
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly recyclable polymer supported ionic liquids as efficient heterogeneous catalysts for batch and flow conversion of CO2 to cyclic carbonates†
Tao Wangac,
Wenlong Wanga,
Yuan Lyu*a,
Xingkun Chenac,
Cunyao Liac,
Yan Zhangac,
Xiangen Songa and
Yunjie Ding *ab
*ab
aDalian National Laboratory for Clean Energy, Dalian, 116023, P. R. China. E-mail: dyj@dicp.ac.cn; luyuan@dicp.ac.cn; Fax: +86-411-84379143; Tel: +86-411-84379143
bState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, P. R. China
cUniversity of Chinese Academy of Sciences, Beijing 100039, P. R. China
First published on 12th January 2017
Abstract
A series of cross-linked polymer supported ionic liquids based on quaternary ammonium salts have been prepared using a facile method of radical polymerization. Styrene-functionalized quaternary ammonium chloride salts, which are derived from imidazole, trimethylamine, triethylamine and triethanolamine, were copolymerized with divinyl benzene (DVB) under solvothermal conditions. These polymer supported ionic liquids displayed high efficiency in catalytic conversion of CO2 to cyclic carbonates. Notably, to the best of our knowledge, it was the first time that the polymeric ionic liquid catalyst was applied in a continuous flow fixed-bed reactor. During the study period of 130 hours, the polymer supported ionic liquids exhibited high stability.
Introduction
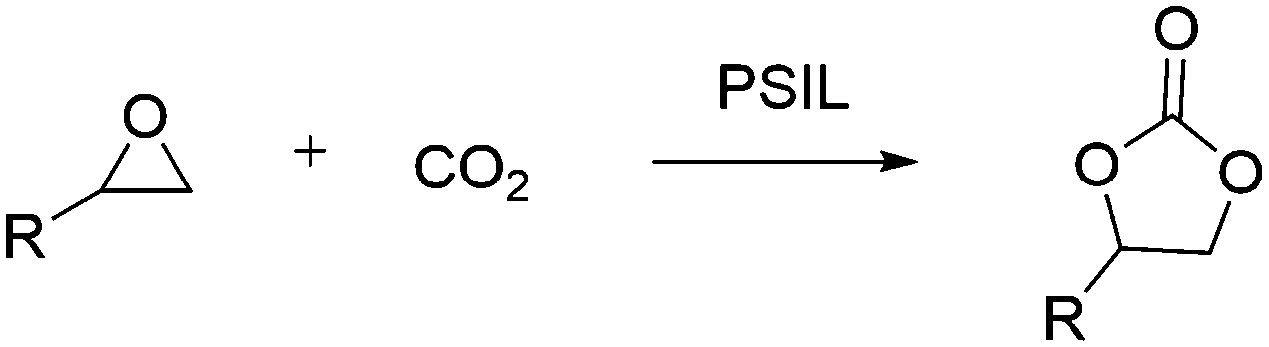
Nowadays, the influence of global climate change on the human living environment has attracted more and more people's attention. Carbon dioxide (CO2) is the main greenhouse gas produced by human activities, on the other hand, it is also one of the most abundant carbon sources.1 Conversion of CO2 into valuable chemicals facilitates the carbon cycle in nature, which is an important goal of catalysis.2 In the field of organic solvents, green reagents and engineering plastics, the widely used organic carbonates, like dimethyl carbonate and diphenyl carbonate, can be derived from cyclic carbonates, which has made the cycloaddition of CO2 with epoxides a commonly used strategy.3Considering the thermodynamic and kinetic stability of CO2 and the high electronegativity of the oxygen atom in epoxides, which is easy to be induced by Lewis acids or other oxophilic substances (such as –OH, –COOH, etc.), a suitable catalyst for the reaction is needed for the desired high efficiency. Various catalysts have been developed for the reactions, including metal oxides, alkali metal salts, transition-metal complexes and ionic liquids.4 It's worthwhile to notice that the ionic liquids (such as quaternary ammonium salts and quaternary phosphonium salts), which displayed excellent properties of low volatility, highly thermal stability and non-flammability, exhibited high activity for this reaction.5 However, the homogeneous catalytic ionic liquids still have the problems of catalysts recovery and product purification.6 So, the immobilization of ionic liquids, including covalent immobilization on polymers or inorganic supports, have been extensively evaluated.7 Particularly, the polymer supported imidazole-based ionic liquids (PSIL) are a series of attractive catalysts because of their high activity and excellent stability.8 Nevertheless, the other types of ionic liquids supported on polymers were rarely investigated. Besides, applying this kind of low-cost and robust catalyst to a continuous fixed-bed reactor is also very attractive due to their potential industrial applications.
In this contribution, firstly, we designed two different convenient ways for the synthesis of imidazolium based polymer supported ionic liquids, and then a series of new polymer supported quaternary ammonium chloride salts, which was derived from trimethylamine, triethylamine and triethanolamine, were afforded by using the optimized method. The prepared supported ionic liquids were systematically investigated as catalysts for the batch and flow cycloaddition of CO2 with epoxides.
Results and discussion
The imidazolium based PSIL were afforded by two simple methods (Scheme 1). The vinyl functionalized imidazolium chloride slat, 1,3-bis(4-vinylbenzyl)imidazolium chloride, was synthesized by the reaction of 1-(chloromethyl)-4-vinylbenzene with imidazole in the method of Path A. Then the imidazolium based PSIL was afforded by the solvothermal copolymerization of 1,3-bis(4-vinylbenzyl)imidazolium chloride and DVB, which was denoted as PSIL(IMD)-1 (Fig. 1). It was worth mentioning that the two steps were completed in an autoclave without any separation and purification steps of the intermediate product. Path B copolymerized 1-(chloromethyl)-4-vinylbenzene with DVB to get chloromethylated polymer firstly. Then the polymer was further cross-linked by imidazole to form the imidazolium based PSIL, which was denoted as PSIL(IMD)-2. PSIL containing quaternary ammonium chloride salts were also prepared in a similar manner of Path A. Trimethylamine-based (PSIL(TMA)), triethylamine-based (PSIL(TEA)) and triethanolamine-based (PSIL(TOA)) polymer supported ionic liquids were obtained by replacing imidazole with trimethylamine, triethylamine and triethanolamine, respectively (Fig. 2).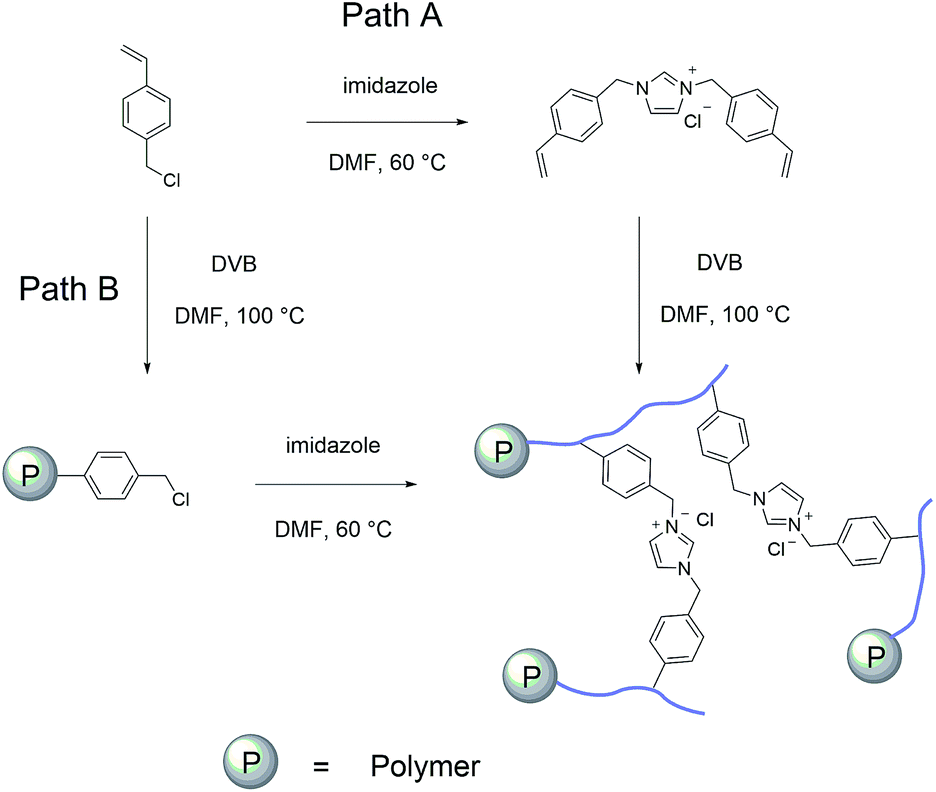 | ||
| Scheme 1 Synthesis of polymer supported ionic liquids of PSIL(IMD)-1 and PSIL(IMD)-2. | ||
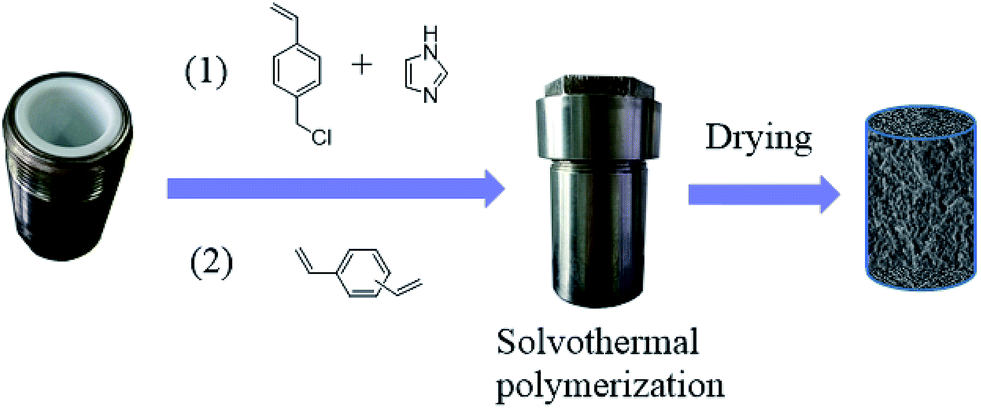 | ||
| Fig. 1 Illustration of solvothermal copolymerization of PSIL(IMD)-1. | ||
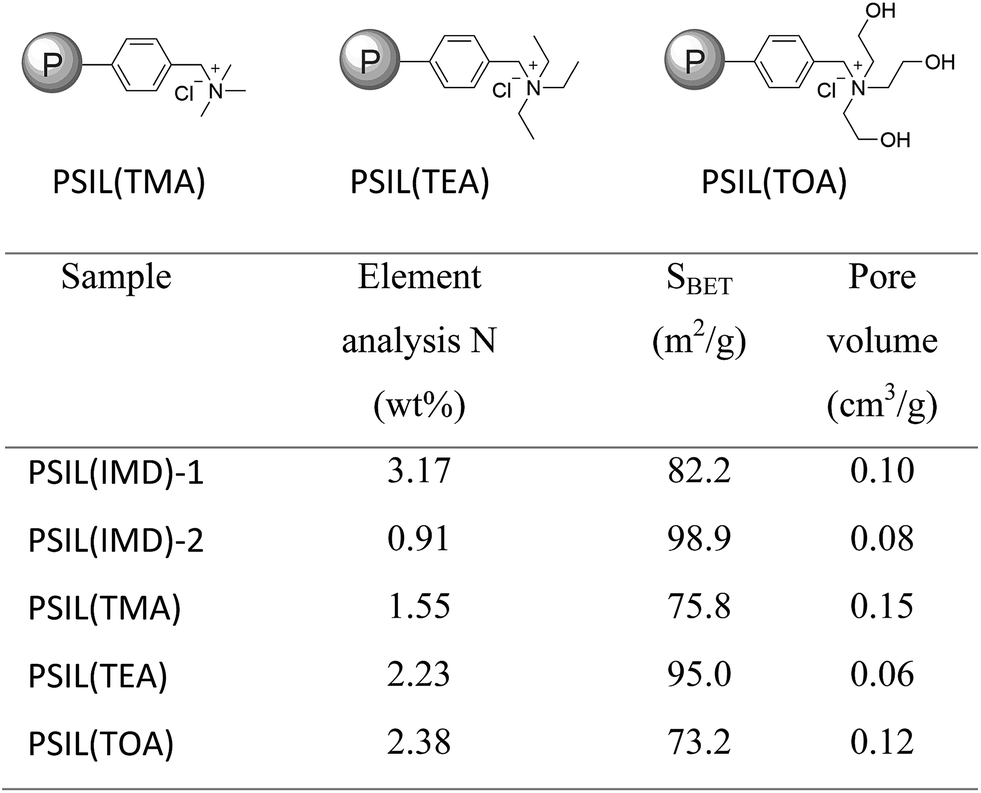 | ||
| Fig. 2 Structures of the polymer supported ionic liquids PSIL(TMA), PSIL(TEA), PSIL(TOA), the element analysis of N contents and textural parameters of the PSIL. | ||
The nitrogen (N) contents of these materials were determined by elemental analysis (Fig. 2). The two imidazolium based PSIL, which were afforded from different preparation methods, gave significant different N contents. PSIL(IMD)-1 gave a nitrogen content of 3.17%, which was just a little lower than the added amount of it for the synthesis of PSIL(IMD)-1 (3.90%). While the nitrogen content of PSIL(IMD)-2 was as little as 0.91%, which indicated that Path A was more feasible than the Path B for the preparation of PSIL. Their pore properties were analysed by N2 physisorption (Fig. 2). They were moderately porous with surface areas from 75.8 m2 g−1 to 98.9 m2 g−1. The pore volumes of all the PSIL were no more than 0.15 cm3 g−1. Since the PSIL essentially maintained the same polarity of the ionic liquids monomer, the relatively low pore volumes of the PSIL were probably due to the strong van der Waals force between ionic liquids in the backbone of the PSIL.9 PSIL(IMD)-1 exhibited a little higher pore volume than PSIL(IMD)-2, which was due to the different synthetic routes. In the Path B, since imidazole was post immobilized on the chloromethylated polymer, a portion of porous space in the polymer was likely to be occupied. The results can be further confirmed by the morphology of the PSIL using scanning electron microscopy (SEM) and transmission electron microscopy (TEM) (Fig. S1–S5†). SEM and TEM images showed that all the PSIL exhibited dense structure. Interestingly, aggregated polymer particles were observed in PSIL(TMA), which was thought to be result from the existence of water (H2O) during the preparation of PSIL(TMA) (Fig. S3†). The addition of the aqueous solution of trimethylamine made the reaction changed from solution polymerization to suspension polymerization. Because of the existence of H2O, the PSIL(TMA) exhibited relatively higher pore volume than the other PSIL.
The structural information of PSIL(IMD)-1 was further characterized by 13C MAS NMR (Fig. 3A). Probably due to the addition of divinyl benzene, all the signals were overlapping and were not very informative. The signals around 0–50 ppm were attributed to the terminal CH2 protons, CH moiety and methylene units that linked the benzene and imidazolium ring. The signals around 100–150 ppm were assigned to carbon atoms of benzene and imidazolium ring. It should be mentioned that there was no significant difference between the 13C MAS NMR spectrum of PSIL(IMD)-1 and PSIL(IMD)-2 (Fig. S6†), which indicated the feasibility of the two strategies for the synthesis of PISL. The 13C MAS NMR spectrum of other PISLs was also presented for the confirmation of their structural information (Fig. S7–S9†). Excellent thermal stability is a crucial requirement for PSIL used as catalyst. As a representative, the thermal stability of PSIL(IMD)-1 was characterized by thermogravimetric analysis (TG) under N2 (Fig. 2B). The curve shows an ideal thermal stability with decomposition temperature at 350 °C.
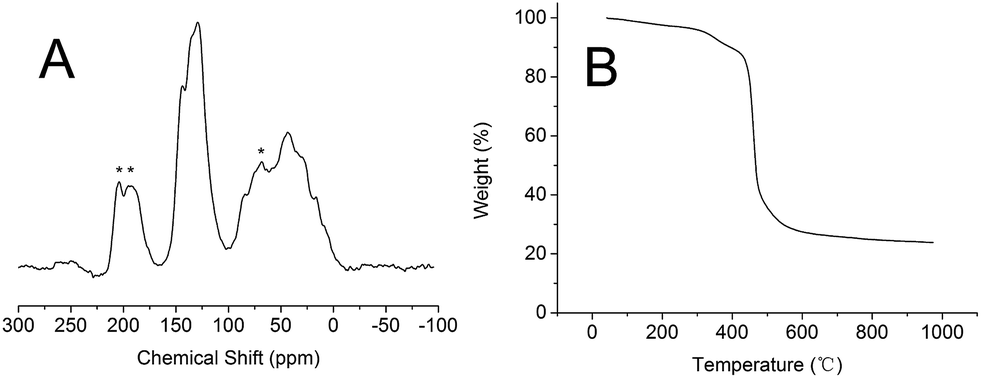 | ||
| Fig. 3 (A) 13C MAS NMR spectra and (B) the TG curves of (a) PSIL(IMD)-1. | ||
The cycloaddition reaction of CO2 with epoxides in the presence of PSIL was firstly conducted in a 30 mL autoclave for the optimization of catalysts and conditions. Then the stability of the optimized catalyst was tested in a continuous fixed-bed. The reaction conditions for the cycloaddition of CO2 with propylene oxide were evaluated on the catalyst of PSIL(IMD)-1. The influence of the pressure of CO2 on the cycloaddition reaction was evaluated at 120 °C with a reaction time of 5 h (Fig. 4). An increase in CO2 pressure from 1 MPa to 2 MPa resulted in a little increase in the yield of propylene carbonate (from 92.4% to 96.0%). The concentration of CO2 in the substrate of propylene oxide increased with the pressure. This facilitated the reaction because CO2 was a reactant. Further increase of CO2 pressure from 2 MPa to 6 MPa leaded to a decrease of the yield of propylene carbonate (from 96.0% to 87.2%). This was probably due to the gas–liquid diffusion between propylene oxide and CO2, which have an effect on the reaction kinetics in the mass transfer. So there are optical ranges of CO2 pressure for the maximum yield of propylene carbonate.8a,10 The reaction temperature was investigated in the range of 100–140 °C at 2 MPa with a reaction time of 5 h (Fig. 5). The reaction temperature was shown to have a positive influence on the catalytic activity. In the relatively low temperature range, the yield of propylene carbonate was drastically increased from 53.9% to 83.3% with temperature from 100 °C to 110 °C. The rising of propylene carbonate yield slowed down in the high temperature range. And the yield of propylene carbonate achieved 99.9% at 130 °C.
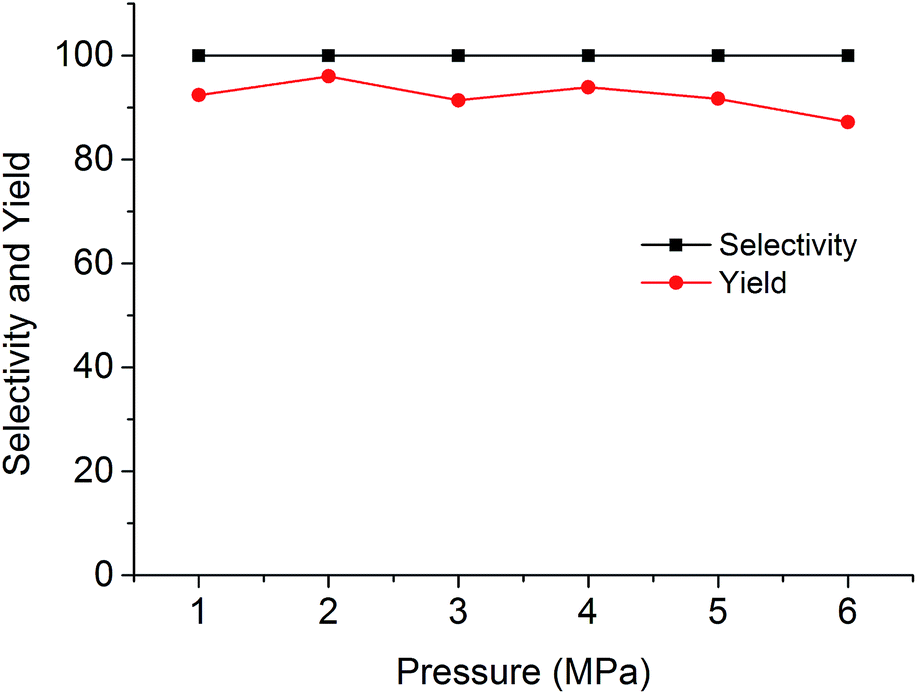 | ||
| Fig. 4 Cycloaddition of CO2 with propylene oxide using the PSIL(IMD)-1 catalyst under different pressures of CO2 (reaction conditions: 0.12 g catalyst, 1.0 g (17.2 mmol) propylene oxide, 120 °C, 5 h). | ||
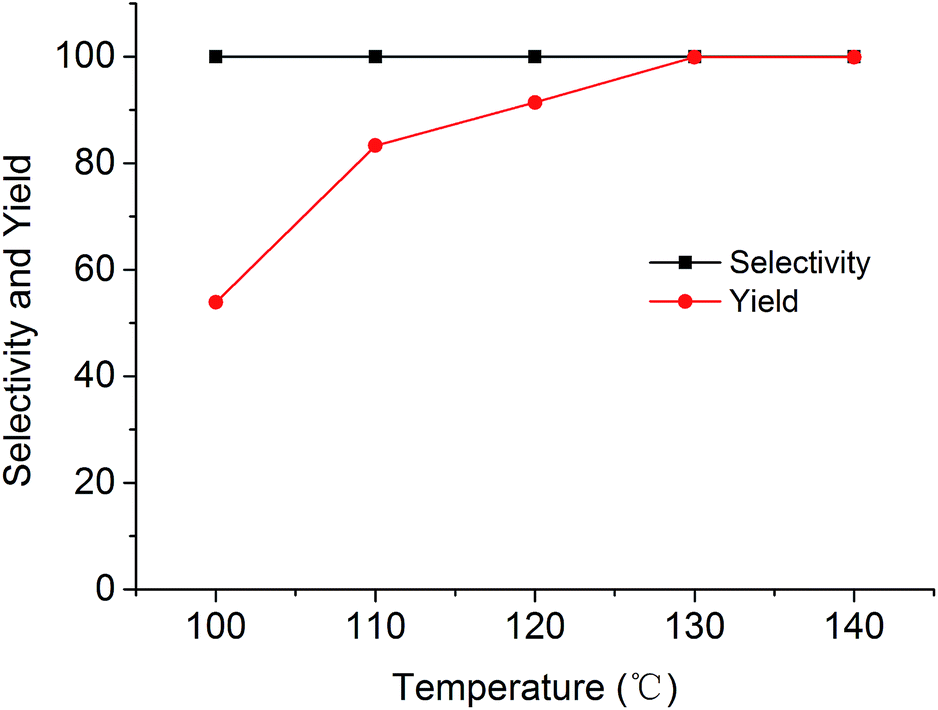 | ||
| Fig. 5 Cycloaddition of CO2 with propylene oxide using the PSIL(IMD)-1 catalyst at different temperatures (reaction conditions: 0.12 g catalyst, 1.0 g (17.2 mmol) propylene oxide, 2 MPa CO2, 5 h). | ||
To evaluate the catalytic activities of different PSIL for the cycloaddition of CO2 with propylene oxide, the reaction was carried out at 130 °C for 5 h under the CO2 pressure of 2 MPa (Table 1, entries 1–5). The yield of propylene carbonate catalysed by PSIL(IMD)-1 could reach as high as 99.9% (entry 1). While, the catalyst of PSIL(IMD)-2 only gave the yield of 80.2% (entry 2). Considering the similar pore structures of the two PSIL, the high activity of PSIL(IMD)-1 can be assigned to its abundant nitrogen content. It was confirmed by the elemental analysis of nitrogen contents. The other three PSIL also exhibited satisfactory results with the yield of product higher than 95% (entries 3–5). It was worth noted that the catalytic activity of the three PSIL decreased in the order PSIL(TMA) > PSIL(TEA) > PSIL(TOA), which was exactly the opposite order of the nitrogen contents in the three PSIL. These indicated that the species of quaternary ammonium salts in the backbone of the PSIL had different influences on the reaction. In order to verify the catalytic efficiency of the PSIL, we also polymerized the divinyl benzene itself, which was denoted as poly(DVB). The poly(DVB) did not show catalytic activity in the reaction (entry 6). It was also notable that when the amount of substrate was increased from 1.0 g to 3.0 g, the PSIL(IMD)-1 system still gave an excellent yield of 96.4% (entry 7). By evaluating various epoxides under the optimized conditions, the PSIL(IMD)-1 exhibited high yields for the target cyclic carbonates with the S/C (substrate/catalyst) ratio of 127 (entries 8–11). In the reported work of Dyson et al., a catalyst of poly[bvbim]Cl was synthesized by direct polymerization of 1,3-bis(4-vinylbenzyl)imidazolium chloride without the cross-linker DVB.11 And the poly[bvbim]Cl catalyst gave the conversions of allyl glycidyl ether and styrene oxide no more than 81% with the S/C ratio of 100, which were conducted at 140 °C and 2 MPa with a reaction time of 24 h. The catalyst of PSIL(IMD)-1 exhibited high conversions of these two substrates with 98% (entry 9 and entry 11). The main reason was because the density of the ionic liquids in poly[bvbim]Cl was much larger than that in the PSIL(IMD)-1, which resulted in deeply imbedded and insufficient use of the active sites.
|
|||
|---|---|---|---|
| Entrya | Catalyst | Epoxides R | Yieldc |
| a Reaction conditions: 0.12 g catalyst, 17.2 mmol epoxides, 2 MPa CO2, 130 °C, 5 h.b 3.0 g (51.6 mmol) propylene oxide.c The yield for propylene carbonate was determined by GC, the yields for other cyclic carbonates were determined by 1H NMR. | |||
| 1 | PSIL(IMD)-1 | Me | 99.9 |
| 2 | PSIL(IMD)-2 | Me | 80.2 |
| 3 | PSIL(TMA) | Me | 97.5 |
| 4 | PSIL(TEA) | Me | 96.3 |
| 5 | PSIL(TOA) | Me | 95.4 |
| 6 | Poly(DVB) | Me | None |
| 7b | PSIL(IMD)-1 | Me | 96.4 |
| 8 | PSIL(IMD)-1 | Bu | 89.1 |
| 9 | PSIL(IMD)-1 | Ph | 98.3 |
| 10 | PSIL(IMD)-1 | CH2Cl | 98.5 |
| 11 | PSIL(IMD)-1 | CH2OCH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
97.5 |
Since the catalytic stability and reusability was of primal interest for the design of heterogeneous catalyst, the stability examination of PSIL(IMD)-1 was conducted in a fixed-bed reactor (Scheme 2 and Fig. 6). 0.3 g of PSIL(IMD)-1 was used in the reaction. The substrate of propylene oxide was continuously fed (15 μL min−1) without any solvents. The gas hourly space velocity (GHSV) was 4000 h−1 and the pressure of CO2 was 2 MPa. The PSIL(IMD)-1 maintained stable activity through the study period of 130 hours with the yield of propylene carbonate slightly ranging between 41% and 45%. This was because the comonomers of 1,3-bis(4-vinylbenzyl)imidazolium chloride and DVB both had two styrene arms, and the ionic liquids could be firmly incorporated into the backbone of PSIL. Han and co-workers reported a polymer supported ionic liquids (PSIL 4), which was afforded by the copolymerization of single vinyl functionalized imidazole salt (3-butyl-1-vinylimidazolium chloride) and DVB.8a The experiment of recyclability exhibited their PSIL 4 catalyst was used five runs with no considerable decrease in the yield of propylene carbonate (from 97.4% to 95.8%), since the ionic liquid was grafted onto a highly cross-linked polymer matrix with one arm. In addition, comparing with the metal existed catalysis systems (metal oxides, alkali metal salts and transition-metal complexes, etc.), there was no trouble of metal leaching in the PSIL catalysts. So the catalyst of PSIL(IMD)-1 could show excellent catalytic stability.
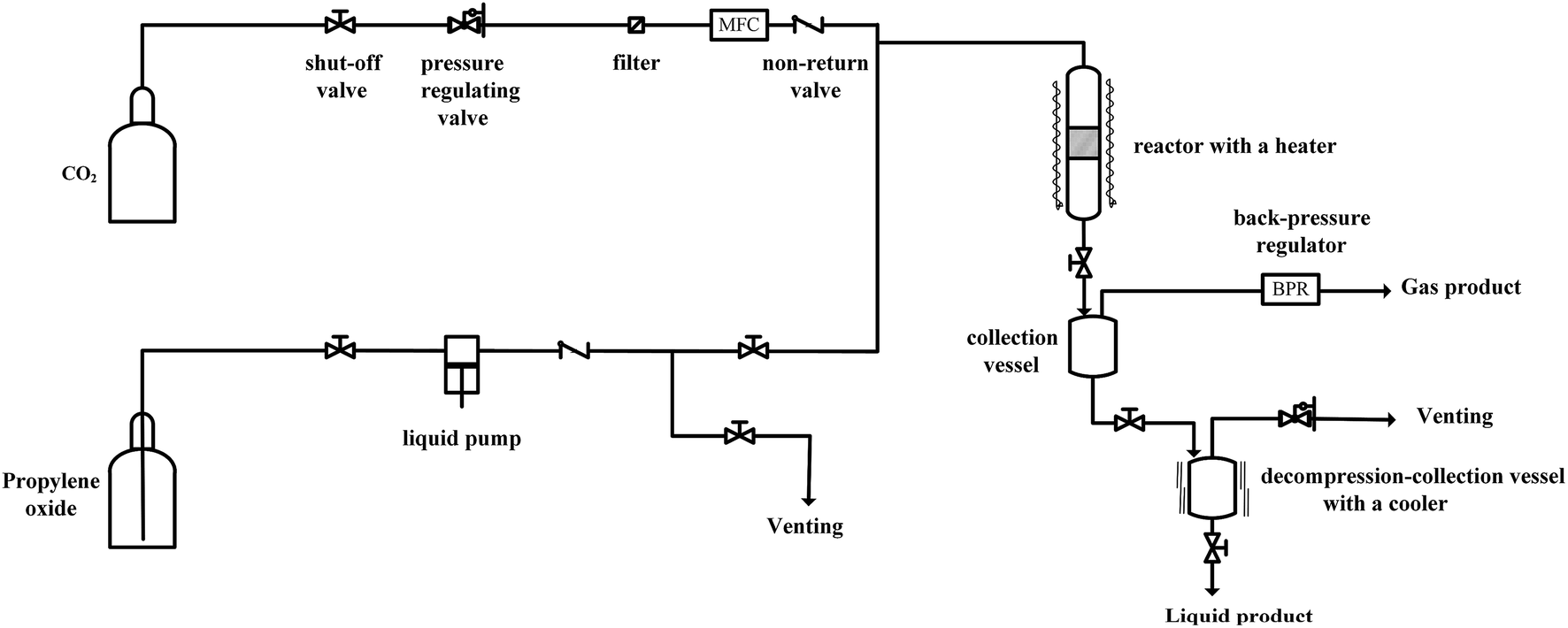 | ||
| Scheme 2 The simple flow chart of the reaction equipment. | ||
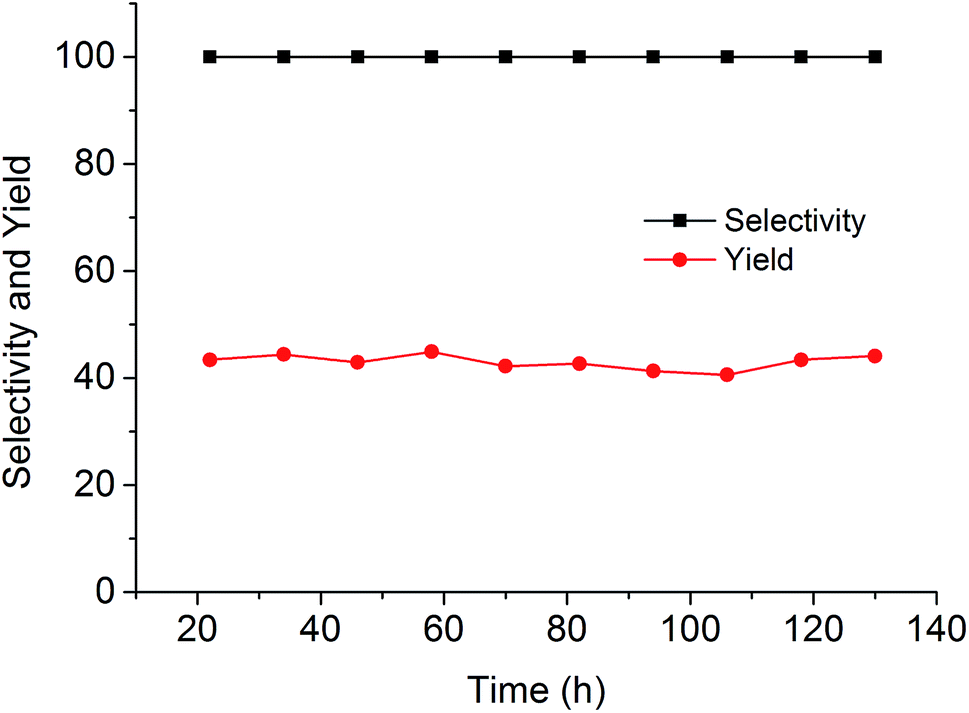 | ||
| Fig. 6 Stability test of PISL(IMD)-1 in a fixed-bed reactor (reaction conditions: 0.30 g catalyst, 15 μL min−1 propylene oxide, 130 °C, 2 MPa CO2, GHSV = 4000 h−1). | ||
Conclusions
Two simple strategies were presented for the synthesis of PSIL. Through the comparison of the nitrogen contents and the catalytic activity of PSIL(IMD)-1 and PSIL(IMD)-2, the Path A was proved as a more useful way than Path B. Using the method of Path A, a series of polymer supported quaternary ammonium chloride ionic liquids were afforded by the radical copolymerization of 4-vinylbenzyl functionalized quaternary ammonium chloride salts with DVB. The prepared PSIL exhibited high catalytic activities in the cycloaddition of CO2 with epoxides. Especially for the PSIL(IMD)-1 catalyst, which exhibited higher catalytic activity and stability than some reported polymer supported imidazole-based ionic liquids. Considering that the ionic liquids was firmly embedded in the polymer backbone, the PSIL(IMD)-1 catalyst was applied in a fixed-bed reactor for the first time. As expected, the PSIL(IMD)-1 exhibited stable performance in the cycloaddition of CO2 with propylene oxide after continuous flow reaction for 130 hours.Experimental
CO2 was supplied by Zhonghao Guangming research & design institute of chemical industry Corp. with a purity of 99.99%. Imidazole (99%), 1-(chloromethyl)-4-vinylbenzene (90%) and AIBN (99%) were obtained from J&K chemical. Divinylbenzene (80%) was also provided by J&K chemical as a mixture of 1,2-, 1,3-, and 1,4-divinylbenzene. Trimethylamine (33% in H2O), triethylamine (99%), triethanolamine and other reagents were obtained from Tianjin Kemiou Chemical Reagent Co., Ltd and used as received. The N element analysis was performed by organic element analyzer of Flash 2000, Thermo scientific. Nitrogen isotherms at 77.3 K were measured using Quantachrome Autosorb-1. The sample was outgassed at 120 °C for 10 h. Thermogravimetric analysis (TG) was measured using NETZSCH STA 449F3, and the samples were heated from 40 °C to 1000 °C at the rate of 10 °C min−1 under N2. Transmission electron microscope (TEM) images were taken on a JEM-2100 with an accelerating voltage of 200 kV. Polymer morphologies were investigated on a JSM-7800F scanning electron microscope (SEM). 13C MAS NMR spectra were obtained on a VARIAN infinity plus 400 spectrometer under a magic angle spinning rate of 6 kHz.Synthesis of PSIL
Typical procedure for the synthesis of propylene carbonate via an autoclave
As a typical run for the cycloaddition of CO2 and propylene oxide, PISL(IMD)-1 (0.12 g) and propylene oxide (1.0 g, 17.2 mmol) were added into a 30 mL autoclave. The reactor was purged 2 times with CO2 and heated from room temperature to the reaction temperature of 130 °C, stirred for 5 h. During the reaction, the pressure in the reactor was held constant (2.0 MPa) by the injection of more CO2 with a pressure regulator. The catalyst was separated by centrifugation. The product was analysed by an Agilent 7890A gas chromatograph with a HP-5 column using a FID detector and 1-butanol as an internal standard. It was worth noting that the yields for other cyclic carbonates were determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard.Typical procedure for the synthesis of propylene carbonate via a fixed-bed reactor
Acknowledgements
This work was supported by the Strategic priority research program of the Chinese Academy of Sciences, Grant No. XDB17020400 and the National Natural Science Foundation of China (21303190).Notes and references
- R. Quadrelli and S. Peterson, Energy Policy, 2007, 35, 5938–5952 CrossRef.
- (a) Y. J. Zhou, Z. W. Fu, S. J. Wang, M. Xiao, D. M. Han and Y. Z. Meng, RSC Adv., 2016, 6, 40010–40016 RSC; (b) P. Lanzafame, G. Centi and S. Perathoner, Chem. Soc. Rev., 2014, 43, 7562–7580 RSC; (c) N. von der Assen, P. Voll, M. Peters and A. Bardow, Chem. Soc. Rev., 2014, 43, 7982–7994 RSC.
- (a) J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS; (b) M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC; (c) M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- (a) S. J. Zhang, Y. H. Chen, F. W. Li, X. M. Lu, W. B. Dai and R. Mori, Catal. Today, 2006, 115, 61–69 CrossRef CAS; (b) V. Calo, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561–2563 CrossRef CAS PubMed; (c) Y. Tsutsumi, K. Yamakawa, M. Yoshida, T. Ema and T. Sakai, Org. Lett., 2010, 12, 5728–5731 CrossRef CAS PubMed; (d) S. S. Yu, X. H. Liu, J. G. Ma, Z. Niu and P. Cheng, J. CO2 Util., 2016, 14, 122–125 CrossRef CAS; (e) L. Wang, G. Y. Zhang, K. Kodamaa and T. Hirose, Green Chem., 2016, 18, 1229–1233 RSC; (f) N. Tenhumberg, H. Buttner, B. Schaffner, D. Kruse, M. Blumenstein and T. Werner, Green Chem., 2016, 18, 3775–3788 RSC; (g) S. Roy, B. Banerjee, A. Bhaumik and S. M. Islam, RSC Adv., 2016, 6, 31153–31160 RSC; (h) C. A. Montoya, C. F. Gomez, A. B. Paninho, A. V. M. Nunes, K. T. Mahmudov, V. Najdanovic-Visak, L. M. D. R. S. Martins, M. F. C. G. da Silva, A. J. L. Pombeiro and M. N. da Ponte, J. Catal., 2016, 335, 135–140 CrossRef CAS.
- V. I. Parvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef CAS PubMed.
- J. Q. Wang, K. Dong, W. G. Cheng, J. Sun and S. J. Zhang, Catal. Sci. Technol., 2012, 2, 1480–1484 CAS.
- (a) K. Motokura, S. Itagaki, Y. Iwasawa, A. Miyaji and T. Baba, Green Chem., 2009, 11, 1876–1880 RSC; (b) J. Tharun, Y. Hwang, R. Roshan, S. Ahn, A. C. Kathalikkattil and D. W. Park, Catal. Sci. Technol., 2012, 2, 1674–1680 RSC; (c) D. Wei-Li, J. Bi, L. Sheng-Lian, L. Xu-Biao, T. Xin-Man and A. Chak-Tong, Catal. Today, 2014, 233, 92–99 CrossRef; (d) B. Mousavi, S. Chaemchuen, B. Moosavi, Z. X. Luo, N. Gholampour and F. Verpoort, New J. Chem., 2016, 40, 5170–5176 RSC; (e) X. C. Wang, Y. Zhou, Z. J. Guo, G. J. Chen, J. Li, Y. M. Shi, Y. Q. Liu and J. Wang, Chem. Sci., 2015, 6, 6916–6924 RSC.
- (a) Y. Xie, Z. F. Zhang, T. Jiang, J. L. He, B. X. Han, T. B. Wu and K. L. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255–7258 CrossRef CAS PubMed; (b) X. H. Wu, M. P. Wang, Y. Z. Xie, C. Chen, K. Li, M. M. Yuan, X. G. Zhao and Z. S. Hou, Appl. Catal., A, 2016, 519, 146–154 CrossRef CAS.
- M. I. Burguete, F. Galindo, E. Garcia-Verdugo, N. Karbass and S. V. Luis, Chem. Commun., 2007, 3086–3088 RSC.
- R. C. Luo, X. T. Zhou, W. Y. Zhang, Z. X. Liang, J. Jiang and H. B. Ji, Green Chem., 2014, 16, 4179–4189 RSC.
- S. Ghazali-Esfahani, H. B. Song, E. Paunescu, F. D. Bobbink, H. Z. Liu, Z. F. Fei, G. Laurenczy, M. Bagherzadeh, N. Yan and P. J. Dyson, Green Chem., 2013, 15, 1584–1589 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: SEM, TEM images and 13C NMR spectra. See DOI: 10.1039/c6ra26780g |
| This journal is © The Royal Society of Chemistry 2017 |