
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermo-reversible sol–gel transition of aqueous solutions of patchy polymers†
Indravadan A. Parmar,
Aarti S. Shedge,
Manohar V. Badiger,
Prakash P. Wadgaonkar and
Ashish K. Lele *
*
Polymer Science and Engineering Division, CSIR-National Chemical Laboratory, Pune 411 008, India. E-mail: ak.lele@ncl.res.in
First published on 17th January 2017
Abstract
While aqueous solutions of several amphiphilic thermo-reversible polymers show gelation upon heating, there are fewer examples of polymer solutions that exhibit gelation when cooled. This paper reports an interesting phenomenon of abrupt thermoreversible gelation of aqueous solutions of a hydrophobically modified polymer upon cooling. A high molecular weight precursor copolymer (PCP, 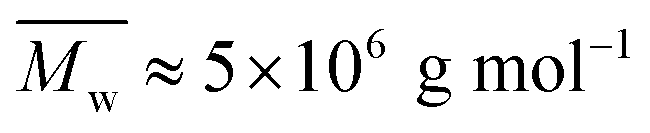 ) of N,N-dimethylacrylamide (70 mol%) and acrylic acid (30 mol%) was modified by reacting 10 mol% of the acrylic acid groups with n-dodecyl amine to form a hydrophobically modified copolymer (HMCP). The composition of the copolymer was ascertained using NMR spectroscopy. Cooling the solution of PCP at a controlled rate resulted in a gradual increase in its low shear viscosity as dictated by the flow activation energy. In contrast, cooling the solution of HMCP under identical conditions resulted in an abrupt and large non-Arrhenius increase in viscosity at a specific transition temperature, which decreased with decrease in polymer concentration. Fluorescence measurements and dynamic light scattering data showed that abrupt gelation happened upon cooling, when polymer coils percolate accompanied with concomitant transition in chain conformation from compact micellar coils formed by intra-chain hydrophobic associations to swollen polymer coils connected by inter-chain hydrophobic interactions.
) of N,N-dimethylacrylamide (70 mol%) and acrylic acid (30 mol%) was modified by reacting 10 mol% of the acrylic acid groups with n-dodecyl amine to form a hydrophobically modified copolymer (HMCP). The composition of the copolymer was ascertained using NMR spectroscopy. Cooling the solution of PCP at a controlled rate resulted in a gradual increase in its low shear viscosity as dictated by the flow activation energy. In contrast, cooling the solution of HMCP under identical conditions resulted in an abrupt and large non-Arrhenius increase in viscosity at a specific transition temperature, which decreased with decrease in polymer concentration. Fluorescence measurements and dynamic light scattering data showed that abrupt gelation happened upon cooling, when polymer coils percolate accompanied with concomitant transition in chain conformation from compact micellar coils formed by intra-chain hydrophobic associations to swollen polymer coils connected by inter-chain hydrophobic interactions.
Introduction
Over the past few decades, the phenomenon of thermally induced reversible gelation has been used for designing smart materials.1–8 An interesting example is that of a thermo-responsive injectable system, which finds application in controlled release technology and regenerative medicine.9–11 Among the materials that show thermoreversible gelation, the most prevalent ones are aqueous solutions of linear/graft copolymers having the thermodynamic property of lower critical solution temperature (LCST) or upper critical solution temperature (UCST). In either case, gelation is driven by phase separation of the polymer rich phase upon heating or cooling to an association temperature close to the LCST or UCST, respectively.12–14 The amphiphilic nature of the copolymers prevents macroscopic phase separation of the polymer phase so that the mixture exhibits visual homogeneity of a soft gel.Of the two types of polymer solutions viz. LCST and UCST, there are several examples of the former type showing thermo-reversible gelation.15–34 Various combinations of synthetic and natural water-soluble polymers have been shown to exhibit LCST type thermoreversible gelation. For example, thermoreversible polymers made by grafting LCST polymers like poly(N-isopropylacrylamide) (PNIPAM), polyethylene oxide (PEO) and poly(ethylene oxide-co-propylene oxide) (PEPO) onto water soluble polymers such as polyacrylic acid (PAA)19,20 and carboxymethyl cellulose (CMC)16,18 have been reported. Sui et al.35 reported rapid responsive thickening and self-assembly of a brush copolymer viz., poly(ethylene oxide)-graft-poly(N,N-dimethylaminoethyl methacrylate) in aqueous solution. Many hydrophobically modified polymers when dissolved in water at concentrations in the semidilute unentangled regime in the presence or absence of surfactants show gelation either at room temperature36–49 or upon increasing temperature.50
In comparison, the examples of polymer solutions that gel upon cooling are fewer. These include a few hydrophobically modified polymers having UCST-type phase behavior,13,51–53 natural polymers,54–56 diblock copolymer brush grafted silica nanoparticles,57 poly(PEO-co-styrene) dissolved in an ionic liquid,58 solutions of PNIPAM microgels59 and host–guest interactions.60 In these examples, the mechanism of gelation is either phase separation of UCST type polymers upon cooling or physical gelation/jamming of colloidal particles. An excellent review of the fundamental aspects and applications of thermo-responsive copolymers is available in the literature.61
Besides thermally induced gelation, shear induced gelation has also been observed in polymeric systems having a few localized, energetically favored interactions such as in ionomers,62–64 worm-like micellar solutions,65,66 colloidal solutions67,68 and aqueous solutions of hydrophobically modified polymers (HMPs).64,69–71 The molecular cause for shear thickening phenomenon in polymer solutions was suggested to be a shear induced transformation of intramolecular to intermolecular interactions.69,72
The present work reports a sol–gel transition under quiescent or near quiescent conditions upon cooling an aqueous solution of a hydrophobically modified poly(N,N-dimethylacrylamide-co-acrylic acid). The transition is manifested as a large and abrupt non-Arrhenius increase in low shear viscosity of the polymer solution when cooled below a transition temperature, which decreases with decrease in copolymer concentration. The change in viscosity with temperature is reversible albeit with large hysteresis. The main objective of this work is to obtain insights into the mechanism of this gelation phenomenon. We show that abrupt gelation of the copolymer solution is not caused by phase separation upon cooling. Rather, it occurs because of simultaneous change in chain conformation from a compact micellar state stabilized by intra-chain hydrophobic associations to a swollen coil state in which hydrophobic patches on the coils become accessible for inter-chain interactions. Copolymers such as these can possibly be used as rheology modifiers in several consumer products.
The paper is structured as follows. The Experimental section describes synthesis of the precursor poly(N,N-dimethylacrylamide-co-acrylic acid) copolymer and its hydrophobic modification. The salient results of the work from structural characterization experiments, rheology, dynamic light scattering and fluorescence measurements are presented in the Results section. The mechanistic interpretation of the data is presented in the Discussions section, while the Conclusions section summarizes the salient findings.
Experimental
Materials
N,N-Dimethylacrylamide (DMA), acrylic acid (AA), n-dodecyl amine (DDA), sodium metabisulfite (SMBS) and ammonium persulfate (APS) were purchased from Aldrich Chemicals. DMA and AA were purified by distillation before use. Dicyclohexylcarbodiimide (DCC), chloroform and diethyl ether were purchased from Loba Chemicals, India and were used as received. N-Methyl-2-pyrrolidinone (NMP, Loba Chemicals) was dried over calcium hydride and distilled under reduced pressure.Synthesis
The synthesis of hydrophobically modified copolymer (HMCP) was carried out as per procedures given by Lele et al.69 and Cadix et al.,70 which involves two steps: (a) synthesis of precursor copolymer (PCP) viz. poly(N,N-dimethylacrylamide-co-acrylic acid), and (b) modification of some of the acrylic acid units in the precursor co-polymer with n-dodecyl amine. This two-step synthesis is outlined in Scheme 1. Detailed experimental protocols for the synthesis of PCP and HMCP are provided in ESI.†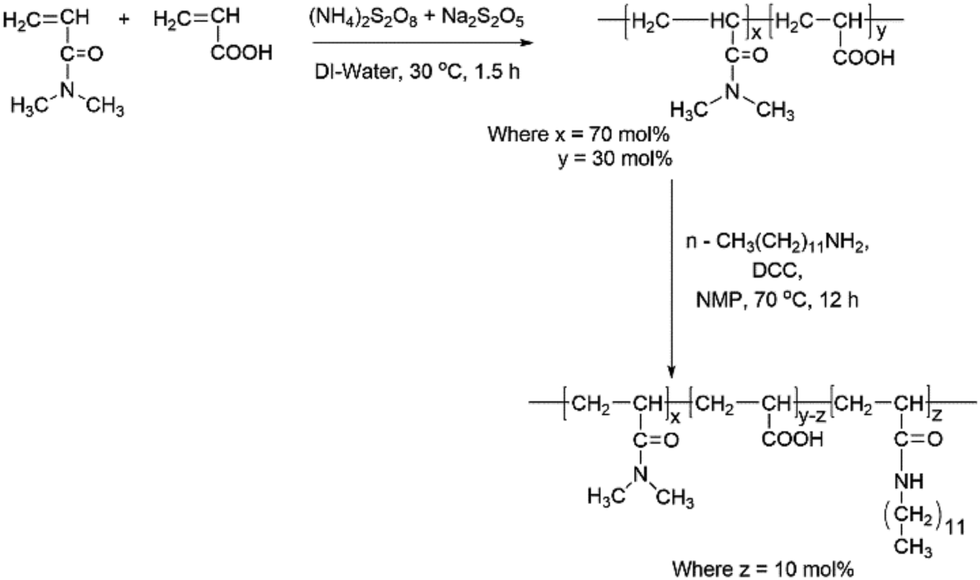 | ||
| Scheme 1 Synthetic pathway for HMCP. | ||
Characterization
The molecular weight of PCP was determined by aqueous gel permeation chromatography with reference to polyacrylamide standards. Compositions of PCP and HMCP were confirmed by 13C and 1H nuclear magnetic resonance spectroscopy. Thermal study of aqueous solution of HMCP was carried out using differential scanning calorimetry. The details of instrumentations and protocols are provided in ESI.†Rheology
Rheological studies were performed on aqueous solutions of PCP and HMCP, which were prepared by dissolving the polymers in deionized water and adjusting the pH to 8.5 by addition of appropriate amounts of aqueous sodium hydroxide. At this pH the copolymers exist as sodium salts.Specific viscosity of polymer solution of concentration CP < 20 mg mL−1 was measured using Ubbelohde viscometer under isothermal conditions. For concentrated solution (CP > 20 mg mL−1), a drag-flow rheometer (MCR-301, Anton Paar) was used to measure zero shear viscosity, and the specific viscosity was calculated from these measurement. Details of measurements of specific viscosity are provided in the ESI.†
Sol–gel transition of HMCP solutions was quantified using low shear viscosity measurements as a function of temperature. Experiments were performed using the cup and bob geometry of the MCR 301 rheometer. Polymer solutions of 25, 45 and 58 mg mL−1 were sheared at a small constant stress of 0.5 Pa while simultaneously cooling at a rate of 0.5 °C min−1 from 35 °C to 0 °C followed by holding the solutions at 0 °C for 20 minutes and then heating them at the same rate to 35 °C. These experiments were repeated for temperature rate ramps of 1 and 5 °C min−1. Care was taken to minimize evaporation loss of water during these experiments by placing wet tissues inside the lid of the cup and bob geometry.
Dynamic light scattering
Dynamic light scattering (DLS) experiments were performed on a dilute aqueous solution of HMCP of concentration CP = 0.2 mg mL−1 containing 1 mM NaCl to screen electrostatic interactions. Measurements were done using a 3D-LS goniometer system from LS instruments (Switzerland) equipped with a 632.8 nm He–Ne laser with a maximum power of 20 mW. The sample was placed in a quartz cuvette, which was kept in a constant temperature toluene bath maintained at temperatures ranging from 40 °C to 15 °C. Scattering was measured at right angle to the direction of the source. Data was collected for two minutes at each temperature. The intensity autocorrelation function g2(τ) was analysed by cumulant method. The apparent diffusion coefficient Do was determined according to Γ(q) = 2Doq2, where Γ(q) is decay rate of correlation function at the scattering wave vector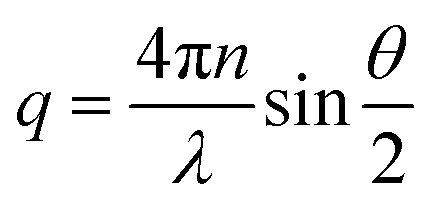 , n being the refractive index of water and λ is the laser wavelength. The relaxation time τ were obtained by inverse Laplace transformation (τ = 1/Γ). From the Do values, the hydrodynamic radius Rh of chains was determined using the Stokes–Einstein relationship for the translation diffusion.73
, n being the refractive index of water and λ is the laser wavelength. The relaxation time τ were obtained by inverse Laplace transformation (τ = 1/Γ). From the Do values, the hydrodynamic radius Rh of chains was determined using the Stokes–Einstein relationship for the translation diffusion.73| Rh = KBT/6πηDo | (1) |
Temperature dependence of solvent viscosity, η in eqn (1) was taken into account when calculating the hydrodynamic radius. The intensity autocorrelation function was also analysed by the CONTIN (constrained regularization method for inverting DATA) method, which gave information about relaxation modes.
Photophysical studies
Fluorescence studies were performed using Horiba Jobin Yvon Fluorolog 3 spectrophotometer having a 450 W xenon lamp for steady state fluorescence and nano LED of 560 nm for fluorescence decay time. Steady state emission and excitation were measured at different temperatures for 25 mg mL−1 aqueous solutions of PCP and HMCP to which pyrene was added externally at a concentration of 10−7 M. Emission and excitation slit width was maintained at 1 nm throughout the experiments, and the data was obtained in “S1/R1” mode (to account for the variation in lamp intensity). Temperature dependent fluorescence was recorded using a Peltier sample compartment having Peltier Sample Cooler F-3004 attached with a thermoelectric temperature controller (model no. LFI-3751) having an autotune PID supplied by wavelength electronics. Samples were equilibrated for 10 minutes at each temperature before recording a spectrum. The tolerance range for each set temperature was ±0.5 °C.Results
Molecular characteristics of PCP and HMCP
Fig. 1 shows quantitative 13C NMR spectrum of PCP recorded in DMSO-d6. The peaks in the range 35–39 ppm correspond to methylene and methine carbons of the backbone. The peaks at 173 ppm and 176 ppm, which are also shown in the inset, correspond to carbonyl of amide and acid, respectively. The integrated intensity ratio of carbonyl signals of amide and acid was used to determine the ratio of DMA and AA units in PCP and it was found to be 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 mol%. The rate of polymerization of DMA is higher than that of AA, and this is expected to result in the formation of a blocky structure of the PCP backbone.68,69,74
:30 mol%. The rate of polymerization of DMA is higher than that of AA, and this is expected to result in the formation of a blocky structure of the PCP backbone.68,69,74
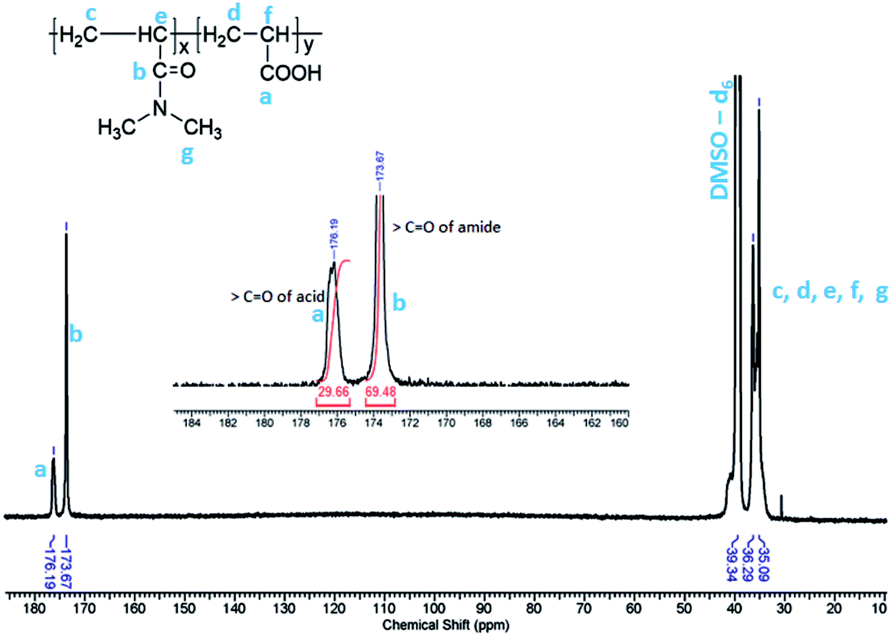 | ||
| Fig. 1 125 MHz 13C NMR spectrum of poly(N,N dimethylacryl amide-co-acrylic acid) in DMSO-d6. | ||
The weight-average molecular weight of PCP, as determined by GPC, was found to be Mw ∼ 5 × 106 g mol−1 (polyacrylamide standard) with a dispersity of 2.7 as shown in Fig. S1 of ESI.†
Fig. 2 shows quantitative 1H NMR spectra of PCP and HMCP recorded in DMSO-d6. Methylene (–CH2) protons of n-dodecyl chain appeared as overlapping peaks on the peaks due to methylene and methine protons of the polymer backbone (1.1 to 1.8 ppm). Therefore, these are not useful to quantify the content of n-dodecyl side chains in the copolymer. However, the terminal methyl (–CH3) protons of n-dodecyl side chain appeared at 0.87 ppm, which is sufficiently different than the chemical shift of methyl (–CH3) protons of DMA group (2.81 ppm). Thus the methyl protons offer clear evidence for the incorporation of n-dodecyl chain onto the copolymer. Comparing the areas under peaks at 0.87 ppm and 2.81 ppm confirmed that the hydrophobic modification was about 10 mol%.
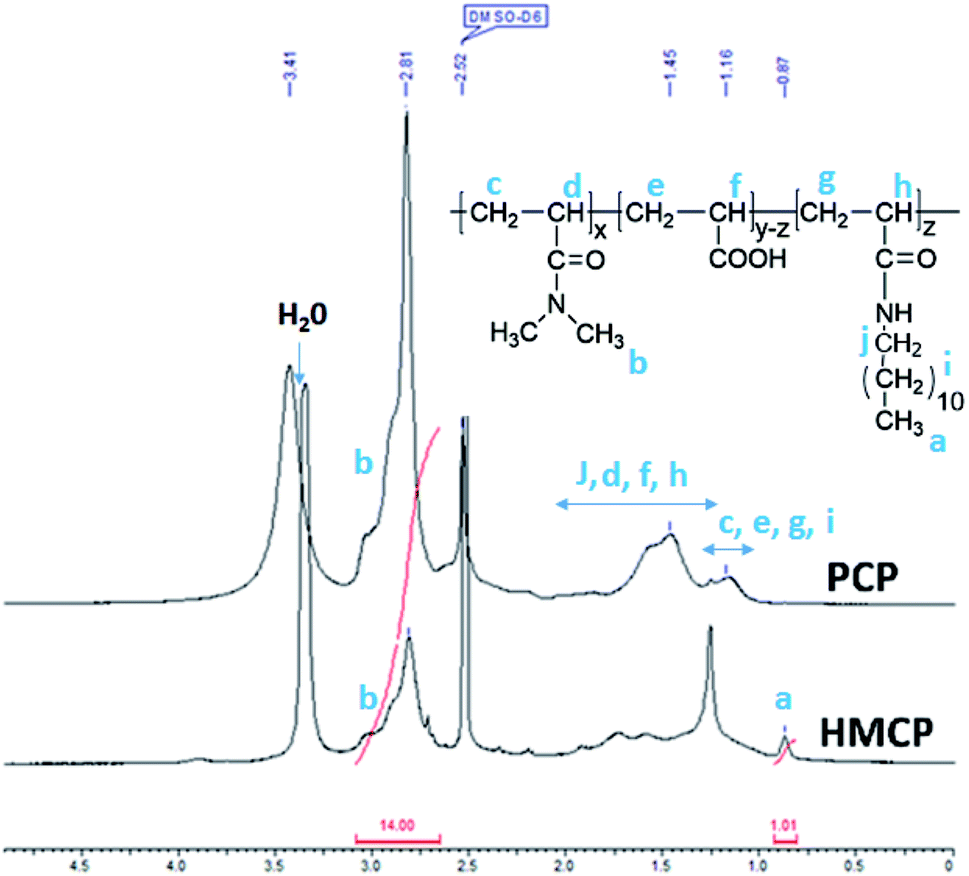 | ||
| Fig. 2 500 MHz 1H NMR spectra of PCP and HMCP in DMSO-d6. | ||
Sol–gel transition
A representative example of sol–gel transition upon cooling under quiescent conditions is shown in Fig. 3(a) for HMCP solution of CP = 45 mg mL−1 maintained at 35 °C and 0 °C. This can also be seen in the video SV1 in the ESI.† The solution is in a liquid state at 35 °C but forms a self-supporting gel at 0 °C. This sol–gel transition is reversible and can be repeated several times. In contrast, PCP solution of the same concentration does not show sol–gel transition as seen in Fig. 3(b). It is important to note here that the molecular weight, the comonomer composition of PCP and the extent of hydrophobic modification are critical parameters in order to observe the abrupt sol–gel transition phenomenon. This is elaborated in Table T1, and Fig. S1–S3 of ESI.† | ||
| Fig. 3 (a) An aqueous solution of HMCP (CP = 45 mg mL−1) gels upon cooling to 0 °C and liquefies upon heating to 35 °C. (b) PCP solution of the same concentration does not show sol–gel transition. | ||
Rheology
Fig. 4 shows the dependence of specific viscosity, ηsp on polymer concentration, CP for aqueous solutions of PCP and HMCP at 5 °C. In the lower concentration regime (CP < 20 mg mL−1), the specific viscosity of HMCP solution was lower than that of PCP solution, which indicates compaction of polymer chains due to intramolecular association of n-dodecyl hydrophobes. For both polymers in this regime of concentration, the scaling exponent was observed to be 0.8, in agreement with previous report.70 For polymer concentration CP > 20 mg mL−1, the viscosities of both polymers increased substantially with concentration. However, the viscosity of HMCP solution showed substantially higher concentration dependence and exceeded that of PCP solution beyond 50 mg mL−1. The substantial dependence of viscosity on concentration indicates a regime in which network structure is formed through chain entanglements and/or intermolecular associations. Similar trends were observed at higher temperatures. In general, the specific viscosity of both polymers decreased with increasing temperature indicating contraction of chains.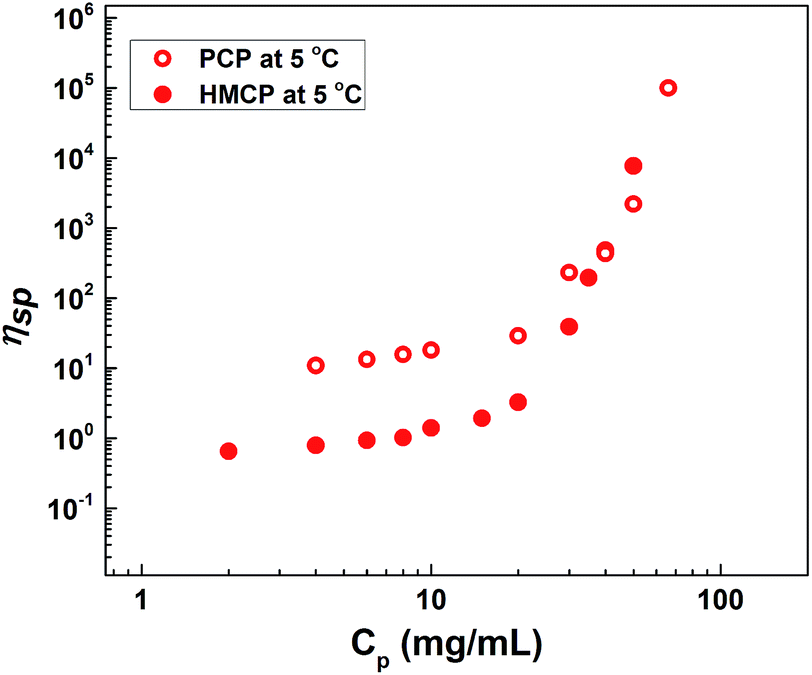 | ||
| Fig. 4 Dependence of specific viscosity on concentration at 5 °C. | ||
The cooling induced sol–gel transition shown in Fig. 3 was further quantified using low shear rheological measurements. Fig. 5(a) shows steady shear viscosity (closed symbols) of PCP solution of concentration CP = 25 mg mL−1 obtained by imposing constant shear stress of 0.5 Pa while simultaneously cycling the temperature (open symbols) of the solution between 25 °C and 0 °C at ramp rates of 0.5, 1 and 5 °C min−1 with hold time of 10 min at the extreme temperatures. The viscosity of the solution follows the temperature with a small lag time. The same data is displayed in Fig. 5(b) in the form of Arrhenius plot of log(η/ηo) vs. (1/T − 1/To) where ηo is the solution viscosity at a reference temperature To = 25 °C. The linearity of the data suggests that change in the viscosity of PCP solution with temperature follows Arrhenius law and is therefore dictated by thermal energy alone. The activation energy (∼28.3 kJ mol−1) calculated from the Arrhenius plot is similar to that of polyacrylamide solutions reported earlier.75 Weak hysteresis behavior is seen in Fig. 5(b) especially at the higher ramp rates of temperature cycles. This is likely due to the finite relaxation time of the PCP solution implying that for heating rates faster than the rate of relaxation of polymer chains, the solution viscosity will lag behind temperature as can be seen from the data.
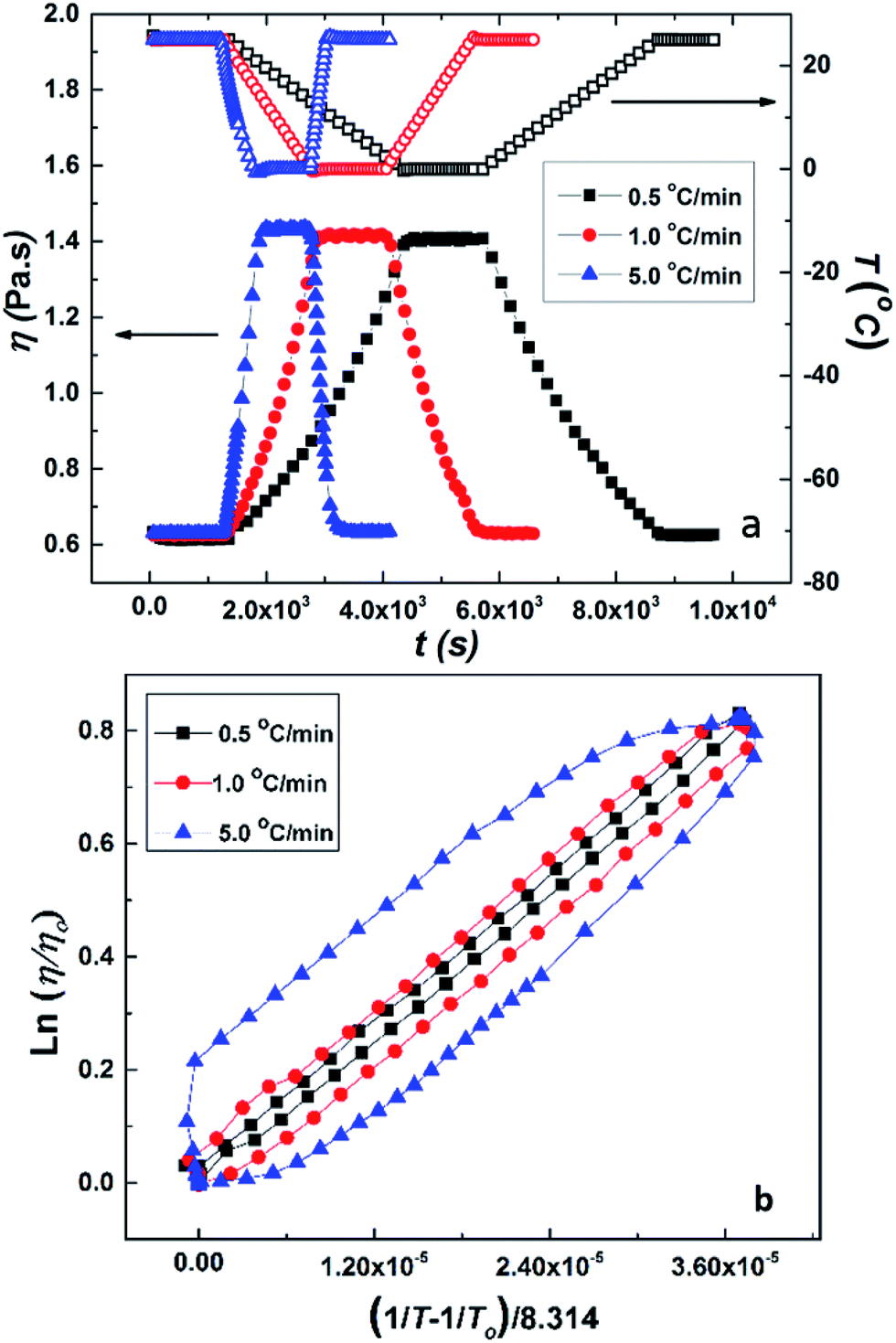 | ||
| Fig. 5 (a) Temperature ramp experiment on 25 mg mL−1 of aqueous solution of PCP. Closed symbols show viscosity data and open symbols show temperature data. (b) Arrhenius plot (i.e., dependency of viscosity on temperature) for aqueous solution of PCP (25 mg mL−1). | ||
It is interesting to compare the above results with the data for HMCP solution of the same concentration (CP = 25 mg mL−1) subjected to the same rheological tests. This is shown in Fig. 6(a) and (b). Comparing Fig. 6(a) and 5(a), it can be distinctly seen that over the same temperature range the change in viscosity of the HMCP solution is orders of magnitude greater than the change in viscosity of PCP solution. The data in Fig. 6(a) also shows that the viscosity of HMCP solution changes abruptly at certain temperatures during the heating and cooling cycles. Like PCP, the HMCP solution also shows time lag between the change in viscosity and temperature. Fig. 6(b) shows the rheology data for HMCP solution in the form of an Arrhenius plot. It can be seen that the data is non-linear, especially in the lower temperature range, and therefore does not obey Arrhenius law. This suggests that the large increase in viscosity is caused by intermolecular attraction, which dominates thermal energy. Indeed as demonstrated later, the large rise in viscosity is caused by hydrophobic associations of the n-dodecyl groups. The other distinct feature of the data in Fig. 6(b) is the strong hysteresis. As explained later, the observed hysteresis is a result of time required for HMCP chains to change their conformations.
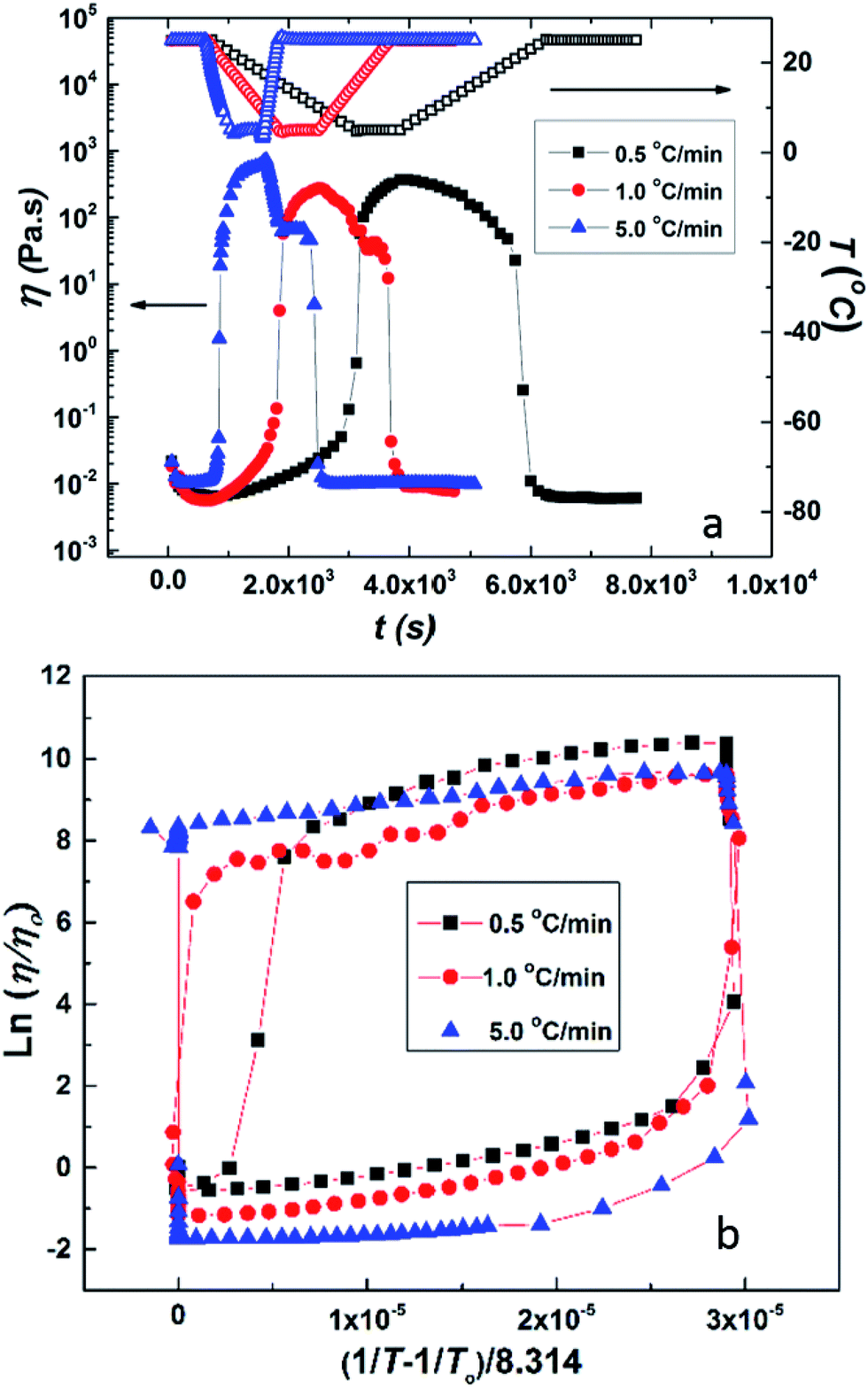 | ||
| Fig. 6 (a) Temperature ramp experiment on 25 mg mL−1 of aqueous solution of HMCP. Closed symbols show viscosity data and open symbols show temperature data. (b) Arrhenius plot (temperature dependence of viscosity) for the same HMCP solution. | ||
The large increase in viscosity of HMCP solutions upon cooling is also accompanied by an increase in elasticity. This can be seen from isothermal frequency sweep experiments at various temperatures as shown in Fig. S4(a) of ESI.† The HMCP solution of CP = 25 mg mL−1 shows predominantly viscous behavior at temperatures above 25 °C whereas its behavior is largely elastic at temperatures below 6 °C. This trend is also seen for HMCP solutions of CP = 58 mg mL−1 shown in Fig. S4(b) of ESI.† Thus upon cooling, HMCP solutions convert from sol to gel state. The gel state is stable as long as temperature is held constant (see Fig. S5 of ESI,† which shows viscoelastic moduli of HMCP gel at 0 °C for 10 h).
It is important to note that the observed sol–gel transition of the HMCP solution is not caused by applied shear. This is obvious in quiescent experiments shown in Fig. 3. In the low shear rheology experiments, the applied probe shear stress of 0.5 Pa is well below the critical shear stress (∼10 Pa) at which shear-induced gelation is seen in the same system.69 Similarly, the large change in viscosity of HMCP solution at low temperature is not caused by freezing. This was ascertained by differential scanning calorimetry measurements, which were performed on water and HMCP solution of CP = 25 mg mL−1 by cooling and heating the samples between 50 °C and −50 °C at 5 °C min−1 (see Fig. S6 of ESI†). It was observed that the onset of freezing for both samples was close to −20 °C (the large supercooling is because of the high cooling rate of 5 °C min−1). The rheology data shown in Fig. 6(a) for this HMCP solution when cooled at the same rate of 5 °C min−1 showed that the large change in viscosity occurred at about 6 °C, which is much higher than the observed freezing point.
Fig. 7 shows steady shear data for HMCP solutions of concentrations CP = 58, 45 and 25 mg mL−1 obtained at constant shear stress of 0.5 Pa while cooling from 35 °C to 0 °C at 0.5 °C min−1 followed by holding at 0 °C for about 10 h. In all cases an abrupt and large increase in viscosity was seen at specific transition temperatures, which decreased with decrease in polymer concentration. Also with decreasing concentration, both the magnitude of jump in viscosity at the transition temperature and the abruptness of the transition increased. Interestingly for all three HMCP concentrations, the rapid increase in viscosity started when the sols attained a certain value of viscosity (in this case, ∼0.02 Pa s). This suggests the possibility of percolation driven sol–gel transition in which polymer chains form space filling networks through associations. In order to probe this hypothesis, dynamic light scattering experiments were performed on HMCP solution as discussed below.
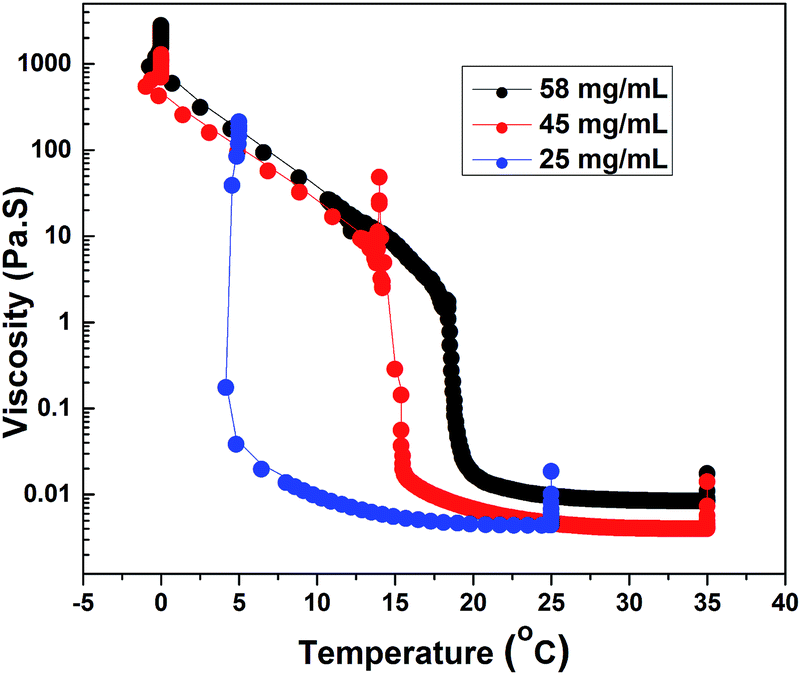 | ||
| Fig. 7 Effect of temperature on viscosity of HMCP solution at different concentrations. | ||
Dynamic light scattering
As mentioned earlier, DLS experiments were done on a dilute aqueous HMCP solution of concentration CP = 0.2 mg mL−1 in presence of 1 mM NaCl at various temperatures. Analysis of DLS data in the form of decorrelation functions [g2(τ) vs. τ] and relaxation time distribution curves [A(τ) vs. Rh] are shown in Fig. S7(a) and (b) of ESI† for five temperatures of 15, 20, 30, 35 and 40 °C. The relaxation spectra of the solution showed one dominant diffusional mode. Even at this low concentration, some amount of intermolecular aggregation cannot be ruled out entirely. The hydrodynamic radius, Rh of the scattering entities was calculated using eqn (1). Fig. 8 shows dependence of hydrodynamic radius Rh on temperature from which it is evident that the polymer chains swell with decrease in temperature. This is in agreement with previous reports.76 The swelling of polymer chains suggests that the polymer–water interactions become more favorable upon cooling. It has been reported that aqueous solutions of homopolymers of N,N-dimethylacrylamide have LCST-type phase behavior in which attractive interactions between the repeat units of the polymer and water molecules increase upon cooling.77 Since N,N-dimethylacrylamide is the major comonomer in PCP and HMCP, these copolymers can also be expected to have the thermodynamic quality of LCST. Hence the behavior seen in Fig. 8 is not unexpected. It must be noted however that the PCP and HMCP solutions do not show macroscopic phase separation in this temperature range. Thus, the sol–gel transition seen for HMCP is not a result of LCST driven macroscopic phase separation.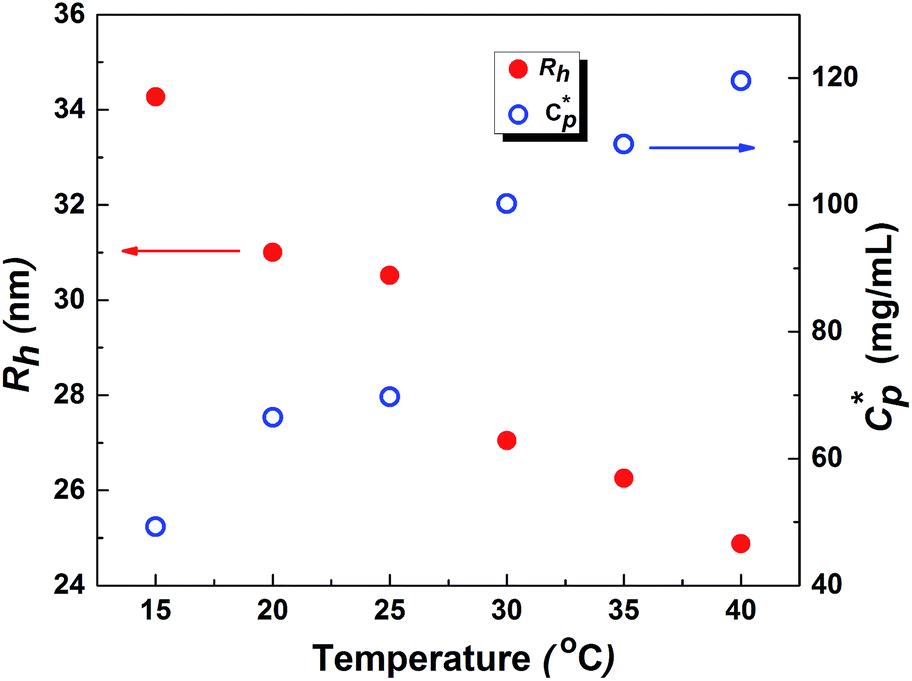 | ||
Fig. 8 Effect of temperature on overlap concentration  and hydrodynamic radius (Rh) of polymer chains of HMCP. Measurements were done on a dilute HMCP solution of CP = 0.2 mg mL−1 in the presence of salt at different temperatures. and hydrodynamic radius (Rh) of polymer chains of HMCP. Measurements were done on a dilute HMCP solution of CP = 0.2 mg mL−1 in the presence of salt at different temperatures. | ||
The critical polymer overlap concentration, 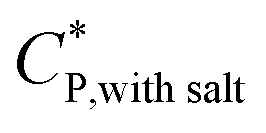 for the HMCP solution containing 1 mM NaCl as a function of temperature can be estimated from the Rh data using eqn (2)78 and is also shown in Fig. 8.
for the HMCP solution containing 1 mM NaCl as a function of temperature can be estimated from the Rh data using eqn (2)78 and is also shown in Fig. 8.
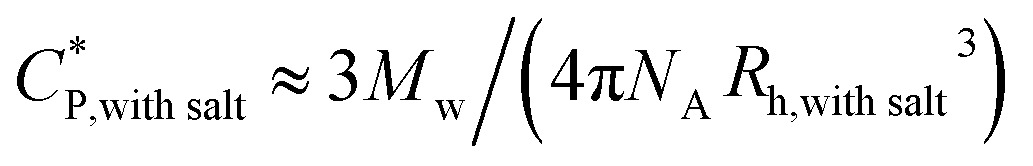 | (2) |
The critical overlap concentration increased from 49 mg mL−1 to 120 mg mL−1 over the temperature interval of 15 °C to 40 °C. The overlap concentration for the HMCP solution in the absence of salt should be lower on account of electrostatic repulsion of carboxylate anions on the copolymer backbone. If we assume, for sake of simplified argument, that 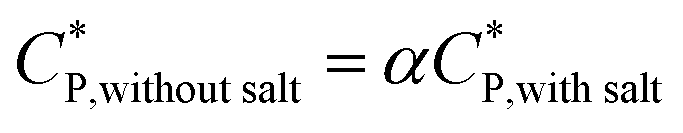 where α is a temperature independent contraction factor, then the temperature dependence of the overlap concentration of HMCP solutions used for rheological measurements would be the same as that shown in Fig. 8 but with a scaling factor of α.
where α is a temperature independent contraction factor, then the temperature dependence of the overlap concentration of HMCP solutions used for rheological measurements would be the same as that shown in Fig. 8 but with a scaling factor of α.
Consequently, the steady shear viscosity vs. temperature data shown earlier in Fig. 7 can now be re-plotted in terms of viscosity vs. scaled concentration 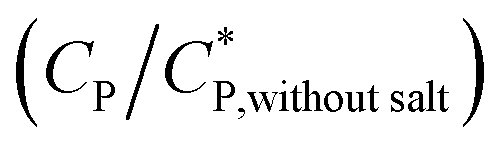 . The overlap concentration at intermediate temperatures was calculated by fitting the data in Fig. 8 with a second order polynomial. The viscosity vs. scaled concentration plot is shown in Fig. 9. It is clear that data for various concentrations in Fig. 7 collapse nearly completely on a master curve. The small deviation from complete collapse could be attributed to perhaps a weak temperature dependence of the contraction factor. The collapse of data on mastercurve supports the hypothesis that when a sample-spanning percolation threshold is reached on account of slightly overlapping polymer coils, the viscosity of solution rapidly increases and the solution converts into a gel. In the concentration regime
. The overlap concentration at intermediate temperatures was calculated by fitting the data in Fig. 8 with a second order polynomial. The viscosity vs. scaled concentration plot is shown in Fig. 9. It is clear that data for various concentrations in Fig. 7 collapse nearly completely on a master curve. The small deviation from complete collapse could be attributed to perhaps a weak temperature dependence of the contraction factor. The collapse of data on mastercurve supports the hypothesis that when a sample-spanning percolation threshold is reached on account of slightly overlapping polymer coils, the viscosity of solution rapidly increases and the solution converts into a gel. In the concentration regime 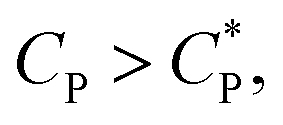 the viscosity increases more gradually compared to the diverging rise in viscosity of hard sphere suspensions. This gradual viscosity enhancement of HMCP solution is likely due to softer repulsion caused by interpenetration of polymer coils.
the viscosity increases more gradually compared to the diverging rise in viscosity of hard sphere suspensions. This gradual viscosity enhancement of HMCP solution is likely due to softer repulsion caused by interpenetration of polymer coils.
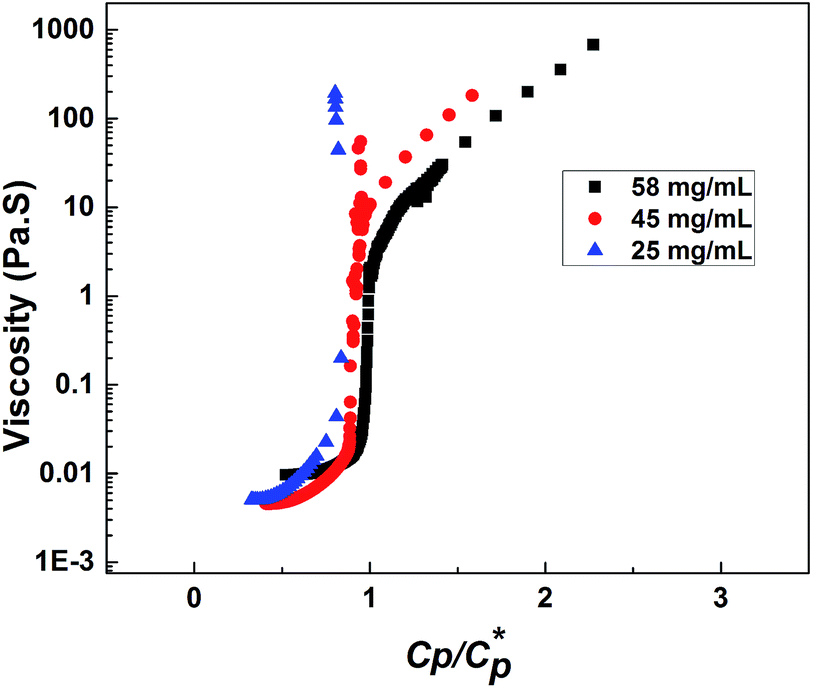 | ||
| Fig. 9 Dependence of viscosity on scaled concentration of HMCP solution. | ||
Percolation alone, however, cannot explain the abrupt sol-to-gel transition observed in HMCP solutions. Thus PCP solutions, which are expected to have lower overlap concentrations than HMCP, do not show any gelation at the same concentrations and over the same temperature range. It is likely that the large increase in viscosity of HMCP solutions at the transition temperature is caused by interpolymer interactions along with percolation. Specifically, it can be hypothesised that when the HMCP coils start overlapping their intra molecular hydrophobic associations convert into intermolecular hydrophobic interactions, which drives the gelation process. To examine this hypothesis photophysical experiments were performed and are discussed below.
Fluorescence measurements
Fig. 10(a) compares the normalized emission spectra of PCP solution containing pyrene (10−7 g mol−1) at different temperatures in the range 0–40 °C. Also shown for sake of comparison is the emission spectrum of pure water containing similar amount of pyrene. In each spectrum, the five peaks below 450 nm are characteristic vibronic bands of pyrene in aqueous solution. It is evident for the PCP solution that the peaks remain unchanged with temperature indicating absence of microstructural changes in the solution upon cooling. Fig. 10(b) shows normalized emission spectra of HMCP solution at different temperatures. In addition to the vibronic bands, a broad peak above 450 nm is clearly visible in these spectra.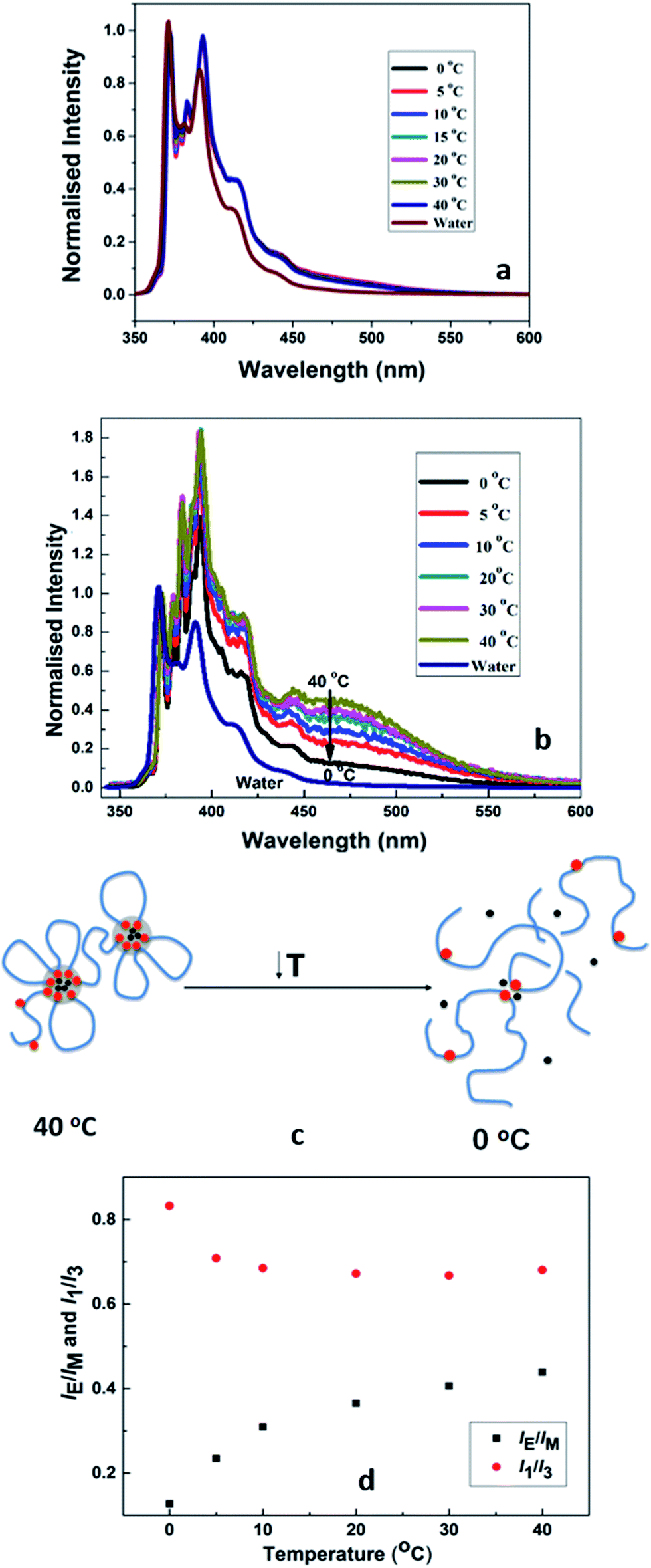 | ||
| Fig. 10 (a) Fluorescence measurements on PCP solution (b) fluorescence measurements on HMCP solution (c) schematic showing conformational changes of HMCP polymer chains upon cooling; red dots indicate hydrophobes, black dots indicate pyrene and blue chains indicate polymer backbone, (d) relative efficiency of excimer fluorescence emission expressed in terms of IE/IM and I1/I3 ratios as a function of temperature. | ||
This peak is characteristic of the pyrene excimer and suggests the entrapment of pyrene molecules in the hydrophobic domains formed by the association of n-dodecyl side chains. The data shows a gradual decrease in the intensity of excimer peak as the temperature was lowered from 40 °C to 0 °C. This suggests that upon cooling the pyrene molecules were released from the hydrophobic domains as shown schematically in Fig. 10(c).
These changes in the pyrene environment in HMCP solutions as a function of temperature can be quantified in terms of relative efficiency of excimer fluorescence emission expressed in terms of IE/IM and I1/I3 ratios as shown in Fig. 10(d). Here, IE and IM are the intensities (peak heights) of the excimer and monomer, respectively, while I1 and I3 are the intensities of the first and third peaks of the emission spectrum. These ratios are sensitive to the solvent microenvironment and serve as a measure of interaction between the pyrene and solvent.79,80 For the PCP solutions the IE/IM ratio was zero at all temperatures, whereas for the HMCP solution the IE/IM showed a concomitant decrease from 0.43 at 40 °C to 0.12 at 0 °C. Also for PCP solutions, the value of I1/I3 remained 1.4 at all temperatures, where as for HMCP solutions the I1/I3 ratio remained almost constant at 0.67 between temperatures 40 °C and 5 °C and increased to 0.82 below 5 °C. A value of I1/I3 above 1.5 is an indication of pyrene experiencing a hydrophilic environment whereas a value of 0.67 suggests a hydrophobic environment. Thus in case of PCP solution, the pyrene molecules experience only hydrophilic environment at all temperatures. But for the HMCP solution, pyrene molecules experience hydrophobic environment above 5 °C and increasingly hydrophilic environment below 5 °C.
From the fluorescence measurements it can be expected that above 5 °C the pyrene molecules are trapped inside micelles formed by intramolecular condensation of n-dodecyl groups. As the temperature is reduced below 5 °C the pyrene molecules are gradually released from the hydrophobic cores in to the aqueous medium.
Discussion
The following scenario regarding changes in the microstructure of the HMCP solutions as a function of temperature emerges when the data from rheology, dynamic light scattering and fluorescence measurements are considered together. At higher temperatures the HMCP chains have relatively compact configuration caused by unfavorable polymer–water interactions coupled with intra-chain association of the hydrophobic n-dodecyl side chains, which form micelles that trap pyrene molecules. The loss in entropy during micelle formation is balanced by enthalpic interactions between the hydrophobes. Upon cooling, entropy is gained by breaking the intra-chain hydrophobic associations. The enthalpic penalty is overcome to some extent by polymer–solvent interactions, which become increasingly favorable at lower temperature resulting into swelling of polymer coils. Below a critical temperature, the polymer coils start overlapping thereby bringing the exposed hydrophobic n-dodecyl groups into proximity. Now the hydrophobic groups from different polymer coils associate and bridge the overlapping coils to form a sample-spanning percolating network.Given that the copolymer backbone structure is likely to be blocky as discussed earlier, the copolymer chains can be considered to have sticky patches of hydrophobic groups that are buried inside the coils at higher temperatures. Upon cooling, the coils swell and expose the sticky patches. At a critical temperature the swollen coils percolate and the sticky groups bind them into a transient network (Fig. 11). This is manifested as rapid increase in sample viscosity. The absence of such sticky groups on PCP coils prevents the sol-to-gel transformation. Upon re-heating the solution, the polymer coils start to shrink. However, since the shrinking force has to overcome the binding force of the hydrophobic interactions in the entire network, a significant increase in temperature (super-heating) is required before the change in conformation can occur. Hence, a strong hysteresis is observed for the sol–gel–sol transition.
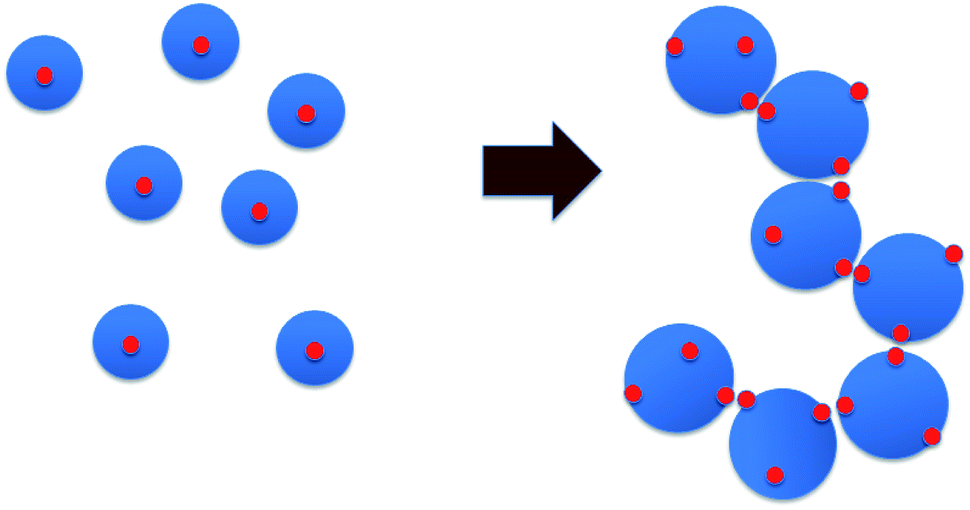 | ||
| Fig. 11 Schematic of percolation mechanism. Blue circles are polymer coils, red dots are hydrophobic patches. | ||
Conclusions
In this work, an unusual abrupt sol–gel transition of aqueous solutions of a hydrophobically modified copolymer upon cooling under quiescent or near-quiescent conditions has been demonstrated. The polymer has three important features built in its chemical structure: (i) large coil size caused by high molecular weight and repulsive ionic interactions in the chain, (ii) tendency to swell upon cooling, and (iii) presence of patches of hydrophobic side-groups attached to the main chain. The sol–gel transition was probed rheologically by changing the solution temperature while applying a weak shear flow, and was manifested as a large and abrupt rise in viscosity (and elasticity) at a critical temperature. The temperature dependence of viscosity was observed to have non-Arrhenius dependence indicating the presence of inter polymer energetic interactions that are at least as strong, if not more, than thermal energy. The sol–gel transition is reversible but with strong hysteresis. The critical temperature for sol-to-gel transition was found to be a function of polymer concentration. Dynamic light scattering measurements showed that the polymer coils swell upon cooling. The collapse of viscosity–temperature data for various concentrations on a master curve when the temperature axis was replaced with scaled concentration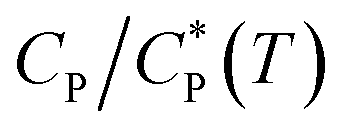 clearly showed that gelation happened when the coils started to overlap. However, this alone was insufficient to cause gelation. Fluorescence spectroscopy data showed that cooling-induced swelling of the polymer coils was responsible for driving a change in polymer conformation in which at lower temperatures inter-chain association of the hydrophobic patches on the polymer coils were formed at the expense of intra-chain associations. The inter-chain associations together with coil overlap resulted in the formation of a percolating transient network that drove the gelation process.
clearly showed that gelation happened when the coils started to overlap. However, this alone was insufficient to cause gelation. Fluorescence spectroscopy data showed that cooling-induced swelling of the polymer coils was responsible for driving a change in polymer conformation in which at lower temperatures inter-chain association of the hydrophobic patches on the polymer coils were formed at the expense of intra-chain associations. The inter-chain associations together with coil overlap resulted in the formation of a percolating transient network that drove the gelation process.
Acknowledgements
I. P. acknowledges CSIR, New Delhi for the Senior Research Fellowship (SRF). We would like to acknowledge Dr S. K. Asha for fluorescence spectroscopy data.Notes and references
- J. A. Syrett, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 978–987 RSC
.
- J. L. Hu, H. P. Meng, G. Q. Li and S. I. Ibekwe, Smart Mater. Struct., 2012, 21, 053001 CrossRef
- J. B. Beck and S. J. Rowan, J. Am. Chem. Soc., 2003, 125, 13922–13923 CrossRef CAS PubMed
- O. Roubeau, A. Colin, W. Schmitt and R. Clerac, Angew. Chem., Int. Ed., 2004, 43, 3283–3286 CrossRef CAS PubMed
- K. Kataoka and N. Huh, Journal of Stem Cells & Regenerative Medicine, 2010, 6, 10–14 CAS
- S. Chaterji, I. K. Kwon and K. Park, Prog. Polym. Sci., 2007, 32, 1083–1122 CrossRef CAS PubMed
- L. E. Bromberg and E. S. Ron, Adv. Drug Delivery Rev., 1998, 31, 197–221 CrossRef CAS
- C. D. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC
- J. D. Kretlow, L. Klouda and A. G. Mikos, Adv. Drug Delivery Rev., 2007, 59, 263–273 CrossRef CAS PubMed
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS
- S. B. Lowe, V. T. G. Tan, A. H. Soeriyadi, T. P. Davis and J. J. Gooding, Bioconjugate Chem., 2014, 25, 1581–1601 CrossRef CAS PubMed
- B. V. K. J. Schmidt, J. Elbert, C. Barner-Kowollik and M. Gallei, Macromol. Rapid Commun., 2014, 35, 708–714 CrossRef CAS PubMed
- S. Glatzel, N. Badi, M. Paech, A. Laschewsky and J.-F. Lutz, Chem. Commun., 2010, 46, 4517–4519 RSC
- W. X. Fu and B. Zhao, Polym. Chem., 2016, 7, 6980–6991 RSC
- K. N. Plunkett, X. Zhu, J. S. Moore and D. E. Leckband, Langmuir, 2006, 22, 4259–4266 CrossRef CAS PubMed
- N. R. Gupta, P. P. Ghute and M. V. Badiger, Carbohydr. Polym., 2011, 83, 74–80 CrossRef CAS
- H. Cong and S. Zheng, Colloid Polym. Sci., 2014, 292, 2633–2645 CAS
- N. R. Gupta, A. T. A. Torris, P. P. Wadgaonkar, P. R. Rajamohanan, G. Ducouret, D. Hourdet, C. Creton and M. V. Badiger, Carbohydr. Polym., 2015, 117, 331–338 CrossRef CAS PubMed
- A. Prasannan, H. C. Tsai, Y. S. Chen and G. H. Hsiue, J. Mater. Chem. B, 2014, 2, 1988–1997 RSC
- J. Zhang, L. Y. Chu, C. J. Cheng, D. F. Mi, M. Y. Zhou and X. J. Ju, Polymer, 2008, 49, 2595–2603 CrossRef CAS
- C. Karakasyan, S. Lack, F. Brunel, P. Maingault and D. Hourdet, Biomacromolecules, 2008, 9, 2419–2429 CrossRef CAS PubMed
- G. Bokias, Y. Mylonas, G. Staikos, G. G. Bumbu and C. Vasile, Macromolecules, 2001, 34, 4958–4964 CrossRef CAS
- H. W. Chen, W. W. Li, H. Zhao, J. G. Gao and Q. J. Zhang, J. Colloid Interface Sci., 2006, 298, 991–995 CrossRef CAS PubMed
- R. Liu, F. Cellesi, N. Tirelli and B. R. Saunders, Polymer, 2009, 50, 1456–1462 CrossRef CAS
- H. H. Lin and Y. L. Cheng, Macromolecules, 2001, 34, 3710–3715 CrossRef CAS
- T. Aubry, F. Bossard, G. Staikos and G. Bokias, J. Rheol., 2003, 47, 577–587 CrossRef CAS
- I. Koonar, C. Zhou, M. A. Hillmyer, T. P. Lodge and R. A. Siegel, Langmuir, 2012, 28, 17785–17794 CrossRef CAS PubMed
- C. Zhou, M. A. Hillmyer and T. P. Lodge, J. Am. Chem. Soc., 2012, 134, 10365–10368 CrossRef CAS PubMed
- R. R. Taribagil, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2010, 43, 5396–5404 CrossRef CAS
- H. H. Nguyen, B. Payre, J. Fitremann, N. L. D. Viguerie and J. D. Marty, Langmuir, 2015, 31, 4761–4768 CrossRef CAS PubMed
- H. Y. Tai, D. Howard, S. Takae, W. X. Wang, T. Vermonden, W. E. Hennink, P. S. Stayton, A. S. Hoffman, A. Endruweit, C. Alexander, S. M. Howdle and K. M. Shakesheff, Biomacromolecules, 2009, 10, 2895–2903 CrossRef CAS PubMed
- M. A. Ward and T. K. Georgiou, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2850–2859 CrossRef CAS
- A. P. Constantinou and T. K. Georgiou, Polym. Chem., 2016, 7, 2045–2056 RSC
- S. T. Hemp, A. E. Smith, W. C. Bunyard, M. H. Rubinstein and T. E. Long, Polymer, 2014, 55, 2325–2331 CrossRef CAS PubMed
- K. Y. Sui, X. Zhao, Z. M. Wu, Y. Z. Xia, H. C. Liang and Y. J. Li, Langmuir, 2012, 28, 153–160 CrossRef CAS PubMed
- T. K. Wang, I. Iliopoulos and R. Audebert, ACS Symp. Ser., 1991, 467, 218–231 CrossRef CAS
- S. Biggs, J. Selb and F. Candau, Polymer, 1993, 34, 580–591 CrossRef CAS
- P. Kujawa, A. Audibert-Hayet, J. Selb and F. Candau, Macromolecules, 2006, 39, 384–392 CrossRef CAS
- H. Niu, F. Wang and R. A. Weiss, Macromolecules, 2015, 48, 645–654 CrossRef CAS
- J. Yang, F.-K. Shi, C. Gong and X.-M. Xie, J. Colloid Interface Sci., 2012, 381, 107–115 CrossRef CAS PubMed
- K. Xu, H. An, C. Lu, Y. Tan, P. Li and P. Wang, Polymer, 2013, 54, 5665–5672 CrossRef CAS
- R. J. English, J. H. Laurer, R. J. Spontak and S. A. Khan, Ind. Eng. Chem. Res., 2002, 41, 6425–6435 CrossRef CAS
- Y. Yang, D. Schulz and C. A. Steiner, Langmuir, 1999, 15, 4335–4343 CrossRef CAS
- Z. Liang, T. Gao, J. Xu, Z. Li, X. Liu and F. Liu, Chem. Res. Chin. Univ., 2015, 31, 633–639 CrossRef CAS
- U. Gulyuz and O. Okay, Soft Matter, 2013, 9, 10287–10293 RSC
- D. C. Tuncaboylu, A. Argun, M. Sahin, M. Sari and O. Okay, Polymer, 2012, 53, 5513–5522 CrossRef CAS
- U. Gulyuz and O. Okay, Macromolecules, 2014, 47, 6889–6899 CrossRef CAS
- H. Hussain, B. H. Tan, G. L. Seah, Y. Liu, C. B. He and T. P. Davis, Langmuir, 2010, 26, 11763–11773 CrossRef CAS PubMed
- D. Hourdet, J. Gadgil, K. Podhajecka, M. V. Badiger, A. Brulet and P. P. Wadgaonkar, Macromolecules, 2005, 38, 8512–8521 CrossRef CAS
- K. Loyen, I. Iliopoulos, R. Audebert and U. Olsson, Langmuir, 1995, 11, 1053–1056 CrossRef CAS
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC
- J. Seuring and S. Agarwal, ACS Macro Lett., 2013, 2, 597–600 CrossRef CAS
- J. Seuring and S. Agarwal, Macromol. Rapid Commun., 2012, 33, 1898–1920 CrossRef CAS PubMed
- H. Yoshioka, Y. Mori, S. Tsukikawa and S. Kubota, Polym. Adv. Technol., 1998, 9, 155–158 CrossRef CAS
- J. L. Doublier, I. Cote, G. Llamas and G. Charlet, in Physics of Polymer Networks, ed. S. Wartewig and G. Helmis, 1992, vol. 90, pp. 61–65 Search PubMed
- M. H. Nordby, A. L. Kjoniksen, B. Nystrom and J. Roots, Biomacromolecules, 2003, 4, 337–343 CrossRef CAS PubMed
- R. A. E. Wright, B. Hu, D. M. Henn and B. Zhao, Soft Matter, 2015, 11, 6808–6820 RSC
- Y. He and T. P. Lodge, Macromolecules, 2008, 41, 167–174 CrossRef CAS
- Y. Zhao, Y. Cao, Y. L. Yang and C. Wu, Macromolecules, 2003, 36, 855–859 CrossRef CAS
- Z. S. Ge, J. M. Hu, F. H. Huang and S. Y. Liu, Angew. Chem., Int. Ed., 2009, 48, 1798–1802 CrossRef CAS PubMed
- R. X. Liu, M. Fraylich and B. R. Saunders, Colloid Polym. Sci., 2009, 287, 627–643 CAS
- X. Y. Qiao and R. A. Weiss, Abstr. Pap. Am. Chem. Soc., 2012, vol. 243 Search PubMed
- R. A. Weiss, J. J. Fitzgerald and D. Kim, Macromolecules, 1991, 24, 1064–1070 CrossRef CAS
- G. Bokias, D. Hourdet and I. Iliopoulos, Macromolecules, 2000, 33, 2929–2935 CrossRef CAS
- Y. T. Hu, P. Boltenhagen and D. J. Pine, J. Rheol., 1998, 42, 1185–1208 CrossRef CAS
- Y. T. Hu, P. Boltenhagen, E. Matthys and D. J. Pine, J. Rheol., 1998, 42, 1209–1226 CrossRef CAS
- B. Cabane, K. Wong, P. Lindner and F. Lafuma, J. Rheol., 1997, 41, 531–547 CrossRef CAS
- D. C. Pozzo and L. M. Walker, Colloids Surf., A, 2004, 240, 187–198 CrossRef CAS
- A. Lele, A. Shedge, M. Badiger, P. Wadgaonkar and C. Chassenieux, Macromolecules, 2010, 43, 10055–10063 CrossRef CAS
- A. Cadix, C. Chassenieux, F. Lafuma and F. Lequeux, Macromolecules, 2005, 38, 527–536 CrossRef CAS
- S. Suzuki, T. Uneyama, T. Inoue and H. Watanabe, Macromolecules, 2012, 45, 888–898 CrossRef CAS
- J. Wang, L. Benyahia, C. Chassenieux, J.-F. Tassin and T. Nicolai, Polymer, 2010, 51, 1964–1971 CrossRef CAS
- M. Rubinstein and R. H. Colby, Polymer Physics, OUP Oxford, 2003, pp. 403–406 Search PubMed
- B. L. Rivas, S. A. Pooley, M. Soto, H. A. Maturana and K. E. Geckeler, J. Appl. Polym. Sci., 1998, 67, 93–100 CrossRef CAS
- K. Lewandowska, J. Appl. Polym. Sci., 2007, 103, 2235–2241 CrossRef CAS
- C. C. A. Cadix, Synthese et caracterisation des proprietes de polymeres amphiphiles rheo-epaississants, Universite Pierre et Marie Curie, Paris, 2002 Search PubMed
- F. Fischer, D. Zufferey and R. Tahoces, Polym. Int., 2011, 60, 1259–1262 CrossRef CAS
- Q. C. Ying and B. Chu, Macromolecules, 1987, 20, 362–366 CrossRef CAS
- K. Kaushlendra and S. K. Asha, Langmuir, 2012, 28, 12731–12743 CrossRef CAS PubMed
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039–2044 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27030a |
| This journal is © The Royal Society of Chemistry 2017 |