
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of nanoparticles based on PDMMLA derivative copolymers and study of warfarin encapsulation and controlled release†
Inès Msolliab,
Rima Belibela,
Frédéric Chaubeta,
Raoui Mounir Maaroufib and
Christel Barbaud *a
*a
aINSERM U1148, Laboratory for VascularTranslational Science (LVTS), Institut Galilée, Université Paris 13, Sorbonne Paris Cité, 99 Avenue Jean-Baptiste Clément, Villetaneuse, F-93430, France. E-mail: barbaud@univ-paris13.fr; Fax: +33 149403083; Tel: +33 149403357
bLaboratory of Genetic, Biodiversity, and Bioresources Valorisation, Université de Monastir, Institut Supérieur de Biotechnologie de Monastir, Avenue Taher Haddad (Bp 74), Monastir 5000, Tunisia
First published on 20th January 2017
Abstract
Poly((R,S)-3,3-dimethylmalic acid) (PDMMLA) copolymers were synthesized by anionic ring opening polymerization of benzylic and hexylic lactones. The PDMMLA copolymer family has biodegradable and biocompatible properties of interest as nanoparticles (NPs) can be formed from PDMMLA derivatives. For this purpose, three statistical copolymers (PDMMLAH30-co-Hex70, PDMMLAH50-co-Hex50 and PDMMLAH70-co-Hex30) and three block copolymers (PDMMLAH30-b-Hex70, PDMMLAH50-b-Hex50 and PDMMLAH70-co-Hex30) were synthesized and characterized. The main objective of the present study was to formulate new PDMMLA derivative NPs with defined size and shape that can efficiently be used as a novel controlled delivery system for a hydrophobic antithrombotic drug: warfarin. Polymeric NPs were prepared by nanoprecipitation method, optimized and characterized by Transmission Electronic Microscopy (TEM) for morphology investigation and Dynamic Light Scattering (DLS) for size and surface charge analysis. Controlled release of the drug is followed by reverse phase High Performance Liquid Chromatography (HPLC) for four weeks in phosphate buffer at pH = 7.4.
Introduction
Cardiovascular diseases (CVD) are the main cause of mortality worldwide with more than 17 million annual deaths. In addition, global cardiovascular deaths are expected to raise to more than 23 million by 2030.1 Cardiovascular death is often related to acute myocardial infarction or strokes that are mostly the consequence of an underlying atherosclerotic process.2 Atherosclerosis is a chronic inflammatory response in the arterial vessel wall leading to thrombosis formation which is an inappropriate clot formation within intact vascular structure.3Two classes of anticoagulants agents were deeply investigated: antithrombotic and antiplatelet molecules that inhibit the formation and growth of thrombi. Most of the anticoagulant drugs are characterized by a limited therapeutic efficacy because of their hydrophobic nature.4 Coumarinic derivatives are a part of these drugs, they are natural5 or synthetic molecules6 belonging to the benzopyrones family7 and recognized for their great importance in medicine.8 However they present two major limitations: narrow therapeutic index and a highly fluctuating dose–response relation.9,10
Warfarin is an oral anticoagulant drug, also known as coumadin representing the most common coumarinic derivatives in medicine.11 It's the most widely common anticoagulant as it effectively prevents thrombosis and thromboembolism formation in many diseases.12 It acts as a vitamin K antagonist that inhibit reduction of vitamin K-2,3-epoxide to vitamin K thus disrupting synthesis of vitamin K dependent coagulation factors: II, VII, IV and X. Warfarin suffers from difficult clinical management because of a narrow therapeutic range and considerable interpatient variability. This may lead to severe bleeding that can be life-threatening.11,13,14 The controlled delivery of warfarin performed with adequately build nanosystems would greatly limit these drawbacks.
Poly(lactic acid) (PLA) and poly(lactic-co-glycolic acid) (PLGA) and poly(ε-caprolactone) (PCL) are among the most successfully developed polymers in the field of medicine due to their attractive properties: (i) biodegradability and biocompatibility, (ii) Food and Drug Administration (FDA) and European Medicines Agency (EMA) approval in drug delivery systems for parenteral administration, (iii) well described formulations and methods of production adapted to various types of drugs, (iv) protection of drug from degradation, (v) possibility of sustained release, (vi) possibility to modify surface properties to provide stealth, better interaction with specific organs or cells.15–17 PLA/PLGA-based NPs are currently studied to increase the efficacy of treatments of cancer, inflammatory diseases, cerebral and cardiovascular diseases, infection and diabetes, because of the sustained release of the therapeutic agent from stable NP by improving pharmacokinetic and pharmacodynamic profiles.18
Anyway, the design of a nanosystem based on biocompatible functional polymers dedicated to the delivery of a particular drug still remains challenging.19
In this study, we have synthesized and characterized poly((R,S)-3,3-dimethylmalic acid) (PDMMLA), new functional polyesters shaped as nanoparticles for the delivery of warfarin.
PDMMLA derivates are obtained by anionic ring opening polymerization of racemic lactones prepared in few steps from diethyl oxalpropionate.20 PDMMLA derivates based NPs are formed by nanoprecipitation method and were characterized to determine its shape, size and surface charge properties (see ESI Fig. S1†). Warfarin which is a hydrophobic drug was encapsulated and its controlled release was followed by RP-HPLC.
Experimental
Chemicals
Warfarin and phenylbutazone were purchased from Sigma Aldrich Chemicals (StQuentin Fallavier, France).All other solvents and chemical products were purchased from Carlo Erba (Chaussée du Vexin, France) and Sigma-Aldrich Analytical France (St Quentin Fallavier, France) and were used as received. Anhydrous THF was prepared by the distillation of THF on sodium-benzophenone and under N2 atmosphere. Absolute EtOH was distilled over sodium under nitrogen atmosphere in order to prepare the initiator solutions. The later was dried under vacuum and N2 atmosphere. All polymerization reactions were realized in anhydrous THF under nitrogen atmosphere. Acetonitrile (ACN) and phosphate buffer saline (PBS) (which prepared fresh daily) were HPLC grades. Deionised water was purified by using a Millipore Milli-Q®Integral System (Molsheim, France).
Physicochemical analysis
The Reverse Phase High Performance Liquid Chromatography (RP-HPLC) analytical system was constituted with a Purospher®STAR endcapped RP-18 column (250 × 4.6 mm i.d., 5 μm, Merck, Darmstadt, Germany) used at room temperature with a mixture of ACN and PBS (40/60, v/v) at a flow rate of 0.5 mL min−1. The system included a Waters 2695 HPLC system (Milford, MA, USA) integrated with a column oven, an auto sampler, and a vacuum degasser. The detector was a Waters 2996 PDA detector (Milford, MA, USA). Empower Pro®5.0 software (Waters Corporation) monitored the equipment and allowed the treatment of the data.
PDMMLA synthesis and characterization
P1 (PDMMLABn30-co-Hex70), P2 (PDMMLABn50-co-Hex50), P3 (PDMMLABn70-co-Hex30) and block copolymers: P4 (PDMMLABn30-b-Hex70), P5 (PDMMLABn50-b-Hex50), and P6 (PDMMLABn70-b-Hex30), those copolymers were deprotected and benzylic groups were removed to form finally P1′ (PDMMLAH30-co-Hex70), P2′ (PDMMLAH50-co-Hex50), P3′ (PDMMLAH70-co-Hex30) and block copolymers: P4′ (PDMMLAH30-b-Hex70), P5′ (PDMMLAH50-b-Hex50), and P6′ (PDMMLAH70-b-Hex30).
Polymerization was started by tetraethylammonium benzoate C6H5COO− +N(Et)4 (36 mg mL−1 in distilled absolute ethanol) as initiator by a living anionic ring opening polymerization.
For statistical copolymers we have synthesized a long chain with a monomer/initiator ratio [m]/[a] = 103 and for block copolymers, short chains were synthesized with a ratio of [m]/[a] = 102. The initiator solution of each copolymer was dried under vacuum to remove absolute ethanol and then kept under nitrogen atmosphere at room temperature.
Benzylic and hexylic lactones already weighed and diluted in anhydrous THF were added to the initiator under nitrogen atmosphere. For statistical copolymers P1, P2 and P3 benzylic lactones were added, followed by the addition of the hexylic lactones whereas for block copolymers P4, P5 and P6, benzylic lactones were first added, polymerization process is instantaneously started to form the first block, after complete polymerization of benzylic lactones, hexylic lactones were added and polymerization process is continued to form the second block. All polymerizations were followed by FT-IR spectroscopy with the disappearance of the lactone band at 1835 cm−1 in favor of the ester band at 1754 cm−1. The polymerization process was stopped by the addition of 2 drops of glacial acetic acid in the reaction mixture. Then THF was evaporated and crude polymers were diluted in pure acetone before precipitation in absolute ethanol and left to stand overnight in order to allow polymers precipitation. After complete precipitation, ethanol supernatant was manually eliminated and polymers pellet was diluted in acetone and transferred into a crystallizer to be dried and weighed.
The proportions between benzylic and hexylic groups in the copolymers were determined from 1H NMR spectrum, by calculating the integration ratio of the “O–CH2–Ph” of the benzylic group against the integration of the “O–CH2–(CH2)4–CH3” of the hexylic group (see ESI Fig. S2†).
All copolymers were then deprotected by catalytic hydrogenolysis in the presence of palladium on charcoal (20% of PDMMLA mass) in acetone to remove the benzylic group.
Formation of the nanoparticles
NPs obtained from deprotected statistical copolymers (P1′, P2′ and P3′) and block copolymers (P4′, P5′ and P6′), were prepared thanks to the nanoprecipitation method already described by Fessi et al.23 NPs were formed by the adding of 100 mg of PDMMLA copolymers and a suitable percentage of warfarin to a 10 mL of acetone. This mixture was added dropwise to 20 mL of pure water under moderate agitation at room temperature. NPs were formed instantaneously by rapid solvent diffusion during dropping and the medium got a milky aspect. Nanosuspensions were kept under agitation. Weight ratios of warfarin/polymer of 3%, 6% and 10% have been assayed. Warfarin free PDMMLA NPs were prepared according to the same procedure omitting the drug.NPs were separated from non-encapsulated warfarin by ultracentrifugation at 40.000 rpm for 30 min followed by pellet resuspension. This procedure was repeated 3 times to remove the maximum of the non-encapsulated and surface bound warfarin.
PDMMLA NPs were characterized by TEM to determine their shape and by DLS to determine their size and surface charge.
Release experiments
To start the warfarin controlled release, pellets recovered from the ultracentrifugation were resuspended in 10 mL of PBS (pH = 7.4), the flask being maintained under a continuous gentle agitation in a temperature-controlled water bath kept at 37 °C. The released warfarin was checked over 4 weeks. Samples (1 mL) were collected twice a week, centrifuged at 4.500 rpm for 15 min. The supernatants were freeze-dried overnight at −80 °C, lyophilized, resuspended in 1 mL of acetonitrile (ACN) and analyzed by Reverse Phase High Performance Liquid Chromatography (RP-HPLC). Standard solutions were freshly prepared by dilution of the stock solution of warfarin (1000 μg mL−1) and phenylbutazone (100 μg mL−1). The amounts of warfarin and phenylbutazone were determined from the peak area correlated with the standard curve. All analysis was performed in triplicate.Results and discussion
We have synthesized statistical and block amphiphilic PDMMLA derivatives (with n: benzylic monomer proportion = 30%, 50% or 70% and m: hexylic monomer proportion = 70%, 50% or 30%) shaped as NPs for the controlled delivery of warfarin. PDMMLA derivatives have the assets of ester cleavable bonds in their backbones which make their degradation possible by simple hydrolysis. Besides, these synthetic copolymers with a stereogenic center in the repetitive unit for controlling the macromolecular structure include lateral chemical functions which could be adapted to carry on further chemical modifications.24 Moreover, it was previously demonstrated that PDMMLA degraded to give (R,S)-3,3-dimethylmalic acid,24 a non-toxic product involved in the biosynthesis of the pantothenate and the coenzyme A catalyzed by the β,β-dimethyldehydrogenase (EC. 1.1.1.84).25,26The formation of different copolymers was monitored by FTIR with the disappearance of the vibration band at 1835 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the lactonic function) besides the ester band at 1754 cm−1 (see ESI Fig. S3†).
O stretching of the lactonic function) besides the ester band at 1754 cm−1 (see ESI Fig. S3†).
Then lactones underwent an anionic ring-opening polymerization to form the different copolymers.24 Polymerizations were thus followed by FTIR by a progressive decrease of the lactonic band and increase of the ester band.
The formation of the copolymers was confirmed by 1H NMR analysis. The 1H NMR spectrum of the statistical copolymer evidenced the signals of aromatic hydrogen at 7.4 ppm, of the ester group at 5.3 ppm, and of the methylene groups of benzyl at 5.2 ppm and hexyl at 0.8, 1.25, 1.6 and 4.20 ppm respectively (see ESI Fig. S4†).
Copolymer proportions were obtained from 1H NMR spectra, by determining the integration ratio of the methylene groups of the benzylic ester against the integration of some methylene groups of the hexylic ester at (Table 1). P1–P6 were then deprotected by catalytic hydrogenolysis to give P1′–P6′ with free COOH moieties which provide the hydrophilic and acidic characters to the copolymers.27 Yields, molecular weights and glass transition temperatures are gathered in the Table 1.
| Copolymer | Hexa (%) | Bna (%) | Yield (%) | ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) wb (g mol−1) wb (g mol−1) |
nb (g mol−1) |
Dc | Tgd (°C) |
|---|---|---|---|---|---|---|---|
| a Obtained from 1H-NMR integration.b HPSEC-MALLS-dRI in THF, 0.5 mL min−1, n ± 500 g mol−1, w ± 500 g mol−1.c D = w/n.d Determined by DSC. |
|||||||
| P1 | 70.9 | 29.1 | 85 | 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
17800 |
1.07 | 5.6 |
| P2 | 52.0 | 48.0 | 79 | 39000 |
18800 |
2.07 | 19.3 |
| P3 | 31.1 | 68.9 | 85 | 24000 |
13700 |
1.75 | 31.7 |
| P4 | 70.3 | 29.7 | 81 | 34800 |
29900 |
1.16 | −6.2/63.0 |
| P5 | 49.0 | 51.0 | 97 | 28100 |
28000 |
1.00 | −1.3/72.5 |
| P6 | 28.7 | 71.3 | 98 | 22600 |
22600 |
1.00 | −9.7/52.2 |
| P1′ | — | — | — | 141200 |
128500 |
1.10 | 13.4 |
| P2′ | — | — | — | 85400 |
79600 |
1.07 | 11.2 |
| P3′ | — | — | — | 35900 |
33600 |
1.07 | 51.7 |
| P4′ | — | — | — | 109200 |
44500 |
2.45 | −9.8/56.2 |
| P5′ | — | — | — | 139600 |
123200 |
1.13 | −9.0/57.2 |
| P6′ | — | — | — | 53700 |
30700 |
1.75 | −9.1/60.3 |
One Tg was measured for each of the statistical polymers: 5.6, 19.3 and 31.7 for P1, P2 and P3 respectively. This reflects merely the statistical character of the copolymers which destroy the regularity of the PDMMLA chain. Whereas for block copolymers, two Tg have been obtained which indicate that these polymers are constituted by two blocks: one for the benzylic block and another for the hexylic block.28
In fact, when temperature is lower than Tg, PDMMLA derivatives backbone possess only molecular vibrational motion, and copolymers become hard and glassy on its “frozen” state, this state will protect the copolymer from water diffusion and thus from hydrolysis, as temperature increases approaching Tg, PDMMLA derivatives backbones develop rotational motion and as the temperature increases interactions between copolymer chains become looser thus copolymers become on its “elastic” state.
Or in our study, all Tg were lower than body temperature (37 °C) which mean that copolymers are on its elastic state that will facilitate water diffusion and therefore hydrolysis is easier, leading to a faster degradation time. These considerations make them very interesting materials to be used in association with bioactive molecules in the short and medium term.
On another hand, molar mass distribution value expressed by D for the six copolymers were between 1 and 2 reflecting a good homogeneity of copolymers' chains.
After catalytic hydrogenolysis and the removal of the benzylic group, deprotected copolymers were characterized by 1H NMR spectrum (see ESI Fig. S5†).
The deprotected statistical copolymers have higher glass transition temperatures (Tg) (13.4 and 51.7 °C for P1′ and P3′ except P2′ = 11.2 °C) than the corresponding protected copolymers besides the increase of the molecular weights likely due to the formation of intermolecular hydrogen bonds between the carboxyl groups. Hydrogen bonds also hinder the motion of the polymeric chains.29 Whereas for the block copolymers we noticed that both Tg tended to shift to a low temperature region probably due to the effect of crystallization which occurred at lower temperatures.
Statistical and block copolymers were shaped as spherical NPs thanks to the nanoprecipitation method, a widely used technique based on interfacial deposition because of the displacement of a solvent with a non-solvent.16,30 In addition, the formation of the NPs was obtained without any additives. This allowed the fabrication of on demand NPs of controlled shape, size, and charge surface, all crucial factors that have been shown to affect NPs interactions with biological systems in vitro and in vivo.31,32
According to TEM analysis (Fig. 1A), the average size of all NPs was about 50 nm (Table 2), whereas polydispersity index (PDI) was ranging from 0.08 to 0.25.
 | ||
| Fig. 1 TEM analysis of NPs. (A) Free NPs and (B) warfarin loaded NPs (W-NPs). | ||
Nanoparticles have a highly negative zeta potential from −60 to −80 mV (Table 2). This high negative charge, mainly due to the presence of the carboxylate units on the surface, ensures and maintains the stability of the nanosuspensions since NPs with lower charge surface could undergo some aggregation.33,34
TEM analysis evidenced spherical NPs from the six copolymers, but the more regular ones have been obtained with P1′ and P4′. Those two copolymers were then selected for the remainder of the study assuming that a hydrophobic larger proportion and larger hydrophobic domain respectively may provide better encapsulation efficiency and a relatively slow controlled release of the hydrophobic drug warfarin.
Encapsulation efficiencies with 3%, 6% and 10% of warfarin assayed for the release study with NPs prepared from P1′ and P4′ gave the optimal encapsulation efficiencies of about 95%, 90% and 85%, respectively. Consequently, released experiments have been performed with NPs containing 3% of warfarin.
From TEM analysis, free NPs NP1′ were distributed homogeneously (in bulk: Fig. 1A NP1′), whereas free NPs NP4′ were dispersed heterogeneously (Fig. 1A, NP4′). Once drug was added, a slick of NPs with a homogenous size and shape distribution was obtained either with W-NP1′ or W-NP4′ (Fig. 1B).
NPs size strongly influences the degradation of the polymer matrix35 and affects the performance of the nanosystem in many aspects such as circulation time, hepatic filtration, renal clearance, endocytosis and tissue extravasation/diffusion of the carrier.36
NPs NP1′ and NP4′ had an average hydrodynamic diameter in the range of 77 to 93 nm, and a polydispersity index (PDI) of 0.1 for both of them (Fig. 2) emphasizing the monodispersity of NPs formulation charges surface of the loaded NPs were similar to those of unloaded ones i.e. −50 mV and −65 mV for NP1′ and NP4′ respectively and the corresponding nanosuspensions were very stable without observable aggregation for at least 4 weeks.
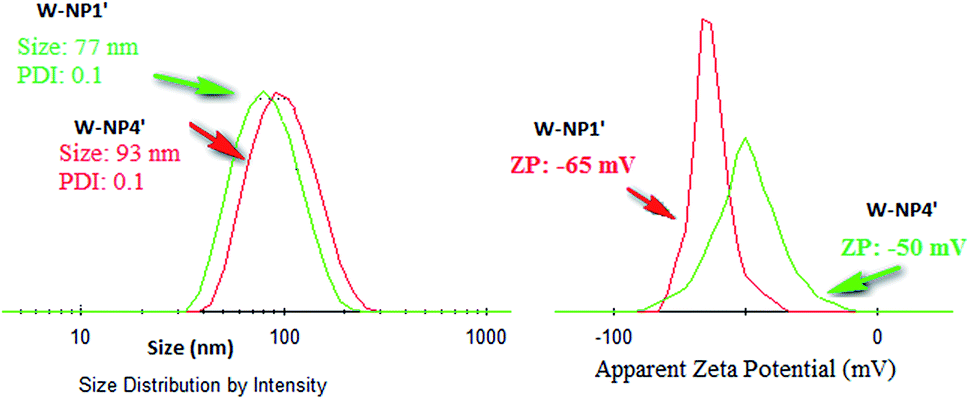 | ||
| Fig. 2 Size and zeta potential distribution of nanoparticles W-NP1′ and W-P4′N. | ||
Controlled release of warfarin
A short term release of warfarin was investigated for 4 weeks to explore warfarin release profile in vitro in a similar way as in physiological conditions. From release results carried out by HPLC for the 4 weeks, 0.83 mg and 1.12 mg corresponds to 38% and 47% for respectively W-NP1′ and W-NP4′ of the total introduced warfarin quantity. From DLS analysis and for almost two weeks, no apparent change was observed either in size or in zeta potential. From the fourteenth day some modifications were noticed: size tends to slightly increase by a flattening of the size distribution. These modifications continued until the end of the study to obtain finally a larger NPs size: 120 nm for W-NP1′ and 145 nm for W-NP4′ as well as the increase of surface charge up to −21 and −28 mV respectively (Fig. 3).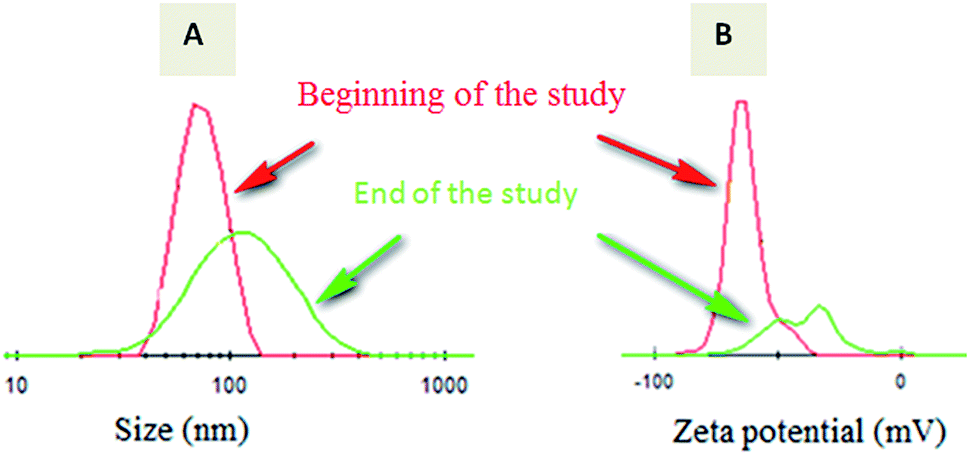 | ||
| Fig. 3 Evolution of the size distribution (A) and zeta potential (B) of loaded NPs from the beginning (red curve) to the end (green curve) of the study. | ||
These profiles evolution could be due to some limited aggregation of NPs. A preferential degradation of hydrophilic chain would lead to a relative increase of hydrophobic parts of the polymers and a decrease of the amount of carboxylate groups. As a consequence, the surface charge decreased.
A steady slow profile of the warfarin during the first two weeks and the warfarin release accentuation from the twelfth day were observed similarly for W-NP1′ and W-NP4′ (Fig. 4). It is noteworthy that the release profile of warfarin is done in a sustained and progressive way, in the absence of the burst release generally characterizing a surface-bound drug. This may emphasize the advantages of the PDMMLA derivatives as drug carriers and delivery systems comparing to other polymers.
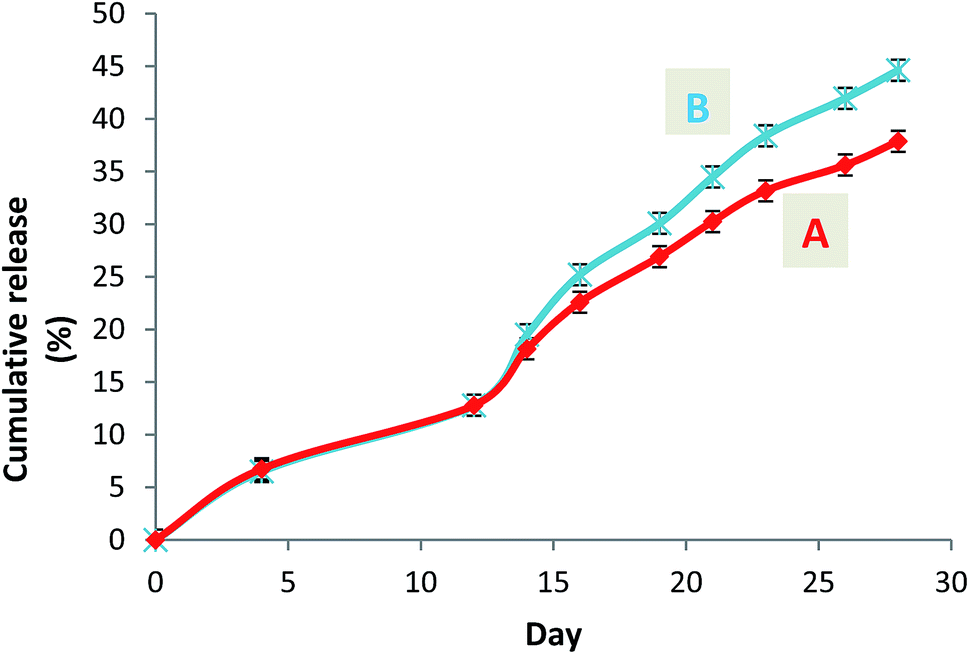 | ||
| Fig. 4 Cumulative release curves of warfarin from the loaded nanoparticles W-NP1′ (A) and W-NP4′ (B). | ||
The release of the therapeutic agents can proceed gradually over days to months depending on the polymer structure, and thus maintain the plasma drug concentrations at therapeutic levels for prolonged periods of time. This restricts the administration frequency of polymeric drug carriers.37
Indeed, release kinetics of drugs from such drug delivery systems are controlled by erosion and/or diffusion mechanisms that have been demonstrated to be dependent on the polymer characteristics35 since these release mechanisms are affected by the degradation rate of polymers.35,38
The architecture of polymers has clearly a strong influence on the release behavior. First, we must note that both polymers have glass transition temperatures allowing the diffusion of water molecules within the particles. However, we could consider that statistical copolymers based nanoparticles behave as matrix systems. The structure of the macromolecules allows a more important concentration of warfarin in the core of the particles. Block copolymers lead to core–shell architecture with a hydrophilic shell surrounding a hydrophobic core with a kind of ‘chemical composition gradient’ from the outer area to the inner one. In the first case, the release of the drug would be driven by the erosion of the particles into smaller hydrophobic structures. A straight release of the drug is the consequence of the increase of the specific surface of the particles. In the second case, the outer hydrophilic shell would act as a rate controlled membrane (dissolution controlled reservoir).
In a nutshell, thanks to the anionic ring opening polymerization we could synthesize well-defined and size-controlled copolymers without the drawback of copolymer large size. In addition to the versatility of PDMMLA derivatives by adding several types of pendant groups (hydrophilic, neutral, hydrophobic and reactive),24,39,40 the hydrophilic/hydrophobic balance is adjustable to enable the modulation of a slower or quicker degradation. At this level we assume that a higher proportion of hexylic group and a lower acid proportion result consequently in a slower degradation kinetic of copolymers and vice versa, which in turn, makes possible the monitoring of the encapsulation efficiency and release of the drug. PLGA, PLA and PCL, the main polymers used for controlled drug delivery present some shortcomings: PLGA main drawbacks are essentially due to the drug stability during preparation and storage steps, this instability mainly due to the acid-catalyzed nature of its degradation products, leading to the incomplete release of such drug,41 besides PLGA drug release profile which proceeds in three phases (triphasic): high burst effect in the beginning, lag phase and finally release phase imposed by polymer erosion. Because of its high hydrophobicity, many efforts have been made to adjust PLGA structure in order to increase its hydrophilicity to enhance drug stability that influence the biodegradation and also drug release kinetics.42,43
In turn, poly(lactic acid) (PLA): poly(D,L-lactide) demands about 12 to 16 months to be completely biodegraded, and poly(L-lactide), is weakly hydrophilic and more crystalline, and could be found in vivo even after 1.5 to 2 years.44
Conclusions
PDMMLA and its derivatives are promising polyesters that have shown an interesting biocompatibility and in vivo degradation giving the (R,S)-3,3-dimethylmalic acid as a non-toxic molecule.45 PDMMLA derivatives seem to be appealing water soluble aliphatic polyesters with a pendant carboxylic acid groups that could be used in several temporary applications such as engineering technology, medical imaging, diagnostics and drug delivery system in the aim to improve hydrophobic drugs pharmacokinetics.28,46,47Drug delivery system is as important as the therapeutic impact itself. Polymer drug delivery system has had a tremendous impact on medical technology, improving therapy performance of many existing drugs. These drug carriers are tailored to overcome the weakness of treatment with free drugs that often lead to poor therapeutic responses.48 In this study, we present an example of application of synthetic poly(dimethylmalic acid) derivatives in the biomedical field.
Warfarin, a hydrophobic drug was successfully encapsulated in NPs made out of PDMMLAH30-co-Hex70 statistical copolymer and PDMMLAH30-b-Hex70 block copolymer, respectively. This was achieved using the nanoprecipitation method with high encapsulation efficiency. In fact the delivery of such therapeutic agents by nanocarriers aiming to improve their bioavailability and efficacy also minimize their toxicity and side effects, ensure their sustained release, overcome biological barriers to target specific action sites and control their release, have been and remain a tremendous challenge.49,50 NPs formed by amphiphilic statistical and block copolymers derived from PDMMLA copolymers represent potentially relevant drug delivery systems for hydrophobic molecules, thanks to their biodegradation properties and their ability to encapsulate such drug in their hydrophobic moieties or domains, besides the possibility to modulate their hydrophobic/hydrophilic balance to make NPs degradation rate faster or slower.
References
- C. D. Mathers and D. Loncar, PLoS Med., 2006, 3, 2011–2030 Search PubMed
.
- G. S. Getz and C. A. Reardon, Arterioscler., Thromb., Vasc. Biol., 2012, 32, 1104–1115 CrossRef CAS PubMed
- A. G. Zaman, G. Helft, S. G. Worthley and J. J. Badimon, Atherosclerosis, 2000, 149, 251–266 CrossRef CAS PubMed
- T. Brighton, Aust. Prescr., 2010, 33, 1–4 Search PubMed
- Iarc Monographs, Some Industrial Chemicals, 2000, 77, 193–225 Search PubMed
- T. Nasr, S. Bondock and M. Youns, Eur. J. Med. Chem., 2014, 76, 539–548 CrossRef CAS PubMed
- C. Kontogiorgis, A. Detsi and D. Hadjipavlou-litina, Expert Opin. Ther. Pat., 2012, 22, 437–454 CrossRef CAS PubMed
- S. P. K. Rohini, J. Biofertil. Biopestic., 2014, 5, 5–7 Search PubMed
- J. I. Weitz and J. Hirsh, Lancet, 1999, 353, 1431–1436 CrossRef
- J. Hirsh, N. Engl. J. Med., 1991, 324, 1865–1875 CrossRef CAS PubMed
- S. Weigt, N. Huebler, R. Strecker, T. Braunbeck and T. H. Broschard, Reprod. Toxicol., 2012, 33, 133–141 CrossRef CAS PubMed
- J. W. Little, J. Oral Med. Oral Surg. Oral Pathol. Oral Radiol., 2012, 113, 575–580 CrossRef PubMed
- N. Bozina, Biochem. Med., 2010, 20, 33–44 CrossRef CAS
- R. Autar, J. Orthop. Nurs., 2009, 13, 165–171 CrossRef
- R. Srikar, A. Upendran and R. Kannan, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2014, 6, 245–267 CrossRef CAS PubMed
- R. Gref, Y. Minamitake, M. T. Peracchia, V. Trubetskoy, V. Torchilin and R. Langer, Science, 1994, 263, 1600–1603 CAS
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed
- D. Pandita, S. Kumar and V. Lather, Drug Discovery Today, 2015, 20, 95–104 CrossRef CAS PubMed
- Z. W. Huang, V. Laurent, G. Chetouani, J. Y. Ljubimova, E. Holler, T. Benvegnu, P. Loyer and S. Cammas-Marion, Int. J. Pharm., 2012, 423, 84–92 CrossRef CAS PubMed
- C. Barbaud, M. Guerrouache and P. Guérin, Tetrahedron Lett., 2002, 43, 9513–9515 CrossRef CAS
- F. E. Kohn, J. W. A. van den Berg and G. van de Ridder, J. Appl. Polym. Sci., 1984, 29, 4265–4277 CrossRef CAS
- R. Belibel, I. Azzouz and C. Barbaud, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1495–1507 CrossRef CAS
- H. Fessi, F. Puisieux, J. P. Devissaguet, N. Ammoury and S. Benita, Int. J. Pharm., 1989, 55, R1–R4 CrossRef CAS
- C. Barbaud, F. Faÿ, F. Abdillah, S. Randriamahefa and P. Guérin, Macromol. Chem. Phys., 2004, 205, 199–207 CrossRef CAS
- C. T. Goodhue and E. E. Snell, Biochemistry, 1966, 5, 403–408 CrossRef CAS PubMed
- B. Acta, Biochim. Biophys. Acta, 1980, 613, 266–274 Search PubMed
- M. J. Gaunt, J. Yu and J. B. Spencer, J. Org. Chem., 1998, 63, 4172–4173 CrossRef CAS
- F. Ouhib, S. Randriamahefa, P. Guérin and C. Barbaud, Des. Monomers Polym., 2005, 8, 25–35 CrossRef CAS
- B. He, J. Bei and S. Wang, Polymer, 2003, 44, 989–994 CrossRef CAS
- S. Hornig, T. Heinze, C. R. Becer and U. S. Schubert, J. Mater. Chem., 2009, 19, 3838–3840 RSC
- A. Albanese, P. S. Tang and W. C. W. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS PubMed
- E. C. Wang and A. Z. Wang, Integr. Biol., 2014, 6, 9–26 RSC
- J. Wang, S. Tian, R. A. Petros, M. E. Napier and N. Carolina, J. Am. Chem. Soc., 2010, 132, 11306–11313 CrossRef CAS PubMed
- H. Jin, D. A. Heller, R. Sharma and M. S. Strano, ACS Nano, 2009, 3, 149–158 CrossRef CAS PubMed
- M. Dunne, O. I. Corrigan and Z. Ramtoola, Biomaterials, 2000, 21, 1659–1668 CrossRef CAS PubMed
- C. Dhand, M. P. Prabhakaran and R. W. Beuerman, RSC Adv., 2014, 4, 32673–32689 RSC
- J. A. Champion, Y. K. Katare and S. Mitragotri, J. Controlled Release, 2007, 121, 3–9 CrossRef CAS PubMed
- R. A. Kenley, M. O. Lee, T. R. Mahoney and L. M. Sanders, J. Am. Chem. Soc., 1987, 20, 2398–2403 CAS
- S. Cammas, I. Renard, V. Langlois and P. Guérin, Polymer, 1996, 37, 4215–4220 CrossRef CAS
- S. Hyon, W. Cha and Y. Ikada, Polym. Bull., 1987, 29, 119–126 Search PubMed
- P. Johansen, Y. Men, R. Audran, G. Corradin, H. P. Merkle and B. Gander, Pharm. Res., 1998, 15, 1103–1110 CrossRef CAS
- A. Breitenbach, Y. X. Li and T. Kissel, J. Controlled Release, 2000, 64, 167–178 CrossRef CAS PubMed
- R. C. Mundargi, V. R. Babu, V. Rangaswamy, P. Patel and T. M. Aminabhavi, J. Controlled Release, 2008, 125, 193–209 CrossRef CAS PubMed
- I. Bala, S. Hariharan and M. N. V. R. Kumar, Crit. Rev. Ther. Drug Carrier Syst., 2004, 21, 387–422 CrossRef CAS PubMed
- P. T. Magee and E. E. Snell, Biochemistry, 1966, 5, 409–416 CrossRef CAS PubMed
- K. L. Aillon, Y. Xie, N. El-Gendy, C. J. Berkland and M. L. Forrest, Adv. Drug Delivery Rev., 2009, 61, 457–466 CrossRef CAS PubMed
- S. D. Caruthers, S. A. Wickline and G. M. Lanza, Curr. Opin. Biotechnol., 2007, 18, 26–30 CrossRef CAS PubMed
- E. Merisko-liversidge, G. G. Liversidge and E. R. Cooper, Eur. J. Pharm. Sci., 2003, 18, 113–120 CrossRef CAS PubMed
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed
- R. Bott, Igarss, 2015, vol. 2014, pp. 1–5 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27015h |
| This journal is © The Royal Society of Chemistry 2017 |