than 150 °C because of the thermal effect from the p–n junction and the phosphor layer. Thus, the thermal stability of the phosphor has a great influence on the light output and color rendering index, and it is a vital factor for the application of w-LEDs. At about 150 °C, lots of the phosphors cannot show good thermal stability compared with those kept at room temperature. Up-to-now, some nitrides or oxynitrides have been proved to be good performance red phosphors because of their good thermal stability.1,2 However, the synthesis of those phosphors requires a very high nitrogen pressure and high sintering temperature, which often results in much higher production costs. Therefore, it is highly desirable to develop new phosphors, especially red emitting phosphors, with excellent thermal stability and lower cost for warm white-light emission.As a kind of rare earth ions, Sm3+ ions generate intense reddish orange emitting light because it can be excited to its 4F7/2 energy level and then relaxed to the 4G5/2 energy level through the non-radiative transition.3–5 Eu3+ ions have been widely studied as an activator for red light emitting materials due to the 5D0 → 7FJ (J = 0, 1, 2, 3, 4) transition of Eu3+.6–8 Presently most of used red phosphors are single doped by Sm3+ or Eu3+, such as Y2O2S:Eu3+,9 Y2O3:Eu3+,10 BaMoO4:Sm3+,11 SrCaMoO6:Sm3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 et al. However, the majority of them show limited thermal stability.
12 et al. However, the majority of them show limited thermal stability.
As for the low-cost phosphors, rare-earth ions doped borates phosphors is a potential candidate because of their low synthesis temperature and high chemical and physical stability,13 such as Ba2Tb(BO3)2Cl:Eu3+,14 and KSr4(BO3)3:Sm3+.15 As Sm3+ and Eu3+ codoped into hosts, energy transfer from Sm3+ to Eu3+ can enhance the emission of the Eu3+ ions and extend the excitation spectrum. Thus energy transfer between Sm3+ and Eu3+ is important to adjust the photoluminescence properties. Up to now, this energy transfer has been investigated in some phosphors.16–18 However, Sm3+ and Eu3+ codoped into suitable borates material, thus appearing energy transfer, especially improving thermal stability of the phosphor has not been reported to our knowledge.
In this study, SrBi2B2O7 was considered as the host. SrBi2B2O7, synthesized by J. Barbier's group in 2006,19 is crystallized in the hexagonal P63 space group. This is a non-centrosymmetric structure containing three crystallographic positions of cations: seven-fold coordinated Sr2+ (6c) sites, eight-fold coordinated Bi(1) (6c) sites, and eight-fold coordinated Bi(2) (6c) sites. Plenty of crystallographic sites are afforded for doped ions to occupy, thus SrBi2B2O7 is a promising host to prepare phosphor with good luminescence properties. In this work, the new red-emitting borate phosphor SrBi2B2O7:Sm3+,Eu3+, which has high thermal stability, is reported for the first time and the high thermal stability mechanism was discussed through defect formation energy calculations. The photoluminescence properties, the energy transfer from Sm3+ to Eu3+ and the preferred occupancy of the dopant were also investigated in detail.
2. Experimental
2.1 Materials and synthesis
The phosphor samples SrBi2B2O7:Sm3+, SrBi2B2O7:Eu3+, and SrBi2B2O7:Sm3+, Eu3+ were prepared by high temperature solid-state reaction method. Analytical reagent SrCO3, Bi2O3, H3BO3, and high purity (99.99%) Sm2O3 and Eu2O3 were used as raw materials. These powders were weighed out, well-mixed and ground thoroughly in an agate mortar. The mixtures were first heated at 500 °C for 24 h to decompose the carbonate and eliminate the water. When cooling down to the room temperature, the presintered samples were ground respectively and heated at 650 °C for 72 h, then cooling down to room temperature with the furnace cooling.2.2 Characterization
X-ray diffraction (XRD) data for phase identification and structural refinement of as-prepared powders were collected by PANalytical powder X-ray diffractometer X'Pert Pro with Cu Kα radiation (40 kV, 40 mA) and the data were collected over a 2θ range from 10° to 140° at an interval of 0.017° with a counting time of 1 s per step. The temperature-dependent XRD patterns were measured on a Rigaku SmartLab X-ray Diffractometer with Cu Kα radiation (40 kV, 180 mA). Those samples to collect XRD data at high temperature are heated from RT to 150 °C with an intermittent heating rate of 3 °C min−1 and then kept for 5 min at 150 °C before XRD data collection. High temperature XRD data were collected with a measurement time of 30 min. Photoluminescence (PL), photoluminescence excitation (PLE) spectra were recorded using a spectrofluorometer (Edinburgh Instruments, FLS920) equipped with a Xe light source and double excitation monochromators. Emitted fluorescence was detected by a photomultiplier (R928P) perpendicular to the excitation beam. A cutoff filter was used to eliminate the second-order emission of the source radiation. The luminescence decay was measured by a μF900 lamp (100 W) as a light source and a photomultiplier (R928P) was used as detector. The temperature-dependent luminescence properties were measured on the spectrofluorometer (Edinburgh Instruments, FLS920), combined with a self-made heating attachment (TAP-02). The internal quantum efficiency of optimized-composition phosphors SrBi2B2O7:0.04Sm3+, SrBi2B2O7:0.06Eu3+ and SrBi2B2O7:0.04Sm3+, 0.06Eu3+ was determined on a standard Edinburgh Instruments FLS920 spectrometer equipped with an integrating sphere attachment.2.3 Computational details
All the calculations were conducted by first-principles calculations, as implemented in the Vienna ab initio simulation package (VASP).20,21 The projector augment wave (PAW) pseudopotential method was used to describe the interactions of elements. Electronic valence configurations of Sr, Bi, B, O, Sm and Eu were set as 5s2, 6s26p3, 2s22p1, 2s22p4, 4f65s25p66s2, 4f75s25p66s2, respectively. The exchange–correlation potential used the Perdew–Burke–Ernzerhof (PBE) functional within the spin-polarized generalized gradient approximation (GGA).22,23 A plane wave basis with cutoff energy of 400 eV was employed. The k-point sampling for Brillouin zone was generated using a 4 × 4 × 2Γ-centered grid. In structural optimized process, the energy change, maximum residual force acting on each ion was set as 1 × 10−4 eV per atom, 0.05 eV Å−1, respectively. As for the formation of defects, they were all obtained through the process that Bi3+ ions are replaced equivalently by Sm3+, Eu3+ ions or both of them (SmBi, EuBi) in the conventional cell of SrBi2B2O7 whose real stoichiometric formula is Sr6Bi12B12O42.24 The formation energies of single defect such as SmBi as well as EuBi and the composite one SmBi + EuBi were calculated in our paper.3. Results and discussion
3.1 Crystal structure
XRD patterns of phosphors SrBi2B2O7:Sm3+, SrBi2B2O7:Eu3+ and SrBi2B2O7:Sm3+, Eu3+ are shown in Fig. 1. All XRD patterns are found to agree well with that reported in the Inorganic Crystal Structure Database (ICSD #245017), indicating that the doped Sm3+ and Eu3+ ions do not generate any impurity or induce significant changes in the host structure.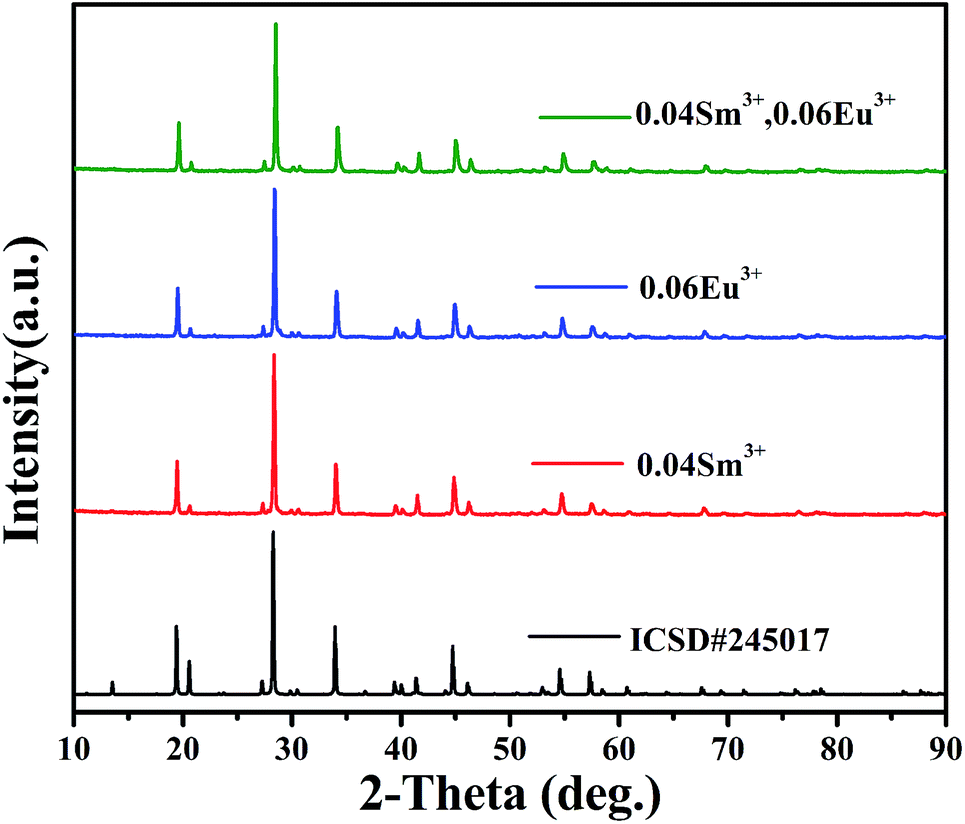 | ||
| Fig. 1 XRD patterns of SrBi2B2O7:Sm3+, SrBi2B2O7:Eu3+, SrBi2B2O7:Sm3+, Eu3+ and the ICSD (#245017) of SrBi2B2O7. | ||
3.2 Luminescence properties of SrBi2B2O7:Sm3+,Eu3+
The PL and PLE spectra of SrBi2B2O7:0.04Sm3+ are shown in Fig. 2(a). The strongest peak located at 403 nm in the excitation spectrum monitored at 598 nm is attributed to 6H5/2 → 4F7/2 transition. Under the 403 nm excitation, the emission spectra of Sm3+ show emission band at 561, 598, 644 and 704 nm due to the 4G5/2 → 6HJ/2 (J = 5, 7, 9, 11) transitions (red curve in Fig. 2(a)). The quenching concentration of SrBi2B2O7:Sm3+ is 4 mol%, as is shown in the inset of Fig. 2(a). Furthermore, the PL and PLE spectra of SrBi2B2O7:0.06Eu3+ are shown in Fig. 2(b). Excited at 393 nm, two main emission bands at 611 nm and 701 nm are attributed to 5D0 → 7F2 and 5D0 → 7F4 transition, and the three weak emission bands of the 5D0 → 7FJ (J = 0, 1, 3) are located at around 578 nm, 587 nm and 653 nm. The excitation spectrum consists of the charge transfer band (CTB) and two excitation peaks at 393 and 464 nm, which are mainly caused by the strong f–f transition of 7F0 → 5L6 and 7F0 → 5D2, respectively. The quenching concentration of SrBi2B2O7:Eu3+ is 6 mol%, as shown in the inset of Fig. 2(b).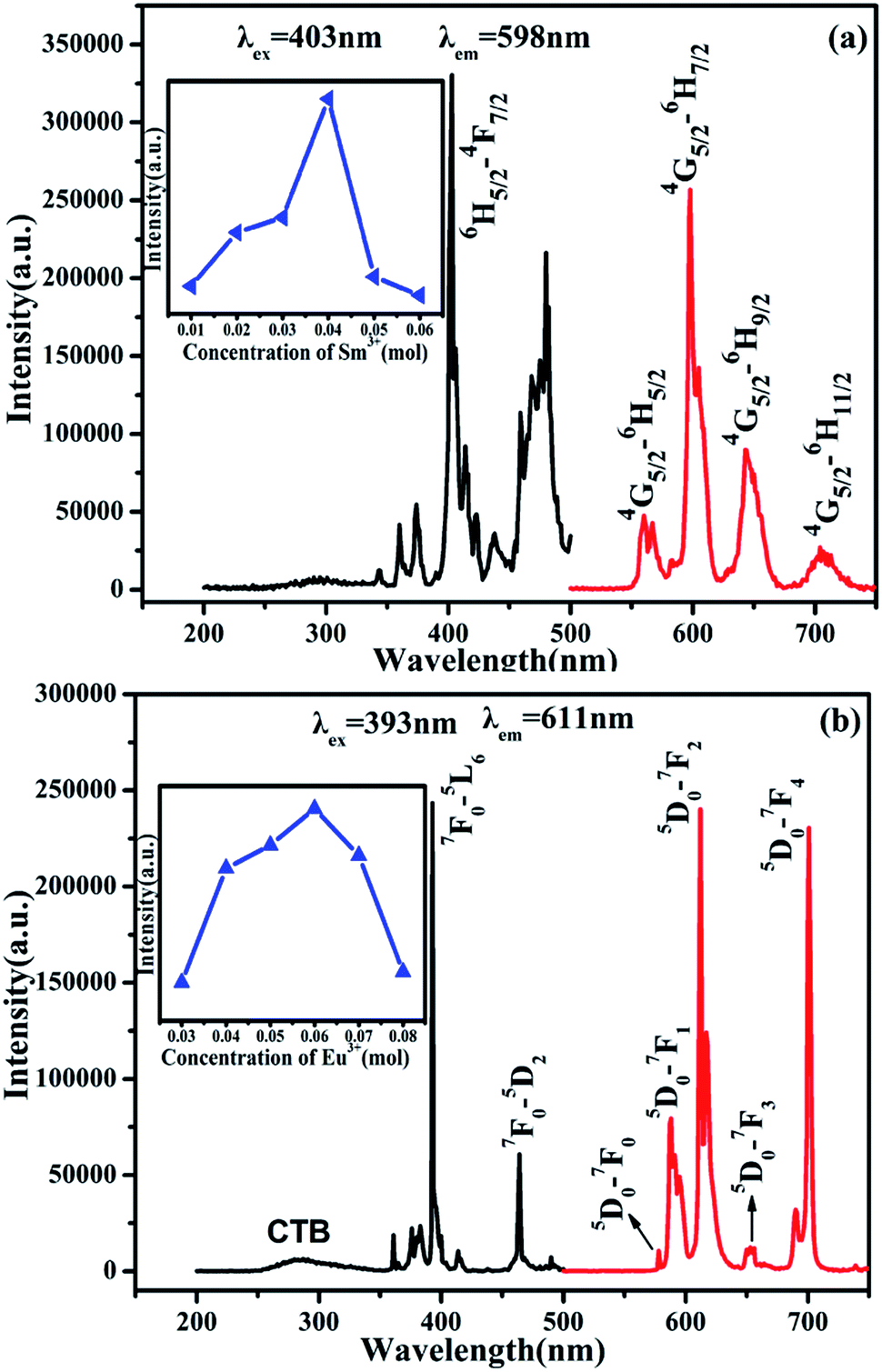 | ||
| Fig. 2 PL and PLE spectra of SrBi2B2O7:0.04Sm3+ (a) and SrBi2B2O7:0.06Eu3+ (b). Inset: the dependence of the intensity on Sm3+ (a) and Eu3+ (b) concentration. | ||
The PL and PLE spectra of Sm3+ and Eu3+-codoped SrBi2B2O7 phosphor are shown in Fig. 3(a). The PLE spectrum monitored at 598 nm shows two strong excitation peaks at 393 nm and 403 nm, which correspond to the 7F0 → 5L6 transition of Eu3+ and 6H5/2 → 4F7/2 transition of Sm3+, respectively. In the PL spectrum excited at 403 nm, seven significant emission peaks located at 561, 598, 644, 578, 587, 611, 701 nm are observed, which are attributed to 4G5/2 → 6H5/2, 4G5/2 → 6H7/2 and 4G5/2 → 6H9/2 transition of Sm3+ and 5D0 → 7F0, 5D0 → 7F1, 5D0 → 7F2 and 5D0 → 7F4 transition of Eu3+, respectively. Fig. 3(b) shows the PL spectra of SrBi2B2O7:0.04Sm3+,yEu3+ (y = 0.01–0.07) phosphors excited at 403 nm. All of the characteristic peaks of Sm3+ and Eu3+ can be observed in the PL spectra of SrBi2B2O7:Sm3+,Eu3+.
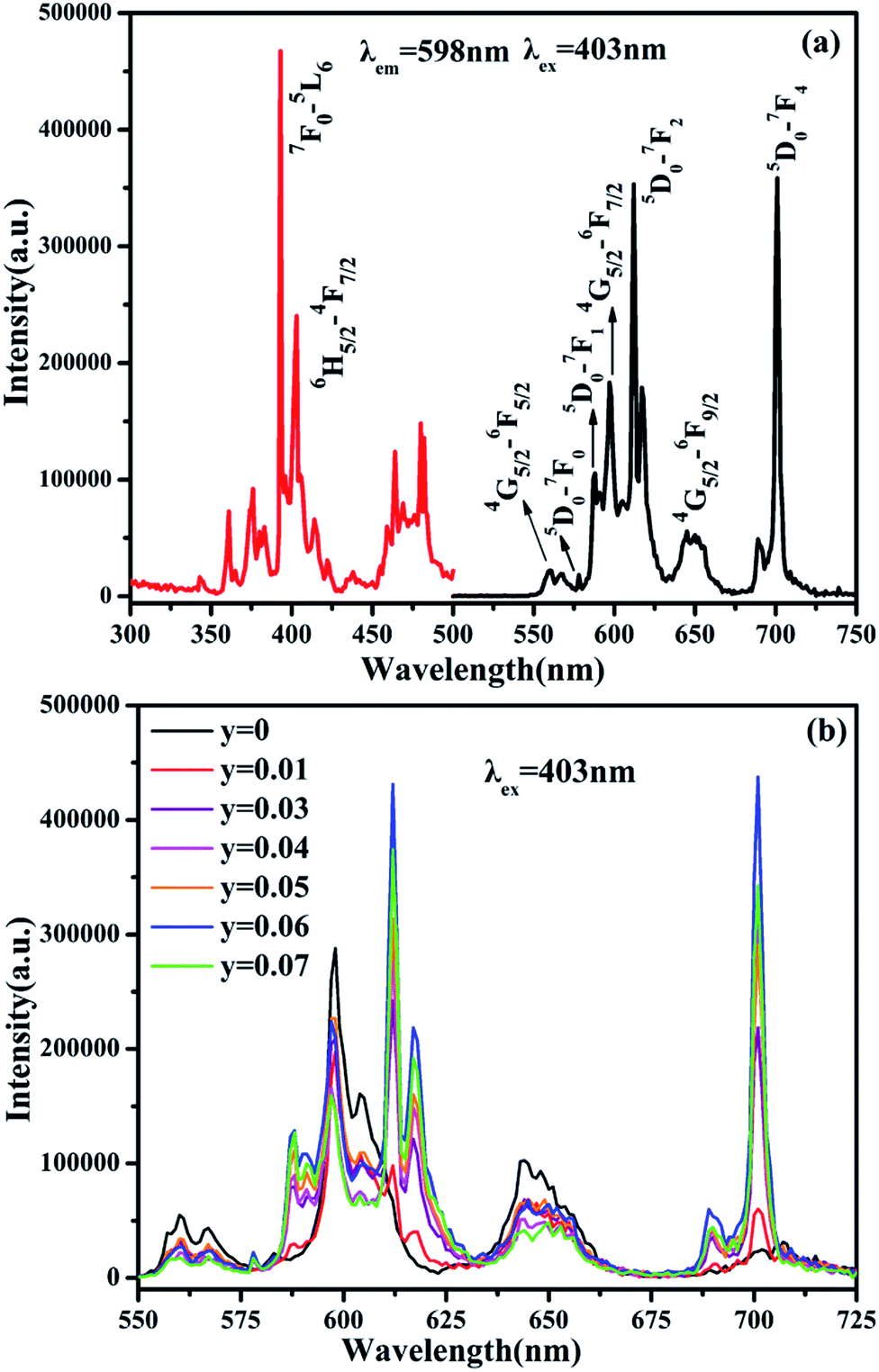 | ||
| Fig. 3 (a) PLE (left) and PL (right) spectra of SrBi2B2O7:0.04Sm3+,0.06Eu3+ phosphor. (b) PL spectra of SrBi2B2O7:0.04Sm3+, yEu3+ phosphor excited at 403 nm. | ||
3.3 Thermal stability of SrBi2B2O7:Sm3+,Eu3+
Thermal stability of the phosphors is very important for the application of w-LEDs, especially for the high-power one. Temperature-dependent PL spectra (excited with λex = 403 nm) of SrBi2B2O7:0.04Sm3+, 0.06Eu3+ at the temperature range of 25–300 °C are shown in Fig. 4(a). Compared to the emission intensity at room temperature, the integrated emission intensities of the characteristic peaks of Sm3+ and Eu3+ under the excitation of 403 nm at 150 °C were still remained about 87.9% and 83.4% of the initial value, which indicates that the thermal stability of SrBi2B2O7:0.04 Sm3+, 0.06Eu3+ phosphor is good. The temperature dependent PL spectra of Sm3+ doped SrBi2B2O7 and Eu3+ doped SrBi2B2O7 were also measured. Fig. 4(b) is the normalized PL intensities of SrBi2B2O7:Sm3+, Eu3+, SrBi2B2O7:Sm3+, and SrBi2B2O7:Eu3+. The PL intensities of the commercial red phosphor SrS:Eu2+ is also shown in Fig. 4(b). It is very interesting that the thermal stability of Sm3+ and Eu3+ codoped phosphor is better than those of Sm3+/Eu3+ single doped phosphors, which are all superior to that of SrS:Eu2+.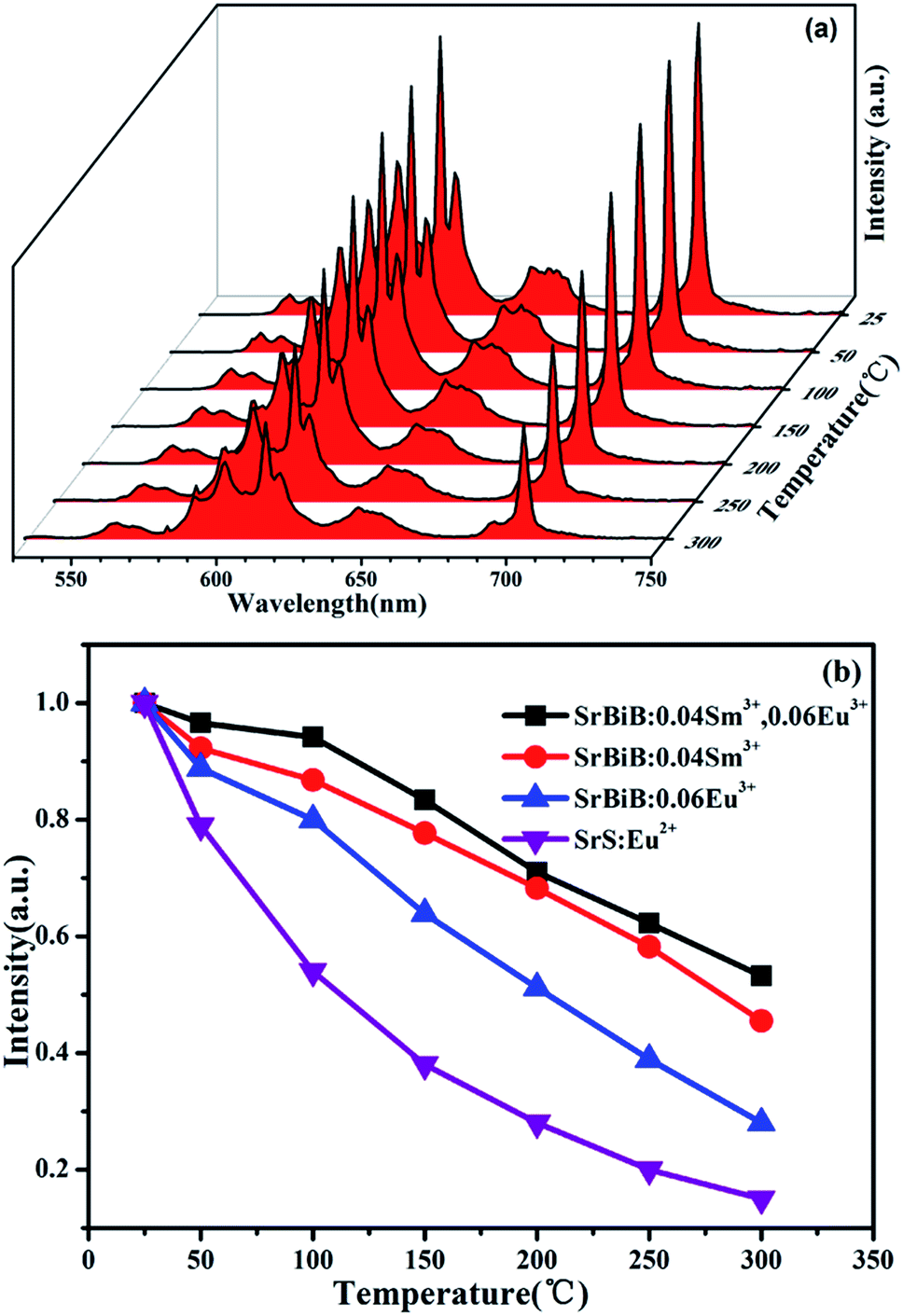 | ||
| Fig. 4 (a) Temperature-dependent PL spectra of SrBi2B2O7:0.04Sm3+, 0.06Eu3+ phosphor (λex = 403 nm). (b) The comparison results of PL intensities of SrBi2B2O7 (SrBiB):0.04Sm3+, 0.06Eu3+ (λex = 403 nm), SrBi2B2O7:0.04Sm3+ (λex = 403 nm), SrBi2B2O7:0.06Eu3+ (λex = 393 nm) as a function of temperature. As a comparison, thermal quenching data of SrS:Eu2+ excited at 460 nm are also measured. | ||
Why the thermal stability is enhanced as Sm3+ and Eu3+ codoped into the SrBi2B2O7? Up to date, the mechanism of high thermal stability enhanced by rare ions codoping is rarely investigated.
To clarify this question, we first study the local crystal environment of Sm3+ and Eu3+ in SrBi2B2O7. The host SrBi2B2O7 crystallizes in the hexagonal space group P63 with a = 9.1404(4) Å, c = 13.0808(6) Å, V = 946.44(7) Å3 and Z = 6. In the non-centrosymmetric structure, the Bi atoms are weakly bonded to the O atoms with strongly asymmetric eight-fold coordination environment.
The BiO8 coordination polyhedra and alternating BO3 triangles are in one layer and linked by edge-sharings, while Sr atoms are in the adjacent layer, as is shown in Fig. 5. There are three crystallographic positions of cations in the unit cell: seven-fold coordinated Sr2+ (6c) sites, eight-fold coordinated Bi(1) (6c) sites, and eight-fold coordinated Bi(2) (6c) sites. As reported by Shannon,25 the effective ionic radius (r) of Sm3+, Eu3+ and Bi3+ is 1.08 Å, 1.07 Å and 1.17 Å, respectively as the coordination number (CN) equals 8, whereas rSm3+ = 1.02 Å, rEu3+ = 1.01 Å and rSr2+ = 1.21 Å as CN = 7. Considering the r and CN, it seems that all the three sites can be occupied by Sm3+ and Eu3+.
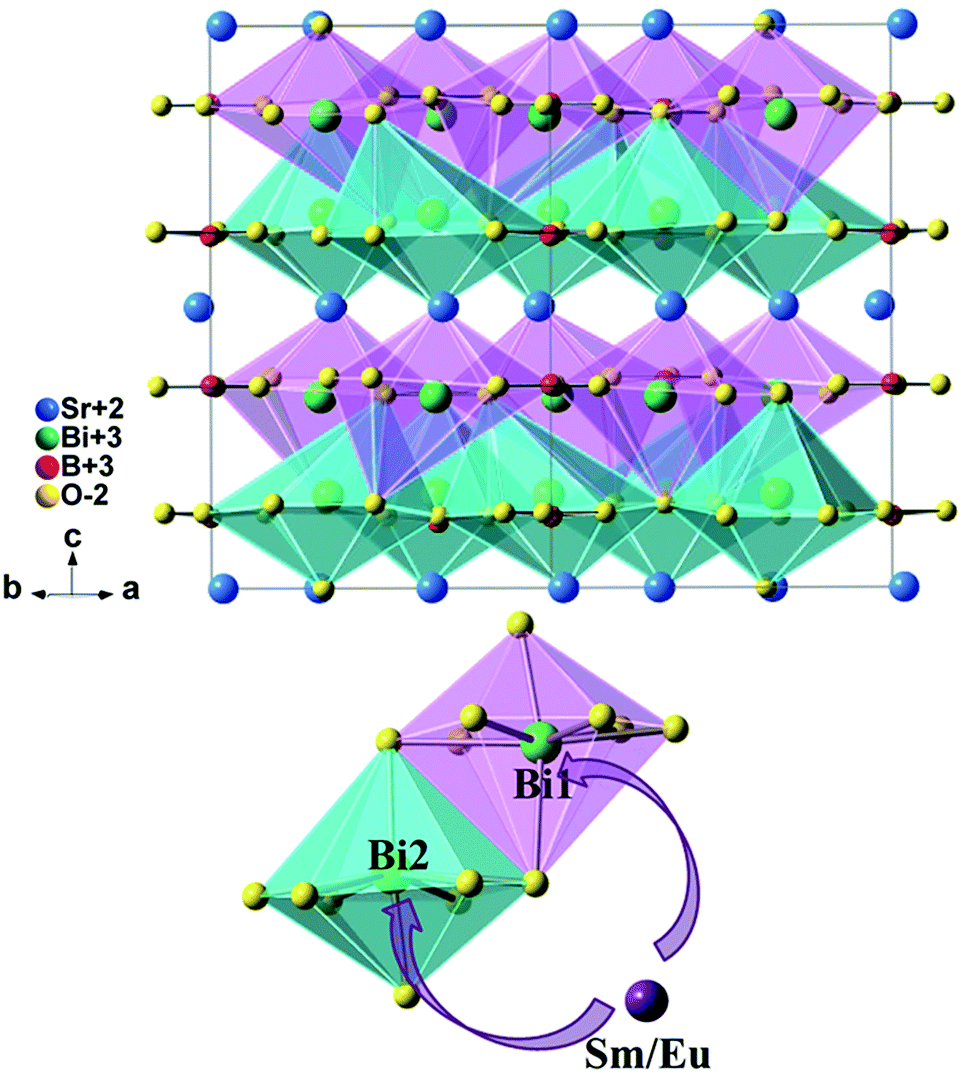 | ||
| Fig. 5 Crystal structure of SrBi2B2O7 and site occupancy preference of Sm3+/Eu3+ ions to Bi1 and Bi2 in SrBi2B2O7. | ||
To clarify the structure and the local crystal environment of Sm3+ and Eu3+ in SrBi2B2O7, the refinement on the XRD patterns of SrBi1.96Sm0.04B2O7, SrBi1.94Eu0.06B2O7, and SrBi1.90Sm0.04Eu0.06B2O7 are performed by Rietveld method26,27 within the Fullprof Program.28 The final agreement factors converged to Rp = 8.50%, Rwp = 9.60%, and Rexp = 3.90% for SrBi1.96Sm0.04B2O7, Rp = 7.40%, Rwp = 9.79%, and Rexp = 4.03% for SrBi1.94Eu0.06B2O7, and Rp = 7.39%, Rwp = 9.64%, and Rexp = 3.97% for SrBi1.90Sm0.04Eu0.06B2O7. Lattice parameters are refined to a = 9.1215(2) Å, c = 13.0550(9) Å, and V = 940.67(4) Å3 for SrBi1.96Sm0.04B2O7; a = 9.1128(3) Å, c = 13.0492(5) Å, and V = 938.47(6) Å3 for SrBi1.94Eu0.06B2O7; a = 9.1084(7) Å, c = 13.0452(6) Å, and V = 937.28(6) Å3 for SrBi1.90Sm0.04Eu0.06B2O7.
Fig. 5 shows the crystal structure of SrBi2B2O7. The final refinement patterns are given in Fig. 6. The crystallographic data, fractional atomic coordinates and occupancy are listed in Tables 1 and S1.†
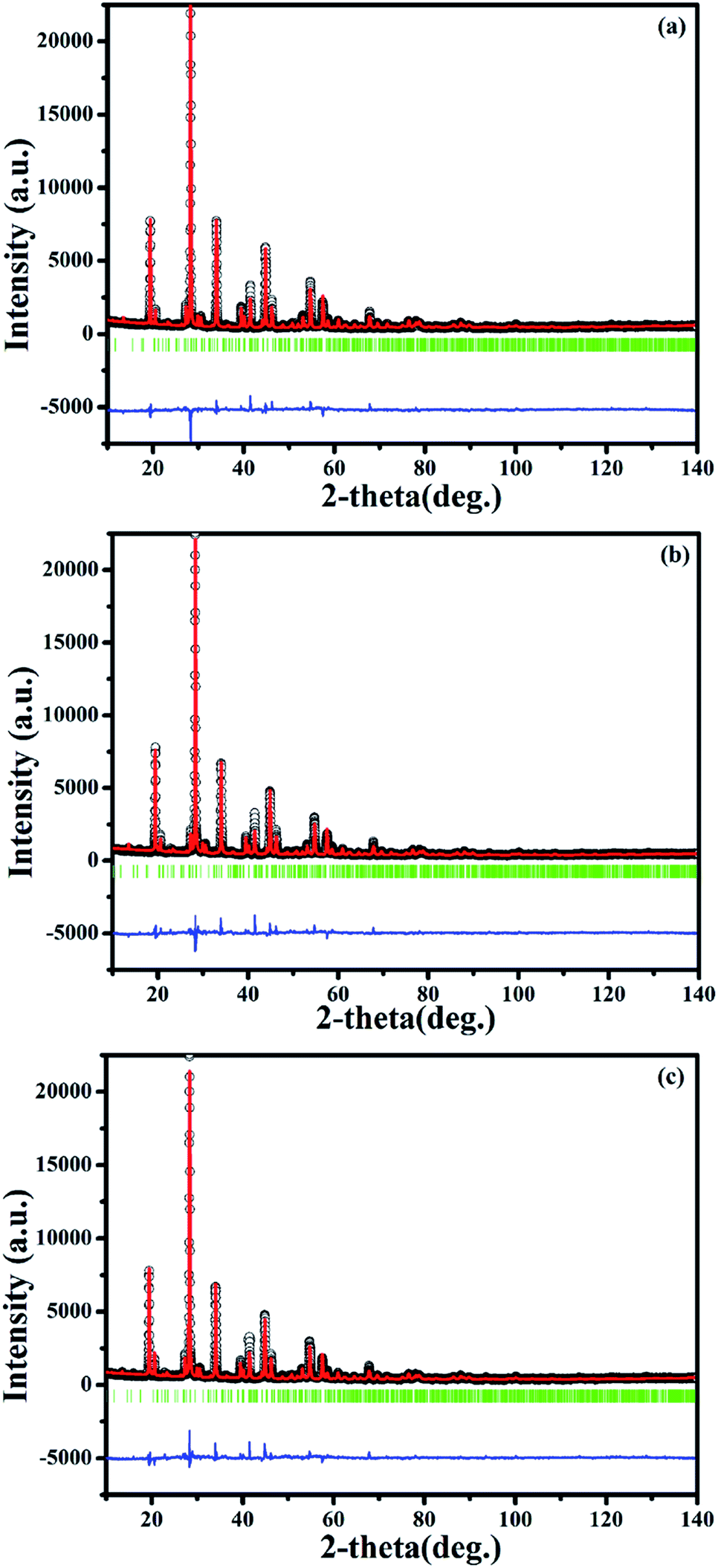 | ||
| Fig. 6 Final Rietveld refinement of the XRD profiles of SrBi1.96Sm0.04B2O7 (a), SrBi1.94Eu0.06B2O7 (b) and SrBi1.90Sm0.04Eu0.06B2O7 (c) at room temperature. Small black circles and the red continuous lines represent the experimental and the calculated values respectively; vertical bars (|) indicate the position of Bragg peaks. The blue bottom trace depicts the corresponding residuals between the experimental and the calculated intensity values. | ||
| Chemical formula | x = 0.04, y = 0 | x = 0, y = 0.06 | x = 0.04, y = 0.06 |
| Crystal system | Hexagonal | Hexagonal | Hexagonal |
| Space group | P63 | P63 | P63 |
| a/Å | 9.1215(2) | 9.1128(3) | 9.1084(7) |
| c/Å | 13.0550(9) | 13.0492(5) | 13.0452(6) |
| V/Å3 | 940.67(4) | 938.47(6) | 937.28(6) |
| Z | 6 | 6 | 6 |
| Rp (%) | 8.50 | 7.40 | 7.39 |
| Rwp (%) | 9.60 | 9.79 | 9.64 |
| Rexp (%) | 3.90 | 4.03 | 3.97 |
| RBragg (%) | 8.61 | 9.52 | 9.86 |
The final compositions determined by refinements are in good agreement with the nominal composition of the starting materials, as shown in Table S1.† It can be seen that the doped cations are preferred to occupy the Bi(1) (6c) sites and Bi(2) (6c) sites simultaneously for all of the Sm3+/Eu3+ single doped samples or codoped samples. This site occupancy is quite different from that of ZnBi2B2O7:Eu3+,6 in which the Eu3+ ions are preferred to only occupy the Zn site instead of Bi sites. ZnBi2B2O7 and SrBi2B2O7 are nearly the same in the chemical formula. However, their structures are different. ZnBi2B2O7 adopts an orthorhombic structure with space group Pba2 and Z = 4. The Bi3+ cations in ZnBi2B2O7 occupy two distinct interlayer sites with asymmetric six-fold coordination environments. The bond valence sum for Bi(1) and Bi(2) in ZnBi2B2O7 is 2.93 and 2.99, respectively,29 which is close to the theoretical value of 3. However, the Bi3+ cations and O2− anions are weakly bonded in SrBi2B2O7. The weak bonding interactions can result in the fragility and easy cleavage. The bond valence sum for Bi(1) and Bi(2) in SrBi2B2O7 is 2.39 and 2.29, respectively,19 which is much lower than those in ZnBi2B2O7 and indicates a significant underbonding. Thus, the Bi3+ cations in SrBi2B2O7 are much more unstable than that in ZnBi2B2O7. Then the different occupancy preferences of the dopants are observed in ZnBi2B2O7 and SrBi2B2O7.
On this basis, the refinement on the XRD patterns of SrBi1.96Sm0.04B2O7, SrBi1.94Eu0.06B2O7 and SrBi1.90Sm0.04Eu0.06B2O7 at 150 °C are further performed by Rietveld refinement within the Fullprof Program to clarify the change of the local crystal environment of Sm3+ and Eu3+ in SrBi2B2O7. The final agreement factors converged to Rp = 7.91%, Rwp = 12.10% and Rexp = 3.96% for SrBi1.96Sm0.04B2O7, Rp = 8.90%, Rwp = 12.90% and Rexp = 4.03% for SrBi1.94Eu0.06B2O7, and Rp = 9.06%, Rwp = 13.10% and Rexp = 3.97% for SrBi1.90Sm0.04Eu0.06B2O7. Lattice parameters are refined to a = 9.1194(3) Å, c = 13.0520(4) Å, and V = 940.02(5) Å3 for SrBi1.96Sm0.04B2O7, a = 9.1100(3) Å, c = 13.0440(5) Å, and V = 937.51(5) Å3 for SrBi1.94Eu0.06B2O7, and a = 9.1057(3) Å, c = 13.0406(5) Å, and V = 936.40(6) Å3 for SrBi1.90Sm0.04Eu0.06B2O7. The final refinement patterns are given in Fig. 7. The crystallographic data, fractional atomic coordinates and occupancy are listed in Tables 2 and S2;† selected average bond lengths (Å) of Bi–O are reported in Table S3.†
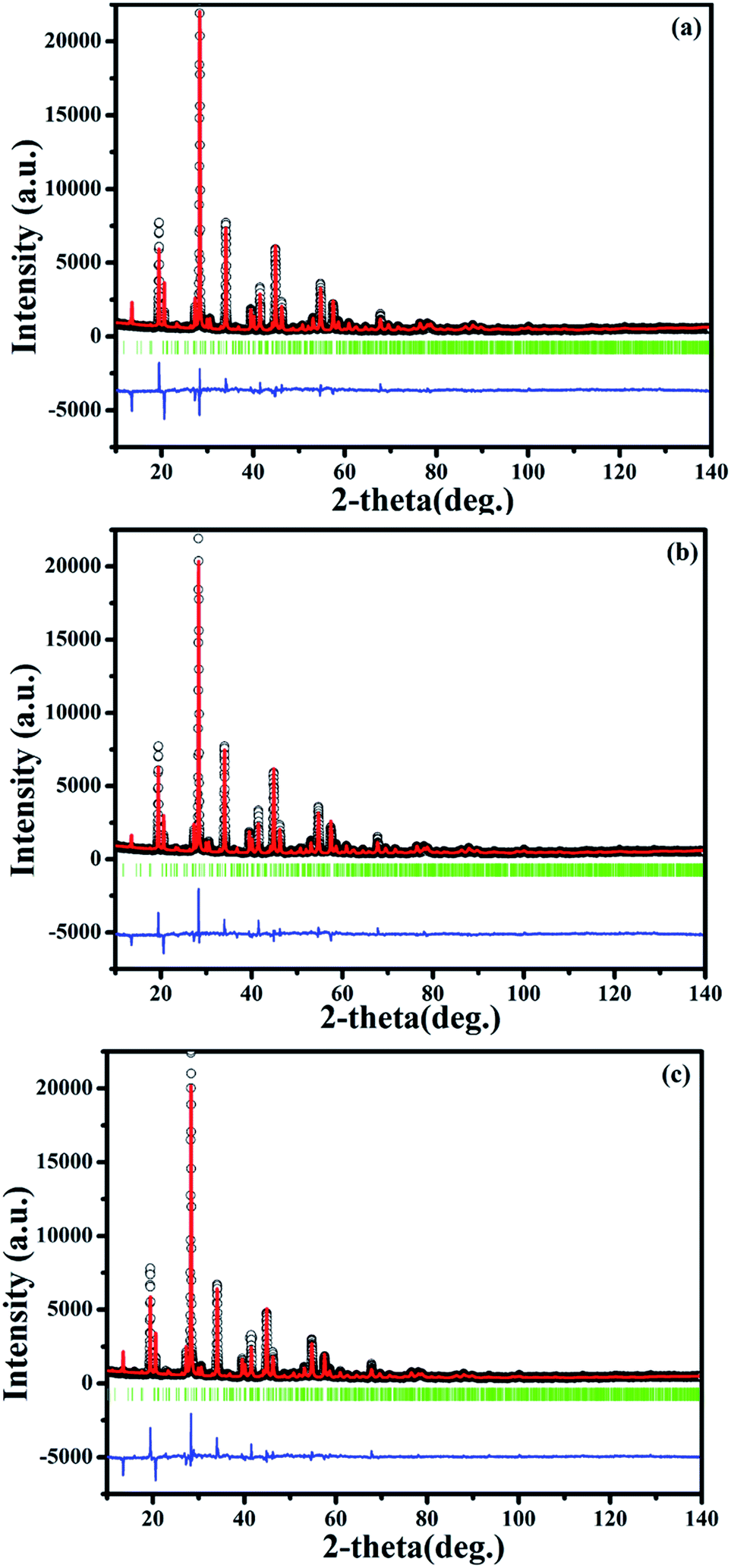 | ||
| Fig. 7 Final Rietveld refinement of the XRD profiles of SrBi1.96Sm0.04B2O7 (a), SrBi1.94Eu0.06B2O7 (b) and SrBi1.90Sm0.04Eu0.06B2O7 (c) at 150 °C. | ||
| Chemical formula | x = 0.04, y = 0 | x = 0, y = 0.06 | x = 0.04, y = 0.06 |
| Crystal system | Hexagonal | Hexagonal | Hexagonal |
| Space group | P63 | P63 | P63 |
| a/Å | 9.1194(3) | 9.1100(3) | 9.1057(3) |
| c/Å | 13.0520(4) | 13.0440(5) | 13.0406(5) |
| V/Å3 | 940.02(5) | 937.51(5) | 936.40(6) |
| Z | 6 | 6 | 6 |
| Rp (%) | 7.91 | 8.90 | 9.06 |
| Rwp (%) | 12.10 | 12.90 | 13.10 |
| Rexp (%) | 3.96 | 4.03 | 3.97 |
| RBragg (%) | 9.87 | 10.10 | 10.35 |
As comparing the refined lattice parameters at 150 °C with those at room temperature, it is very interesting that the lattice parameters a, c, and V become smaller for both codoped and single doped samples. This unusual phenomenon is contrary to the rule of thermal expansion and contraction. Furthermore, as indicated in Table S3,† for the codoped and single doped phosphors, the average bond lengths between Bi (6c) and O (6c) sites at 150 °C is shorter than that at room temperature. Also, the average bond lengths of Bi–O in codoped phosphors at high temperature or RT is shorter than those in single doped phosphors at the same temperature. The above results indicates that the crystal structure of the codoped and single doped phosphors at high temperature is more compact than those of the phosphors at room temperature, which should contribute to the improved thermal stability as Sm3+ and Eu3+ codoped into SrBi2B2O7.
In addition, to investigate whether the high thermal stability is related with the electronic band gap, the electronic band structures of Sm3+/Eu3+ singled doped SrBi2B2O7, Sm3+ and Eu3+ codoped SrBi2B2O7 and undoped SrBi2B2O7 are computed with VASP and shown in Fig. 8. The electronic band gap of SrBi2B2O7:Eu3+, SrBi2B2O7:Sm3+, SrBi2B2O7:Sm3+, Eu3+ and SrBi2B2O7 are 2.57, 2.78, 2.60 and 2.81 eV, respectively. They are different somewhat. However, the difference among these electronic band gaps are small, which means that the electronic band structure should not the main reason to enhance the thermal stability as Sm3+ and Eu3+ codoped into SrBi2B2O7.
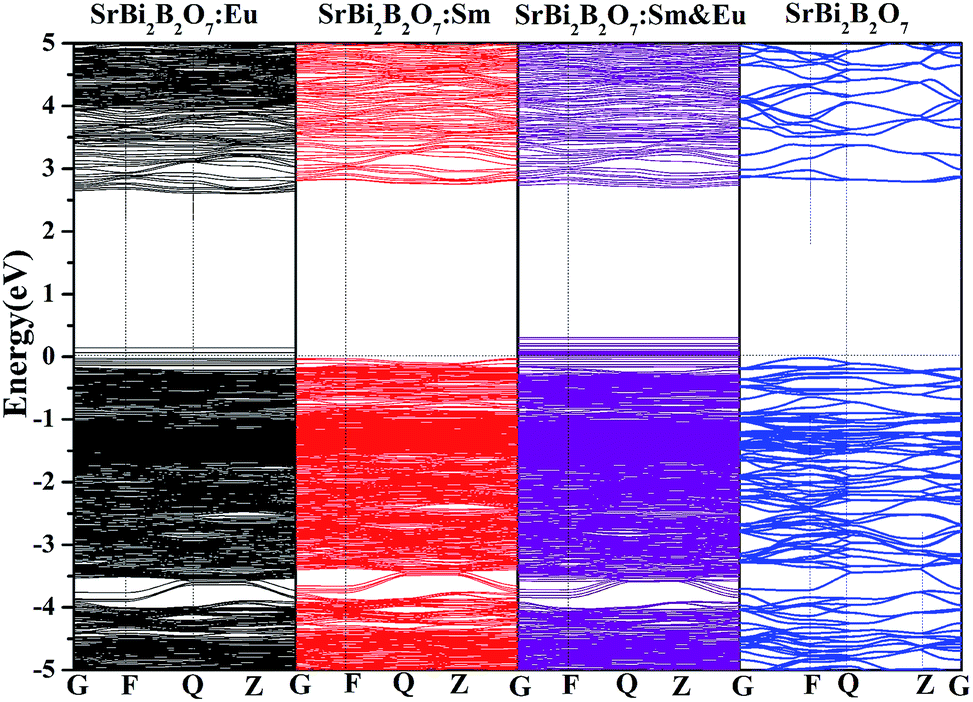 | ||
| Fig. 8 Electronic band structures of undoped and doped SrBi2B2O7. The Fermi level is set to 0 eV. | ||
As rare earth ions doped into the host, the corresponding defects will be formed. As studied on the site occupancy of the doped ions in this study, Sm3+ and Eu3+ ions are preferred to occupy Bi sites, thus the formed defects should mainly SmBi and EuBi. For the formed defects, the defect formation energy (Ef) will be different because of the different doped ions. In this study, to clarify the difference of Ef among Sm3+, Eu3+ single doped and codoped SrBi2B2O7. Ef of SrBi1.90Sm0.04Eu0.06B2O7, SrBi1.94Sm0.06B2O7 and SrBi1.96Eu0.04B2O7 defined as the following formula,30,31 are calculated with VASP.
| Ef = E(Sr6Bi12−x−yB12O42SmxEuy) − E(Sr6Bi12B12O42) − xμSm − yμEu + (x + y)μBi | (1) |
| Sample compositions | Etot (eV) | Ef (eV) |
|---|---|---|
| SrBi1.90Sm0.04Eu0.06B2O7 | −530.84 | −5.47 |
| SrBi1.94Sm0.06B2O7 | −517.67 | −5.20 |
| SrBi1.96Eu0.04B2O7 | −510.15 | 0.30 |
The active energy of Sm3+ and Eu3+ codoped SrBi2B2O7 phosphor was also studied. It is well known that the decrease of the emission intensity at different temperature can be described by the Arrhenius equation:32
| IT = I0/[1 + exp(−Ea/kT)] | (2) |
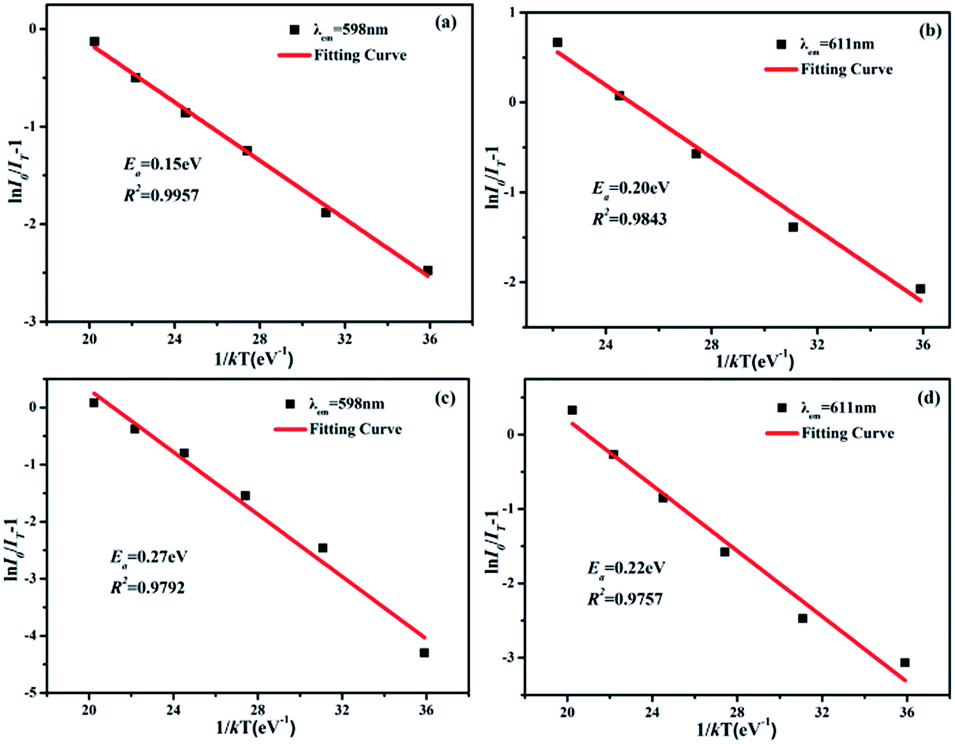 | ||
| Fig. 9 The ln(I0/IT − 1) vs. 1/kT activation energy graph for thermal quenching of the characteristic emission of Sm3+ (a) in SrBi2B2O7:0.04Sm3+ phosphor, Eu3+ (b) in SrBi2B2O7:0.06Eu3+ phosphor, Sm3+ (c) and Eu3+ (d) in SrBi2B2O7:0.04Sm3+,0.06Eu3+ phosphor. | ||
3.4 Quantum efficiency
Quantum efficiency is an important parameter for LED phosphor. To determine the absolute quantum efficiency of photo-conversion for the SrBi2B2O7:Sm3+, Eu3+ phosphor, the optical absorbance (A) and internal quantum efficiency (ηint) was measured using the integrated sphere method. The absorbance can be calculated according to the equation:
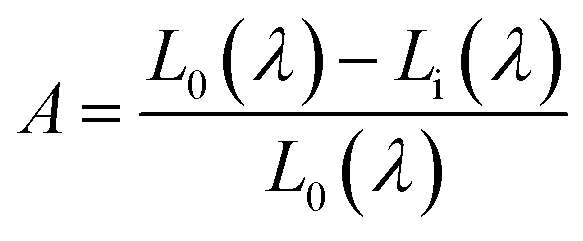 | (3) |
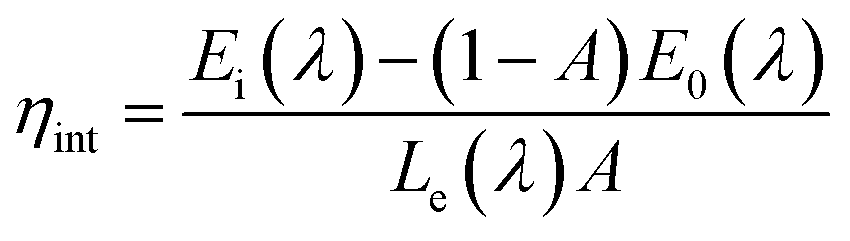 | (4) |
4. Conclusions
In conclusion, a red-emitting SrBi2B2O7:Sm3+,Eu3+ phosphor with improved thermal stability was synthesized through solid state reactions. The origin of the reason why thermal stability is improved is studied. The structural study results indicate that the doped Sm3+ and Eu3+ ions are inclined to occupy Bi(1) (6c) and Bi(2) (6c) sites simultaneously and the crystal structure of the SrBi2B2O7:Sm3+, Eu3+ was more compact at high temperature than that at room temperature. According to the first-principles calculation, the lowest defect formation energy −5.47 eV is obtained in the Sm3+ and Eu3+ codoped SrBi2B2O7 phosphor, which discloses that the defect formation energy should be another intrinsically responsible for the thermal stable luminescence of red-emitting SrBi2B2O7:Sm3+, Eu3+ phosphor.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (51372121, 61274053, U1460201, 51572132), Natural Science Foundation of Tianjin (14JCYBJC17800) and 111 Project (No. B07013) and YangFan Innovative & Entrepreneurial Research Team Project (2014YT02N037). We thank Mrs Xu of A02 group, Institute of Physics, Chinese Academy of Science for her great help in collecting the powder X-ray diffraction data.References
- R. J. Xie, N. Hirosaki, N. Kimura, K. Sakuma and M. Mitomo, Appl. Phys. Lett., 2007, 90, 191101 CrossRef.
- Y. Q. Li, J. E. J. Van Steen, J. W. H. Van Krevel, G. Botty, A. C. A. Delsing, F. J. Disalvo, G. De With and H. T. Hintzen, J. Alloys Compd., 2007, 417, 273 CrossRef.
- J. Xu, Z. H. Ju, X. P. Gao, Y. Q. An, X. L. Tang and W. S. Liu, Inorg. Chem., 2013, 52, 13875 CrossRef CAS PubMed.
- S. Kamimura, H. Yamada and C. N. Xu, Appl. Phys. Lett., 2012, 101, 091113 CrossRef.
- C. C. Lin, Z. R. Xiao, G. Y. Guo, T. S. Chan and R. S. Liu, J. Am. Chem. Soc., 2010, 132, 3020 CrossRef CAS PubMed.
- L. W. Wu, F. X. Zhang, L. Wu, H. Yi, H. R. Wang and Y. Zhang, J. Alloys Compd., 2015, 648, 500 CrossRef CAS.
- H. Yi, F. Li, L. Wu, L. W. Wu, H. R. Wang, B. Wang, Y. Zhang, Y. F. Kong and J. J. Xu, RSC Adv., 2014, 6, 64244 RSC.
- Y. Zhang, L. Wu, M. Y. Ji, B. Wang, Y. F. Kong and J. J. Xu, Opt. Mater. Express, 2012, 1, 92 CrossRef.
- Y. L. Huang, Y. Nakai, T. Tsuboi and H. J. Seo, Opt. Express, 2011, 19, 6303 CrossRef CAS PubMed.
- J. G. Li, X. D. Li, X. D. Sun and T. Ishigaki, J. Phys. Chem. C, 2008, 112, 11707 CAS.
- Z. G. Xia and D. M. Chen, J. Am. Ceram. Soc., 2010, 93, 1397 CrossRef CAS.
- L. L. Wang, H. M. Noh, B. K. Moon, S. H. Park, K. H. Kim, J. Shi and J. H. Jeong, J. Phys. Chem. C, 2015, 119, 15517 CAS.
- W. R. Liu, C. H. Huang, C. P. Wu, Y. C. Chiu, Y. T. Yeh and T. M. Chen, J. Mater. Chem., 2011, 21, 6869 RSC.
- Z. G. Xia, J. Q. Zhuang and L. B. Liao, Inorg. Chem., 2012, 51, 7202 CrossRef CAS PubMed.
- L. Wu, M. Y. Ji, H. R. Wang, Y. F. Kong and Y. Zhang, Opt. Mater. Express, 2014, 8, 1535 CrossRef.
- D. Kang, H. S. Yoo, S. H. Jung, H. Kim and D. Y. Jeon, J. Phys. Chem. C, 2011, 115, 24334 CAS.
- X. Min, Z. H. Huang, M. H. Fang, Y. G. Liu, C. Tang and X. W. Wu, Inorg. Chem., 2014, 53, 6060 CrossRef CAS PubMed.
- J. H. Chen, W. R. Zhao, J. Q. Wang, N. H. Wang, Y. J. Meng, J. He and X. Zhang, Ceram. Int., 2015, 41, 11945 CrossRef CAS.
- J. Barbier and L. M. D. Cranswick, J. Solid State Chem., 2006, 179, 3958 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- B. Y. Qu, B. Zhang, L. Wang, R. L. Zhou and X. C. Zeng, Chem. Mater., 2015, 27, 2195 CrossRef CAS.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- H. M. Rietveld, Acta Crystallogr., 1967, 22, 151 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65 CrossRef CAS.
- J. Rodriguez-Carvajal, M. T. Fernadez-Diaz and J. L. Martinez, J. Phys.: Condens. Matter, 1991, 3, 3215 CrossRef CAS.
- J. Barbier, N. Penin and L. M. D. Cranswick, Chem. Mater., 2005, 17, 3130 CrossRef CAS.
- A. F. Kohan, G. Ceder, D. Morgan and C. G. Van de Walle, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 15019 CrossRef CAS.
- B. Liu, M. Gu and X. L. Liu, Appl. Phys. Lett., 2010, 97, 122101 CrossRef.
- H. Jing, C. F. Guo, G. G. Zhang, X. Y. Su, Z. Yang and J. H. Jeong, J. Mater. Chem., 2012, 22, 13612 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Sm3+ and Eu3+ codoped SrBi2B2O7: a red-emitting phosphor with improved thermal stability. See DOI: 10.1039/c6ra26752a |
| This journal is © The Royal Society of Chemistry 2017 |