DOI:
10.1039/C6RA26971K
(Paper)
RSC Adv., 2017,
7, 2342-2350
Identification of 3-amidoquinoline derivatives as PI3K/mTOR dual inhibitors with potential for cancer therapy†
Received
18th November 2016
, Accepted 19th December 2016
First published on 12th January 2017
Abstract
A new series of 3-amidoquinoline derivatives were designed, synthesized and evaluated as PI3K/mTOR dual inhibitors. Among them, five compounds showed potent PI3Kα inhibitory activities (IC50 < 10 nM) and anti-proliferative activities (IC50 < 1 μM). The representative compound 15a can significantly inhibit other class I PI3Ks, mTOR and phosphorylation of pAkt(Ser473) at low nanomolar level, suggesting that 15a was a potent PI3K/mTOR dual inhibitor. Moreover, 15a displayed favorable pharmacokinetic properties in vivo.
1 Introduction
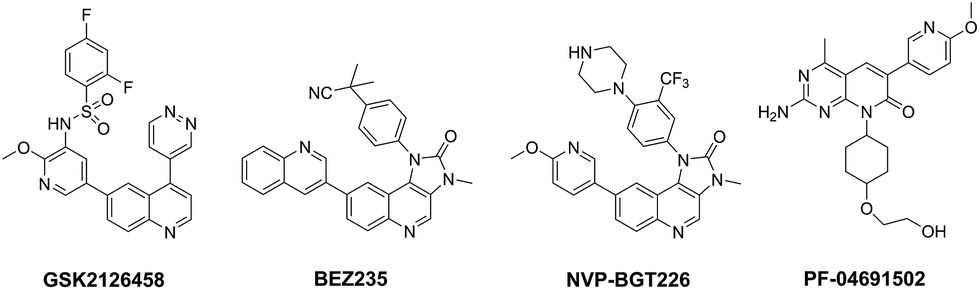
Hyperactivation of the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signalling pathway is common in cancer.1–4 There is abundant evidence demonstrating that genomic aberrations of PI3K and PTEN (PI3K negative regulator) are closely linked to the development and progression of a wide range of human cancers, including breast, colon, ovarian, prostate cancer and glioblastoma etc.5–8 In addition, mTOR-related multiple negative feedback loops, especially mTOR–S6K1–PI3K signalling, have been thought to contribute directly to the reactivation of this pathway, which results in the recrudescence of cancer.9,10 Given the pivotal role of PI3K and mTOR in cancer biology, combined targeting of PI3K and mTOR have has been considered as an attractive anti-cancer strategy and is expected to effectively block signal transduction, overcome negative feedbacks and reduce possibility of drug resistance.11–16 To date, several of PI3K and mTOR dual inhibitors have been advanced into clinical trials, such as GSK2126458, BEZ235, NVP-BGT226 and PF-06491502 (Fig. 1).9,17–21
 |
| | Fig. 1 Structures of clinical PI3K/mTOR dual inhibitors. | |
2 Rational design of PI3K/mTOR inhibitors
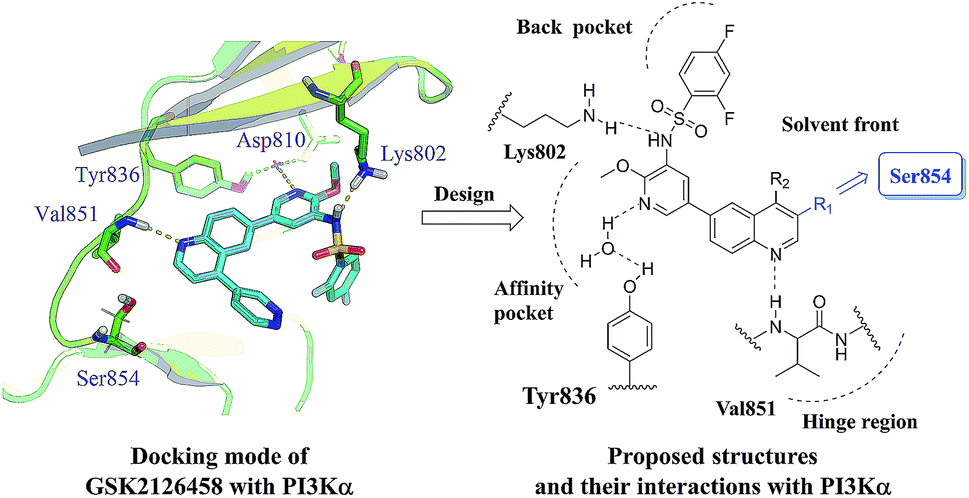
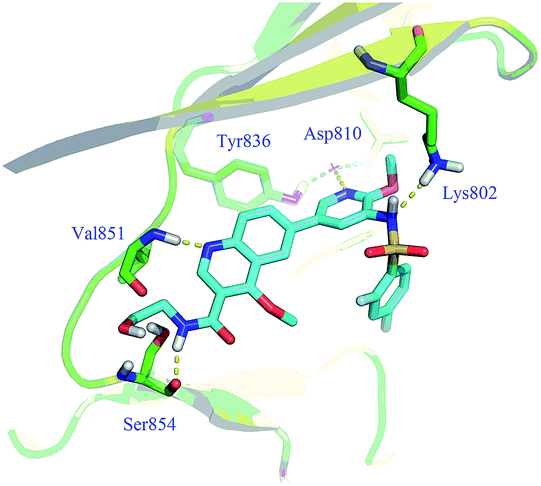
In our previous work, quinoline derivatives were identified as mTOR inhibitors and PI3K/mTOR dual inhibitors with high potency against cancer cells.22,23 Herein, we describe continued efforts in this field to pursue the novel PI3K/mTOR dual inhibitors through structural modification of GSK2126458.18 As a PI3K/mTOR dual inhibitor under clinical trials, GSK2126458 displayed remarkable in vitro and in vivo potency. To guide these efforts, we docked GSK2126458 into PI3Kα protein (PDB code 4JPS) which is currently available.24 As illustrated in Fig. 1, three hydrogen bonds are formed between the molecule and residues (Val851, Lys802, Asp810 and Tyr836). However, the pyridazine ring is not involved in the hydrogen-bonding interaction as it in the crystal structure of GSK2126458 binds to PI3Kγ. The C-3 position of the quinoline ring pointed to the residue Ser854 in space. Therefore, our strategy is removal of pyridazine ring and introduction of various substituents onto the C-3 position of the quinoline ring to explore potential interactions with the residue Ser854. Based on these assumptions, a series of 3-amidoquinoline derivatives were designed and prepared (Fig. 2).
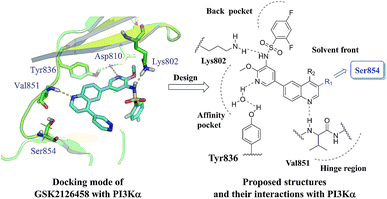 |
| | Fig. 2 The design concept based on the docking study. | |
3 Results and discussion
Synthesis
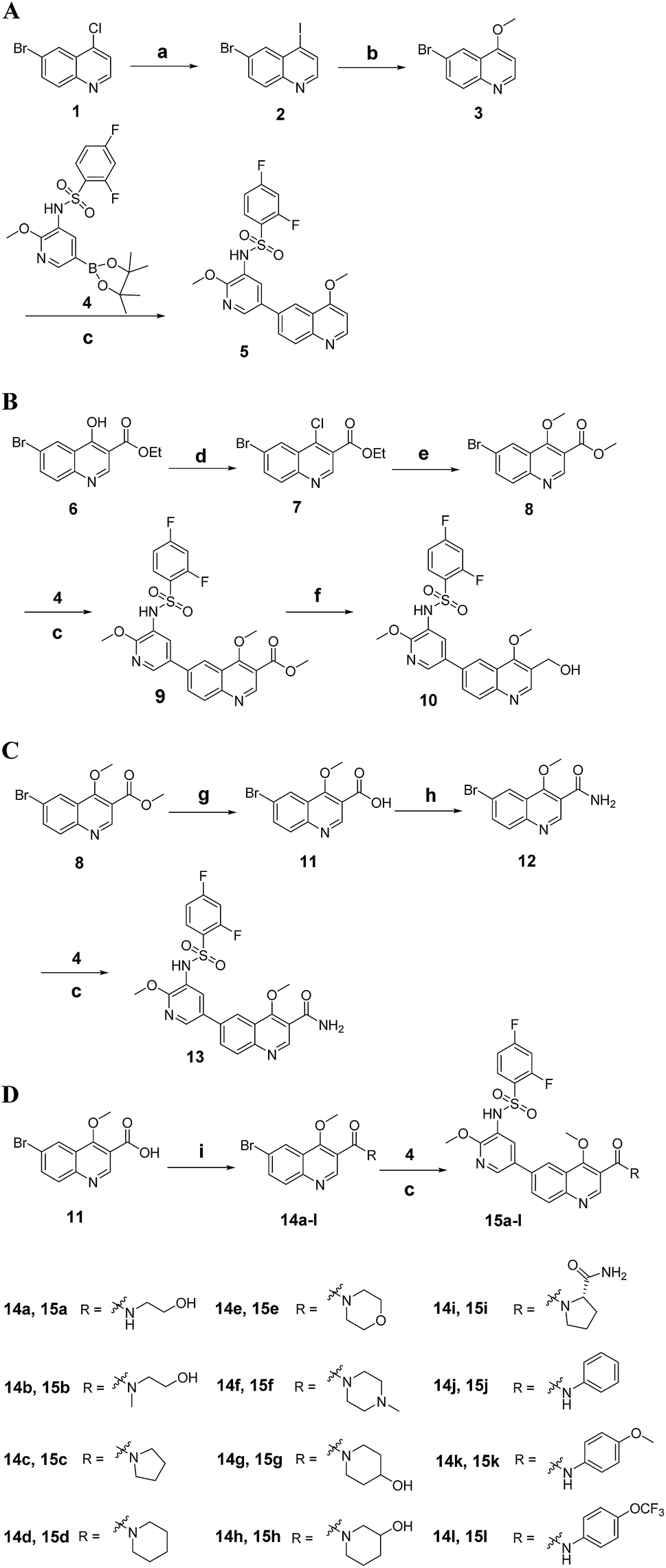
The synthetic route for 3-substituted quinolines 5, 10, 13 and 15a–l is outlined in Scheme 1. Reaction of 4-chloroquinoline 1 with KI provided 4-iodoquinoline 2, which was treated with CH3ONa to give 4-methoxyquinoline 3. Coupling 3 with boric acid ester 4 via Suzuki reaction yielded corresponding target compound 5. Following halogenation of 4-hydroxyquinoline derivative 6 with POCl3, the newly formed 4-chloroquinoline derivative 7 was treated with CH3ONa, leading to the generation of 4-methoxyquinoline derivative 8. Coupling 8 with 4 afforded compound 9, then reduction of 9 in the presence of DIBAL gave target compound 10. Hydrolysis of 8 with NaOH produced quinoline-3-carboxylic acid derivative 11, which was treated with ethyl chloroformate and NH3·H2O successively to afford the quinoline-3-carboxamide derivative 12. Coupling 12 with 4 gave target compound 13. Compound 11 can also be treated with amines in the presence of EDCI and HOBt to give other quinoline-3-carboxamide derivatives 14a–l. Coupling 14a–l with 4 yielded corresponding target compounds 15a–l.
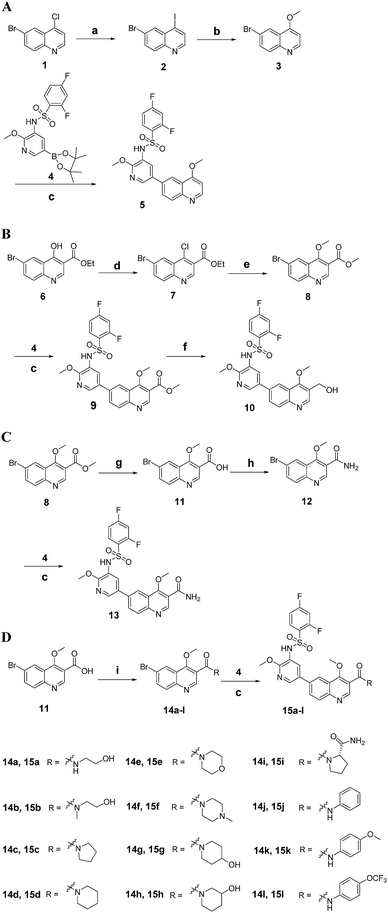 |
| | Scheme 1 (A) The synthetic route for target compounds 5, 10, 13 and 15a–l. Reagents and conditions (A–D): (a) HCl/EA, rt, 30 min, then KI, MeCN, reflux, 48 h; (b) NaOCH3, CH3OH, 50 °C, 12 h; (c) Pd(dppf)2Cl2, K2CO3, dioxane/H2O, 100 °C, 10 h; (B) reagents and conditions: (d) POCl3, 120 °C, 6 h; (e) NaOCH3, CH3OH, rt, 24 h; (f) DIBAL, rt, 6 h; (g) 2 N NaOH, rt, 2 h, then 2 N HCl; (h) ethyl chloroformate, NMM, THF, 30 min, then NH3·H2O, rt, 4 h; (i) amine, EDCI, HOBt, rt, 2 h. | |
Biological evaluation
Enzymatic and anti-proliferative assays in vitro. Firstly, all derivatives were evaluated for their PI3Kα inhibitory activities and exhibited moderate to potent activities (Table 1). Among them, compounds 10, 13 and 15a were more potent in PI3Kα enzymatic assay than compounds 5 and 15b–i. To illustrate their structure–activity relationships (SARs), docking analysis of 15a bound to PI3Kα (PDB code 4JPS) was performed utilizing the Discovery Studio 2.1 software package. As speculated, compound 15a formed an additional hydrogen bond with the residue Ser854 via its hydrogen on the amide bond at the C-3 position of quinoline (Fig. 3).
Table 1 Enzymatic activities and anti-proliferative activities of compounds 5, 10, 13 and 15a–l
| Compd |
IC50a |
| PI3Kα |
mTOR |
PC3 |
HCT116 |
| IC50 values (nM) for PI3Kα and mTOR inhibitory activities; IC50 values (μM) for anti-proliferative activities; values are means of three experiments. |
| 5 |
8.6 |
116 |
3.41 |
4.46 |
| 10 |
1.0 |
8.9 |
0.90 |
1.16 |
| 13 |
2.6 |
1.5 |
1.29 |
0.15 |
| 15a |
1.6 |
1.8 |
0.42 |
1.35 |
| 15b |
7.9 |
13 |
3.54 |
5.90 |
| 15c |
26 |
30 |
2.80 |
2.43 |
| 15d |
27 |
36 |
0.93 |
3.67 |
| 15e |
33 |
27 |
0.57 |
2.51 |
| 15f |
60 |
46 |
0.55 |
2.13 |
| 15g |
12 |
13 |
0.27 |
2.08 |
| 15h |
13 |
15 |
0.79 |
5.55 |
| 15i |
28 |
27 |
16.24 |
10.07 |
| 15j |
5.4 |
189 |
0.96 |
5.05 |
| 15k |
11 |
279 |
1.39 |
6.70 |
| 15l |
72 |
246 |
1.44 |
10.41 |
| BEZ235 |
35 |
21 |
0.51 |
0.22 |
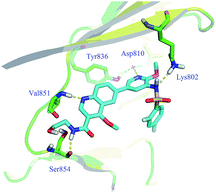 |
| | Fig. 3 Docking mode of 15a with PI3Kα. | |
In contrast, compounds 15b–i without hydrogen proton on the amide bond lost the ability to form hydrogen bond with Ser854, leading to the reduction in enzymatic activities. As for 15j–l, compounds 15k and 15l with methoxyl or trifluoromethoxyl group at C-4 position of phenyl group displayed 2–14 fold drop in enzymatic potency compared to 15j. It suggested that substitution at C-4 position of phenyl is sensitive to the inhibitory activity and relatively large substituents had negative impact on enzymatic potency. Similarly, the evaluated derivatives showed moderate to potent mTOR activities as well.
The results of anti-proliferative assay showed that these derivatives displayed moderate to potent activities against PC-3 cell line (Table 1). Half of them with cellular activities below 1.0 μM displayed better or equivalent anti-proliferation activities compared to that of BEZ235. In particular, compound 15a with potent activities in both enzymatic and anti-proliferative assays, can be used as a promising lead compound for further biological evaluation.
Class I PI3Ks enzymatic assays in vitro. Compound 15a was further screened for its activities against other class I PI3Ks. As shown in Table 2, compound 15a displayed significant inhibitory activities with IC50 values at low nanomolar level, which was more potent than that of the positive control BEZ235, suggesting that 15a was a potent PI3K/mTOR dual inhibitor.
Table 2 Enzymatic activities of compound 15a against class I PI3Ks and mTOR (IC50, nM)a
| Enzyme |
Compd |
| 15a |
BEZ235 |
| Values are means of three experiments. |
| PI3Kα |
1.6 |
35 |
| PI3Kβ |
2.8 |
16 |
| PI3Kγ |
4.4 |
26 |
| PI3Kδ |
1.3 |
89 |
| mTOR |
1.8 |
21 |
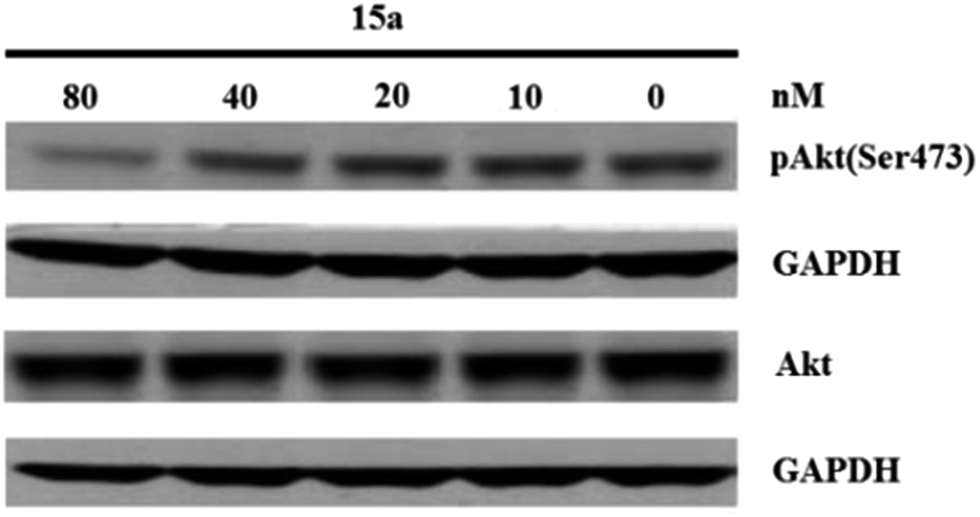
Western blot assay in vitro. PC-3 prostate carcinoma cell have a mutation in PTEN that results in a constitutively activated PI3K signaling pathway. These cells were used to examine if 15a could inhibit pAkt(Ser473) as a measure of inhibition of PI3K signaling. GAPDH was used as the internal control. After western blot assay, it demonstrates that 15a significantly inhibits PI3K/Akt/mTOR signaling in PC-3 cells at the concentration of 80 nM (Fig. 4).
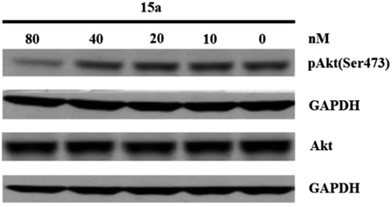 |
| | Fig. 4 The suppressive effect of 15a on pAkt(Ser473) in PC-3 cells. | |
Pharmacokinetics study in vivo. On the basis of this promising profile, 15a was further characterized through PK studies conducted in fasted male mice. PK parameters obtained in mice after oral administration at 5 mg kg−1 as a crystalline suspension in 0.5% methylcellulose. Compound 15a showed favorable in vivo plasma clearance (CL, 0.13 L h−1 kg−1), volume of distribution (Vd, 0.94 L kg−1), mean residence time (MRT, 7.5 h), exposure (AUC, 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 722 h ng−1 mL−1), peak plasma concentration (Cmax, 4624 ng mL−1), and plasma terminal half-life (t1/2, 5.2 h).
722 h ng−1 mL−1), peak plasma concentration (Cmax, 4624 ng mL−1), and plasma terminal half-life (t1/2, 5.2 h).
4 Conclusion
A new series of 3-amidoquinoline derivatives were designed by the docking analysis. Several synthesized target compounds exhibited strong enzymatic activities and anti-proliferative activities. Through the biological evaluation, 15a was identified as a potentially interesting lead molecule, which significantly inhibited other class I PI3Ks and mTOR, as well as the phosphorylation of pAkt(Ser473) at low nanomolar level. Furthermore, compound 15a exhibited favorable pharmacokinetic properties in the established mice model. These findings strongly support our hypothesis that incorporation of suitable substituents at the C-3 position of the quinoline ring could suppress PI3K/AKT/mTOR pathway effectively and achieve potent PI3K/mTOR dual inhibitors for cancer therapy.
5 Experimental section
Chemistry and chemical methods
1H NMR and 13C NMR spectra were recorded on the Brüker 500 and 400 NMR instruments. Chemical shifts are given in ppm (δ) relative to TMS as internal standard, coupling constants (J) are in hertz (Hz), and signals are using the following abbreviations: s, singlet; d, doublet; dd, doublet of doublets; t, triplet; td, doublet of triplets; q, quartet; m, multiplet, etc. Mass spectra (MS) and high resolution mass spectra (HRMS) were measured on Esquire-LC-00075 spectrometer and Waters GCT Premier spectrometer, respectively. IR (KBr disks) spectra were recorded on a Brüker Tensor 27 spectrometer. Melting points were determined with a B-540 Büchi melting-point apparatus. The purity of compounds was determined by Agilent 1260 HPLC system. Column chromatography and thin layer chromatography (TLC) were carried out using silica gel ZCX-3 and GF-254 (Qingdao Haiyang Chemical Co., Ltd.), respectively. Reagents and solvents were commercially available without further purification.
6-Bromo-4-iodoquinoline (2). To a solution of 6-bromo-4-chloroquinoline (1) (3.50 g, 14.46 mmol) in anhydrous EtOAc (20 mL) was added HCl-saturated EtOAc (40 mL) and a white precipitate formed immediately. After stirring for 30 min, the suspension was concentrated under vacuum to afford 6-bromo-4-chloroquinoline hydrochloride as an off white solid (3.91 g, 14.14 mmol).A two-neck flask was charged with 6-bromo-4-chloroquinoline hydrochloride (3.91 g, 14.14 mmol), anhydrous potassium iodide (9.76 g, 70.70 mmol) and anhydrous acetonitrile (100 mL). The resulting slurry was stirred at reflux for 48 h and allowed to cool to room temperature. Saturated aqueous NaHCO3 solution (40 mL) was added to the mixture, followed by 20 mL of a 5% sodium sulfite solution. The reaction mixture was extracted with CH2Cl2 (200 mL × 2). The combined organic extracts were dried over magnesium sulfate and concentrated in vacuo to give the crude product, which was further purified by silica gel column chromatography (25% ethyl acetate/petroleum ether) to give the title compound (4.42 g, 13.27 mmol, 94% yield) as an off-white solid.18 1H NMR (500 MHz, DMSO-d6) δ 8.51 (d, J = 4.5 Hz, 1H, Ar-H), 8.21 (d, J = 4.5 Hz, 1H, Ar-H), 8.11 (t, J = 1.5 Hz, 1H, Ar-H), 7.97–7.91 (m, 2H, Ar-H). ESI-MS: m/z = 334 [M + H]+.
6-Bromo-4-methoxyquinoline (3). To a solution of 2 (200 mg, 0.60 mmol) in anhydrous methanol (10 mL) was added sodium methoxide (65 mg, 1.20 mmol) at 0 °C. The reaction mixture was then heated to 50 °C and stirred for 12 h. After the completion of reaction, the mixture was filtered and the precipitate was washed with water. The obtained solids were then dried under reduced pressure to give the title compound (120 mg, 0.51 mmol, 85% yield) as a white solid.25 1H NMR (500 MHz, DMSO-d6) δ 8.78 (d, J = 5.0 Hz, 1H, Ar-H), 8.25 (d, J = 2.0 Hz, Ar-H), 7.90 (d, J = 9.0 Hz, 1H, Ar-H), 7.86 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H), 7.09 (d, J = 5.0 Hz, 1H, Ar-H), 4.05 (s, 3H, OCH3). ESI-MS: m/z = 238 [M + H]+.
2,4-Difluoro-N-(2-methoxy-5-(4-methoxyquinolin-6-yl)pyridin-3-yl)benzenesulfonamide (5). To a three-neck round bottom flask was added 3 (36 mg, 0.15 mmol), commercially available 2,4-difluoro-N-(2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)benzenesulfonamide (4) (64 mg, 0.15 mmol),18 Pd(dppf)2Cl2 (11 mg, 0.015 mmol) and K2CO3 (62 mg, 0.45 mmol) in dioxane/H2O (3/1). The flask was fitted with a N2 inlet adaptor and purged with N2 for 15 min. The reaction mixture was then sealed under an atmosphere of N2 and stirred at 100 °C for 10 h. The crude mixture was concentrated under reduced pressure and the residue was dissolved in CH2Cl2, washed with water twice, then the organic phase was dried over magnesium sulfate. The crude product was purified by silica gel column chromatography (2% CH3OH/CH2Cl2) to give the title compound (19 mg, 0.042 mmol, 28% yield) as a white solid. Mp 180–181 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.37 (s, 1H, NH), 8.76 (d, J = 5.0 Hz, 1H, Ar-H), 8.45 (d, J = 2.5 Hz, 1H, Ar-H), 8.24 (d, J = 1.5 Hz, 1H, Ar-H), 8.06–8.01 (m, 2H, Ar-H), 7.95 (d, J = 2.5 Hz, 1H, Ar-H), 7.79 (m, 1H, Ar-H), 7.60 (m, 1H, Ar-H), 7.24 (td, J = 8.5, 2.5 Hz, 1H, Ar-H), 7.08 (d, J = 5.0 Hz, 1H, Ar-H), 4.10 (s, 3H, OCH3), 3.69 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ 165.6 (dd, JC–F = 252.5, 11.5 Hz), 162.1, 159.9 (dd, JC–F = 256.2, 16.6 Hz), 158.0, 152.4, 148.4, 142.9, 134.3, 133.9, 132.4 (d, JC–F = 10.6 Hz), 130.1, 129.6, 128.8, 125.5 (dd, JC–F = 14.0, 3.5 Hz), 121.4, 120.3, 118.9, 112.4 (d, JC–F = 21.7, 3.7 Hz), 106.3 (t, JC–F = 26.0 Hz), 102.0, 56.7, 53.9. IR (KBr) ν 3422, 3262, 2928, 2851, 1601, 1488, 1468, 1384, 1308, 1160, 1145 cm−1. MS (ESI) m/z = 458 [M + H]+, HRMS (ESI) m/z calcd for C22H18F2N3O4S [M + H]+ 458.0980, found 458.0986. HPLC purity = 97.8%.
Ethyl 6-bromo-4-chloroquinoline-3-carboxylate (7). To a 100 mL round-bottomed flask was added ethyl 6-bromo-4-hydroxyquinoline-3-carboxylate (6) (10.0 g, 33.90 mmol), POCl3 (100 mL) and DMF (2 mL). The mixture was stirred at reflux for 6 h. After cooling to room temperature, the reaction mixture was poured into ice water (100 mL) and stirred for 1 h. Then the pH of the mixture was adjusted to 8 using saturated aqueous NaHCO3. The mixture was extracted with EtOAc and the organic phase was dried over sodium sulfate and concentrated in vacuo to give the title compound (8.82 g, 28.18 mmol, 83% yield) as a brown solid.22 ESI-MS: m/z = 314 [M + H]+.
Methyl 6-bromo-4-methoxyquinoline-3-carboxylate (8). To a solution of 7 (8.0 g, 25.56 mmol) in anhydrous methanol (200 mL) was added sodium methoxide (2.76 g, 51.12) at 0 °C. The reaction mixture was stirred for 24 h at room temperature. After the completion of reaction, the mixture was filtered and the precipitate was washed with water. The obtained solids were then dried under reduced pressure to give the title compound (6.53 g, 22.14 mmol, 87% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 9.04 (s, 1H, Ar-H), 8.40 (d, J = 2.0 Hz, 1H, Ar-H), 8.02 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H), 7.99 (d, J = 9.0 Hz, 1H, Ar-H), 4.10 (s, 3H, OCH3), 3.96 (s, 3H, OCH3). ESI-MS: m/z = 296 [M + H]+.
Methyl 6-(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-4-methoxyquinoline-3-carboxylate (9). This compound was prepared from 8 (221 mg, 0.75 mmol) and 4 (320 mg, 0.75 mmol) according to the general synthesis procedure of 5 to afford the title compound (90 mg, 0.18 mmol, 23% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 10.37 (s, 1H, NH), 9.03 (s, 1H, Ar-H), 8.47 (s, 1H, Ar-H), 8.36 (s, 1H, Ar-H), 8.14 (m, 2H, Ar-H), 7.99 (s, 1H, Ar-H), 7.80 (m, 1H, Ar-H), 7.57 (m, 1H, Ar-H), 7.22 (m, 1H, Ar-H), 4.14 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 3.70 (s, 3H, OCH3). ESI-MS: m/z = 516 [M + H]+.
2,4-Difluoro-N-(5-(3-(hydroxymethyl)-4-methoxyquinolin-6-yl)-2-methoxypyridin-3-yl)benzenesulfonamide (10). DIBAL (300 μL, 0.48 mmol) was added to a solution of 9 (60 mg, 0.12 mmol) in CH2Cl2 under an atmosphere of N2 and stirred for 6 h at room temperature. After the completion of reaction, 1 N NaOH (4 mL) was added to the mixture and then stirred for 10 min. The mixture was extracted with CH2Cl2 and the organic phase was dried over sodium sulfate. The crude product was purified by silica gel column chromatography (5% CH3OH/CH2Cl2) to give the title compound (9 mg, 0.018 mmol, 15% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 10.36 (s, 1H, NH), 8.89 (s, 1H, Ar-H), 8.42 (s, 1H, Ar-H), 8.19 (d, J = 1.5 Hz, 1H, Ar-H), 8.09 (d, J = 9.0 Hz, 1H, Ar-H), 7.99 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H), 7.94 (s, 1H, Ar-H), 7.80 (m, 1H, Ar-H), 7.55 (m, 1H, Ar-H), 7.22 (td, J = 8.5, 2.0 Hz, 1H, Ar-H), 5.42 (t, J = 7.0 Hz, 1H, OH), 4.77 (d, J = 7.0 Hz, 2H, CH2), 4.09 (s, 3H, OCH3), 3.70 (s, 3H, OCH3). MS (ESI) m/z = 488 [M + H]+.
6-Bromo-4-methoxyquinoline-3-carboxylic acid (11). Methyl 6-bromo-4-methoxyquinoline-3-carboxylate 8 (6.0 g, 19.42 mmol) and 2 N NaOH (200 mL) were charged in a 500 mL round-bottomed flask. The mixture was stirred at reflux for 2 h. After cooling to room temperature, the pH of the mixture was adjusted to 5 using 2 N HCl and the resulting solid was filtered and washed with water. The filter cake was then dried under reduced pressure to afford the title compound (5.12 g, 18.22 mmol, 94% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 13.69 (s, 1H, COOH), 9.03 (s, 1H, Ar-H), 8.38 (m, Ar-H), 7.97 (m, 2H, Ar-H), 4.11 (s, 3H, OCH3). ESI-MS: m/z = 282 [M + H]+.
6-Bromo-4-methoxyquinoline-3-carboxamide (12). A solution of 11 (200 mg, 0.71 mmol), ethyl chloroformate (85 mg, 0.78 mmol) and N-methylmorpholine (79 mg, 0.78 mmol) in dry THF was stirred at room temperature for 30 min. Then, NH3·H2O (0.3 mL) was added and stirred for 4 h. The mixture was washed with NaHSO3 and water, respectively. The organic phase was dried with magnesium sulfate and concentrated in vacuo to afford the crude product, which was further purified by silica gel column chromatography (50% ethyl acetate/petroleum ether) to give the title compound (155 mg, 0.55 mmol, 77% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.80 (s, 1H, Ar-H), 8.37 (d, J = 1.0 Hz, 1H, Ar-H), 8.19 (s, 1H, NH2), 7.96–7.93 (m, 2H, Ar-H), 7.91 (s, 1H, NH2), 4.15 (s, 3H, OCH3). ESI-MS: m/z = 281 [M + H]+.
6-(5-((2,4-Difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-4-methoxyquinoline-3-carboxamide (13). This compound was prepared from 12 (42 mg, 0.15 mmol) and 4 (64 mg, 0.15 mmol) according to the general synthesis procedure of 5 to afford the title compound (12 mg, 0.024 mmol, 16% yield) as a white solid. Mp 207–208 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.39 (s, 1H, NH), 8.82 (s, 1H, Ar-H), 8.47 (s, 1H, Ar-H), 8.33 (s, 1H, Ar-H), 8.16 (s, 1H, NH2), 8.10 (s, 2H, Ar-H), 7.99 (d, J = 2.0 Hz, 1H, Ar-H), 7.89 (s, 1H, NH2), 7.80 (m, 1H, Ar-H), 7.59 (m, 1H, Ar-H), 7.24 (td, J = 8.5, 2.0 Hz, 1H, Ar-H), 4.19 (s, 3H, OCH3), 3.70 (s, 3H, OCH3). IR (KBr) ν 3458, 3389, 3254, 3190, 2938, 2853, 1660, 1602, 1509, 1475, 1421, 1433, 1172, 1153 cm−1. MS (ESI) m/z = 501 [M + H]+, HRMS (ESI) m/z calcd for C23H19F2N4O5S [M + H]+ 501.1044, found 501.1042. HPLC purity = 95.3%.
General procedure A for synthesis of intermediates (14a–l)
A solution of 11 (1.0 equiv.), EDCI (1.5 equiv.) and HOBt (1.0 equiv.) in dry CH2Cl2 was stirred at room temperature for 2 h. Triethylamine (3.0 equiv.) and amine (2.0 equiv.) were then added and stirred for 1 h. The mixture was washed with 1 N NaOH and water, respectively. The organic phase was dried with magnesium sulfate and concentrated in vacuo to afford the crude product, which was further purified by silica gel column chromatography to give the desired compounds.
6-Bromo-N-(2-hydroxyethyl)-4-methoxyquinoline-3-carboxamide (14a). This compound was prepared from 11 (100 mg, 0.36 mmol) and 2-aminoethan-1-ol (44 mg, 0.72 mmol) according to the general synthesis procedure A to afford the title compound (92 mg, 0.28 mmol, 79% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.74 (s, 1H, Ar-H), 8.71 (t, J = 5.5 Hz, 1H, NH), 8.33 (m, 1H, Ar-H), 7.94–7.90 (m, 2H, Ar-H), 4.78 (t, J = 5.5 Hz, 1H, OH), 4.10 (s, 3H, OCH3), 3.55 (q, J = 5.5 Hz, 2H, CH2), 3.37 (q, J = 5.5 Hz, 2H, CH2). ESI-MS: m/z = 325 [M + H]+.
6-Bromo-N-(2-hydroxyethyl)-4-methoxy-N-methylquinoline-3-carboxamide (14b). This compound was prepared from 11 (100 mg, 0.36 mmol) and 2-(methylamino)ethan-1-ol (54 mg, 0.72 mmol) according to the general synthesis procedure A to afford the title compound (88 mg, 0.26 mmol, 72% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.62 (s, 1H, Ar-H), 8.37–8.33 (m, 1H, Ar-H), 7.95–7.91 (m, 2H, Ar-H), 4.87 (t, J = 5.0 Hz, 0.5H, OH), 4.80 (t, J = 5.0 Hz, 0.5H, OH), 4.08 (s, 3H, OCH3), 3.69 (d, J = 5.0 Hz, 1H, CH2), 3.57–3.41 (m, 2H, CH2), 3.33–3.26 (m, 1H, CH2), 3.09 (s, 1.5H, CH3), 2.97 (s, 1.5H, CH3). ESI-MS: m/z = 339 [M + H]+.
(6-Bromo-4-methoxyquinolin-3-yl)(pyrrolidin-1-yl)methanone (14c). This compound was prepared from 11 (100 mg, 0.36 mmol) and pyrrolidine (51 mg, 0.72 mmol) according to the general synthesis procedure A to afford the title compound (86 mg, 0.26 mmol, 72% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.64 (s, 1H, Ar-H), 8.33 (t, J = 1.0 Hz, 1H, Ar-H), 7.91 (m, 2H, Ar-H), 4.03 (s, 3H, OCH3), 3.52 (t, J = 6.5 Hz, 2H, CH2), 3.24 (t, J = 6.5 Hz, 2H, CH2), 1.89 (q, J = 6.5 Hz, 2H, CH2), 1.84 (q, J = 6.5 Hz, 2H, CH2). ESI-MS: m/z = 335 [M + H]+.
(6-Bromo-4-methoxyquinolin-3-yl)(piperidin-1-yl)methanone (14d). This compound was prepared from 11 (100 mg, 0.36 mmol) and piperidine (61 mg, 0.72 mmol) according to the general synthesis procedure A to afford the title compound (78 mg, 0.22 mmol, 62% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.59 (s, 1H, Ar-H), 8.32 (brs, 1H, Ar-H), 7.92 (m, 2H, Ar-H), 4.04 (s, 3H, OCH3), 3.80–3.71 (m, 1H, CH2), 3.62–3.54 (m, 1H, CH2), 3.27–3.22 (m, 2H, CH2), 1.64–1.55 (m, 4H, CH2 × 2), 1.46 (m, 2H, CH2). ESI-MS: m/z = 349 [M + H]+.
(6-Bromo-4-methoxyquinolin-3-yl)(morpholino)methanone (14e). This compound was prepared from 11 (100 mg, 0.36 mmol) and morpholine (63 mg, 0.72 mmol) according to the general synthesis procedure A to afford the title compound (89 mg, 0.25 mmol, 69% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.63 (s, 1H, Ar-H), 8.35–8.30 (m, 1H, Ar-H), 7.92 (m, 2H, Ar-H), 4.06 (s, 3H, OCH3), 3.71 (m, 4H, CH2 × 2), 3.54 (q, J = 5.0 Hz, 2H, CH2), 3.32 (q, J = 5.0 Hz, 2H, CH2). ESI-MS: m/z = 351 [M + H]+.
(6-Bromo-4-methoxyquinolin-3-yl)(4-methylpiperazin-1-yl)methanone (14f). This compound was prepared from 11 (100 mg, 0.36 mmol) and N-methylpiperazine (72 mg, 0.72 mmol) according to the general synthesis procedure A to afford the title compound (95 mg, 0.26 mmol, 72% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.59 (s, 1H, Ar-H), 8.32 (m, 1H, Ar-H), 7.92 (m, 2H, Ar-H), 4.04 (s, 3H, OCH3), 3.70 (t, J = 5.0 Hz, 2H, CH2), 3.33–3.29 (m, 2H, CH2), 2.40 (m, 2H, CH2), 2.27 (t, J = 5.0 Hz, 2H, CH2), 2.20 (s, 3H, CH3). ESI-MS: m/z = 364 [M + H]+.
(6-Bromo-4-methoxyquinolin-3-yl)(4-hydroxypiperidin-1-yl)methanone (14g). This compound was prepared from 11 (100 mg, 0.36 mmol) and 4-hydroxypiperidine (72 mg, 0.72 mmol) according to the general synthesis procedure A to afford the title compound (99 mg, 0.27 mmol, 75% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.62 (d, J = 3.0 Hz, 1H, Ar-H), 8.34 (d, J = 4.0 Hz, 1H, Ar-H), 7.97–7.90 (m, 2H, Ar-H), 4.82 (t, J = 3.5 Hz, 1H, OH), 4.15 (m, 0.5H, CH), 4.06 (m, 3.5H, OCH3 + CH), 3.84–3.73 (m, 1H, CH2), 3.54–3.39 (m, 2H, CH2), 3.17 (m, 1H, CH2), 1.85 (m, 1H, CH2), 1.69 (m, 1H, CH2), 1.53–1.41 (m, 1H, CH2), 1.41–1.30 (m, 1H, CH2). ESI-MS: m/z = 365 [M + H]+.
(6-Bromo-4-methoxyquinolin-3-yl)(3-hydroxypiperidin-1-yl)methanone (14h). This compound was prepared from 11 (100 mg, 0.36 mmol) and 3-hydroxypiperidine (72 mg, 0.72 mmol) according to the general synthesis procedure A to afford the title compound (105 mg, 0.29 mmol, 81% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.63 (s, 0.27H, Ar-H), 8.59–8.55 (m, 0.74H, Ar-H), 8.33 (s, 0.32H, Ar-H), 8.32 (s, 0.65H, Ar-H), 7.91 (s, 2H, Ar-H), 5.02 (m, 0.48H, OH), 4.83 (d, J = 3.5 Hz, 0.33H, OH), 4.80 (d, J = 3.5 Hz, 0.25H, OH), 4.06 (m, 3H, OCH3), 3.78 (s, 0.39H, CH), 3.65 (m, 0.61H, CH), 3.58 (m, 1H, CH2), 3.46 (m, 0.39H, CH2), 3.36 (m, 0.73H, CH2), 3.23 (m, 0.34H, CH2), 3.20–3.15 (m, 0.27H, CH2), 3.10 (m, 0.65H, CH2), 3.04 (m, 0.63H, CH2), 1.80 (m, 1.56H, CH2), 1.63 (m, 0.54H, CH2), 1.52–1.35 (m, 2H, CH2). ESI-MS: m/z = 365 [M + H]+.
(S)-1-(6-Bromo-4-methoxyquinoline-3-carbonyl)pyrrolidine-2-carboxamide (14i). This compound was prepared from 11 (100 mg, 0.36 mmol) and (S)-pyrrolidine-2-carboxamide (82 mg, 0.72 mmol) according to the general synthesis procedure A to afford the title compound (95 mg, 0.25 mmol, 69% yield) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.69 (s, 1H, Ar-H), 8.40 (d, J = 2.0 Hz, 1H, Ar-H), 7.94 (d, J = 9.0 Hz, 1H, Ar-H), 7.82 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H), 6.81 (s, 1H, NH2), 5.52 (s, 1H, NH2), 4.77 (dd, J = 7.5, 3.5 Hz, 1H, CH), 3.48 (m, 1H, CH2), 3.31 (m, 1H, CH2), 2.49–2.39 (m, 1H, CH2), 2.17–2.12 (m, 2H, CH2), 1.95–1.89 (m, 1H, CH2). ESI-MS: m/z = 378 [M + H]+.
6-Bromo-4-methoxy-N-phenylquinoline-3-carboxamide (14j). This compound was prepared from 11 (100 mg, 0.36 mmol) and aniline (67 mg, 0.72 mmol) according to the general synthesis procedure A to afford the title compound (76 mg, 0.21 mmol, 58% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 10.77 (s, 1H, NH), 8.88 (s, 1H, Ar-H), 8.41 (d, J = 2.0, 1H, Ar-H), 8.03–7.94 (m, 2H, Ar-H), 7.75 (d, J = 7.5 Hz, 2H, Ar-H), 7.39 (t, J = 8.0 Hz, 2H, Ar-H), 7.15 (t, J = 7.5 Hz, 1H, Ar-H), 4.14 (s, 3H, OCH3). ESI-MS: m/z = 357 [M + H]+.
6-Bromo-4-methoxy-N-(4-methoxyphenyl)quinoline-3-carboxamide (14k). This compound was prepared from 11 (100 mg, 0.36 mmol) and 4-methoxyaniline (89 mg, 0.72 mmol) according to the general synthesis procedure A to afford the title compound (72 mg, 0.19 mmol, 53% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 10.62 (s, 1H, NH), 8.86 (s, 1H, Ar-H), 8.40 (d, J = 2.0, 1H, Ar-H), 8.00–7.91 (m, 2H, Ar-H), 7.70–7.62 (d, J = 9.0 Hz, 2H, Ar-H), 7.00–6.92 (d, J = 9.0 Hz, 2H, Ar-H), 4.14 (s, 3H, OCH3), 3.76 (s, 3H, OCH3). ESI-MS: m/z = 387 [M + H]+.
6-Bromo-4-methoxy-N-(4-(trifluoromethoxy)phenyl)quinoline-3-carboxamide (14l). This compound was prepared from 11 (100 mg, 0.36 mmol) and 4-(trifluoromethoxy)aniline (127 mg, 0.72 mmol) according to the general synthesis procedure A to afford the title compound (80 mg, 0.18 mmol, 50% yield) as a white solid. ESI-MS: m/z = 441 [M + H]+.
6-(5-((2,4-Difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-N-(2-hydroxyethyl)-4-methoxyquinoline-3-carboxamide (15a). This compound was prepared from 14a (49 mg, 0.15 mmol) and 4 (64 mg, 0.15 mmol) according to the general synthesis procedure of 5 to afford the title compound (21 mg, 0.039 mmol, 26% yield) as a white solid. Mp 215–216 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.36 (s, 1H, NH), 8.75 (s, 1H, Ar-H), 8.69 (t, J = 5.5 Hz, 1H, NH), 8.47 (d, J = 2.0 Hz, 1H, Ar-H), 8.31 (s, 1H, Ar-H), 8.07 (s, 2H, Ar-H), 7.98 (d, J = 2.0 Hz, 1H, Ar-H), 7.76 (m, 1H, Ar-H), 7.58 (m, 1H, Ar-H), 7.21 (td, J = 8.5, 2.0 Hz, 1H, Ar-H), 4.79 (t, J = 5.5 Hz, 1H, OH), 4.14 (s, 3H, OCH3), 3.66 (s, 3H, OCH3), 3.57 (q, J = 5.5 Hz, 2H, CH2), 3.39 (q, J = 5.5 Hz, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 166.1, 165.6 (dd, JC–F = 252.8, 11.6 Hz), 160.2, 159.9 (dd, JC–F = 256.4, 14.0 Hz), 158.1, 151.7, 148.9, 143.0, 135.0, 134.3, 132.3 (d, JC–F = 10.6 Hz), 130.2, 129.8, 129.4, 125.6 (dd, JC–F = 15.0, 3.5 Hz) 122.8, 120.0, 118.3, 112.4 (dd, JC–F = 21.1, 3.4 Hz), 106.3 (t, JC–F = 26.1 Hz), 61.4, 60.0, 53.9, 42.7. IR (KBr) ν 3356, 3050, 2953, 2851, 2785, 1651, 1602, 1487, 1163, 1073 cm−1. MS (ESI) m/z = 545 [M + H]+, HRMS (ESI) m/z calcd for C25H22F2N4O6NaS [M + Na]+ 567.1126, found 567.1120. HPLC purity = 97.5%.
6-(5-((2,4-Difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-N-(2-hydroxyethyl)-4-methoxy-N-methylquinoline-3-carboxamide (15b). This compound was prepared from 14b (51 mg, 0.15 mmol) and 4 (64 mg, 0.15 mmol) according to the general synthesis procedure of 5 to afford the title compound (23 mg, 0.041 mmol, 27% yield) as a white solid. Mp 224–225 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.38 (s, 1H, NH), 8.60 (s, 1H, Ar-H), 8.46 (s, 1H, Ar-H), 8.31 (d, J = 5.5 Hz, 1H, Ar-H), 8.07 (d, J = 5.0 Hz, 2H, Ar-H), 7.97 (s, 1H, Ar-H), 7.79 (m, 1H, Ar-H), 7.59 (m, 1H, Ar-H), 7.23 (t, J = 9.0 Hz, 1H, Ar-H), 4.88 (t, J = 5.0 Hz, 0.5H, OH), 4.81 (t, J = 5.0 Hz, 0.5H, OH), 4.13 (s, 3H, OCH3), 3.70 (m, 1H, CH2), 3.69 (s, 3H, OCH3), 3.50 (m, 2H, CH2), 3.32–3.26 (m, 1H, CH2), 3.10 (s, 1.5H, CH3), 3.00 (s, 1.5H, CH3). 13C NMR (100 MHz, DMSO-d6) δ 167.8, 167.6, 165.6 (dd, JC–F = 251.5, 12.8 Hz), 159.9 (dd, JC–F = 255.6, 13.3 Hz), 158.4, 158.1, 158.1, 151.3, 150.7, 148.4, 148.3, 143.0, 134.9, 134.9, 134.3, 132.3 (d, JC–F = 10.8 Hz), 130.1, 129.6, 129.5, 129.4, 125.6 (dd, JC–F = 13.8, 3.7 Hz), 122.5, 119.9, 116.8, 116.7, 112.3 (dd, JC–F = 22.1, 3.3 Hz), 106.3 (t, JC–F = 23.0 Hz), 60.0, 58.6, 58.3, 53.9, 53.3, 49.9, 38.3, 32.8. IR (KBr) ν 3384, 3102, 2949, 2850, 1650, 1602, 1488, 1342, 1176, 1148 cm−1. MS (ESI) m/z = 559 [M + H]+, HRMS (ESI) m/z calcd for C26H24F2N4O6NaS [M + Na]+ 581.1282, found 581.1273. HPLC purity = 97.9%.
2,4-Difluoro-N-(2-methoxy-5-(4-methoxy-3-(pyrrolidine-1-carbonyl)quinolin-6-yl)pyridin-3-yl)benzenesulfonamide (15c). This compound was prepared from 14c (50 mg, 0.15 mmol) and 4 (64 mg, 0.15 mmol) according to the general synthesis procedure of 5 to afford the title compound (31 mg, 0.056 mmol, 37% yield) as a white solid. Mp 213–214 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.36 (s, 1H, NH), 8.64 (s, 1H, Ar-H), 8.47 (d, J = 2.0 Hz, 1H, Ar-H), 8.32 (brs, 1H, Ar-H), 8.08 (brs, 2H, Ar-H), 7.98 (d, J = 2.0 Hz, 1H, Ar-H), 7.79 (m, 1H, Ar-H), 7.59 (m, 1H, Ar-H), 7.23 (td, J = 10.5, 2.5 Hz, 1H, Ar-H), 4.10 (s, 3H, OCH3), 3.69 (s, 3H, OCH3), 3.56 (t, J = 6.5 Hz, 2H, CH2), 3.28 (t, J = 6.5 Hz, 2H, CH2), 1.93 (q, J = 6.5 Hz, 2H, CH2), 1.88 (q, J = 6.5 Hz, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 165.6 (dd, JC–F = 252.2, 11.4 Hz), 165.5, 159.8 (dd, JC–F = 253.6, 11.2 Hz), 158.5, 158.1, 150.7, 148.5, 143.1, 134.9, 134.4, 132.4, 132.3, 130.1, 129.6, 129.4, 125.5 (dd, JC–F = 13.5, 2.8 Hz), 122.6, 120.3, 119.9, 117.7, 112.4 (dd, JC–F = 22.6, 3.5 Hz), 106.3 (t, JC–F = 25.8 Hz), 74.0, 60.3, 53.9, 48.4, 46.2, 25.8, 25.4, 24.5. IR (KBr) ν 3331, 3102, 3080, 2974, 2876, 1626, 1602, 1487, 1434, 1342, 1177, 1147 cm−1. MS (ESI) m/z = 555 [M + H]+, HRMS (ESI) m/z calcd for C27H24F2N4O5NaS [M + Na]+ 577.1333, found 577.1327. HPLC purity = 95.5%.
2,4-Difluoro-N-(2-methoxy-5-(4-methoxy-3-(piperidine-1-carbonyl)quinolin-6-yl)pyridin-3-yl)benzenesulfonamide (15d). This compound was prepared from 14d (52 mg, 0.15 mmol) and 4 (64 mg, 0.15 mmol) according to the general synthesis procedure of 5 to afford the title compound (20 mg, 0.035 mmol, 23% yield) as a white solid. Mp 225–226 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.35 (s, 1H, NH), 8.62 (s, 1H, Ar-H), 8.45 (d, J = 2.0 Hz, 1H, Ar-H), 8.32 (brs, 1H, Ar-H), 8.08 (brs, 2H, Ar-H), 7.98 (d, J = 2.0 Hz, 1H, Ar-H), 7.81 (m, 1H, Ar-H), 7.56 (m, 1H, Ar-H), 7.22 (td, J = 10.5, 2.5 Hz, 1H, Ar-H), 4.10 (s, 3H, OCH3), 3.68 (s, 3H, OCH3), 3.78–3.66 (m, 2H, CH2), 3.65–3.55 (m, 2H, CH2), 1.65 (m, 2H, CH2), 1.58 (m, 4H, CH2 × 2). 13C NMR (100 MHz, DMSO-d6) δ 165.6 (dd, JC–F = 253.6, 12.0 Hz), 165.6, 159.8 (dd, JC–F = 255.5, 15.0 Hz), 158.6, 158.1, 150.5, 148.4, 143.1, 135.0, 134.4, 132.4 (d, JC–F = 10.9 Hz), 130.2, 129.6, 129.4, 125.6 (dd, JC–F = 14.2, 2.9 Hz), 122.5, 120.4, 119.9, 116.8, 112.4 (dd, JC–F = 22.3, 3.6 Hz), 106.3 (t, JC–F = 26.1 Hz), 66.8, 60.4, 53.9, 48.3, 42.5, 26.0, 25.5, 24.4. IR (KBr) ν 3417, 3102, 3080, 2974, 2877, 1625, 1602, 1487, 1435, 1342, 1177, 1147 cm−1. MS (ESI) m/z = 569 [M + H]+, HRMS (ESI) m/z calcd for C28H26F2N4O5NaS [M + Na]+ 591.1490, found 591.1485. HPLC purity = 94.1%.
2,4-Difluoro-N-(2-methoxy-5-(4-methoxy-3-(morpholine-4-carbonyl)quinolin-6-yl)pyridin-3-yl)benzenesulfonamide (15e). This compound was prepared from 14e (53 mg, 0.15 mmol) and 4 (64 mg, 0.15 mmol) according to the general synthesis procedure of 5 to afford the title compound (25 mg, 0.044 mmol, 29% yield) as a white solid. Mp 219–220 °C. 1H NMR (500 MHz, CDCl3) δ 8.68 (s, 1H, Ar-H), 8.27 (d, J = 2.0 Hz, 1H, Ar-H), 8.21 (d, J = 2.0 Hz, 1H, Ar-H), 8.15 (d, J = 9.0 Hz, 1H, Ar-H), 8.07 (d, J = 2.0 Hz, 1H, Ar-H), 7.87 (m, 1H, Ar-H), 7.29 (s, 1H, Ar-H), 6.95 (m, 2H, Ar-H), 4.18 (s, 3H, OCH3), 3.98 (m, 2H, CH2), 3.97 (s, 3H, OCH3), 3.83 (m, 2H, CH2), 3.68 (m, 2H, CH2), 3.43–3.34 (m, 2H, CH2). IR (KBr) ν 3442, 3104, 2958, 2923, 2856, 1636, 1604, 1488, 1463, 1434, 1347, 1178, 1152, 1119, 1017 cm−1. MS (ESI) m/z = 571 [M + H]+, HRMS (ESI) m/z calcd for C27H24F2N4O6NaS [M + Na]+ 593.1282, found 591.1279. HPLC purity = 95.7%.
2,4-Difluoro-N-(2-methoxy-5-(4-methoxy-3-(4-methylpiperazine-1-carbonyl)quinolin-6-yl)pyridin-3-yl)benzenesulfonamide (15f). This compound was prepared from 14f (54 mg, 0.15 mmol) and 4 (64 mg, 0.15 mmol) according to the general synthesis procedure of 5 to afford the title compound (13 mg, 0.022 mmol, 15% yield) as a white solid. Mp 231–233 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.34 (s, 1H, NH), 8.57 (s, 1H, Ar-H), 8.43 (d, J = 2.5 Hz, 1H, Ar-H), 8.31–8.24 (m, 1H, Ar-H), 8.10–8.04 (m, 2H, Ar-H), 7.94 (d, J = 2.5 Hz, 1H, Ar-H), 7.76 (m, 1H, Ar-H), 7.61–7.55 (m, 1H, Ar-H), 7.20 (td, J = 8.5, 2.5 Hz, 1H, Ar-H), 4.08 (s, 3H, OCH3), 3.73 (m, 2H, CH2), 3.66 (s, 3H, OCH3), 3.34 (m, 2H, CH2), 2.45 (m, 2H, CH2), 2.33 (m, 2H, CH2), 2.23 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ 165.4 (dd, JC–F = 252.5, 11.8 Hz), 165.9, 158.9, 159.8 (dd, JC–F = 255.9, 13.2 Hz), 158.1, 150.5, 148.5, 142.5, 135.1, 133.8, 132.3 (d, JC–F = 11.0 Hz), 130.2, 129.7, 129.4, 125.9 (dd, JC–F = 14.4, 3.6 Hz), 122.5, 121.1, 119.8, 116.4, 112.3 (dd, JC–F = 21.7, 3.0 Hz), 106.25 (t, JC–F = 26.2 Hz), 60.6, 54.7, 54.4, 53.9, 47.2, 45.9, 41.7. IR (KBr) ν 3423, 3071, 2943, 2853, 2796, 1632, 1602, 1486, 1461, 1363, 1344, 1175, 1148, 1119, 1073 cm−1. MS (ESI) m/z = 584 [M + H]+, HRMS (ESI) m/z calcd for C28H27F2N5O5NaS [M + Na]+ 606.1599, found 606.1592. HPLC purity = 97.0%.
2,4-Difluoro-N-(5-(3-(4-hydroxypiperidine-1-carbonyl)-4-methoxyquinolin-6-yl)-2-methoxypyridin-3-yl)benzenesulfonamide (15g). This compound was prepared from 14g (55 mg, 0.15 mmol) and 4 (64 mg, 0.15 mmol) according to the general synthesis procedure of 5 to afford the title compound (24 mg, 0.041 mmol, 27% yield) as a white solid. Mp 241–242 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.38 (s, 1H, NH), 8.59 (d, J = 2.5 Hz, 1H, Ar-H), 8.46 (brs, 1H, Ar-H), 8.31 (brs, 1H, Ar-H), 8.08 (brs, 2H, Ar-H), 7.97 (brs, 1H, Ar-H), 7.79 (m, 1H, Ar-H), 7.59 (m, 1H, Ar-H), 7.23 (t, J = 8.0 Hz, 1H, Ar-H), 4.85 (s, 1H, OH), 4.19 (s, 1H, CH), 4.12 (s, 1.5H, OCH3), 4.08 (s, 1.5H, OCH3), 3.79 (s, 1H, CH2), 3.69 (s, 3H, OCH3), 3.46 (m, 2H, CH2), 3.24–3.14 (m, 1H, CH2), 1.87 (m, 1H, CH2), 1.71 (s, 1H, CH2), 1.43 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 166.3 (dd, JC–F = 252.8, 12.0 Hz), 165.7, 165.6, 159.8 (dd, JC–F = 255.9, 13.3 Hz), 158.8, 158.6, 158.1, 150.5, 150.4, 148.5, 143.1, 135.0, 134.4, 132.4 (d, JC–F = 11.0 Hz), 130.2, 129.6, 129.4, 125.5 (dd, JC–F = 14.9, 4.1 Hz), 122.6, 122.5, 120.3, 119.9, 116.8, 116.7, 112.4 (dd, JC–F = 22.2, 3.6 Hz), 106.3 (t, JC–F = 26.0 Hz), 65.9, 65.5, 60.5, 60.3, 60.2, 53.9, 45.1, 44.8, 39.1, 34.6, 34.3, 34.1, 33.8, 21.2, 14.5. IR (KBr) ν 3384, 3102, 2997, 2947, 2857, 1646, 1603, 1487, 1460, 1364, 1343, 1176, 1155, 1121, 1073, 1017 cm−1. MS (ESI) m/z = 585 [M + H]+, HRMS (ESI) m/z calcd for C28H26F2N4O6NaS [M + Na]+ 607.1439, found 607.1430. HPLC purity = 99.1%.
2,4-Difluoro-N-(5-(3-(3-hydroxypiperidine-1-carbonyl)-4-methoxyquinolin-6-yl)-2-methoxypyridin-3-yl)benzenesulfonamide (15h). This compound was prepared from 14h (55 mg, 0.15 mmol) and 4 (64 mg, 0.15 mmol) according to the general synthesis procedure of 5 to afford the title compound (28 mg, 0.048 mmol, 32% yield) as a white solid. Mp 238–239 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.36 (s, 1H, NH), 8.61 (s, 0.25H, Ar-H), 8.57–8.52 (m, 0.73H, Ar-H), 8.46 (m, 1H, Ar-H), 8.30 (m, 1H, Ar-H), 8.06 (m, 2H, Ar-H), 7.96 (m, 1H, Ar-H), 7.76 (m, 1H, Ar-H), 7.61–7.52 (m, 1H, Ar-H), 7.21 (td, J = 8.5, 2.0 Hz, 1H, Ar-H), 5.03 (m, 0.44H, OH), 4.86 (d, J = 3.5 Hz, 0.29H, OH), 4.81 (d, J = 3.5 Hz, 0.24H, OH), 4.10 (m, 3H, OCH3), 3.86 (m, 0.32H, CH), 3.68 (m, 0.77H, CH), 3.66 (s, 3H, OCH3), 3.62–3.52 (m, 1H, CH2), 3.40 (m, 1H, CH2), 3.26 (m, 0.30H, CH2), 3.19 (m, 0.28H, CH2), 3.13 (m, 0.62H, CH2), 3.05 (m, 0.62H, CH2), 1.81 (m, 1.61H, CH2), 1.66 (m, 0.50H, CH2), 1.53–1.35 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 166.2, 166.1, 165.6 (dd, JC–F = 252.4, 12.1 Hz), 159.8 (dd, JC–F = 268.3, 13.2 Hz), 158.6, 158.1, 150.6, 148.4, 143.0, 135.0, 134.4, 132.4 (d, JC–F = 10.7 Hz), 130.2, 129.6, 129.4, 125.6 (dd, JC–F = 15.4, 2.4 Hz), 122.5, 120.4, 119.9, 117.0, 116.5, 115.7, 112.3 (dd, JC–F = 21.8, 3.1 Hz), 106.3 (t, JC–F = 25.9 Hz), 74.0, 65.4, 65.2, 65.2, 64.8, 60.6, 60.5, 60.2, 59.7, 54.4, 54.1, 53.9, 48.8, 48.6, 47.6, 41.9, 41.9, 32.9, 32.6, 32.4, 25.4, 24.9, 22.9, 22.8, 22.1, 21.7. IR (KBr) ν 3373, 3232, 3097, 2944, 2859, 1646, 1602, 1488, 1464, 1364, 1341, 1177, 1147, 1119, 1072, 1013 cm−1. MS (ESI) m/z = 585 [M + H]+, HRMS (ESI) m/z calcd for C28H26F2N4O6NaS [M + Na]+ 607.1439, found 607.1430. HPLC purity = 98.4%.
(S)-1-(6-(5-((2,4-Difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-4-methoxyquinoline-3-carbonyl)pyrrolidine-2-carboxamide (15i). This compound was prepared from 14i (57 mg, 0.15 mmol) and 4 (64 mg, 0.15 mmol) according to the general synthesis procedure of 5 to afford the title compound (38 mg, 0.064 mmol, 43% yield) as a white solid. Mp 242–243 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.32 (s, 1H, NH), 8.66 (s, 0.69H, Ar-H), 8.50 (s, 0.30H, Ar-H), 8.43 (d, J = 2.0 Hz, 1H, Ar-H), 8.29 (m, 1H, Ar-H), 8.08–8.00 (m, 2H, Ar-H), 7.94 (m, 1H, Ar-H), 7.81–7.73 (m, 1H, Ar-H), 7.60–7.52 (m, 1H, Ar-H), 7.51 (s, 0.65H, NH2), 7.20 (t, J = 8.5 Hz, 1H, Ar-H), 7.12 (s, 0.32H, NH2), 6.99 (s, 0.65H, NH2), 6.76 (s, 0.30H, NH2), 4.48 (dd, J = 8.5, 4.0 Hz, 0.70H, CH), 4.20 (m, 0.35H, CH), 4.15 (s, 2.1H, OCH3), 4.12 (s, 0.9H, OCH3), 3.67 (s, 3H, OCH3), 3.41 (m, 1H, CH2), 3.37–3.31 (m, 1H, CH2), 2.31–2.16 (m, 1H, CH2), 1.96–1.89 (m, 2H, CH2), 1.87–1.77 (m, 1H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 173.7, 173.5, 165.5 (dd, JC–F = 252.1, 11.3 Hz), 166.3, 166.2, 165.9, 165.8, 159.8 (dd, JC–F = 256.5, 12.8 Hz), 158.7, 158.6, 158.1, 158.1, 150.9, 150.7, 150.6, 149.1, 148.4, 143.0, 134.9, 134.3, 132.4 (d, JC–F = 10.0 Hz), 131.0, 130.9, 130.1, 129.6, 129.5, 129.4, 129.1, 127.1, 127.0, 125.6 (dd, JC–F = 15.4, 4.6 Hz), 123.0, 123.0, 122.5, 122.5, 122.2, 122.2, 120.5, 120.0, 116.8, 116.4, 112.4 (dd, JC–F = 22.3, 3.0 Hz), 106.3 (t, JC–F = 26.4 Hz), 61.3, 61.3, 60.7, 60.6, 60.1, 53.9, 49.5, 47.3, 32.0, 30.4, 24.8, 23.0, 23.07. IR (KBr) ν 3389, 3197, 2953, 2877, 1684, 1618, 1603, 1487, 1450, 1430, 1377, 1343, 1175, 1156 cm−1. MS (ESI) m/z = 598 [M + H]+, HRMS (ESI) m/z calcd for C28H25F2N5O6NaS [M + Na]+ 620.1391, found 620.1397. HPLC purity = 95.7%.
6-(5-((2,4-Difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-4-methoxy-N-phenylquinoline-3-carboxamide (15j). This compound was prepared from 14j (53 mg, 0.15 mmol) and 4 (64 mg, 0.15 mmol) according to the general synthesis procedure of 5 to afford the title compound (22 mg, 0.038 mmol, 25% yield) as a white solid. Mp 230–231 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.76 (s, 1H, NH), 10.37 (s, 1H, NH), 8.87 (s, 1H, Ar-H), 8.49 (d, J = 2.0, 1H, Ar-H), 8.38 (s, 1H, Ar-H), 8.13 (s, 2H, Ar-H), 8.01 (d, J = 2.0 Hz, 1H, Ar-H), 7.79 (m, 3H, Ar-H), 7.58 (m, 1H, Ar-H), 7.40 (t, J = 8.0 Hz, 2H, Ar-H), 7.24 (d, J = 7.0 Hz, 1H, Ar-H), 7.16 (t, J = 7.5 Hz, 1H, Ar-H), 4.18 (s, 3H, OCH3), 3.70 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ 165.6 (dd, JC–F = 254.5, 10.4 Hz), 164.8, 160.1, 159.8 (dd, JC–F = 255.8, 13.4 Hz), 158.1, 151.3, 149.0, 143.1, 139.3, 135.1, 134.4, 132.3 (d, JC–F = 10.4 Hz), 130.3, 130.0, 129.5, 129.4, 125.6 (dd, JC–F = 14.6, 4.0 Hz), 124.6, 122.8, 120.4, 120.3, 120.0, 118.7, 112.4 (dd, JC–F = 20.8, 3.6 Hz), 106.3 (t, JC–F = 25.9 Hz), 61.5, 54.0. IR (KBr) ν 3364, 3261, 3104, 2952, 2837, 1656, 1603, 1511, 1486, 1243, 1176, 1149 cm−1. MS (ESI) m/z = 577 [M + H]+, HRMS (ESI) m/z calcd for C29H22F2N4O5NaS [M + Na]+ 599.1177, found 599.1177. HPLC purity = 94.0%.
6-(5-((2,4-Difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-4-methoxy-N-(4-methoxyphenyl)quinoline-3-carboxamide (15k). This compound was prepared from 14k (58 mg, 0.15 mmol) and 4 (64 mg, 0.15 mmol) according to the general synthesis procedure of 5 to afford the title compound (20 mg, 0.033 mmol, 22% yield) as a white solid. Mp 235–236 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.61 (s, 1H, NH), 10.37 (s, 1H, NH), 8.85 (s, 1H, Ar-H), 8.50 (d, J = 2.0 Hz, 1H, Ar-H), 8.38 (s, 1H, Ar-H), 8.12 (s, 2H, Ar-H), 8.01 (d, J = 2.0 Hz, 1H, Ar-H), 7.80 (m, 1H, Ar-H), 7.69 (d, J = 9.0 Hz, 2H, Ar-H), 7.62–7.55 (m, 1H, Ar-H), 7.24 (td, J = 8.5, 2.0 Hz, 1H, Ar-H), 6.97 (d, J = 9.0 Hz, 2H, Ar-H), 4.18 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 3.70 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ 165.6 (dd, JC–F = 252.1, 11.2 Hz), 164.3, 160.0, 158.1, 159.9 (dd, JC–F = 255.7, 12.7 Hz), 156.3, 151.4, 148.9, 143.1, 135.0, 134.4, 132.4, 132.3 (d, JC–F = 11.2 Hz), 130.2, 129.9, 129.4, 125.6 (dd, JC–F = 12.7, 4.6 Hz), 122.8, 121.8, 120.4, 120.0, 118.8, 114.5, 112.4 (dd, JC–F = 22.8, 26.1 Hz), 106.3 (t, JC–F = 26.1 Hz), 61.3, 55.7, 54.0. IR (KBr) ν 3363, 3256, 3072, 2951, 2837, 1658, 1603, 1541, 1511, 1343, 1242, 1176, 1149 cm−1. MS (ESI) m/z = 607 [M + H]+, HRMS (ESI) m/z calcd for C30H24F2N4O6NaS [M + Na]+ 629.1282, found 629.1284. HPLC purity = 94.3%.
6-(5-((2,4-Difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-4-methoxy-N-(4-(trifluoromethoxy)phenyl)quinoline-3-carboxamide (15l). This compound was prepared from 14l (66 mg, 0.15 mmol) and 4 (64 mg, 0.15 mmol) according to the general synthesis procedure of 5 to afford the title compound (25 mg, 0.038 mmol, 25% yield) as a white solid. Mp 249–251 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.96 (s, 1H, NH), 10.37 (s, 1H, NH), 8.88 (s, 1H, Ar-H), 8.51 (d, J = 2.5 Hz, 1H, Ar-H), 8.39 (s, 1H, Ar-H), 8.13 (d, J = 1.0 Hz, 2H, Ar-H), 8.02 (d, J = 2.5 Hz, 1H, Ar-H), 7.89 (d, J = 8.5 Hz, 2H, Ar-H), 7.80 (m, 1H, Ar-H), 7.63–7.56 (m, 1H, Ar-H), 7.42 (d, J = 8.5 Hz, 2H, Ar-H), 7.24 (td, J = 8.5, 2.0 Hz, 1H, Ar-H), 4.18 (s, 3H, OCH3), 3.69 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ 165.6 (dd, JC–F = 252.4, 11.7 Hz), 165.0, 160.3, 159.9 (dd, JC–F = 256.1, 13.5 Hz), 158.2, 151.2, 149.0, 144.6, 143.2, 138.5, 135.1, 134.4, 132.3 (d, JC–F = 10.7 Hz), 130.3, 130.1, 129.3, 125.6 (dd, JC–F = 13.6, 2.9 Hz), 122.7, 122.3, 121.7, 120.4 (d, JC–F = 206.2 Hz), 120.4, 120.0, 118.4, 112.4 (dd, JC–F = 22.0, 3.6 Hz), 106.3 (t, JC–F = 25.6 Hz), 61.6, 54.0. IR (KBr) ν 3358, 3106, 2952, 2854, 1675, 1604, 1509, 1483, 1345, 1251, 1175, 1151 cm−1. MS (ESI) m/z = 661 [M + H]+, HRMS (ESI) m/z calcd for C30H21F5N4O6NaS [M + Na]+ 683.1000, found 683.0995. HPLC purity = 94.6%.
6 Pharmacokinetic study
Fasted male mice were dosed orally at 5 mg kg−1 using a dosing formulation consisting of 0.5% methylcellulose. Blood samples were collected and placed into chilled tubes containing EDTA as the anticoagulant. The samples were centrifuged at 10000 rpm for 10 min, and plasma was collected and stored frozen until analysis. 15a was extracted from the plasma samples by protein precipitation, and the plasma concentration of 15a was assessed by UPLC-MS/MS (Waters). Pharmacokinetic analysis was conducted using Winnolin software. Mean plasma concentration values for each time point were used to generate plasma clearance [CL (L h−1 kg−1)], mean residence time [MRT (h)], peak plasma concentration [Cmax (ng mL−1)], plasma terminal half-life [t1/2 (h)], volume of distribution [Vd (L kg−1)], and exposure [AUC (h ng−1 mL−1)]. Animal study was approved by the Animal Research Committee at Jiaxing University (log number JXU2015120812), and animal care was provided in accordance with institutional guidelines.
Acknowledgements
The authors thank the project supported by the Public Welfare Technology Application Projects of Zhejiang Province (2016C33089) for financial support.
References
- L. C. Cantley, Science, 2002, 296, 1655–1657 CrossRef CAS PubMed.
- B. Vanhaesebroeck, L. Stephens and P. Hawkins, Nat. Rev. Mol. Cell Biol., 2012, 13, 195–203 CrossRef CAS PubMed.
- J. U. Flanagan and P. R. Shepherd, Biochem. Soc. Trans., 2014, 42, 120–124 CrossRef CAS PubMed.
- J. A. Engelman, Nat. Rev. Cancer, 2009, 9, 550–562 CrossRef CAS PubMed.
- K. Okkenhaug and R. Roychoudhuri, Sci. Signaling, 2015, 8, pe3 CrossRef PubMed.
- A. E. Yueh, S. N. Payne, A. A. Leystra, D. R. Van De Hey, T. M. Foley, C. A. Pasch, L. Clipson, K. A. Matkowskyj and D. A. Deming, PLoS One, 2016, 11, e0148730 Search PubMed.
- M. S. Song, L. Salmena and P. P. Pandolfi, Nat. Rev. Mol. Cell Biol., 2012, 13, 283–296 CAS.
- C. H. Huang, D. Mandelker, O. Schmidt-Kittler, Y. Samuels, V. E. Velculescu, K. W. Kinzler, B. Vogelstein, S. B. Gabelli and L. M. Amzel, Science, 2007, 318, 1744–1748 CrossRef CAS PubMed.
- A. Carracedo, L. Ma, J. Teruya-Feldstein, F. Rojo, L. Salmena, A. Alimonti, A. Egia, A. T. Sasaki, G. Thomas, S. C. Kozma, A. Papa, C. Nardella, L. C. Cantley, J. Baselga and P. P. Pandolfi, J. Clin. Invest., 2008, 118, 3065–3074 CAS.
- X. Lv, X. Ma and Y. Hu, Expert Opin. Drug Discovery, 2013, 8, 991–1012 CrossRef CAS PubMed.
- W. Peng, Z. C. Tu, Z. J. Long, Q. Liu and G. Lu, Eur. J. Med. Chem., 2016, 108, 644–654 CrossRef CAS PubMed.
- F. Lei, C. Sun, S. Xu, Q. Wang, Y. OuYang, C. Chen, H. Xia, L. Wang, P. Zheng and W. Zhu, Eur. J. Med. Chem., 2016, 116, 27–35 CrossRef CAS PubMed.
- T. Saurat, F. Buron, N. Rodrigues, M. L. de Tauzia, L. Colliandre, S. Bourg, P. Bonnet, G. Guillaumet, M. Akssira, A. Corlu, C. Guillouzo, P. Berthier, P. Rio, M. L. Jourdan, H. Benedetti and S. Routier, J. Med. Chem., 2014, 57, 613–631 CrossRef CAS PubMed.
- M. M. Stec, K. L. Andrews, Y. Bo, S. Caenepeel, H. Liao, J. McCarter, E. L. Mullady, T. San Miguel, R. Subramanian, N. Tamayo, D. A. Whittington, L. Wang, T. Wu, L. P. Zalameda, N. Zhang, P. E. Hughes and M. H. Norman, Bioorg. Med. Chem. Lett., 2015, 25, 4136–4142 CrossRef CAS PubMed.
- F. Han, S. Lin, P. Liu, J. Tao, C. Yi and H. Xu, Bioorg. Med. Chem. Lett., 2014, 24, 4538–4541 CrossRef CAS PubMed.
- H. Cheng, C. Li, S. Bailey, S. M. Baxi, L. Goulet, L. Guo, J. Hoffman, Y. Jiang, T. O. Johnson, T. W. Johnson, D. R. Knighton, J. Li, K. K. Liu, Z. Liu, M. A. Marx, M. Walls, P. A. Wells, M. J. Yin, J. Zhu and M. Zientek, ACS Med. Chem. Lett., 2012, 4, 91–97 CrossRef PubMed.
- Y. N. Liu, R. Z. Wan and Z. P. Liu, Mini-Rev. Med. Chem., 2013, 13, 2047–2059 CrossRef CAS PubMed.
- S. D. Knight, N. D. Adams, J. L. Burgess, A. M. Chaudhari, M. G. Darcy, C. A. Donatelli, J. I. Luengo, K. A. Newlander, C. A. Parrish, L. H. Ridgers, M. A. Sarpong, S. J. Schmidt, G. S. Van Aller, J. D. Carson, M. A. Diamond, P. A. Elkins, C. M. Gardiner, E. Garver, S. A. Gilbert, R. R. Gontarek, J. R. Jackson, K. L. Kershner, L. Luo, K. Raha, C. S. Sherk, C. M. Sung, D. Sutton, P. J. Tummino, R. J. Wegrzyn, K. R. Auger and D. Dhanak, ACS Med. Chem. Lett., 2010, 1, 39–43 CrossRef CAS PubMed.
- S. M. Maira, F. Stauffer, J. Brueggen, P. Furet, C. Schnell, C. Fritsch, S. Brachmann, P. Chene, A. De Pover, K. Schoemaker, D. Fabbro, D. Gabriel, M. Simonen, L. Murphy, P. Finan, W. Sellers and C. Garcia-Echeverria, Mol. Cancer Ther., 2008, 7, 1851–1863 CrossRef CAS PubMed.
- K. Y. Chang, S. Y. Tsai, C. M. Wu, C. J. Yen, B. F. Chuang and J. Y. Chang, Clin. Cancer Res., 2011, 17, 7116–7126 CrossRef CAS PubMed.
- H. M. Cheng, S. Bagrodia, S. Bailey, M. Edwards, J. Hoffman, Q. Y. Hu, R. Kania, D. R. Knighton, M. A. Marx, S. Ninkovic, S. X. Sun and E. Zhang, Med. Chem. Commun., 2010, 1, 139–144 RSC.
- X. Ma, X. Lv, N. Qiu, B. Yang, Q. He and Y. Hu, Bioorg. Med. Chem., 2015, 23, 7585–7596 CrossRef CAS PubMed.
- X. Lv, H. Ying, X. Ma, N. Qiu, P. Wu, B. Yang and Y. Hu, Eur. J. Med. Chem., 2015, 99, 36–50 CrossRef CAS PubMed.
- P. Furet, V. Guagnano, R. A. Fairhurst, P. Imbach-Weese, I. Bruce, M. Knapp, C. Fritsch, F. Blasco, J. Blanz, R. Aichholz, J. Hamon, D. Fabbro and G. Caravatti, Bioorg. Med. Chem. Lett., 2013, 23, 3741–3748 CrossRef CAS PubMed.
- W. Kevin, A. Michael, L. Kathryn, W. Catherine and D. Matthew, Patent WO2012009194A1, 2012.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26971k |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Ming Lia,
Yanmei Zhaob,
Guoqiang Liua and
Shuyu Zhana
*a,
Ming Lia,
Yanmei Zhaob,
Guoqiang Liua and
Shuyu Zhana