
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CuI promoted sulfenylation of organozinc reagents with arylsulfonyl chlorides†
Ying Fu *,
Yuhu Su,
Qin-shan Xu,
Zhengyin Du,
Yulai Hu,
Ke-Hu Wang and
Danfeng Huang
*,
Yuhu Su,
Qin-shan Xu,
Zhengyin Du,
Yulai Hu,
Ke-Hu Wang and
Danfeng Huang
College of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou, Gansu 730070, P. R. China. E-mail: fuying@iccas.ac.cn; Fax: +86-931-7971989; Tel: +86-931-7971533
First published on 18th January 2017
Abstract
A CuI promoted sulfenylation of organozinc reagents with arylsulfonyl chlorides/PPh3 has been explored. This reaction proceeded smoothly through an alkyl/aryl radical (generated from organometallics) under mild conditions and produced the desired sulfide products in excellent yields.
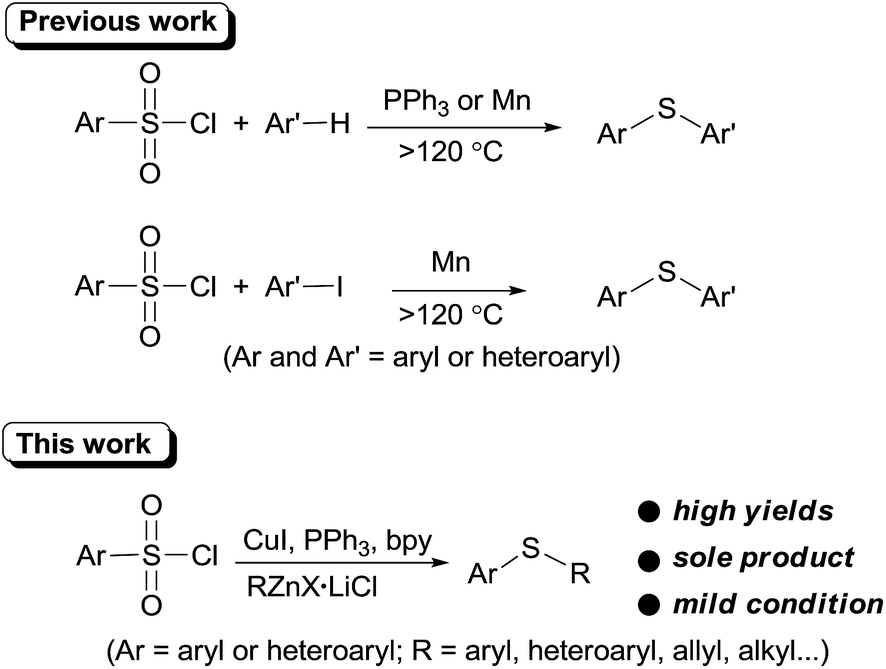
Thioether is a very important structural motif in numerous natural products and bioactive molecules, and is widely used as a versatile building block in organic molecules. As a consequence, novel synthetic protocols have been continuously developed1 and much of recent attention has focused on exploitation environmental friendly, thiol-free materials such as Bunte Salts,2 potassium ethyl xanthogenate,3 NaS2O3,4 KSCN,5 CS2,6 sulfonyl hydrazides,7 DMSO8 and N-(aryl/alkylthio)succinimides9 etc. as the sulfur source.
Recently, arylsulfonyl chloride, considering its abundance and inexpensiveness, has emerged as a promising thiol-free sulfur source in thioether synthesis. In 2011, You et al. first demonstrated that arylsulfonyl chlorides can be employed as sulfur source for sulfenylation of electron-rich arenes or heteroarenes.10 This promising sulfenylation protocol was quickly recognized and was further enlarged in sulfenylation of (hetero)arenes in PEG-400,11 quinones12 and iodoarenes.13 However, these methodologies are limited to diaryl sulfide synthesis and require high temperature.
Organozinc reagents are mild organometallics and were used extensively in organic synthesis.14 However, these privileged organometallics have rarely been employed in C–S bond formation reactions unless a reactive sulfur electrophile such as sulfenyl chlorides15 or SO2 (ref. 16) etc. were introduced as the substrates. Previously, we developed a CuI catalyzed synthesis of arylsulfones from organozinc reagents and arylsulfonyl chlorides.17 During this study, we accidentally found that when PPh3 was employed as the ligand, thioether can be formed unexpectedly. Owing to our interests on the synthesis and application of organozinc reagents in organic synthesis,18 we here report an CuI promoted sulfenylation of organozinc reagents employing commercially available arylsulfonyl chlorides as the sulfur source under mild reaction (Scheme 1).
 | ||
| Scheme 1 Approaches toward transformation of sulfonyl chlorides into thioethers. | ||
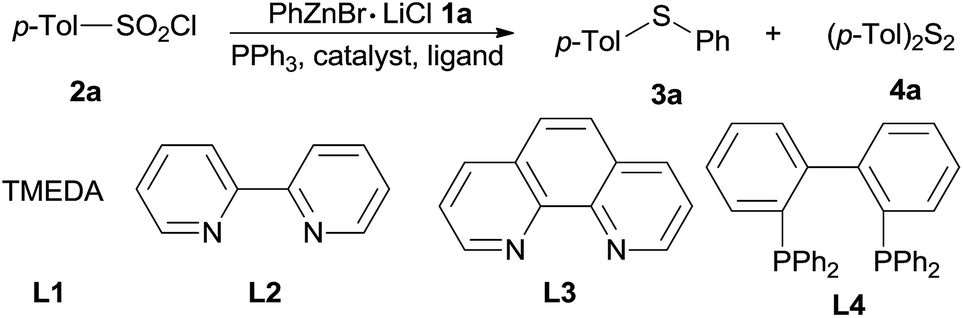
At the outset of this investigation, optimization of the reaction parameters was performed using phenylzinc bromide 1a and p-tolylsulfonyl chloride 2a as the model substrates (Table 1). When the reaction was conducted by adding p-tolylsulfonyl chloride 2a into phenylzinc bromide 1a in the presence of CuI (1.0 equiv.) and PPh3 (2.2 equiv.) in THF at room temperature, phenyl p-tolyl sulfone was formed in 68% isolated yield without formation of any sulfides product. Alternatively, when phenylzinc bromide 1a was added into a mixture of p-tolylsulfonyl chloride 2a and PPh3 (2.2 equiv.) in the presence of CuI (1.0 equiv.) in THF at room temperature, p-tolyl disulfide 4a was obtained in nearly quantitative yield (95%, entry 1). The appearance of disulfide 4a can be ascribed to immediate reduction of p-tolylsulfonyl chloride 2a by PPh3.19 However, this experimental result also illustrates the fact that organozinc reagents are inert organometallic species towards organodisulfides. To further improve the reactivity of organozinc reagents, two equivalents of TMEDA (tetramethylethylenediamine, L1) was added and heating to reflux overnight, the yield of 3a was improved to 35% (entry 2). Replacement of CuI with other cuprous salts such as CuBr, CuCl, CuCN and Cu(OAc)2 are all effective, albeit without obvious yield improvement (entries 3–6). Ligands L1–L4 screening showed that bipyridine (L2) was the best one, enhancing the yield of 3a to 63% (entries 7–9). Organozinc reagents exhibit enhanced reactivity in a polar aprotic solvent, e.g. DMF.20 Gratifyingly, the use of THF–DMF (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) as a solvent dramatically improved the yield of 3a to 88% yield and disulfides 4a was cleanly consumed at room temperature after 12 hours (entry 10).
:1, v/v) as a solvent dramatically improved the yield of 3a to 88% yield and disulfides 4a was cleanly consumed at room temperature after 12 hours (entry 10).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Cat./ligand | Temp. | 3ab [%] | 4ab [%] |
| a Reaction conditions: 1a (2 mmol) in THF (4 mL) was added into a THF (4 mL) solution containing catalyst (1.0 mmol), ligand (2.0 mmol), 2a (1.0 mmol) and PPh3 (2.2 mmol) under argon atmosphere and was then stirred overnight.b Isolated yields.c 1.0 mL of DMF was added. | |||||
| 1 | THF | CuI/— | RT | 0 | 95 |
| 2 | THF | CuI/L1 | Reflux | 35 | 61 |
| 3 | THF | CuBr/L1 | Reflux | 29 | 61 |
| 4 | THF | CuCl/L1 | Reflux | 32 | 62 |
| 5 | THF | CuCN/L1 | Reflux | 28 | 64 |
| 6 | THF | Cu(OAc)2/L1 | Reflux | 31 | 67 |
| 7 | THF | CuI/L2 | Reflux | 63 | 31 |
| 8 | THF | CuI/L3 | Reflux | 56 | 32 |
| 9 | THF | CuI/L4 | Reflux | 42 | 51 |
| 10 | THF/DMF | CuI/L2 | RT | 88c | 0 |
Although classic reactive organometallics such as Grignards,21 organolithium reagents22 and some mild organometallic species such as aryltrimethoxysilanes23 and triarylbismuthanes24 are able to convert disulfides into sulfides, there are some apparent problems relating to these protocols. Grignards and organolithium reagents will simultaneously cleave both C–S and S–S bonds of disulfides,25 thus are limited in practical sulfides synthesis. On the other hand, triarylbismuthanes26 and aryltrimethoxysilanes27 themselves were prepared from corresponding Grignards, therefore, precludes existence of some important functional groups on these reagents. In this respect, our organozinc protocol28 is advantageous both on their structural diversity and wide spectrum of functional groups tolerance.
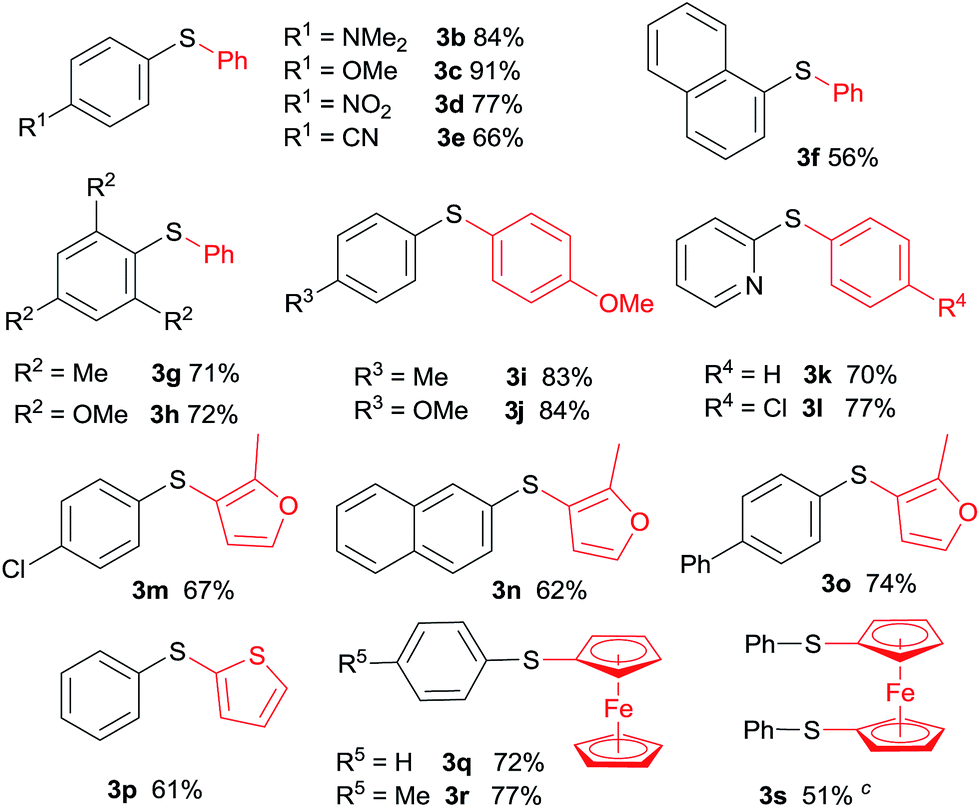
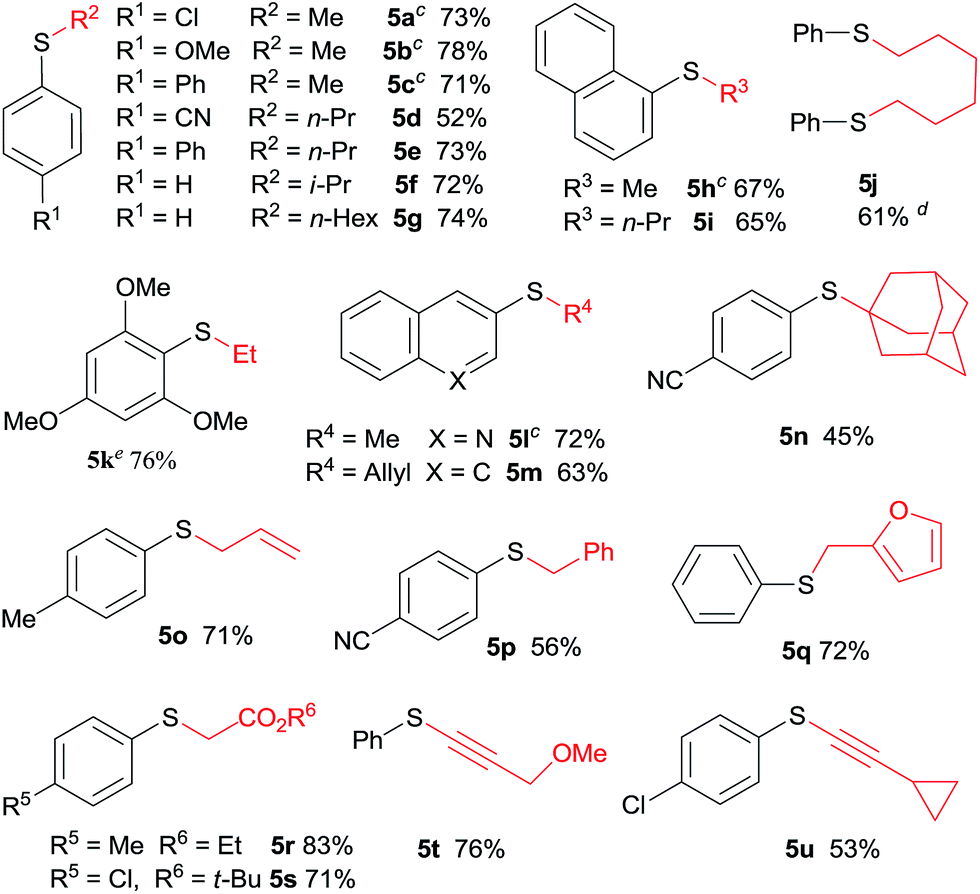
The scope and generality of this CuI promoted sulfenylation of various aryl and heteroarylzinc bromides with aromatic sulfonyl chlorides/PPh3 couple was investigated under the optimized conditions (Table 2). To PhZnBr·LiCl 1a, arylsulfonyl chlorides containing either electron-donating or electron-withdrawing groups were smoothly converted into diaryl sulfides (3a–h) in good to excellent yields. A variety of important functional groups, including nitro (3d) and cyano (3e) were well tolerated under this optimized reaction conditions. The steric hindrance effect of this reaction was not obvious; 2,6-disubstituted arylsulfonyl chlorides could participate this transformation, giving the sulfide products (3g, 3h) in good yields. Heteroaromatic sulfides containing furan (3m–3o), thiophene (3p) and ferrocene (3q–3s) moieties can be easily prepared from corresponding organozinc bromides in good isolated yields.
|
|---|
| a Reaction conditions: 1 (2 mmol) in THF (4 mL) was added into a THF–DMF (5 mL, 4:1, v/v) solution containing CuI (1.0 mmol), bpy (2.0 mmol), 2a (1.0 mmol) and PPh3 (2.2 mmol) under argon atmosphere and was then stirred at room temperature overnight.b Isolated yields.c Biszincation of ferrocene (1.0 mmol) was performed using n-butyllithium (2.2 mmol) and ZnCl2 (2.0 mmol). |
 |
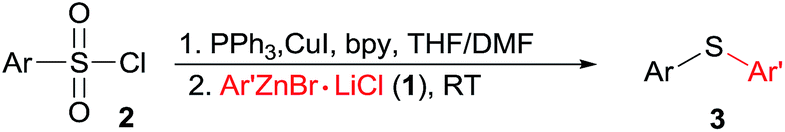
The application of aliphatic organozinc reagents with various aromatic sulfonyl chlorides was then investigated (Table 3). Gratifyingly, a range of aliphatic organozinc reagents, both commercial available dialkylzinc reagents (5a–c, 5h, 5k and 5l) and aliphatic organozinc halides, prepared by ZnCl2 mediated transmetalation of Grignards or organolithium reagents, were compatible with the developed conditions. Notably, adamantyl thioether 5n was also successfully prepared using adamantylzinc bromide in 45% yield. Allylic and benzylic thioethers (5m, 5o–5q) were easily prepared using allyl or benzylzinc bromides in good yields. Also of note are thioethers 5r and 5s were readily prepared in high yields via Reformasky type reaction. Zinc acetylides (in situ formed via zincation of terminal akynes by Et2Zn) were successfully employed to form alkynylsulfides in good yields (5t and 5u).
|
|---|
| a 1 (2 mmol) in THF (4 mL) was added into a THF–DMF (5 mL, 4:1, v/v) solution containing CuI (1.0 mmol), bpy (2.0 mmol), 2a (1.0 mmol) and PPh3 (2.2 mmol) under argon atmosphere and was then stirred at room temperature overnight.b Isolated yields.c Me2Zn (4 mmol) was used.d 1,6-Dibromozinc hexane (0.5 mmol) was used.e Et2Zn (4 mmol) was used. |
 |
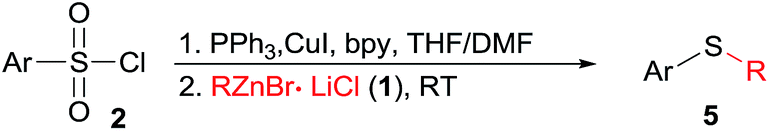
The halogen/magnesium exchange reaction29 and the direct deprotonative metalation30 of arenes or heteroarenes with strong bases are now recognized as powerful method for highly functionalized organometallic reagents preparation. In this respect, iodobenzene bearing an electron-withdrawing CF3 substituent found difficulty in direct magnesium insertion.31 Nevertheless, treatment of 4-iodotrifluorobenzene 6 by turbo Grignards (i-PrMgCl·LiCl)32 and then transmetalated with ZnCl2 afforded the corresponding organozinc reagents, which reacted with 4-methoxybenzenesulfonyl chloride/PPh3, giving sulfide 7a in 74% yield (Scheme 2). Similarly, dimethyl 4,4′-thiodibenzoate 7b was prepared in 82% yield. Furthermore, zincation of benzo[d]oxazole 8 with TMPZnCl·LiCl33 and then reaction with 3,4,5-trimethoxybenzenesulfonyl chloride/PPh3 couple yielded the sulfide 9 in 71% yield.
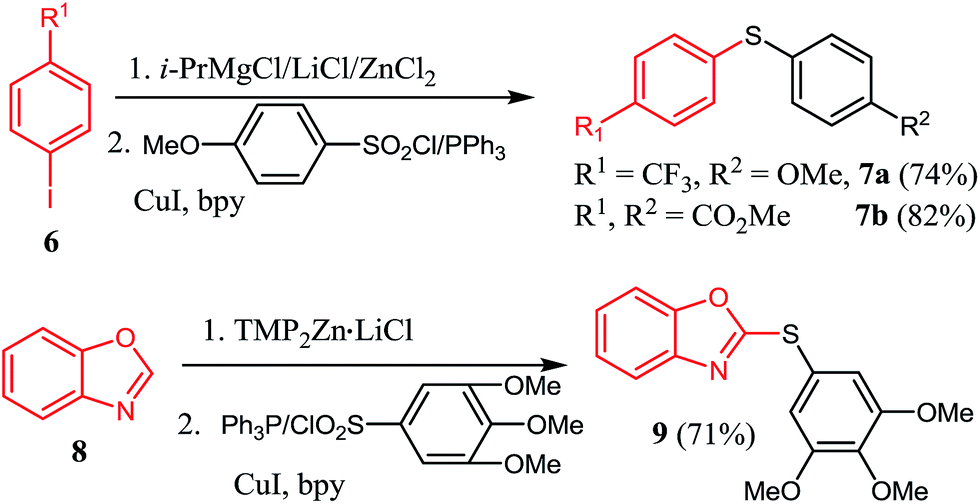 | ||
| Scheme 2 Reaction of organozinc reagents delivered via Mg/I exchange reaction and deprotonation method. | ||
To illustrate a possible mechanism for this transformation, some control experiments were conducted (Scheme 3). When p-tolylsulfonyl chloride 2a was treated with PhZnBr·LiCl 1a at the same reaction without PPh3, sulfone 10 was obtained in 63% isolated yield and sulfide 3a was not detected at all (Scheme 3a), indicating that PPh3 was the only reductant. However, PPh3 could not reduce sulfone 10 at room temperature in THF/DMF (5:1, v/v). When diphenyl disulfide 11 (1 mmol) instead of phenylsulfonyl chloride/PPh3 was used in reaction with 4-methoxyphenylzinc bromide 1b, sulfide 3c was obtained in 94% isolated yield, indicating that diaryl disulfide in situ formed by reduction of sulfonyl chlorides and PPh3 were the reactive intermediates (Scheme 3b). Interestingly, treatment of 4-chlorophenylzinc iodide 1c (1 equiv.) with mixed disulfide 12 (1 equiv.) gave sulfides 3m and 5a in exactly 1:1 ratio along with quantitative remaining of 12 according to crude 1H NMR analysis, addressing a radical mechanism of this reaction as in a nucleophilic displacement reaction, 3m and 5a will be formed in different ratio owing to the unsymmetric structure nature of the mixed disulfide 12. The significant difference between organozinc reagents and Grignard reagents was also highlighted here as Grignards normally nucleophilically cleave S–S bond of disulfides and leaving another part of disulfide as thiol by-product. Furthermore, when a radical scavenger, 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO, 2 equivalent) was added into the sulfenylation reaction of 4-methoxyphenylzinc bromide 1b (2 equiv.) with phenylsulfonyl chloride (1 equiv.), sulfide 3c was not produced (Scheme 3d). Meanwhile, adduct (13) of the thiyl radical34 with TEMPO was also not obtained. TEMPO was totally decomposed by organozinc reagents, leaving disulfide 4a untouched, whereas adduct (14) of 1b with TEMPO was obtained in 23% yield (GC-MS analysis).
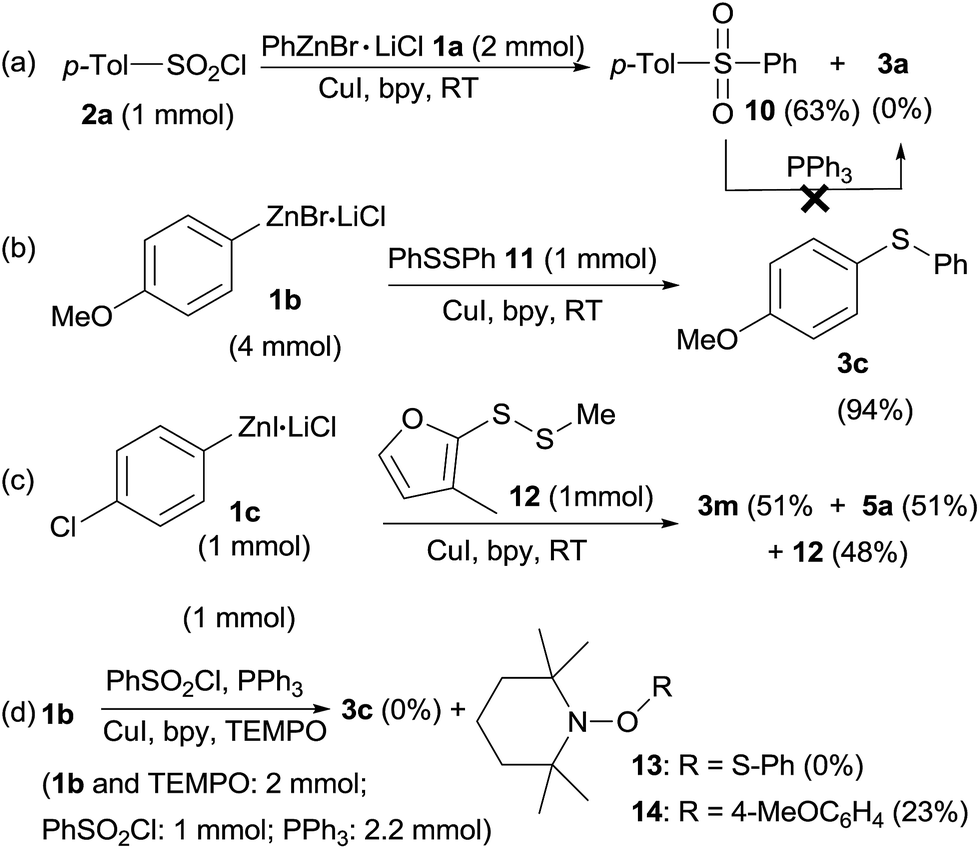 | ||
| Scheme 3 Some control experiments. | ||
Based on aforementioned experimental facts, a plausible mechanism was proposed (Scheme 4). Arylsulfonyl chloride (I) was reduced by PPh3 to give diaryl disulfide (II).19 Transmetalation of RZnX with CuI gave the organocopper reagents RCu35 which underwent a homolytic dissociation to generate a R˙ radical.36 It should be noted here that R˙ radical can also be generated from organozinc reagents in presence of trace amount of oxygen.37 Disulfide (II) captured R˙ radical to form thioether (III). Meanwhile, a thiyl radical (IV) was produced which either underwent homocoupling to regenerate the disulfide (II) or was captured by another R˙ radical to give thioether (III).
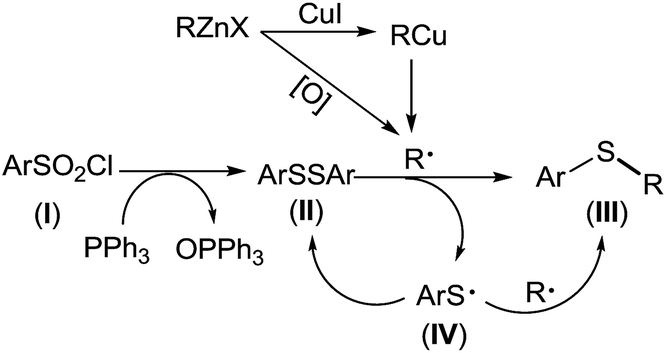 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusion
In summary, we have developed an efficient and practical method for the preparation of aromatic sulfides based on CuI promoted reaction of organozinc reagents with aromatic sulfonyl chlorides. This reaction initiated via a alkyl/aryl radical generated from organozinc reagents rather than thiyl radical generated from diaryl disulfides. A plausible reaction mechanism has been given on the basis of the control experiments.Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (No. 21262030, 20962017).References and Notes
- For recent reviews: (a) A. Ghaderi, Tetrahedron, 2016, 72, 4758 CrossRef CAS; (b) X.-Q. Pan, J.-P. Zou, W.-B. Yi and W. Zhang, Tetrahedron, 2015, 71, 7481 CrossRef CAS; (c) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. Andy Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291 RSC; (d) X. Zhu and S. Chiba, Chem. Soc. Rev., 2016, 45, 4504 RSC.
- J. T. Reeves, K. Camara, Z. S. Han, Y. Xu, H. Lee, C. A. Busacca and C. H. Senanayake, Org. Lett., 2014, 16, 1196 CrossRef CAS PubMed.
- D. J. C. Prasad and G. Sekar, Org. Lett., 2011, 13, 1008 CrossRef CAS PubMed.
- (a) Z. Qiao, J. Wei and X. Jiang, Org. Lett., 2014, 16, 1212 CrossRef CAS PubMed; (b) X.-Q. Chu, X.-P. Xu and S.-J. Ji, Chem.–Eur. J., 2016, 22, 14181 CrossRef CAS PubMed; (c) A. Rostami, A. Rostami and A. Ghaderi, J. Org. Chem., 2015, 80, 8694 CrossRef CAS PubMed.
- M. Cai, R. Xiao, T. Yan and H. Zhao, J. Organomet. Chem., 2014, 749, 55 CrossRef CAS.
- (a) P. Zhao, H. Yin, H. Gao and C. Xi, J. Org. Chem., 2013, 78, 5001 CrossRef CAS PubMed; (b) H. Firouzabadi, N. Iranpoor and A. Samadi, Tetrahedron Lett., 2014, 55, 1212 CrossRef CAS.
- (a) N. Singh, R. Singh, D. S. Raghuvanshi and K. Nand Singh, Org. Lett., 2013, 15, 5874 CrossRef CAS PubMed; (b) F.-L. Yang, F.-X. Wang, T.-T. Wang, Y.-J. Wang and S.-K. Tian, Chem. Commun., 2014, 50, 2111 RSC.
- F. Luo, C. Pan, L. Li, F. Chen and J. Cheng, Chem. Commun., 2011, 47, 5304 RSC.
- T. Hostier, V. Ferey, D. G. Pardo and J. Cossy, Org. Lett., 2015, 17, 3898 CrossRef CAS PubMed.
- Q. Wu, D. Zhao, X. Qin, J. Lana and J. You, Chem. Commun., 2011, 47, 9188 RSC.
- D. Wang, S. Guo, R. Zhang, S. Lin and Z. Yan, RSC Adv., 2016, 6, 54377 RSC.
- X. Yu, Q. Wu, H. Wan, Z. Xu, X. Xu and D. Wang, RSC Adv., 2016, 6, 62298 RSC.
- Y. Wang, X. Zhang, H. Liu, H. Chen and D. Huang, Org. Chem. Front., 2017, 4, 31 RSC.
- (a) Z. Rappoport and I. Marek, The Chemistry of Organozinc Compounds: R-Zn, Wiley, Chichester, UK, 2006 Search PubMed; (b) P. Knochel and P. Jones, Organozinc Reagents: A Practical Approach, Oxford University Press, Oxford, 1999 Search PubMed; (c) E. Erdik, Organozinc Reagents in Organic Synthesis, CRC, New York, 1996 Search PubMed; (d) A. D. Dilman and V. V. Levin, Tetrahedron Lett., 2016, 57, 3986 CrossRef CAS.
- I. M. Yonova, C. A. Osborne, N. S. Morrissette and E. R. Jarvo, J. Org. Chem., 2014, 79, 1947 CrossRef CAS PubMed.
- (a) B. N. Rocke, K. B. Bahnck, M. Herr, S. Lavergne, V. Mascitti, C. Perreault, J. Polivkova and A. Shavnya, Org. Lett., 2014, 16, 154 CrossRef CAS PubMed; (b) N. Margraf and G. Manolikakes, J. Org. Chem., 2015, 80, 2582 CrossRef CAS PubMed.
- Y. Fu, W. Zhu, X. Zhao, H. Hügel, Z. Wu, Y. Su, Z. Du, D. Huang and Y. Hu, Org. Biomol. Chem., 2014, 12, 4295 CAS.
- (a) Y. Fu, Y. Liu, Y. Chen, H. M. Hügel, M. Wang, D. Huang and Y. Hu, Org. Biomol. Chem., 2012, 10, 7669 RSC; (b) Y. Fu, X. Hu, Y. Chen, Y. Yang, H. Hou and Y. Hu, Synthesis, 2012, 44, 1030 CrossRef CAS; (c) Y. Fu, X.-L. Zhao, H. Hügel, B. Hou, D. Huang and Z. Du, Curr. Org. Chem., 2015, 19, 2324 CrossRef CAS; (d) Y. Fu, X. L. Zhao, H. Hügel, D. Huang, Z. Du, K. Wang and Y. Hu, Org. Biomol. Chem., 2016, 14, 9720 RSC.
- (a) G. W. Kabalka, M. S. Reddy and M.-L. Yao, Tetrahedron Lett., 2009, 50, 7340 CrossRef CAS; (b) D. Wan, Y. Yang, X. Liu, M. Li, S. Zhao and J. You, Eur. J. Org. Chem., 2016, 55 CrossRef CAS.
- K. Kobayashi and Y. Kondo, Org. Lett., 2009, 11, 2035 CrossRef CAS PubMed.
- (a) H. Wuyts, Bull. Soc. Chim. Fr., 1906, 166 CAS; (b) A. J. Parker and N. Kharasch, Chem. Rev., 1959, 59, 583 CrossRef CAS; (c) S. Munavalli, D. I. Rossman, D. K. Rohrbaugh and C. P. Ferguson, J. Fluorine Chem., 1993, 61, 147 CrossRef CAS.
- S. Munavalli, D. I. Rossman, D. K. Rohrbaugh and C. P. Ferguson, J. Fluorine Chem., 1993, 61, 155 CrossRef CAS.
- P.-S. Luo, M. Yu, R.-Y. Tang, P. Zhong and J.-H. Li, Tetrahedron Lett., 2009, 50, 1066 CrossRef CAS.
- S. Yasuike, M. Nishioka, N. Kakusawa and J. Kurita, Tetrahedron Lett., 2011, 52, 6403 CrossRef CAS.
- D. I. Rossman, D. K. Rohrbaugh, C. Parker Ferguson and L. J. Szafraniec, J. Fluorine Chem., 1992, 59, 91 CrossRef.
- Y. Matano, T. Miyamatsu and H. Suzuki, Organometallics, 1996, 15, 1951 CrossRef CAS.
- A. S.-Y. Lee, Y.-T. Chang, S.-F. Chu and K.-W. Tsao, Tetrahedron Lett., 2006, 47, 7085 CrossRef CAS.
- S. S. Ashirbaev, V. V. Levin, M. I. Struchkova and A. D. Dilman, J. Fluorine Chem., 2016, 191, 143 CrossRef CAS.
- (a) A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 3333 CrossRef CAS PubMed; (b) R. L. Y. Bao, R. Zhao and L. Shi, Chem. Commun., 2015, 51, 6884 RSC.
- (a) B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794 CrossRef CAS PubMed; (b) D. Seyferth, Organometallics, 2009, 28, 2 CrossRef CAS.
- J. L. Leazer Jr, R. Cvetovich, F.-R. Tsay, U. Dolling, T. Vickery and D. Bachert, J. Org. Chem., 2003, 68, 3695 CrossRef PubMed.
- D. Hauk, S. Lang and A. Murso, Org. Process Res. Dev., 2006, 10, 733 CrossRef CAS.
- D. Haas, M. Mosrin and P. Knochel, Org. Lett., 2013, 15, 6162 CrossRef CAS PubMed.
- (a) X. Zhu, P. Li, Q. Shi and L. Wang, Green Chem., 2016, 18, 6373 RSC; (b) X. Zhu, X. Xie, P. Li, J. Guo and L. Wang, Org. Lett., 2016, 18, 1546 CrossRef CAS PubMed.
- P. Knochel and R. D. Singer, Chem. Rev., 1993, 93, 2117 CrossRef CAS.
- (a) N. J. Rijs, N. J. Brookes, R. A. J. O'Hair and B. F. Yates, J. Phys. Chem. A, 2012, 116, 8910 CrossRef CAS PubMed; (b) H. Gilman, R. G. Jones and L. A. Woods, J. Org. Chem., 1952, 17, 1630 CrossRef CAS.
- F. Denes, S. Cutri, A. Perez-Luna and F. Chemla, Chem.–Eur. J., 2006, 12, 6506 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27201k |
| This journal is © The Royal Society of Chemistry 2017 |