
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemiluminescent chemodosimetric probes for sulfide based on cyclometalated Ir(III) complexes†
Seo-Yeon Kim,
Hoon Jun Kim and
Jong-In Hong *
*
Department of Chemistry, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 151-747, Korea. E-mail: jihong@snu.ac.kr
First published on 10th February 2017
Abstract
We developed two electrochemiluminescent probes (1, 2) for sulfide (S2−), based on cyclometalated Ir(III) complexes. Addition of sulfide anions greatly increased the ECL signal of probes by cleaving a PET quencher moiety from probe ligands. Probe 2 showed a high turn-on ratio of ECL and good selectivity for sulfide anions over various anions and biothiols.
Hydrogen sulfide (H2S), a well-known toxic gas, has recently been recognized as an important gaseous signalling molecule.1,2 H2S is generated endogenously from L-cysteine by several enzymes such as cystathionine γ-lyase (CSE),3 cystathionine β-synthase (CBS),4 and 3-mercaptopyruvate sulfur transferase (3-MST).5 As a gasotransmitter, H2S regulates various biological processes in the cardiovascular,6 central nervous,7 immune,8 and gastrointestinal9 systems. In blood plasma, 10–100 μM of sulfide is considered the normal level.10,11 However, an abnormal level of H2S is associated with some diseases, including Alzheimer's disease,12 Down's syndrome, diabetes,13 and liver cirrhosis.14 Therefore, simple methods for selective detection of H2S are required in order to diagnose various diseases that increase the plasma H2S concentration to abnormal levels.
So far, various approaches have been studied for the detection of H2S, such as electrochemical analysis,15 gas chromatography,16 and colorimetric17 and fluorescent18 assays. In particular, a large number of fluorescent chemodosimeters for H2S were developed based on the strong reducing19,20 or nucleophilic21,22 properties of sulfide anions. However, fluorescent assays cannot be used for point-of-care (POC) detection due to the requirement of an additional optical source and bulky equipment.
Electrochemiluminescence (ECL) is a light-emitting process caused by the electron-transfer reaction between electrochemically generated radical species at the electrodes.23 In comparison with conventional fluorescent methods, the ECL method has many advantages, including no background optical signal, high sensitivity and no need of extra light sources, providing simple and miniaturized sensing tools.23–25 These features afford strong benefits in the development of POC detection sensors. ECL luminophores were developed using various phosphorescent heavy-metal complexes such as Ru(II),26 Os(II),27 Eu(III),28 Re(I),29 Pt(II),30 and Ir(III).31–33 Among them, Ir(III) complexes have attracted increasing attention because they exhibit high luminescence efficiency, good electrochemical stability, and easy tunability of the luminescent colour by modulating the substitution of ligands.31–33
Herein, we designed two ECL chemodosimetric probes for the sulfide anion based on Ir(III) complexes (Scheme 1). We selected (piq)2Ir(pic) (piq = 1-phenylisoquinoline, pic = picolinate) as a luminophore,34 and the dinitrophenyl (DNP) group as a photo-induced electron transfer (PET) quencher35 and a reaction site,35–37 providing bright emission after the nucleophilic aromatic substitution (SNAr) by the sulfide anion. Probe 1 has a DNP group on the ancillary ligand, while probe 2 has an additional quencher group on the main ligands that would further reduce the phosphorescent intensity of the probe itself and thus produce a high turn-on ratio in response to sulfide anions. The reaction products of probes 1 and 2 were confirmed by MALDI-TOF mass spectra (Fig. S1 and S2†). Synthetic procedures of probes 1 and 2 are described in the ESI (Schemes S1 and S2†).
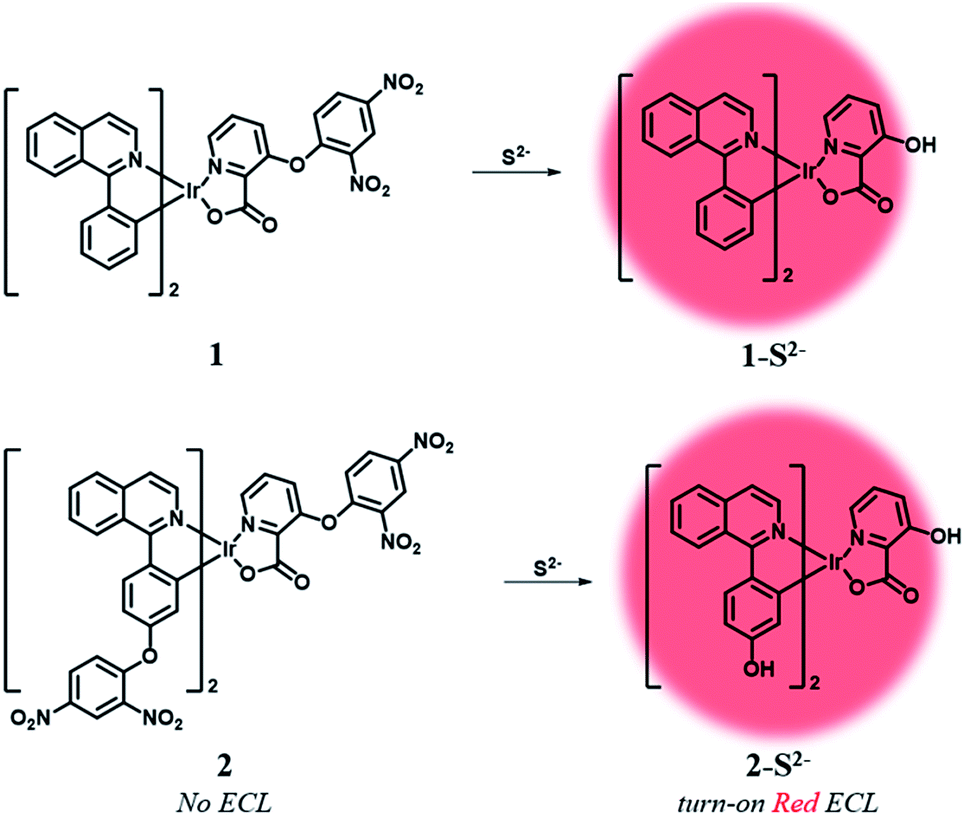 | ||
| Scheme 1 Electrogenerated chemiluminescent sensing mechanism of probes 1 and 2. | ||
Initially, we examined the phosphorescence spectra of probes 1 and 2. As shown in Fig. 1a, the phosphorescence intensity of 1 gradually increased at 606 nm (λex = 460 nm) until 10 equiv. of sulfide anion (100 μM) was added. The phosphorescence intensity of 2 increased more dramatically than 1 at 601 nm (Fig. 1b). Probe 2 required a larger amount of sulfide anion (150 μM, 15 equiv.) than 1 for saturation because the former has more reaction sites for sulfide. The estimated limit of detection (LOD) was calculated to be 1.9 μM for 1 and 0.2 μM for 2 (signal-to-noise (S/N) ratio = 3). Then, we compared the phosphorescence turn-on ratios of 1 and 2 in the presence of 100 μM of sulfide anion (Fig. 1c). The turn-on ratio of 2 was greater than 1, as we expected. UV-vis absorption spectra were also investigated (Fig. 1d). In the presence of sulfide (10 equiv.), the absorption peak around 457 nm increased significantly. Probe 2 solution showed the corresponding colour change from colourless to yellow, enabling colorimetric detection through the naked eye.
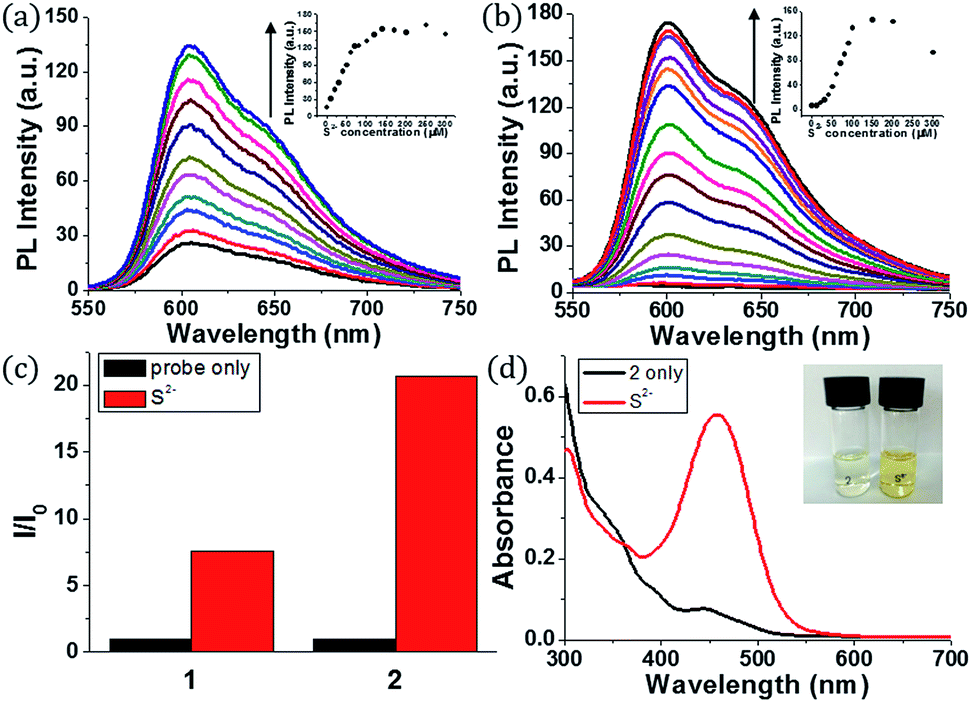 | ||
| Fig. 1 (a) Phosphorescent emission spectra of 1 (10 μM) in the presence of 0–100 μM of sulfide in CH3CN (inset: changes in phosphorescence intensity of 1 at 606 nm upon the addition of sulfide) (b) phosphorescent emission spectra of 2 (10 μM) in the presence of 0–150 μM of sulfide in CH3CN (inset: changes in phosphorescent intensity of 2 upon the addition of sulfide) (c) turn-on ratio of 1 and 2 in the absence (black bar) and presence (red bar) of 100 μM sulfide in CH3CN (d) UV-vis absorption of 2 (10 μM) before and after addition of sulfide (100 μM) in CH3CN. | ||
Density functional theory (DFT) calculations supported the PET sensing mechanism of the probes (Fig. S3†). The HOMOs of 1 and 2 were mainly localized on the Ir(III) centre and phenyl ring of piq and the LUMO is localized on the DNP, whereas the LUMO of Ir(III) complexes is generally localized on the isoquinoline of the main ligands. Hence, we expected that probes 1 and 2 were able to show an “off–on” emission signal in response to sulfide through the PET modulation. We also conducted cyclic voltammetry (CV) measurements and compared the HOMO/LUMO energy levels of luminophores (1-S2− and 2-S2−) with the LUMO of a quencher (1-chloro-2,4-dinitrobenzene) to confirm the PET mechanism experimentally (Fig. 2). As expected, the LUMO (−4.36 eV) of the quencher is located between the HOMO (−5.35 eV) and LUMO (−3.08 eV) of 1-S2− as well as the HOMO (−5.19 eV) and LUMO (−2.87 eV) of 2-S2−. Therefore, the phosphorescence signal of the probes was quenched by the PET process before the cleavage of the DNP moiety from Ir(III) complexes upon the addition of sulfide.
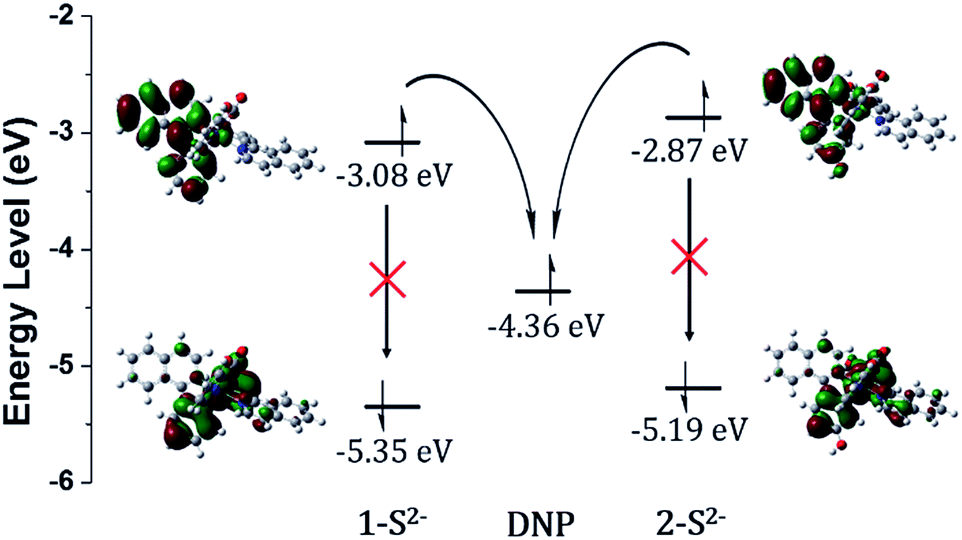 | ||
| Fig. 2 HOMO/LUMO energy levels calculated from CV measurements and electronic distributions of 1-S2− and 2-S2− and photo-induced electron transfer pathway. | ||
The ECL measurements were performed during the CV process. Probe 1 itself showed the initial ECL intensity at around 1.4 V, but further increase in the ECL intensity of 1 was observed in the presence of sulfide (Fig. 3a). A titration curve of 1 was obtained against various concentrations of the sulfide anion (Fig. 3b). A linear relationship between the ECL intensity and the sulfide concentration was observed from 0 to 80 μM of sulfide. The estimated LOD was calculated to be 27 nM, significantly lower than the LOD of phosphorescence. Hence, 1 can be used as a highly sensitive ECL sensor to detect abnormal levels of S2−.
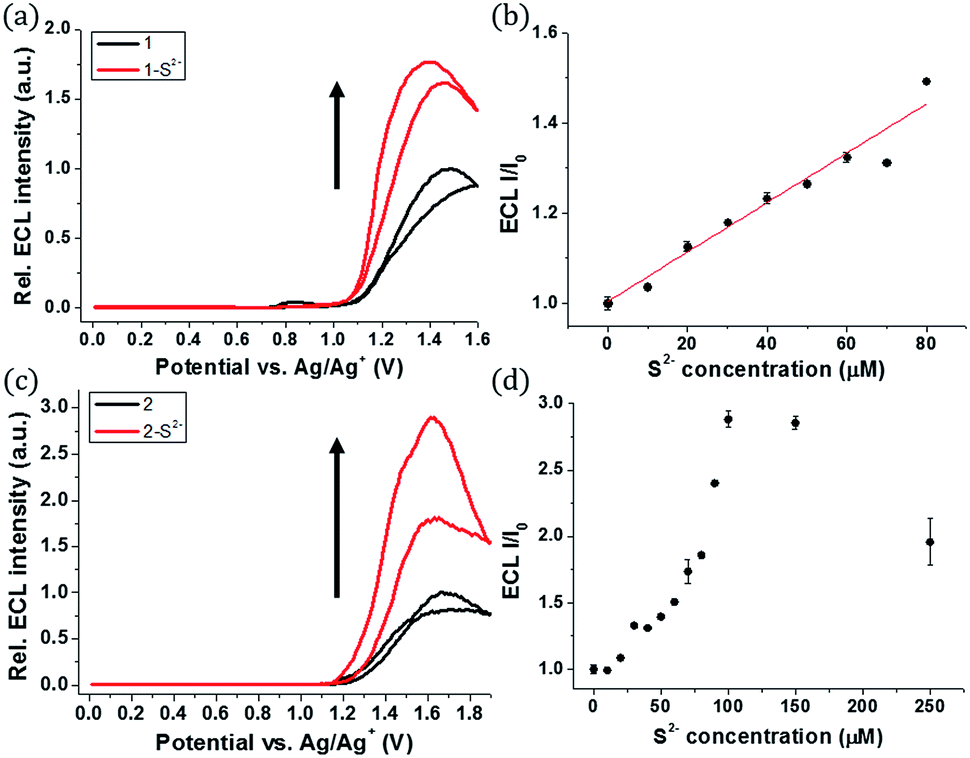 | ||
| Fig. 3 (a) ECL intensity of 1 (10 μM) upon the addition of sulfide (80 μM) in CH3CN (25 mM TPA, and 0.1 M TBAPF6 as the supporting electrolyte) (b) ECL titration curve of 1 (10 μM) upon the addition of sulfide in CH3CN (10 mM TPA, and 0.1 M TBAPF6 as the supporting electrolyte) (c) ECL intensity of 2 (10 μM) upon the addition of sulfide (150 μM) in CH3CN (25 mM TPA, and 0.1 M TBAPF6 as the supporting electrolyte) (d) ECL titration curve of 2 (10 μM) upon the addition of sulfide in CH3CN (10 mM TPA, 0.1 M TBAPF6 as the supporting electrolyte) (the potential is swept at a Pt disk electrode (diameter: 2 mm) vs. Ag/Ag+, scan rate: 0.1 V s−1). | ||
A similar turn-on ECL of 2 was observed in the presence of sulfide (Fig. 3c). Probe 2 showed 3-fold enhancement of the ECL intensity at around 1.6 V in the presence of 15 equiv. of sulfide (150 μM). The emission signal increased greatly when the sulfide concentration was in the range of 0–100 μM (Fig. 3d). Although the ECL turn-on ratio of 2 was greater than that of 1, the absolute ECL intensity of 2 was low even after saturation with sulfide (Fig. S4†). The maximum signal of 2 was only 5.7% of 1 in the presence of sulfide (80 μM, 8 equiv.) (Table S1†). The low ECL intensity of 2 caused low sensitivity toward the sulfide anion, and the estimated LOD of 2 was calculated to be 0.3 μM, which is relatively high compared to that of 1 (Fig. S5†).
The low ECL intensity of 2 can be explained by the CV measurements (Fig. 4). We compared the HOMO/LUMO energy levels of 1-S2− and 2-S2− calculated from CV measurements with the HOMO of tri-n-propylamine (TPA). One of the conditions for the efficient ECL emission is that the HOMO of the emitter should be lower than that of TPA for an efficient generation of TPA+˙.38 The HOMO energy level of 1-S2− (−5.35 eV) is relatively well matched with that of TPA (−5.38 eV), so that 1 can emit a strong ECL in response to the sulfide anion through the relatively smooth electron transfer from TPA HOMO to 1-S2− HOMO. In contrast, the HOMO energy level of 2-S2− (−5.19 eV) is quite higher than that of TPA because the hydroxyl groups on the main ligands destabilized the HOMO level. Thus, the electron transfer from TPA to 2-S2− hardly occurred, causing a weak ECL emission. The CV studies rationalized the low ECL intensity of 2. Furthermore, the energy level of the TPA radical is higher than that of the LUMO of (1-S2−)+˙ or (2-S2−)+˙, so that the excited state of (1-S2−)+˙ or (2-S2−)+ can be easily formed to generate the ECL.39
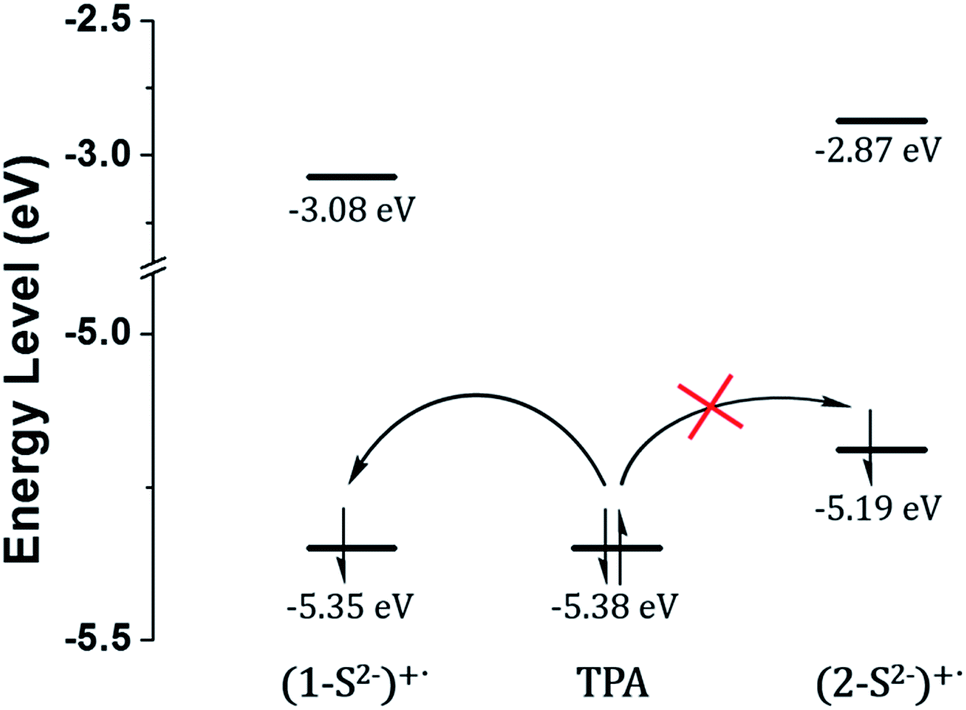 | ||
| Fig. 4 HOMO/LUMO energy levels calculated from CV measurements and generation of TPA+˙ through the catalytic pathway. | ||
Further, we carried out additional experiments with Ru(bpy)32+, the most frequently used ECL luminophore, to confirm the effect of remaining sulfide ions on the ECL signal in solution (Fig. S6†). We confirmed that almost no ECL changes were observed in the presence of excess sulfide (100 μM, 10 equiv.), when compared to the ECL intensity of Ru(bpy)32+ in the absence of sulfide ions. These data could prove that remaining excess sulfide ions in solution have only a little effect on the change of ECL intensities.
The selectivity of 1 and 2 was tested by adding 100 μM of each anion to 10 μM of probe 2 (Fig. 5 and S7–S9†). Only the sulfide anion induced a significant increase in the ECL intensity, whereas almost no ECL changes were observed upon the addition of other anions, including F−, Cl−, Br−, I−, HCO3−, CO32−, C2O42−, SO42−, NO3−, N3−, AcO−, and SCN−. In particular, CN− and biothiols such as cysteine, homocysteine, and glutathione, which are difficult to be distinguished from sulfide, could not increase the emission intensity. These results suggest that probe 2 is a highly selective probe for sulfide (S2−) over other analytes and can be used for biological applications based on ECL analysis.
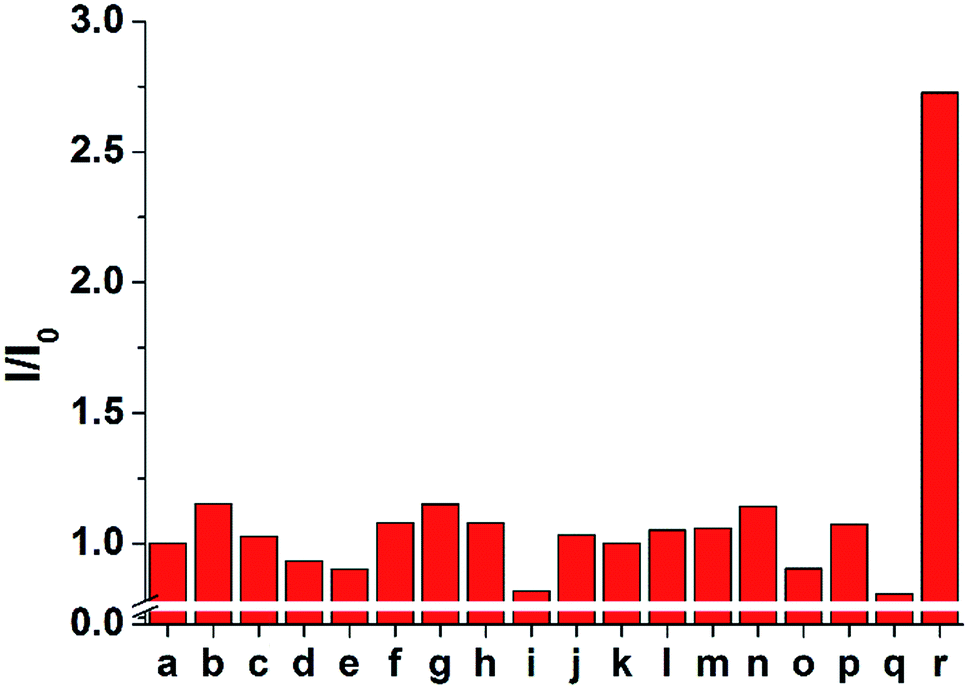 | ||
| Fig. 5 ECL responses of 2 (10 μM) in the presence of various analytes (100 μM) in CH3CN. (25 mM TPA, and 0.1 M TBAPF6 as the supporting electrolyte) (a) probe only, (b) F−, (c) Cl−, (d) Br−, (e) I−, (f) HCO3−, (g) CO32−, (h) C2O42−, (i) SO42−, (j) NO3−, (k) N3−, (l) AcO−, (m) SCN−, (n) CN−, (o) Cys, (p) Hcy, (q) GSH, (r) S2−. | ||
Conclusions
We designed two “off–on” chemodosimetric ECL probes for sulfide (S2−), based on cyclometalated Ir(III) complexes. In the presence of sulfide anions, the ECL intensity of probe 2 increased greatly due to the blocking of the PET quenching process. Furthermore, the probe showed a high turn-on ratio with sulfide only. We expect that our rational sensing approach will pave the way for the development of various ECL-based sensing tools for small biomolecules.Acknowledgements
This work was supported by the National Research Foundation (Grant No. 2015R1A2A1A15055347) funded by the MSIP.Notes and references
- M. M. Gadalla and S. H. Snyder, J. Neurochem., 2010, 113, 14–26 CrossRef CAS PubMed.
- O. Kabil and R. Banerjee, J. Biol. Chem., 2010, 285, 21903–21907 CrossRef CAS PubMed.
- N. Sen, B. D. Paul, M. M. Gadalla, A. K. Mustafa, T. Sen, R. Xu, S. Kim and S. H. Snyder, Mol. Cell, 2012, 45, 13–24 CrossRef CAS PubMed.
- S. Singh, D. Padovani, R. A. Leslie, T. Chiku and R. Banerjee, J. Biol. Chem., 2009, 284, 22457–22466 CrossRef CAS PubMed.
- M. T. Norihiro Shibuya, M. Yoshida, Y. Ogasawara, T. Togawa, K. Ishii and H. Kimura, Antioxid. Redox Signaling, 2009, 11, 703–714 CrossRef PubMed.
- J. W. Calvert, S. Jha, S. Gundewar, J. W. Elrod, A. Ramachandran, C. B. Pattillo, C. G. Kevil and D. J. Lefer, Circ. Res., 2009, 105, 365–374 CrossRef CAS PubMed.
- K. Abe and H. Kimura, J. Neurosci., 1996, 16, 1066–1071 CAS.
- R. C. Zanardo, V. Brancaleone, E. Distrutti, S. Fiorucci, G. Cirino and J. L. Wallace, FASEB J., 2006, 20, 2118–2120 CrossRef CAS PubMed.
- D. R. Linden, Antioxid. Redox Signaling, 2014, 20, 818–830 CrossRef CAS PubMed.
- A. Papapetropoulos, A. Pyriochou, Z. Altaany, G. Yang, A. Marazioti, Z. Zhou, M. G. Jeschke, L. K. Branski, D. N. Herndon, R. Wang and C. Szabo, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21972 CrossRef CAS PubMed.
- Y. H. Chen, W. Z. Yao, B. Geng, Y. L. Ding, M. Lu, M. W. Zhao and C. S. Tang, Chest, 2005, 128, 3205–3211 CrossRef CAS PubMed.
- K. Eto, T. Asada, K. Arima, T. Makifuchi and H. Kimura, Biochem. Biophys. Res. Commun., 2002, 293, 1485–1488 CrossRef CAS PubMed.
- P. Kamoun, M. C. Belardinelli, A. Chabli, K. Lallouchi and B. Chadefaux-Vekemans, Am. J. Med. Genet., Part A, 2003, 116, 310–311 CrossRef PubMed.
- S. Fiorucci, E. Antonelli, A. Mencarelli, S. Orlandi, B. Renga, G. Rizzo, E. Distrutti, V. Shah and A. Morelli, Hepatology, 2005, 42, 539–548 CrossRef CAS PubMed.
- N. S. Lawrence, J. Davis, L. Jiang, T. G. J. Jones, S. N. Davies and R. G. Compton, Electroanalysis, 2000, 12, 1453–1460 CrossRef CAS.
- J. Radford-Knoery and G. A. Cutter, Anal. Chem., 1993, 65, 976–982 CrossRef CAS.
- J. Bae, M. G. Choi, J. Choi and S.-K. Chang, Dyes Pigm., 2013, 99, 748–752 CrossRef CAS.
- F. Yu, P. Li, P. Song, B. Wang, J. Zhao and K. Han, Chem. Commun., 2012, 48, 2852–2854 RSC.
- A. R. Lippert, E. J. New and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 10078–10080 CrossRef CAS PubMed.
- H. Peng, Y. Cheng, C. Dai, A. L. King, B. L. Predmore, D. J. Lefer and B. Wang, Angew. Chem., Int. Ed., 2011, 50, 9672–9675 CrossRef CAS PubMed.
- Y. Chen, C. Zhu, Z. Yang, J. Chen, Y. He, Y. Jiao, W. He, L. Qiu, J. Cen and Z. Guo, Angew. Chem., Int. Ed., 2013, 52, 1688–1691 CrossRef CAS PubMed.
- C. Liu, J. Pan, S. Li, Y. Zhao, L. Y. Wu, C. E. Berkman, A. R. Whorton and M. Xian, Angew. Chem., Int. Ed., 2011, 50, 10327–10329 CrossRef CAS PubMed.
- M. M. Richter, Chem. Rev., 2004, 104, 3003–3036 CrossRef CAS PubMed.
- W. Miao, Chem. Rev., 2008, 108, 2506–2553 CrossRef CAS PubMed.
- X. Yue, Z. Zhu, M. Zhang and Z. Ye, Anal. Chem., 2015, 87, 1839–1845 CrossRef CAS PubMed.
- P. McCord and A. J. Bard, J. Electroanal. Chem. Interfacial Electrochem., 1991, 318, 91–99 CrossRef CAS.
- D. Bruce, M. M. Richter and K. J. Brewer, Anal. Chem., 2002, 74, 3157–3159 CrossRef CAS PubMed.
- M. M. Richter and A. J. Bard, Anal. Chem., 1996, 68, 2641–2650 CrossRef CAS PubMed.
- M. M. Richter, J. D. Debad, D. R. Striplin, G. A. Crosby and A. J. Bard, Anal. Chem., 1996, 68, 4370–4376 CrossRef.
- E. M. Gross, N. R. Armstrong and R. M. Wightman, J. Electrochem. Soc., 2002, 149, E137 CrossRef CAS.
- Y. You and W. Nam, Chem. Soc. Rev., 2012, 41, 7061–7084 RSC.
- Y. Zhou, H. Gao, X. Wang and H. Qi, Inorg. Chem., 2015, 54, 1446–1453 CrossRef CAS PubMed.
- M.-J. Li, P. Jiao, M. Lin, W. He, G.-N. Chen and X. Chen, Analyst, 2011, 136, 205–210 RSC.
- J. M. Fernandez-Hernandez, E. Longhi, R. Cysewski, F. Polo, H.-P. Josel and L. De Cola, Anal. Chem., 2016, 88, 4174–4178 CrossRef CAS PubMed.
- X. Cao, W. Lin, K. Zheng and L. He, Chem. Commun., 2012, 48, 10529–10531 RSC.
- T. Liu, Z. Xu, D. R. Spring and J. Cui, Org. Lett., 2013, 15, 2310–2313 CrossRef CAS PubMed.
- Y. Liu and G. Feng, Org. Biomol. Chem., 2014, 12, 438–445 CAS.
- J. I. Kim, I.-S. Shin, H. Kim and J.-K. Lee, J. Am. Chem. Soc., 2005, 127, 1614–1615 CrossRef CAS PubMed.
- R. Y. Lai and A. J. Bard, J. Phys. Chem. A, 2003, 107, 3335–3340 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional spectroscopic data. See DOI: 10.1039/c6ra26826a |
| This journal is © The Royal Society of Chemistry 2017 |