
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe concise synthesis and biological evaluation of C-glycosyl chalcone analogues inspired by the natural product aspalathin†
Tao Zhang and
Zhijie Fang *
*
Department of Chemical Engineering, Nanjing University of Science & Technology, Nanjing 210094, P. R. China. E-mail: zjfang@njust.edu.cn
First published on 12th January 2017
Abstract
We describe the synthesis and biological evaluation of C-glycosyl chalcone analogues of aspalathin. The DPPH radical scavenging method was used to detect their antioxidant activity and an MTT colorimetric assay was employed to test the anticancer activity against Hep-G2 liver carcinoma cells and MCF-7 human breast carcinoma cells. The antioxidant ability between C-furanosides and C-pyranosides was compared. Compound 3c′ was shown to be the most promising compound with good antioxidant ability and the ability to inhibit Hep-G2 and MCF-7 cells, with IC50 values of 4.9 μM and 19 μM, respectively.
1 Introduction
In Bryophyta, the presence of C-glycosyl flavonoids was reported for the first time in the genera Plagiochila and Plagiochasma.1 They have been shown to possess antiviral,2 cytotoxic3,4 and protective effects against DNA damage.5,6 Aspalathin is a representative natural C-glycosyl flavonoid that was first isolated from the leaves of Aspalathus linearis and used in the manufacture of rooibos tea.7,8 Early enthusiasm for the commercial development of this compound was focused on its use as a potent antioxidant, its radical scavenging activity9–11 towards anti-aging and enhancing the immune system. Subsequent investigation led to the discovery that aspalathin reduced the gene expression of hepatic enzymes related to glucose production and lipogenesis, and as such, was regarded as a potential therapeutic for diabetes.12 Recently, studies show that aspalathin can inhibit the proliferation and infiltration of liver cancer cells.13,14Although the natural product is readily available from fermentation, the levels (typically less than 0.5%) of aspalathin in the fermented rooibos aqueous extract is low15,16 and as such is often the case with natural product derivatization. Currently, aspalathin has been pursued as a target for total synthesis, with two successful syntheses being reported.17,18 However, these accomplishments call for the number of steps to be reduced and the reaction materials used are of high cost, which define a rather substantial barrier to the use of chemical synthesis for the discovery of novel aspalathin analogues. Moreover, there are almost no reports on the analogues of aspalathin. Hence, according to the statement mentioned above, the synthesis of its analogues is significant and meaningful.
Recently, we have systematically explored the one-step synthesis of compound 1 from glucose catalyzed by different Lewis acids.19 The target product C-glycoside-derived tetrahydrofurans comprised of a sugar ring and a rigid furan ring with an acetyl group, which can be used as a reactive part to prepare derivatives of C-glycosyl chalcones. Herein, inspired by the structure of aspalathin and the recent studies, we report a concise synthesis of aspalathin analogues, with glucose as a starting material in only three steps.
2 Experimental
2.1 General experimental procedures
All chemicals were of analytical grade and used without further purification. Cells were supplied by Guang Liang group in Wenzhou Medical University. Reactions were monitored using thin-layer chromatography on silica gel coated TLC plates and detection was performed by UV absorption (254 nm) where applicable and by spraying with 50% sulfuric acid in ethanol, followed by charring at 150 °C. Melting points (m.p.) were determined on a WRS-1B digital melting-point apparatus (Shanghai Shenguang Instrument Co., Ltd). 1H and 13C NMR spectra were recorded on Bruker-500 (500/125 MHz) and Bruker-300 (300/75 MHz) spectrometers. ESI-LCMS spectra were obtained on a Shimadzu LCMS-2010 EV mass spectrometer. Elemental analysis was carried out on a Perkin Elmer 2400 II.2.2 Cytotoxicity assay
An MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) assay was performed. Cells were seeded in a 96-well plate at a concentration of 1.0 × 10−4 cells per well and allowed to adhere overnight. Five replicates were prepared for each treatment and cultured for 48 or 72 h. After 20 μL of MTT (5 mg mL−1) was added to each well, the cells were cultured for another 4 h. The supernatant was discarded. After 150 μL of DMSO was added to each well, the samples were incubated for 30 min and then swirled for 10 min. The absorbance (A) at 570 nm was measured using a microplate reader. Experiments were repeated three times and averaged.2.3 Determination of the antioxidant activity using the DPPH radical scavenging method
The antioxidant activity of the C-glycoside derivatives was measured in terms of their hydrogen donating or radical scavenging ability using the stable radical, DPPH. An 80% aqueous ethanolic solution (0.6 mL) of the antioxidant was placed in a cuvette and 1.8 mL of a 6 × 10−5M aqueous ethanolic solution of DPPH (at 0.25 mol of antioxidant per mol of DPPH) was added. Absorbance measurements commenced immediately. The decrease in absorbance at 517 nm was determined continuously with data captured at 5 s intervals with a Beckman DU-65 spectrophotometer and Data Capture software for 2 h. An 80% aqueous ethanol solution was used to zero the spectrophotometer. The absorbance of the DPPH radical without antioxidant, i.e. the control, was measured daily. All determinations were performed in duplicate. The percentage inhibition of the DPPH radical by the samples was calculated according to the following formula:| % inhibitor = [(A(0) − A(t))/A(0)] × 100 |
2.4 Synthesis of the C-glycosyl flavonoid analogues
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 petroleum ether–ethyl acetate), the reaction mixture was evaporated to dryness under reduced pressure, dissolved in dichloromethane and washed with brine. The organic phase was collected and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the residue was further purified by silica gel column chromatography using 2:1 petroleum ether–ethyl acetate to furnish pure products.:2 petroleum ether–ethyl acetate to furnish pure products.:2 petroleum ether–ethyl acetate), the product was precipitated in the solvent. Then, the product was filtered and washed with cold water. The pure product was then obtained.:2 petroleum ether–ethyl acetate), the product was precipitated in the solvent. Then, the product was filtered and washed with cold water. The pure product was then obtained (6.15 g) in 92.3% yield as a light yellow powder.
:2 petroleum ether–ethyl acetate), the reaction mixture was evaporated to dryness under reduced pressure, dissolved in dichloromethane and washed with brine. The organic phase was collected and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the residue was further purified by silica gel column chromatography using 2:1 petroleum ether–ethyl acetate to furnish pure products.:2 petroleum ether–ethyl acetate to furnish pure products.:2 petroleum ether–ethyl acetate), the product was precipitated in the solvent. Then, the product was filtered and washed with cold water. The pure product was then obtained.:2 petroleum ether–ethyl acetate), the product was precipitated in the solvent. Then, the product was filtered and washed with cold water. The pure product was then obtained (6.15 g) in 92.3% yield as a light yellow powder.3 Results and discussion
3.1 Design
Structurally, aspalathin is comprised of a glycosyl unit and a dihydrochalcone moiety connecting through a β-glycosidic bond (Fig. 1). Usually, Friedel–Crafts and Koenigs–Knorr glycosylation methods are used in the synthesis of aspalathin. However, due to the high steric hindrance of the substrate, the product yields are relatively low.17 Therefore, we designed and synthesized three types of chalcone analogues comprised of a glycosyl unit and dihydrochalcone-like moiety via aldol reaction. In addition, the C-pyranoside chalcones were also synthesized in order to compare their antioxidant activity with C-furanosides. Both the series of compounds have a carbohydrate part and chalcone-like part, which are similar with aspalathin to some extent. The method avoids using the Friedel–Crafts and Koenigs–Knorr glycosylation methods and is much more efficient and economic.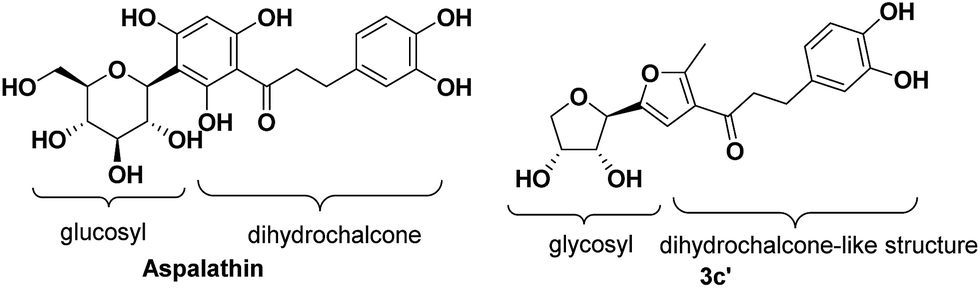 | ||
| Fig. 1 The structure of aspalathin and compound 3c′. | ||
3.2 Synthesis
We have reported that the condensation of glucose with acetylacetone in the presence of a Lewis acid in ethanol afforded compound 1.19 To synthesize compounds 3a′–3c′, first, compound 1 in ethanol was refluxed with aromatic aldehydes using a base-catalyst in order to afford (E)-α,β-unsaturated ketones i.e. 2a–2c. Then, compounds 2a–2c were subjected to hydrogenolysis in the presence of a catalyst, 10% palladium–carbon, under a hydrogen atmosphere in a dichloromethane and ethanol mixture to afford the desired compounds 3a′–3c′ (Scheme 1). The total yield of the three steps was around 44–52%. When compared with the synthetic route of aspalathin, the method for the synthesis of compounds 3a′–3c′ was simpler and the reaction materials were of low cost.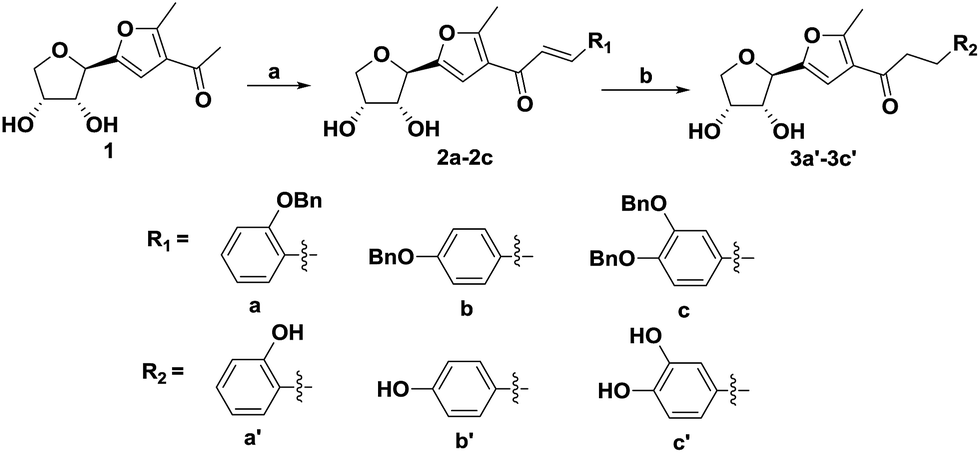 | ||
| Scheme 1 Preparation of the C-furanoside chalcone analogues. Reagents and conditions: (a) 20% NaOH, aromatic aldehyde, 60 °C, 6 h; (b) 10% Pd/C, H2, dichloromethane/methanol (1:1), r.t., 4 h. | ||
Compounds 5a and 5b can be synthesized via a direct aldol condensation,20 which was catalyzed by proline–TEA in the presence of methanol in yields of 95% and 93%, respectively. Due to the high activity and excellent water solubility of compound 6, it is not easy to separate the final product after the one-step direct aldol reaction. Herein, we prepared it via two steps (Scheme 2), compound 4b was selected as the starting material, which lowers the polarity of the intermediate and was helpful for the separation of the final product. In addition, in the 1H NMR spectra of all the synthesized chalcones, the hydrogens on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond have larger coupling constants (furanoside, J = 15.6 Hz; pyranoside, J = 16 Hz), which indicate that the configurations of all synthesized chalcones are the E-isomer.
C bond have larger coupling constants (furanoside, J = 15.6 Hz; pyranoside, J = 16 Hz), which indicate that the configurations of all synthesized chalcones are the E-isomer.
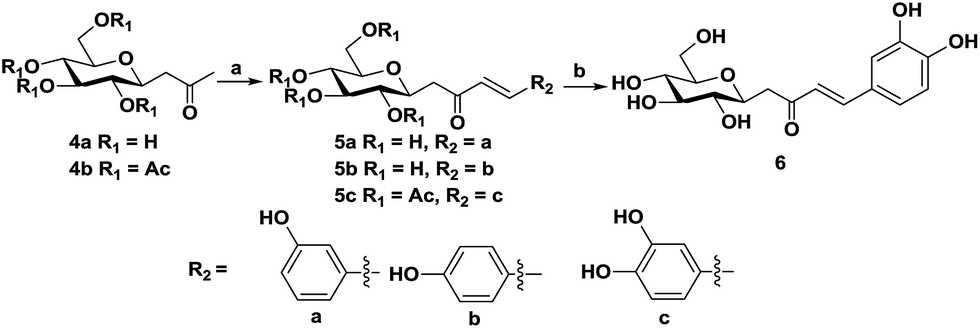 | ||
| Scheme 2 Preparation of the C-pyranoside chalcone analogues. Reagents and conditions: (a) TEA–proline, aromatic aldehyde, r.t., 48 h; (b) MeONa, methanol. | ||
3.3 Bioassay
Naturally occurring C-aryl glycosides exhibit a range of interesting biological properties. Aspalathin shows potent antioxidant and radical scavenging activity, and has recently been found to inhibit the proliferation and infiltration of liver cancer cells.13 Therefore, to further explore the bioactivity of the C-glycosyl chalcone analogues, we studied their antioxidant activity using the DPPH radical scavenging method and their anticancer activity using the MTT experiments.We synthesized polyphenol compounds based on C-glycosides as mentioned above to compare the antioxidant ability. The polyphenols binding with different sugars showed different DPPH radical scavenging ability as seen in Fig. 2, the decrease in absorbance of the DPPH radical (DPPH˙) due to its reduction by different antioxidants was illustrated. The absorbance decreases as a result of a color change from purple to yellow as the radical was scavenged by the antioxidants through donation of hydrogen to form the stable DPPH–H. The more rapidly the absorbance decreases, the more potent the antioxidant activity of the compound is in terms of its hydrogen donating ability.
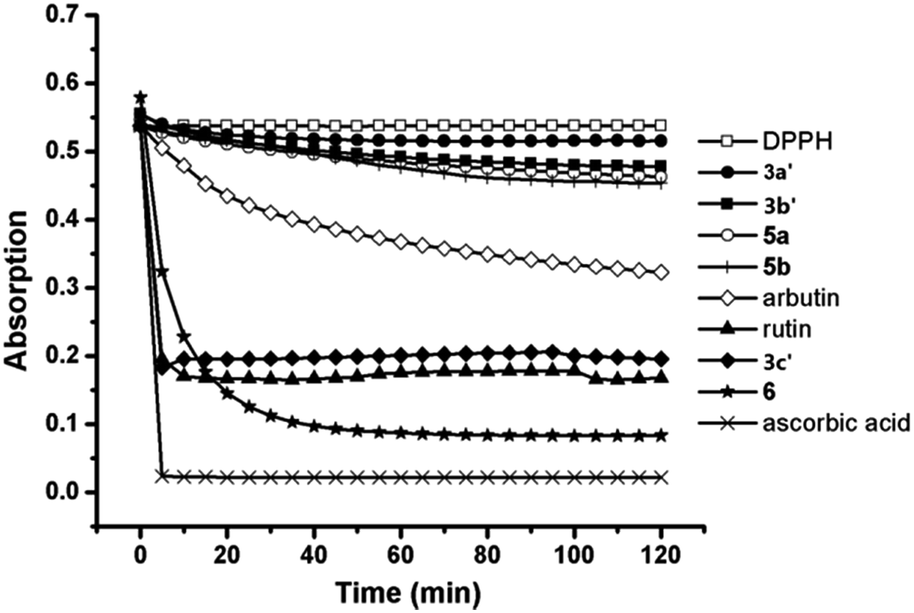 | ||
| Fig. 2 The free radical scavenging effect of the pure C-glycosyl chalcone derivatives using the DPPH method (0.25 mol mol−1). α-Arbutin, ascorbic acid and rutin were used as reference materials. | ||
Compounds 3a′, 3b′, 5a, 5b and α-arbutin showed a slow decrease in absorbance, whereas the other compounds, including ascorbic acid, rutin, compound 3c′ and 6, displayed a rapid decrease in the absorbance when the analysis was performed at 0.25 mol mol−1 DPPH radical. The percentage inhibition of the DPPH radical by the compounds tested is given in Table 1. Joubert's study11 showed that the antioxidant ability of aspalathin was slightly more than rutin; therefore, the antioxidant activity of these compounds, as determined by the DPPH radical scavenging method, decreased in the order ascorbic acid > aspalathin > rutin > 3c′ > α-arbutin > 5b > 5a > 3b′ > 3a′. Even though rutin is more potent than 3c′ as an antioxidant, the value gap was not significant. Compound 3c was found to be a potent DPPH radical scavenger with 63.55% inhibition in terms of these results. In addition, as seen in Table 1, the antioxidant ability of C-pyranosides was higher than that of the C-furanosides in this test.
| Compound | % inhibition | Compound | % inhibition |
|---|---|---|---|
| α-Arbutin | 39.93 | 3a′ | 9.76 |
| Ascorbic acid | 95.91 | 3b′ | 13.3 |
| Rutin | 68.85 | 3c′ | 63.55 |
| 5a | 13.89 | 6 | 85.60 |
| 5b | 15.66 |
As C-glycosyl flavonoids have been recently found to inhibit the proliferation and infiltration of liver cancer cells, Fig. 3 shows the anti-cancer activity of compounds 3a′, 3b′ and 3c′ with two types of cancer cells (i.e. liver cancer cells HepG2 and breast cancer cells MCF-7). Compound 3c′ exhibited significant inhibition against HepG2 and MCF-7 cells and the IC50 values can reach 4.9 μM and 19 μM, respectively.
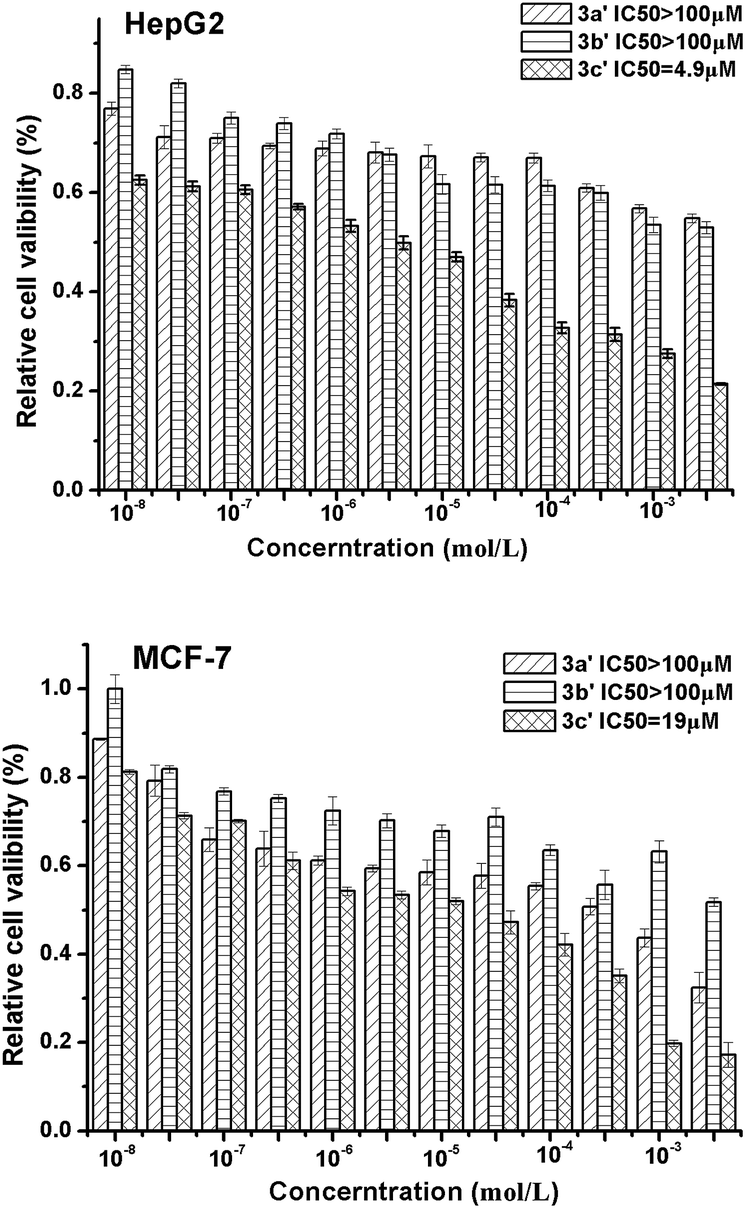 | ||
| Fig. 3 MTT assay of compounds 3a′, 3b′ and 3c′. | ||
4 Conclusion
In summary, we have demonstrated the synthesis of aspalathin analogues and their bioactivities. DPPH radical scavenging experiments showed the antioxidant ability of compound 3c′ was basically the same as rutin and the antioxidant ability of the C-pyranosides was higher than the C-furanosides. The MTT colorimetric assay indicated that compound 3c′ can also inhibit the proliferation of HepG2 cells and MCF-7 cells with IC50 values of 4.9 μM and 19 μM, respectively. The analogues of C-glycosyl chalcone aspalathin are easy to synthesize and deserve further study.Acknowledgements
The authors appreciate the financial support from the prospective joint project of Production, Education & Research in Jiangsu Province, China (Grant No. BY2013004-02).References
- R. Schoeneborn and R. Mues, Phytochemistry, 1993, 34, 1143–1145 CrossRef CAS.
- S. J. Semple, S. F. Nobbs, S. M. Pyke, G. D. Reynolds and R. L. P. Flower, J. Ethnopharmacol., 1999, 68, 283–288 CrossRef CAS PubMed.
- L. Hu, L. Li, D. Xu, X. Xia, R. Pi, D. Xu, W. Wang, H. Du, E. Song and Y. Song, Chem.-Biol. Interact., 2014, 213, 51–59 CrossRef CAS PubMed.
- B. A. Bhat, K. L. Dhar, S. C. Puri, A. K. Saxena, M. Shanmugavel and G. N. Qazi, Bioorg. Med. Chem. Lett., 2005, 15, 3177–3180 CrossRef CAS PubMed.
- P. W. Snijman, S. Swanevelder, E. Joubert, I. R. Green and W. Gelderblom, Mutat. Res., 2007, 631, 111–123 CAS.
- L. Standley, P. Winterton, J. L. Marnewick, W. C. Gelderblom, E. Joubert and T. J. Britz, J. Agric. Food Chem., 2001, 49, 114–117 CrossRef CAS PubMed.
- J. W. Drynan, M. N. Clifford, J. Obuchowicz and N. Kuhnert, Nat. Prod. Rep., 2010, 27, 417–462 RSC.
- B. H. Koeppen and D. G. Roux, Biochem. J., 1966, 99, 604–609 CrossRef CAS PubMed.
- E. Joubert, T. Beelders, D. Beer, C. J. Malherbe and A. J. Villiers, J. Agric. Food Chem., 2012, 60, 9171–9179 CrossRef CAS PubMed.
- J. D. Merwe, E. Joubert, M. Manley, D. Beer, C. J. Malherbe and W. C. A. Gelderblom, J. Agric. Food Chem., 2010, 58, 2214–2220 CrossRef PubMed.
- A. Gadow, E. Joubert and C. F. Hansmann, J. Agric. Food Chem., 1997, 45, 632–638 CrossRef.
- M. J. Son, M. Minakawa, Y. Miura and K. Yagasaki, Eur. J. Nutr., 2013, 52, 1607–1619 CrossRef CAS PubMed.
- S. Kwak, M. S. Han and J. S. Bae, Fitoterapia, 2015, 100, 179–186 CrossRef CAS PubMed.
- Y. Kazumi, M. Yutaka, H. Shuichi and N. Hiromichi, JP Patent, #2007197409, August 09, 2007.
- E. Joubert, P. Winterton, T. J. Britz and W. C. A. Gelderblom, J. Agric. Food Chem., 2005, 53, 10260–10267 CrossRef CAS PubMed.
- M. Pengilly, E. Joubert, W. H. Zyl, A. Botha and M. Bloom, J. Agric. Food Chem., 2008, 56, 4047–4053 CrossRef CAS PubMed.
- A. Yepremyan, B. Salehani and T. G. Minehan, Org. Lett., 2010, 12, 1580–1583 CrossRef CAS PubMed.
- Z. Han, M. C. Achilonu, P. S. Kendrekar, E. Joubert, D. Ferreira, S. L. Bonnet and J. H. Westhuizen, J. Nat. Prod., 2014, 77, 583–588 CrossRef CAS PubMed.
- Y. F. Xiang, Z. J. Fang and L. X. Li, Chem. Res. Appl., 2013, 25, 1426–1429 CAS.
- J. F. Wang, M. Lei, Q. Li, Z. Ge, X. Wang and R. Li, Tetrahedron, 2009, 65, 4826–4833 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26969a |
| This journal is © The Royal Society of Chemistry 2017 |