
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A covalent triazine framework-based heterogenized Al–Co bimetallic catalyst for the ring-expansion carbonylation of epoxide to β-lactone†
Senkuttuvan Rajendiran,
Prakash Natarajan and
Sungho Yoon *
*
Department of Bio & Nano Chemistry, Kookmin University, 861-1 Jeongneung-dong, Seongbuk-gu, Republic of Korea. E-mail: yoona@kookmin.ac.kr
First published on 16th January 2017
Abstract
Difficulties in product separation and ineffective recycling of the homogenous catalyst deter the mass production of β-lactones via carbonylation of epoxides. Herein, we address these issues, for the first time, using a recyclable heterogenized catalyst [bpy-CTF-Al(OTf)2][Co(CO)4] that efficiently converts propylene oxide (PO) to β-butyrolactone with high selectivity.
β-Lactones are important intermediates for ring-opening polymerization to form poly(β-hydroxyalkanoates), a naturally occurring biodegradable1 and thermoplastic polyester.2 Many synthetic methods are available for preparing β-lactones,3 among which catalytic ring-opening carbonylation of epoxides has received considerable attention because of its ability to produce enantiopure β-lactones.4 Over the past several decades, it has been reported that Co2(CO)8 precursors catalyze the carbonylation of epoxides to lactones.5 Particularly, Coates et al. employed homogenous bimetallic Lewis acid–base salts [(salph)M(THF)2][Co(CO)4] (salph = N,N′-o-phenylene bis(3,5-di-tert-butylsalicylideneimine), M = Al3+, Cr3+), as catalysts for the conversion of epoxides to lactones, which are the best catalysts to date for epoxide carbonylation.4a,6 However, difficulty in product separation and poor catalyst recycling may significantly limit their practical utilization. To overcome these drawbacks, heterogenization of the active sites of previously reported homogenous catalysts on the solid support, guarantying high catalytic efficiency with facile separation, is highly desired.
In recent years, covalent triazine frameworks (CTFs) have attracted much attention as heterogeneous catalytic supports,7 because they have a high surface area, stability, large pore volumes and structural tunability.8,9 In particular, bipyridine (bpy)-based CTF (bpy-CTF, represented as 1), which contains bipyridine ligand motifs in pore walls of the framework as shown in Scheme S1 (ESI†), has potential to form complexes with N–N coordination environment.10 Hence, we hypothesized that immobilization of an {Al(OTf)2}[Co(CO)4] (OTf = trifluoromethane sulfonate) motif on the bpy-CTF system, as shown in Fig. 1b, would provide similar N2O2 coordination environment for Al3+ cation and Co anion as those provided by the [(salph)Al(THF)2][Co(CO)4] (4) homogeneous catalyst (Fig. 1a). Herein, we report the synthesis and excellent catalytic conversion of a heterogenized catalyst [bpy-CTF-Al(OTf)2][Co(CO)4] for the carbonylation of PO into β-butyrolactone. In addition, facile separation and sequential recycling of the catalyst was demonstrated.
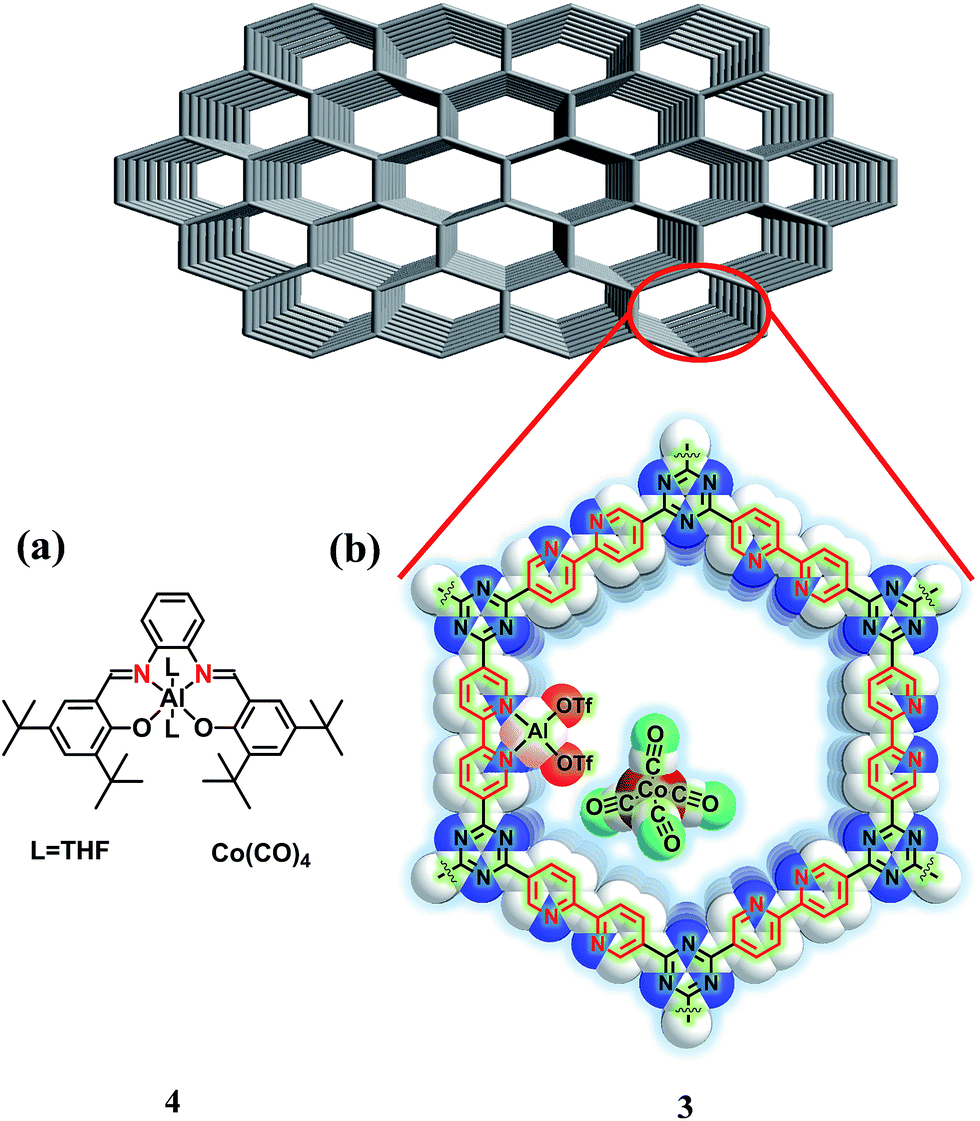 | ||
| Fig. 1 Structural representation of salph-based (a) homogeneous [(salph)Al(THF)2][Co(CO)4] (4) and (b) relevant structural features of the heterogenized catalyst [bpy-CTF-Al(OTf)2][Co(CO)4] (3) (white: carbon, blue: nitrogen, pink: aluminum, red: (OTf), maroon: cobalt, and green: oxygen; hydrogen atoms are omitted for clarity). | ||
The stacked 2D layers of porous 1 material was synthesized by following the reported procedures.9,10 Pore size distribution of as prepared 1 was analyzed using Barrett–Joyner–Halenda method, elucidating a predominant presence of pores with an average pore size of 2.4 nm (Fig. S2 and Table S1 in ESI†). Upon treatment of Al(OTf)3 with 1 in methanol solution and sequential refluxing leads to only 0.57 wt% of metal immobilization (Table S2, ESI†). To increase the amount of Al(OTf)3 immobilization on 1, a hydrothermal method was used for much vigorous reaction condition, and yielded [bpy-CTF-Al(OTf)3] (2) as a black precipitate. Scanning electron microscopy (SEM) images of 2 show irregularly shaped blocks with the mean size of >35 (5) μm (Fig. S3a, ESI†). The energy-dispersive spectroscopy (EDS) mapping demonstrates the uniform distribution of aluminum, sulfur and fluorine atoms throughout the framework while maintaining the ratio of ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:9 (Fig. S3b–d and Table S3, ESI†), which suggested the formation of 2 as expected (Scheme S1, ESI†). In addition, inductively coupled plasma-optical emission spectroscopy (ICP-OES) analysis revealed that the Al content is 3.78 wt%.
:3:9 (Fig. S3b–d and Table S3, ESI†), which suggested the formation of 2 as expected (Scheme S1, ESI†). In addition, inductively coupled plasma-optical emission spectroscopy (ICP-OES) analysis revealed that the Al content is 3.78 wt%.
To exchange OTf− anions with exogenous [Co(CO)4]− anions, a freshly prepared KCo(CO)4 solution was treated with a methanolic solution of 2 at ambient temperature, resulting in a black precipitate (Fig. S4, ESI†). The infrared (IR) spectrum of [bpy-CTF-Al(OTf)2Co(CO)4] (3) showed a CO stretching peak at 1878 cm−1, corresponding to the carbonyl groups in the tetrahedral [Co(CO)4]− (Fig. S5, ESI†), indicating the successful exchange of OTf− with [Co(CO)4]−. The SEM images of the isolated 3 as depicted in Fig. 2a and b, indicates that the irregular block shape of the 1 was maintained. A uniform distribution of aluminum, fluorine, sulfur and cobalt atoms throughout the framework was confirmed via EDS mapping (Fig. 2c and d). Using ICP-OES, the precise contents of Al and Co were measured to be 3.76 and 2.67 wt%, respectively. (The Al and Co contents calculated based on full occupancy of {Al(OTF)2}+ and [Co(CO)4]− units on the 1 are 3.82 and 8.34 wt%, respectively.) Interestingly, these values are lower than the calculated ones. It indicates that each CTF ring was immobilized with one [Al(OTf)2Co(CO)4] and two Al(OTf)3 unit as depicted in Fig. S6 (ESI†).
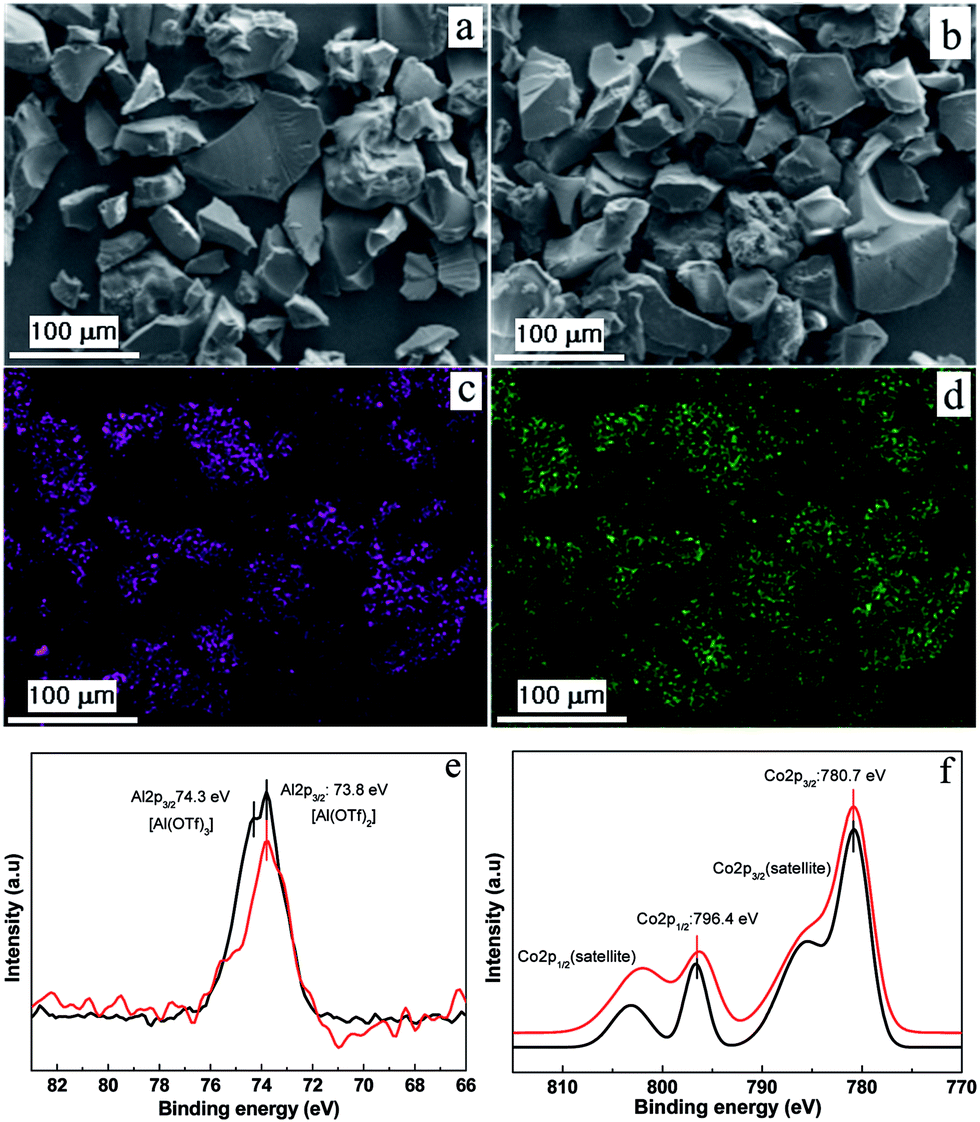 | ||
Fig. 2 SEM images of (a) 1 and (b) complex 3. (c) Al and (d) Co elemental mappings of 3. High-resolution XPS spectra of complex ( ) 4 and ( ) 4 and ( ) 3 (e) Al 2p peaks and (f) Co 2p peaks. ) 3 (e) Al 2p peaks and (f) Co 2p peaks. | ||
To understand the change of pore volume and surface area upon immobilization, nitrogen adsorption/desorption measurements were performed (Fig. S7, Table S1, ESI†). In the case of 1, surface area and pore volume were found to be 684 m2 g−1 and 0.40 cm3 g−1, respectively. Whereas after immobilization, the surface area and the pore volume were reduced to 203 m2 g−1 and 0.14 cm3 g−1, respectively. It possibly originates from the successful immobilization of [Al(OTf)2]− and [Co(CO)4]− unit on the 1, as we planned. It is worth to note that, as depicted in Fig. S6 (ESI†), the presence of [Al(OTf)2]− and [Co(CO)4]− ions did not completely block the cavities of 1 leaving possible channels for small molecules such, as PO and CO, to reach the active catalytic sites.
To confirm whether 3 has similar active site as homogenous catalyst 4, X-ray photoelectron spectroscopy (XPS) analysis was employed with special care to study the coordination environment of Al3+ and Co1− in 3. For comparison, the homogeneous catalyst 4 was analyzed and the binding energies (BEs) corresponding to Al3+ 2p3/2, Co1− 2p1/2 and Co1− 2p3/2 peaks were found to be 73.8, 780.7 and 796.4 eV, respectively (Fig. 2e and f). As shown in the Fig. 2e and f, the heterogeneous catalyst 3 also has the Al3+ 2p3/2, Co1− 2p1/2 and Co1− 2p3/2 ions with BE values of 73.8, 780.7 and 796.4 eV respectively, which was well matched with homogeneous catalyst 4. (The additional Al3+ peak at 74.3 eV in Fig. 2e of catalyst 3 may originate from the Al(OTf)3 species.) These results collectively reiterate that the heterogeneous 3 has similar environment to that of homogeneous catalyst 4.
Carbonylation of PO using catalysts was performed in a stainless steel tube reactor using the weakly coordinating polar solvent (dimethoxy ethane, DME), which is the best solvent reported for carbonylation of an epoxide into a lactone.4,6 Initially, carbonylation of PO was studied using the reference complex 4 (1 mol% to substrate) in DME, at 50 °C under 6.0 MPa of CO which resulted in 91% conversion to β-butyrolactone and acetone in 9:1 ratio, which is very close to that of the reported result (Table 1, entry 1).4 After the initial study with 4, the activity of the as-prepared heterogeneous catalyst 3 was evaluated, using the same reaction conditions of entry 1. The catalyst 3 carbonylated PO to β-butyrolactone with 46% conversion and 83% selectivity (entry 2). When decreasing the ratio of the substrate to catalyst (S/C) to 30, complete conversion was achieved, and interestingly, catalyst 3 was able to convert PO to β-butyrolactone with 90% selectivity (entry 3). It is noteworthy that the selectivity for β-butyrolactone was increased when increasing the catalyst concentration.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Temp (°C) | Pressure (MPa) | Time (h) | Conversiond (%) | Selectivityd (%) | |
| β-Butyrolactone | Acetone | ||||||
| a Reaction performed in 2.5 mL of DME pressurized using CO at room temperature.b S/C = 100.c S/C = 30.d Determined by 1H-NMR spectroscopy with naphthalene as an internal standard.e Remaining were unreacted epoxide.f Hot-filtration test. | |||||||
| 1 | 4b | 50 | 6.0 | 24 | 91e | 90 | 10 |
| 2 | 3b | 50 | 6.0 | 24 | 46e | 83 | 17 |
| 3 | 3c | 50 | 6.0 | 24 | >99 | 90 | 10 |
| 4 | 3c | 70 | 6.0 | 12 | 92e | 26 | 74 |
| 5 | 3c | 90 | 6.0 | 12 | >99 | 17 | 83 |
| 6 | 3c | 34 | 6.0 | 24 | 51e | 48 | 52 |
| 7 | 3c | 34 | 0.1 | 2 | 47e | 27 | 73 |
| 8 | 3c,f | 50 | 6.0 | 24 | <1e | — | — |

To increase the rate of reaction and reduce the acetone formation, the catalytic activity of 3 was studied at different temperatures. Upon increasing the temperature to 70 °C and 90 °C, (Table 1, entry 4 & 5) the acetone formation was more dominant than lactone. This may be because of the facile β-hydride elimination at higher temperatures that facilitates the formation of acetone after rearrangement (vide infra). To enable the commercial applicability of the heterogeneous catalyst 3, PO carbonylation was performed at ambient temperature with various pressure (entry 6 & 7), surprisingly large quantity of acetone was formed.
Based on the proposed mechanism of PO carbonylation (Fig. 3), the catalytic cycle initiates the activation of PO by immobilized Al3+ cation, followed by a nucleophilic attack of [Co(CO)4]− at the less hindered carbon of PO to form both aluminum-alkoxide and cobalt alkyl species b.4a,5f At moderate reaction temperature, the available CO in the pores of 3 may facilitate the migratory insertion of CO into the cobalt–alkyl bond and produces cobalt acyl intermediate c, which further undergo ring closing to form β-butyrolactone. However, at higher temperature the availability of CO in the pores of 3 may be limited, which slow down the CO insertion into the Co–alkyl bond b. The delayed CO insertion may lead to β-hydride elimination, followed by enolate protonation d and tautomerization to form acetone as the major product (Table 1, entry 4 & 5).
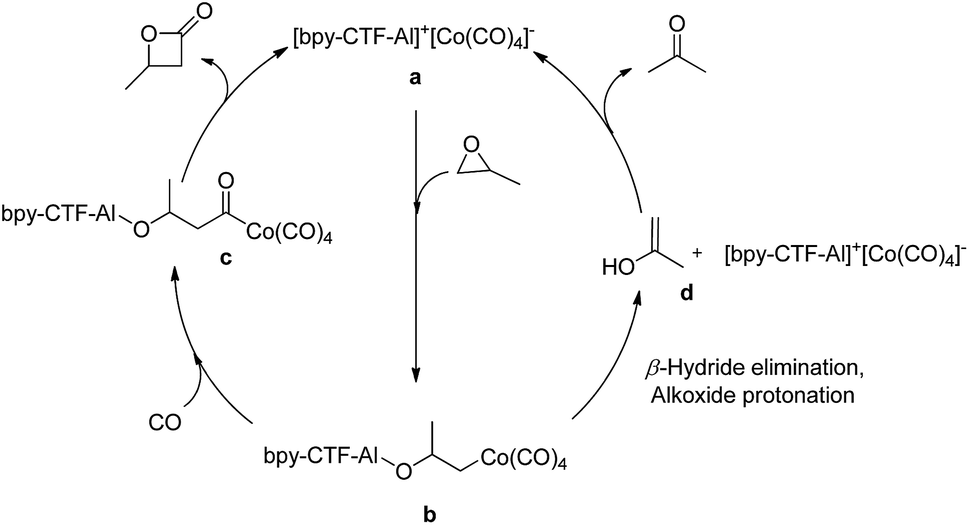 | ||
| Fig. 3 A plausible mechanism of PO carbonylation and acetone formation from a common intermediate. | ||
After the initial run, catalyst 3 was separated from the reaction mixture via simple filtration and was washed with DME. Interestingly, FT-IR analysis of the recovered catalyst shows a carbonyl peak at 1878 cm−1, indicating that the catalyst is still active for further carbonylation (Fig. S8, ESI†). SEM together with EDS analysis, supports the uniform distribution of aluminum, fluorine, sulfur and cobalt atoms throughout the whole sample (Fig. S9, ESI†). However, ICP-OES analysis revealed that Al and Co contents are 3.72 and 2.48 wt%, respectively, indicating the leaching of a small amount of [Co(CO)4]− anion from the catalyst 3 to the filtrate. Hot-filtration experiment was performed to test for homogeneous contributions to the rate; the catalyst 3 was heated in 2.5 mL of DME at 50 °C for 24 h and the resulting mixture was filtered to recover the solution part. Carbonylation of PO was performed with the filtrate at 50 °C under 6.0 MPa of CO which resulted in >99% of PO exists in the solution, indicating that no catalytically active Co species in the filtrate for PO carbonylation (Table 1, entry 8). The recovered catalyst was used at least three times for carbonylation at the same reaction condition as the first cycle and the results are shown in Table 2. Compared to the first cycle, the catalytic activity in recycle runs was maintained at ca. 91%. Slight reduction of activity may originate from partial discharge of the [Co(CO)4]− anion from the catalyst in the course of separation. It is noteworthy that the relative amount of acetone compared to lactone in recycling runs increases. It may originate from previously described discharge of [Co(CO)4]− anion while maintaining the amount of Al3+ cation unaffected, which leads to β-hydride elimination augmented to form more acetone. To check the possibility of the regeneration of the recovered catalyst with comparatively reduced amount of [Co(CO)4]−, it was subsequently treated with fresh K[Co(CO)4] after the third run (see the procedure in ESI†). Interestingly, it is found that the regenerated catalyst converts PO into the β-butyrolactone (90%) with over all conversion efficiency of 96% (entry 4). These results show that the heterogeneous catalyst 3 has an ability to retain their activity for effective carbonylation, which make 3 an attractive catalyst towards industrial application.
| Cycle | Time (h) | Conversionb (%) | Selectivityb (%) | |
|---|---|---|---|---|
| β-Butyrolactone | Acetone | |||
| a Reaction conditions: DME = 2.5 mL, S/C = 30, CO = 6.0 Mpa, temperature = 50 °C.b Determined by 1H-NMR spectroscopy.c After the 3rd run, the catalyst 3 was regenerated with fresh K[Co(CO)4] (see ESI). | ||||
| 1 | 24 | >99 | 90 | 10 |
| 2 | 24 | 92 | 88 | 12 |
| 3 | 24 | 81 | 84 | 16 |
| 4c | 24 | 96 | 90 | 10 |
In summary, the first heterogenized complex [bpy-CTF-Al(OTf)2][Co(CO)4] was synthesized by immobilizing Al(OTf)3 on the covalent organic framework and subsequent exchanging of the [OTf]− anion with the [Co(CO)4]− anion. This heterogenized catalyst 3 exhibited excellent activity under mild conditions (6.0 MPa and 50 °C), with a high selectivity of 90% for the ring expansion carbonylation of PO to β-butyrolactone. More importantly, the activity and selectivity of the heterogeneous catalyst were comparable to those of the homogeneous catalyst. Furthermore, the catalyst was readily separated from the product and repeatedly used for several runs. These results demonstrate that the heterogenized catalyst 3 might pave the way to achieve sustainable epoxide carbonylation on an industrial scale. Further studies focused on increasing the rate, selectivity for β-lactone derivatives using heterogenized catalysts are under progress.
Acknowledgements
We acknowledged financial support by the C1 Gas Refinery Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2015M3D3A1A01064879).Notes and references
- (a) H. M. Muller and D. Seebach, Angew. Chem., Int. Ed., 1993, 32, 477 CrossRef; (b) K. Sudesh, H. Abe and Y. Doi, Prog. Polym. Sci., 2000, 25, 1503 CrossRef CAS.
- (a) T. U. Gerngross and S. C. Slater, Sci. Am., 2000, 283, 36 CrossRef CAS; (b) S. Bronco, G. Consiglio, R. Hutter, A. Batistini and U. W. Suter, Macromolecules, 1994, 27, 4436 CrossRef CAS; (c) Z. Z. Jiang and A. Sen, J. Am. Chem. Soc., 1995, 117, 4455 CrossRef CAS.
- (a) K. Nozaki, N. Sato and H. Takaya, J. Am. Chem. Soc., 1995, 117, 9911 CrossRef CAS; (b) B. H. Chang and J. Kwiatek, Abstr. Pap. Am. Chem. Soc., 1995, 210, 51 Search PubMed; (c) M. Yokouchi, Y. Chatani and H. Tadokoro, J. Polym. Sci., Polym. Phys. Ed., 1976, 14, 81 CrossRef CAS; (d) P. Kurcok, M. Kowalczuk, K. Hennek and Z. Jedlinski, Macromolecules, 1992, 25, 2017 CrossRef CAS; (e) Y. Hori, M. Suzuki, A. Yamaguchi and T. Nishishita, Macromolecules, 1993, 26, 5533 CrossRef CAS.
- (a) Y. D. Y. L. Getzler, V. Mahadevan, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1174 CrossRef CAS PubMed; (b) V. Mahadevan, Y. D. Y. L. Getzler and G. W. Coates, Angew. Chem., Int. Ed., 2002, 41, 2781 CrossRef CAS; (c) J. A. R. Schmidt, V. Mahadevan, Y. D. Y. L. Getzler and G. W. Coates, Org. Lett., 2004, 6, 373 CrossRef CAS PubMed.
- (a) E. Drent and E. Kragtwijk, (Shell Internationale Research Maatschappij B.V., Neth.). Eur. Pat. Appl., EP 577206, 1994, Chem. Abstr., 1994, 120, 191517; (b) J. T. Lee, P. J. Thomas and H. Alper, J. Org. Chem., 2001, 66, 5424 CrossRef CAS PubMed; (c) K. Khumtaveeporn and H. Alper, Acc. Chem. Res., 1995, 28, 414 CrossRef CAS; (d) P. Davoli, I. Moretti, F. Prati and H. Alper, J. Org. Chem., 1999, 64, 518 CrossRef CAS; (e) H. Wolfle, H. Kopacka, K. Wurst, P. Preishuber-Pflugl and B. Bildstein, J. Organomet. Chem., 2009, 694, 2493 CrossRef; (f) M. Allmendinger, M. Zintl, R. Eberhardt, G. A. Luinstra, F. Molnar and B. Rieger, J. Organomet. Chem., 2004, 689, 971 CrossRef CAS.
- J. W. Kramer, E. B. Lobkovsky and G. W. Coates, Org. Lett., 2006, 8, 3709 CrossRef CAS PubMed.
- (a) A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef CAS PubMed; (b) F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478 CrossRef CAS PubMed; (c) S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524 CrossRef CAS PubMed; (d) S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816 CrossRef CAS PubMed.
- (a) D. Beaudoin, T. Maris and J. D. Wuest, Nat. Chem., 2013, 5, 830 CrossRef CAS PubMed; (b) P. Katekomol, J. Roeser, M. Bojdys, J. Weber and A. Thomas, Chem. Mater., 2013, 25, 1542 CrossRef CAS; (c) C. E. Chan-Thaw, M. Bojdys, P. Katekomol, K. Kailasam, R. Palkovits, F. Schuth, A. Villa, L. Prati and A. Thomas, Abstr. Pap. Am. Chem. Soc., 2010, 240 Search PubMed; (d) C. E. Chan-Thaw, A. Villa, P. Katekomol, D. S. Su, A. Thomas and L. Prati, Nano Lett., 2010, 10, 537 CrossRef CAS PubMed; (e) K. Kailasam, Y. S. Jun, P. Katekomol, J. D. Epping, W. H. Hong and A. Thomas, Chem. Mater., 2010, 22, 428 CrossRef CAS.
- S. Hug, M. E. Tauchert, S. Li, U. E. Pachmayr and B. V. Lotsch, J. Mater. Chem., 2012, 22, 13956 RSC.
- K. Park, G. H. Gunasekar, N. Prakash, K. D. Jung and S. Yoon, ChemSusChem, 2015, 8, 3410 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, characterization of 1, 2 & 4 and detailed experimental procedure of PO carbonylation. See DOI: 10.1039/c6ra26828e |
| This journal is © The Royal Society of Chemistry 2017 |