
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Wentinoids A–F, six new isopimarane diterpenoids from Aspergillus wentii SD-310, a deep-sea sediment derived fungus†
Xin Li‡
ab,
Xiao-Dong Li‡ab,
Xiao-Ming Lia,
Gang-Ming Xua,
Yang Liuab and
Bin-Gui Wang *a
*a
aLaboratory of Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Key Laboratory of Experimental Marine Biology, Institute of Oceanology, Chinese Academy of Sciences, Nanhai Road 7, Qingdao 266071, P. R. China. E-mail: wangbg@ms.qdio.ac.cn
bUniversity of Chinese Academy of Sciences, Yuquan Road 19A, Beijing 100049, P. R. China
First published on 16th January 2017
Abstract
Six new isopimarane-type diterpenoid derivatives, wentinoids A–F (1–6), along with a known congener (7), were identified from a culture of Aspergillus wentii SD-310, a fungus isolated from a deep-sea sediment sample. The structures of these compounds were determined by analysis of spectroscopic data, and their absolute configurations were established by single crystal X-ray analysis or TDDFT-ECD calculations. This is the first time to report the isolation of isopimarane analogues from the fungal species Aspergillus wentii. Among these compounds, wentinoid A (1) possesses a unique 20-acetal and a 7,20-oxa-bridged functionality, while wentinoid B (2) contains an unusual 8,20-lactone-bridged scaffold. Compound 1 exhibited potent inhibitory activities against four plant-pathogenic fungi.
Introduction
Diterpenoids of the isopimarane-type comprise a structurally diverse and functionally noteworthy group of natural products, which are much more widely distributed in plants1–3 than in fungi.4 A number of isopimarane analogues have been proven to act as ecologically and biologically functional substances. Typical examples of isopimarane diterpenoids with ecological-related activities are pedinophyllols, which were isolated from the Chinese liverwort Pedinophyllum interruptum and exhibited germination inhibition for Arabidopsis thaliana seeds.5 Some isopimarane derivatives displayed cytotoxicity,6,7 anti-microbial,8,9 anti-malarial,10 anti-inflammatory,11 and anti-virus12 properties as well as inhibitory activities against acetylcholinesterase,13 α-glucosidase,3 and nitric oxide production.2 As part of our ongoing efforts to discover bioactive metabolites from marine-derived fungi,14–16 we recently focused our attention on Aspergillus wentii SD-310, a fungal strain obtained from a deep-sea sediment sample. As a result, a series of 20-nor-isopimarane derivatives were isolated and identified.15,16 Further work on this fungus has now resulted in the isolation of six new isopimarane-type diterpenoid derivatives, wentinoids A–F (1–6), together with a known compound 7. The planar structures of compounds 1–6 were established on the basis of spectroscopic analysis, and the absolute configurations were confirmed by single-crystal X-ray diffraction analysis or ECD calculations. These compounds represents the first example of isopimarane analogues isolated from the fungal species A. wentii, with wentinoid A (1) belongs to a rarely described class of tetracyclic isopimaranes, which contain an oxa-bridge-ring. In contrast to other related congeners, compound 1 possesses a unique 20-acetal moiety and 7,20-oxa-bridged functionality. Meanwhile, wentinoid B (2) represents a tetracyclic isopimarane and contains an unusual 8,20-lactone-bridged scaffold. The antimicrobial activities of the isolated compounds against 11 human-, and aqua-pathogenic bacteria and seven plant-pathogenic fungi were evaluated. Details of the isolation, structure elucidation, and biological activities of compounds 1–7 are described herein.Results and discussion
The mycelia and culture broth of A. wentii SD-310 were extracted with MeOH and EtOAc, respectively, to obtain two extracts, which were combined and purified by a combination of column chromatography (CC) on Si gel, Lobar LiChroprep RP-18, and Sephadex LH-20, to yield compounds 1–7 (Fig. 1).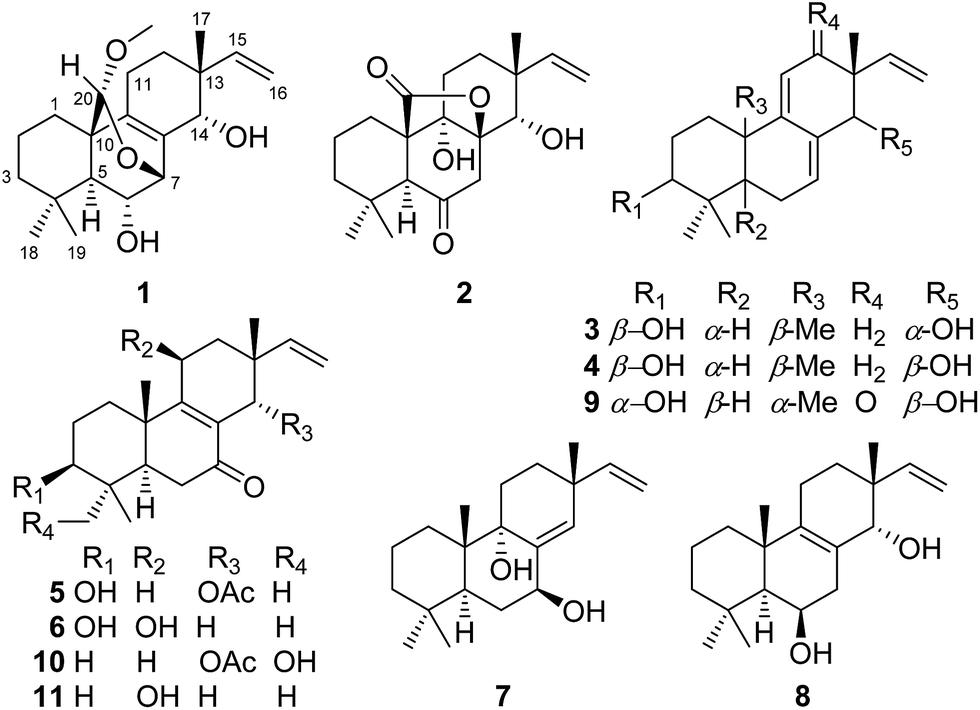 | ||
| Fig. 1 Chemical structures of compounds 1–7 and reference compounds 8–11. | ||
Wentinoid A (1) was initially obtained as colorless oily powder. Its molecular formula was determined to be C21H32O4 by HRESIMS, indicating six degrees of unsaturation. The 1H and 13C NMR, DEPT, and HMQC spectroscopic data (Tables 1 and 2) revealed that 1 contained a tetra-substituted olefin (δC 131.4, C-8; δC 141.6, C-9), a vinyl group (δC 145.8, δH 6.03, dd, J = 17.7, 10.2 Hz, CH-15; δC 111.4, δH 4.98, d, J = 17.7 Hz, 4.96, d, J = 10.2 Hz, CH2-16), an acetal carbon (δC 97.7, C-20), three oxygenated methines (δC/H 68.0/3.68, C-6; δC/H 72.7/4.14, C-7; δC/H 71.1/3.49, C-14), and a methoxy group (δC/H 54.2/3.13, C-21). The protons of methoxy group (H3-21) showed HMBC correlation to C-20, which implied a connection between the methoxy and C-20 (Fig. 2). The attachment of the gem-dimethyl groups (C-18 and C-19) to the quaternary carbon C-4 were identified by their mutual HMBC correlations, along with correlations from H3-18 and H3-19 to C-3, C-4, and C-5. Furthermore, signals for three quaternary carbons, an sp3 methine, five aliphatic methylenes, and a singlet methyl were also observed from the NMR spectra.
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | α 1.49 m | α 1.35 td (13.6, 3.6) | α 1.34 td (12.9, 5.4) | α 1.34 td (12.8, 5.4) | α 1.44 m | α 1.35 td (13.1, 3.6) |
| β 1.86 m | β 1.78 d (13.6) | β 1.78 dt (12.9, 5.4) | β 1.79 dt (12.8, 3.3) | β 1.78 m | β 2.45 m | |
| 2 | 1.45 m | α 1.46 m | 1.54 m | 1.55 m | 1.60 m | α 1.55 m |
| β 1.89 dtt (13.6, 13.6, 3.6) | β 1.61 m | |||||
| 3 | α 1.17 m | α 1.08 td (13.6, 3.6) | 2.99 m | 2.99 m | 3.11 m | 3.05 m |
| β 1.41 m | β 1.30 d (13.6) | |||||
| 5 | 0.75 d (3.5) | 2.84 s | 1.08 dd (11.6, 4.5) | 1.05 dd (11.7, 4.5) | 1.62 dd (14.3, 4.2) | 1.48 m |
| 6 | 3.68 m | — | α 2.13 m | α 2.05 m | α 2.34 dd (18.2, 4.2) | α 2.27 dd (18.6, 3.3) |
| β 2.05 m | β 2.00 m | β 2.43 dd (18.2, 14.3) | β 2.48 dd (18.6, 3.5) | |||
| 7 | 4.14 d (4.2) | α 3.35 d (16.4) | 5.77 brs | 5.70 brs | — | — |
| β 2.28 d (16.4) | ||||||
| 11 | α 2.17 d (17.7) | α 1.69 dt (14.8, 3.6) | 5.26 brs | 5.28 brs | α 2.49 m | 4.38 brs |
| β 1.75 ddd (17.7, 11.6, 5.4) | β 1.50 dd (14.8, 3.6) | β 2.27 m | ||||
| 12 | α 1.67 td (11.6, 5.4) | α 2.08 td (13.6, 3.6) | α 2.20 dd (17.8, 4.3) | α 1.93 dd (18.0, 4.6) | α 1.75 m | α 1.80 dd (12.0, 6.0) |
| β 1.29 m | β 1.18 d (13.6) | β 1.97 dd (17.8, 4.3) | β 2.12 m | β 1.48 m | β 1.46 dd (12.0, 6.0) | |
| 14 | 3.49 s | 3.22 s | 3.71 s | 3.54 s | 5.58 s | α 2.10 d (17.2) |
| β 1.97 d (17.2) | ||||||
| 15 | 6.03 dd (17.7, 10.2) | 5.98 dd (17.5, 11.1) | 5.85 dd (16.9, 10.1) | 5.73 dd (17.6, 10.9) | 5.79 dd (17.2, 11.2) | 5.69 dd (17.5, 10.8) |
| 16 | 4.98 d (17.7) | 4.93 d (17.5) | 4.93 dd (16.9, 1.6) | 4.95 d (17.6) | 4.97 dd (17.2) | 4.88 d (10.8) |
| 4.96 d (10.2) | 4.92 d (11.1) | 4.90 dd (10.1, 1.6) | 4.87 d (10.9) | 4.96 d (11.2) | 4.80 d (17.5) | |
| 17 | 0.87 s | 0.94 s | 0.95 s | 0.91 s | 0.78 s | 0.99 s |
| 18 | 0.94 s | 1.12 s | 0.88 s | 0.88 s | 0.89 s | 0.87 s |
| 19 | 0.92 s | 0.95 s | 0.78 s | 0.78 s | 0.79 s | 0.78 s |
| 20 | 4.80 s | — | 0.88 s | 0.88 s | 1.07 s | 1.25 s |
| 21 | 3.13 s | — | — | — | — | — |
| 22 | — | — | — | — | 1.83 s | — |
| 3-OH | — | — | 4.41 s | 4.42 s | 4.52 d (4.3) | 4.47 brs |
| 6-OH | 4.61 brs | — | — | — | — | — |
| 11-OH | — | — | — | — | — | 4.92 d (6.0) |
| 14-OH | 3.95 brs | — | 4.61 s | 4.56 s | — | — |
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 25.9 CH2 | 24.1 CH2 | 34.4 CH2 | 35.0 CH2 | 33.6 CH2 | 34.6 CH2 |
| 2 | 17.7 CH2 | 16.9 CH2 | 26.8 CH2 | 27.4 CH2 | 27.0 CH2 | 27.0 CH2 |
| 3 | 40.7 CH2 | 41.9 CH2 | 76.3 CH | 76.8 CH | 75.6 CH | 76.1 CH |
| 4 | 32.9 qC | 32.4 qC | 38.0 qC | 38.5 qC | 38.4 qC | 38.5 qC |
| 5 | 54.7 CH | 56.3 CH | 47.5 CH | 48.0 CH | 47.5 CH | 49.8 CH |
| 6 | 68.0 CH | 207.4 qC | 22.5 CH2 | 23.0 CH2 | 34.7 CH2 | 32.7 CH2 |
| 7 | 72.7 CH | 48.1 CH2 | 121.0 CH | 123.8 CH | 196.7 qC | 199.9 qC |
| 8 | 131.4 qC | 85.4 qC | 134.0 qC | 134.7 qC | 127.4 qC | 128.9 qC |
| 9 | 141.6 qC | 74.5 qC | 144.6 qC | 144.4 qC | 170.0 qC | 163.3 qC |
| 10 | 44.0 qC | 53.8 qC | 35.5 qC | 36.0 qC | 39.0 qC | 39.4 qC |
| 11 | 21.3 CH2 | 26.0 CH2 | 113.5 CH | 114.6 CH | 21.4 CH2 | 65.3 CH |
| 12 | 28.1 CH2 | 24.3 CH2 | 35.7 CH2 | 34.2 CH2 | 25.4 CH2 | 44.0 CH2 |
| 13 | 39.1 qC | 39.8 qC | 40.1 qC | 40.3 qC | 38.1 qC | 34.8 qC |
| 14 | 71.1 CH | 72.5 CH | 74.7 CH | 75.1 CH | 67.8 CH | 34.0 CH2 |
| 15 | 145.8 CH | 147.7 CH | 142.4 CH | 145.2 CH | 144.0 CH | 145.5 CH |
| 16 | 111.4 CH2 | 110.4 CH2 | 111.7 CH2 | 111.6 CH2 | 112.4 CH2 | 111.2 CH2 |
| 17 | 20.4 CH3 | 21.5 CH3 | 23.2 CH3 | 21.0 CH3 | 20.9 CH3 | 27.6 CH3 |
| 18 | 33.3 CH3 | 32.4 CH3 | 27.4 CH3 | 27.9 CH3 | 27.2 CH3 | 27.6 CH3 |
| 19 | 22.4 CH3 | 19.1 CH3 | 15.2 CH3 | 15.7 CH3 | 15.3 CH3 | 15.5 CH3 |
| 20 | 97.7 CH | 176.7 qC | 20.6 CH3 | 21.0 CH3 | 18.5 CH3 | 18.5 CH3 |
| 21 | 54.2 CH3 | — | — | — | 168.8 qC | — |
| 22 | — | — | — | — | 20.9 CH3 | — |
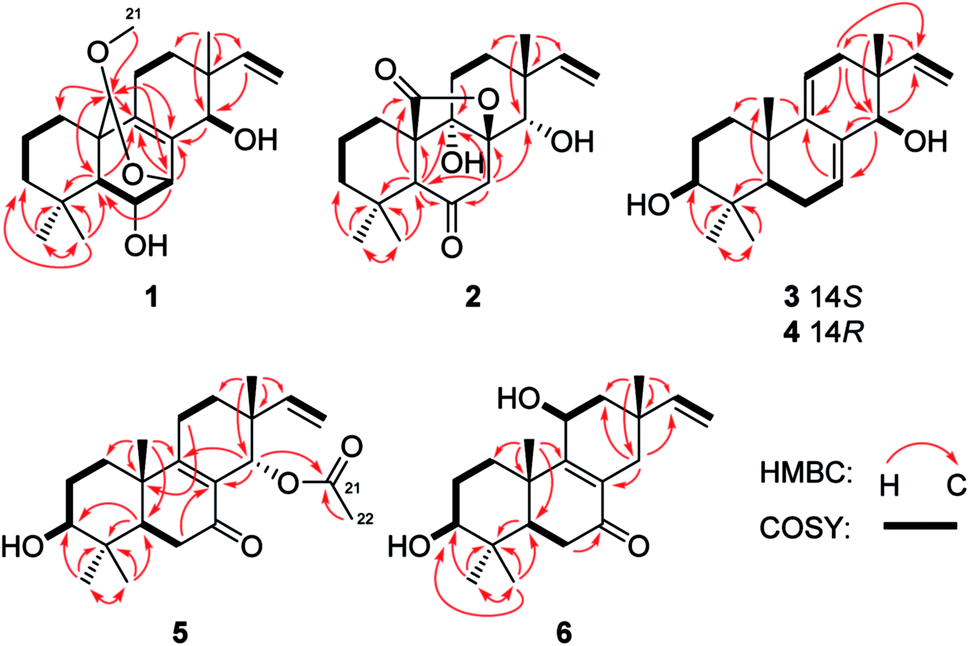 | ||
| Fig. 2 Key COSY and HMBC correlations of compounds 1–6. | ||
Comparison of the NMR data of 1 with that of kaempulchraol C (8),1 an isopimarane-type diterpenoid isolated from rhizomes of Kaempferia pulchra, revealed that the structures of these two compounds are very similar. However, signals for the bridgehead methyl (C-20) and the methylene group (C-7) in 8 were absent in the NMR spectra of 1. Instead, resonances for an acetal carbon (C-20) and an oxygenated methine group (C-7) were observed in the NMR spectra of 1. In addition, C-7 and C-20 connected each other via an oxygen atom, and this deduction was verified by the key HMBC correlations from H-7 to C-5, C-9, and C-20 and from H-20 to C-1, C-5, C-7, and C-9. The planar structure of 1 was fully defined by the HMBC data as shown in Fig. 2.
The relative configuration of 1 was assigned by analysis of the NOESY data (Fig. 3). While NOE correlations from H-5 to H-3α, 6-OH, H-7, H-11α, and H3-18 and from H-15 to H-11α and H-12α indicated the cofacial orientation of these groups, correlations from H-6 to H3-19 and H-20, from H-11β to H3-17, and from H-12β to H-14 placed these groups on the opposite face. To confirm the structure as well as the relative and absolute configurations, crystallization of 1 was performed. After many attempts, single crystals suitable for X-ray analysis were obtained by slow evaporation of a solution of 1 in MeOH. Once the X-ray crystallographic experiment was conducted, the structure and absolute configuration of 1 were confidently assigned as depicted (Fig. 4).
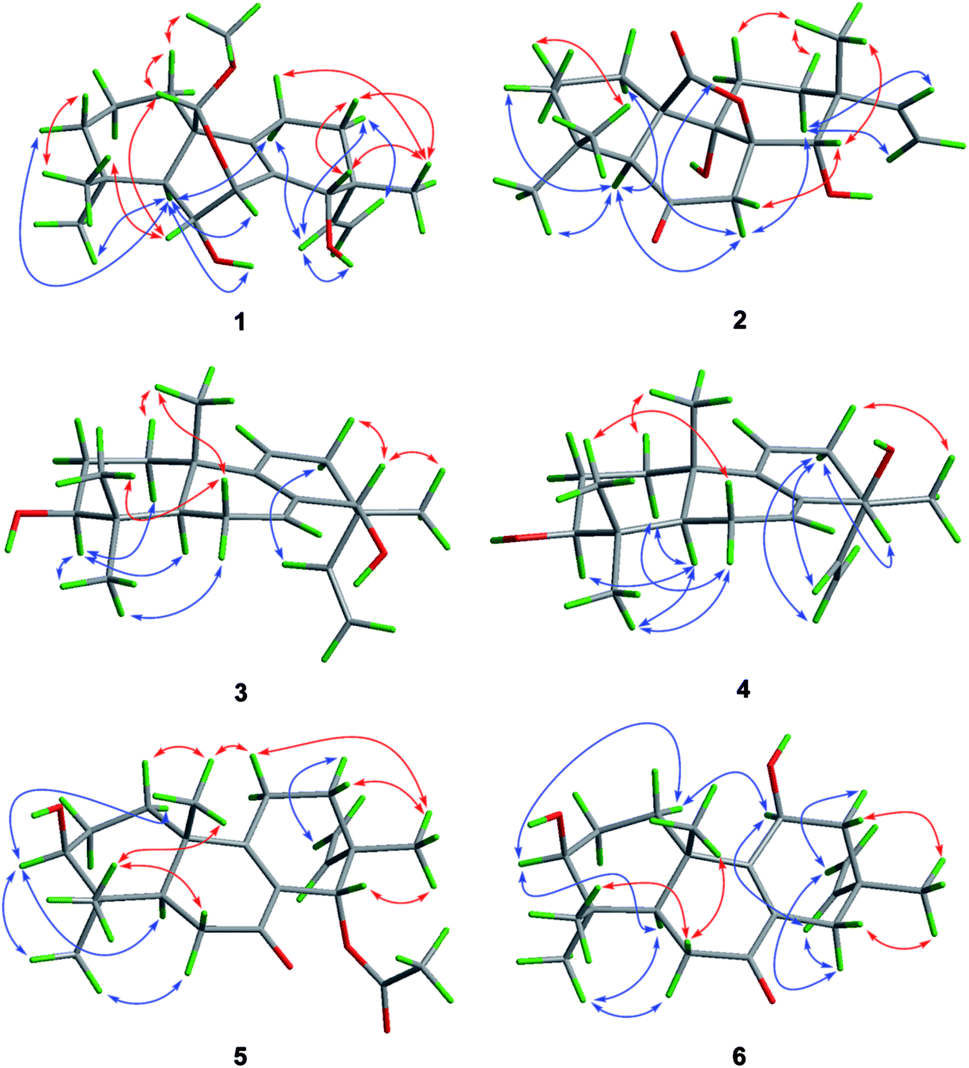 | ||
| Fig. 3 Key NOESY correlations of 1–6. | ||
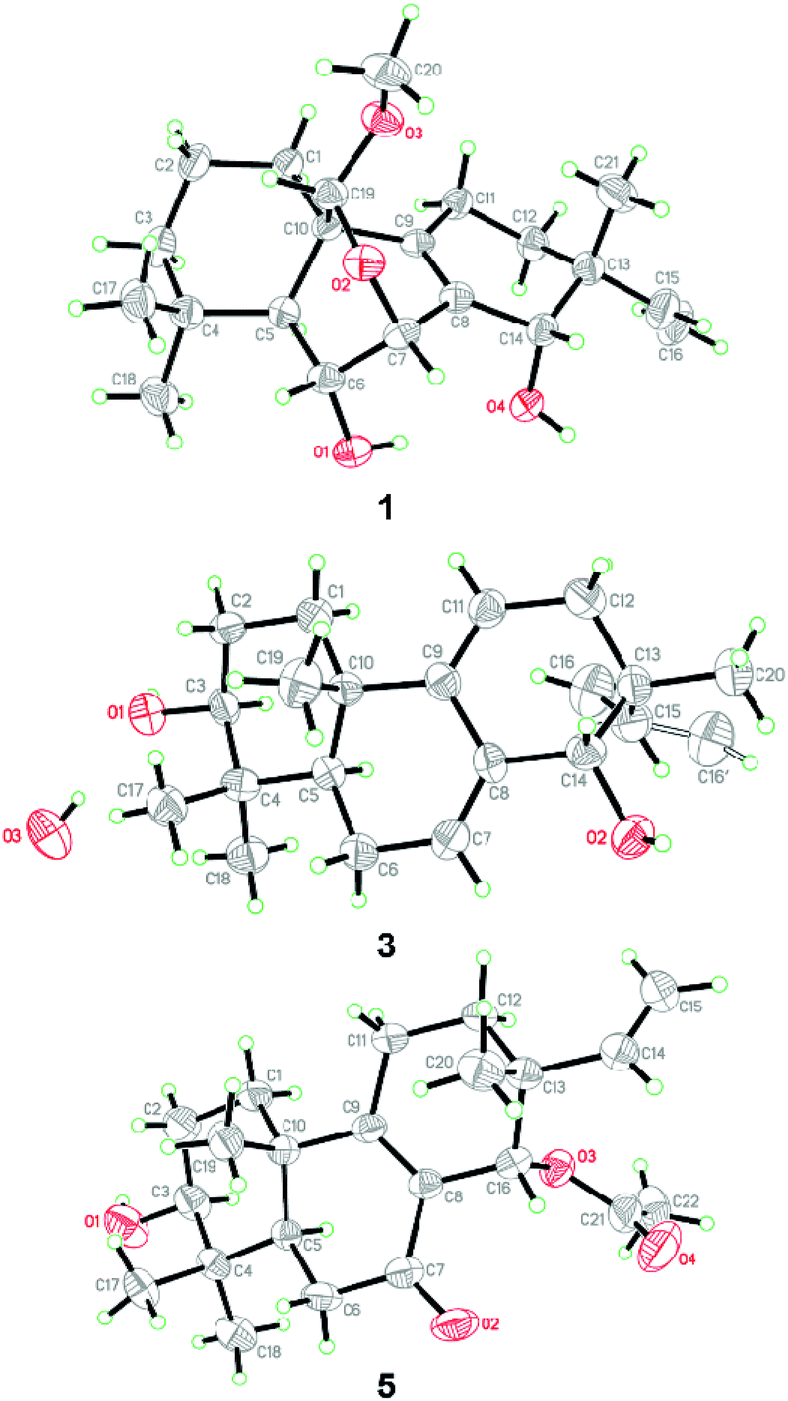 | ||
| Fig. 4 X-ray crystallographic structures of compounds 1, 3, and 5 (note: different numbering systems are used for the structures in the text). | ||
Wentinoid B (2) was obtained as colorless oil. The molecular formula C20H28O5, implying seven degrees of unsaturation, was established by HRESIMS. The 1H NMR spectrum displayed typical signals for a vinyl group (H-15 and H2-16), three singlet methyls (H3-17–H3-19), a pair of methylene protons (H2-7), an oxygenated methine (H-14), an aliphatic methine (H-5), and other ten aliphatic protons (Table 1). Its 13C NMR data (Table 2) exhibited the presence of 20 carbon signals, which were sorted by DEPT and HSQC spectrum into three methyls, seven methylenes (one olefinic), three methines (one oxygenated and one olefinic), and seven quaternary (one lactone, one ketone and one oxygenated aliphatic quaternary) carbons. Interpretation of the COSY and HSQC spectra of 2 resulted in the elucidation of two discrete proton spin-coupling systems corresponding to a –CH2–CH2–CH2– unit (C-1 through C-3) and a –CH2–CH2– unit (C-11 and C-12). Key HMBC correlations shown in Fig. 2 suggested the presence of an isopimarane-type diterpenoid skeleton for 2. The ketone and two hydroxy groups were placed at C-6, C-9 and C-14, respectively, as supported by HMBC correlations from H-5 and H2-7 to C-6 and C-9 and from H2-7 and H3-17 to C-14. The remaining oxygen atom and one degree of unsaturation, together with the consideration of the strongly deshielded nature of C-8 (δC 85.4) suggesting the presence of a lactone bridge between C-8 and C-10 via C-20. Thus, the entire structure of compound 2 was identified as shown in Fig. 1.
The relative configuration of 2 was determined by NOESY experiment and by comparison of 1D NMR data of 2 with related analogues. Key NOE correlations from H-5 to H-7α and H3-18, from H-7α to H-11α and H-12α and from H-12α to H-15 placed these protons on the same face of the molecule. Moreover, NOE correlations from H-7β to H-14 and from H3-17 to H-11β suggested these groups on the opposite face of the molecule. The assignment of the β-orientation of the lactone group was established by analysis of the magnetic anisotropic effect as the method reported by the previous ref. 7. The deshielding effect from the carbonyl at C-20 resulted in downfielded chemical shift of H-2β. The chemical shift of C-9 (δC 74.5) in 13C-NMR was consistent with those of related diterpenoids with an α-OH at C-9 (δC 73.6–75.6).7 In order to determine the absolute configuration of 2, conformational analysis and TDDFT-ECD calculations were performed on the arbitrarily chosen of (5S,8R,9S,10R,13R,14S)-2. The DFT reoptimization of the initial MM + conformers of the selected enantiomer at the B3LYP/6-31G(d) level in gas phase afforded minimum energy conformers. The TDDFT-ECD spectra of the conformers were calculated with B3LYP and the 6-31G(d) basis set. The computed ECD spectra of (5S,8R,9S,10R,13R,14S)-2 matched well to the experimental ECD spectrum, which showed positive cotton effects (CEs) near 228 and 344 nm and negative CE near 299 nm (Fig. 5).
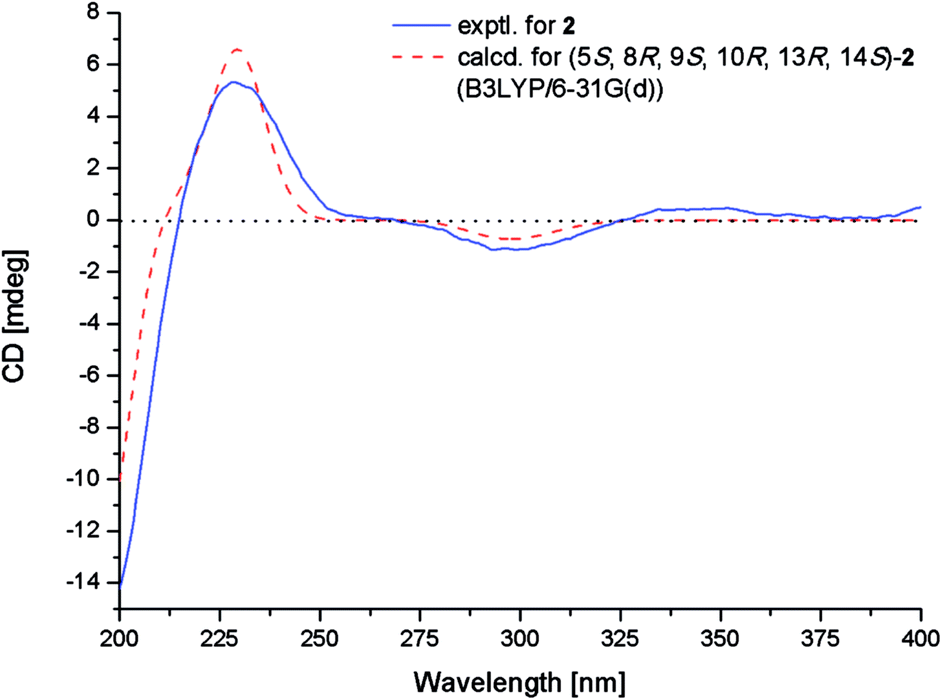 | ||
| Fig. 5 Experimental and calculated ECD spectra of 2. | ||
Wentinoid C (3) was originally obtained as colorless oily powder. The HREIMS data of 3 determined the molecular formula C20H30O2. The 1H and 13C NMR data (Tables 1 and 2) are very similar to those of ent-3β,14α-hydroxypimara-7,9(11),15-triene-12-one (9),17 implying the same carbon skeleton for both compounds. However, resonance at δC 202.2 for the ketone group at C-12 in 9 was not observed in that of 3. Instead, signals for a methylene group at δC 35.7 were found in the 13C-NMR spectra of 3. Compared to 9, obvious upfielded shift for C-11, C-13, and C-15, and downfielded shift for C-17 in the 13C NMR spectrum of 3 were also observed. These data indicated the replacement of a ketone group in 9 by a methylene group in 3, which was consistent with the molecular formula. The COSY correlations from H2-11 to H2-12 as well as key HMBC correlations from H2-12 to C-9, C-14, and C-15 and from H3-17 to C-12 verified the above deduction (Fig. 2).
NOE correlations from H-1β to H3-20, from H-6β to H3-19 and H3-20, and from H-14 to H-12β, H3-17 and H3-20 revealed that these protons are on the same side, while correlations from H-3 to H-1α, H-5, and H3-18 and from H-6α to H3-18 indicated that they are on the other side. To unequivocally determine the absolute configuration, single crystals were cultivated upon slow evaporation of the solvent (MeOH) and a Cu/Kα X-ray diffraction analysis was conducted. The final refinement of the X-ray data resulted in a 0.0(14) Flack parameter, allowing for the unambiguous assignment of the absolute configuration as shown in Fig. 4.
Wentinoid D (4) was obtained as colorless oil. The molecular formula was determined as C20H30O2, same as that of 3, on the basis of HREIMS data. The similar UV absorptions to those of compound 3 implied that 4 was an analogue of 3. The NMR spectra of 4 were also close to that of 3. Inspection of the 1H and 13C NMR data (Tables 1 and 2) suggested that 4 is a diastereomer of 3, epimeric at C-14. This was supported by the minor differences on the chemical shifts for C-7, C-12, C-15, and C-17 in 4, which were from γ-gauche effect caused by the inversion of the absolute configuration at C-14. NOE correlations from H-12β to H3-17 and from H-12α to H-14, H-15, and H2-16 confirmed this deduction (Fig. 3). The structure of 4 was thus assigned as shown in Fig. 1.
Wentinoid E (5), initially obtained as colorless oily powder, had the molecular formula C22H32O4 (seven degrees of unsaturation) as determined by HRESIMS data. Detailed comparison of the NMR data of 5 with those of sorgerolone (10)13 revealed that compound 5 had the same basic structure as 10. However, signals for the methylene group resonating at δC 34.4 and δH 1.35/1.56 (CH2-3) and those of the hydroxymethyl group at δC 70.6 and δH 3.15/3.43 (CH2-18) in 10 disappeared in the NMR spectra of 5. Instead, signals for an oxygenated methine group at δC 75.6 and δH 3.11 (CH-3) and a methyl at δC 27.2 and δH 0.89 (CH3-18) were observed (Tables 1 and 2). The above observation suggested that compound 5 was 3-hydroxy-18-dehydroxy derivative of 10. The cross-peaks from H2-2 to H2-1 and H-3 and from H-3 to 3-OH in the COSY spectrum as well as the correlations from H-5 to C-3, from H3-18 and H3-19 to C-3, C-4, and C-5 in the HMBC spectrum confirmed the above deduction (Fig. 2).
The relative configuration of 5 was deduced from NOESY data. NOE correlations from H-1β to H3-20, from H-11β to H3-17 and H3-20, and from H3-17 to H-14 placed these protons on the same face of the molecule, while H-3, H-5, and H3-18 had α-orientation as confirmed by the correlations from H-3 to H-1α, H-5, and H3-18 (Fig. 3). An X-ray crystallographic experiment confirmed the absolute configuration of 5 as depicted (Fig. 4). The Cu/Kα radiation used for the X-ray diffraction allowed the assignment of the absolute configuration of all of the stereogenic centers in 5 as 3S, 5R, 10S, 13R and 14S.
Wentinoid F (6), was obtained as colorless oil, was assigned the molecular formula C20H30O3 on the basis of HRESIMS data. The 13C NMR spectrum of 6 exhibited signals similar to those presented in 11β-hydroxy-7-oxopimar-8(9),15-dien (11),18 except for the methylene group at δC 35.9 (C-3) in 13C NMR spectrum of 11 was replaced by an oxymethine unit at δC 76.1 (C-3) in that of 6, indicating that 6 was C-3 hydroxylated derivative of 11. The 2D NMR correlations supported this inference by the COSY correlations from H2-2 to H2-1 and H-3 and HMBC correlations from H3-18 and H3-19 to C-3.
The NOE correlations from H-1α to H-3 and H-11, from H-5 to H-3 and H3-18, from H-6α to H3-18, from H-11 to H-15, and from H-14α to H-15 indicated the same orientation of these groups (Fig. 3), whereas correlations from H-6β to H3-20 revealed that these groups were on the other face. The absolute configuration of 6 was established by TDDFT-ECD calculations. The experimental ECD spectrum of 6 matched well with that calculated for (3S,5R,10S,11S,13R)-6 (Fig. 6). The structure and absolute configuration of 6 were thus assigned as shown in Fig. 1.
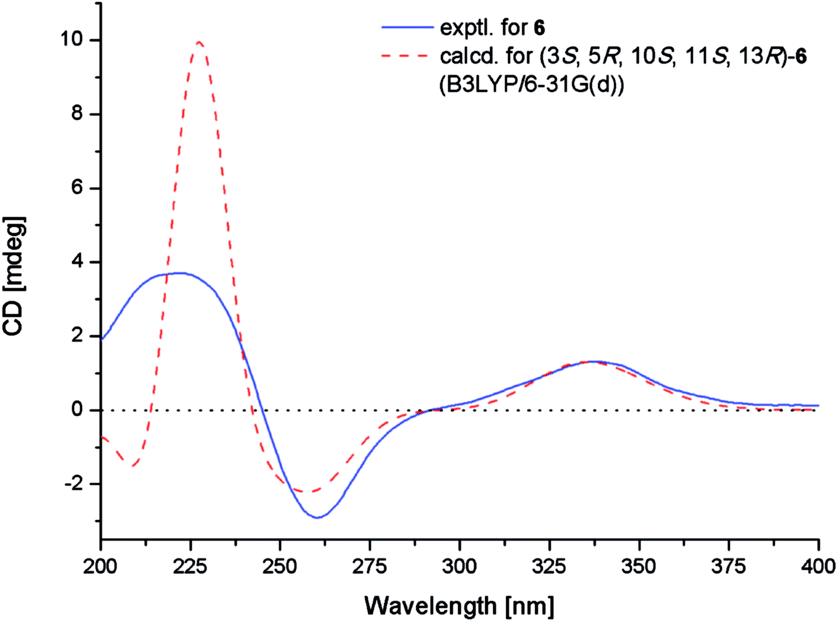 | ||
| Fig. 6 Experimental and calculated ECD spectra of 6. | ||
A plausible biosynthetic pathway for compounds 1–6 is proposed as shown in Scheme 1. In this pathway, compounds 1–6 are produced from geranylgeranylpyrophosphate (GGPP). Intermediate II, produced by reduction of I, is presumed to be a common biosynthetic precursor of compounds 1 and 3–6. After oxidation of II at C-7, C-14, and C-20, C-20 of intermediate III is further oxidized giving aldehyde derivative IV. Aldolization between the aldehyde group at C-20 and 7-OH in IV gives V, and successive methylation of 20-OH yields compound 1. Deprotonation of H-7 and H-11 in II leads to X, which is further oxidized to give isomers 3 and 4. Meanwhile, oxidation of C-3 and C-7 in II gives XI. Further oxidation of C-11 in XI yields 6, whereas oxidation of C-14 in XI gives 5 via XII. After cyclization, GGPP is converted to IX via intermediates VI–IX by multistep oxidation of C-8, C-9, C-14, and C-20. Following esterification between carboxyl at C-20 and 8-OH, compound 2 is formed.
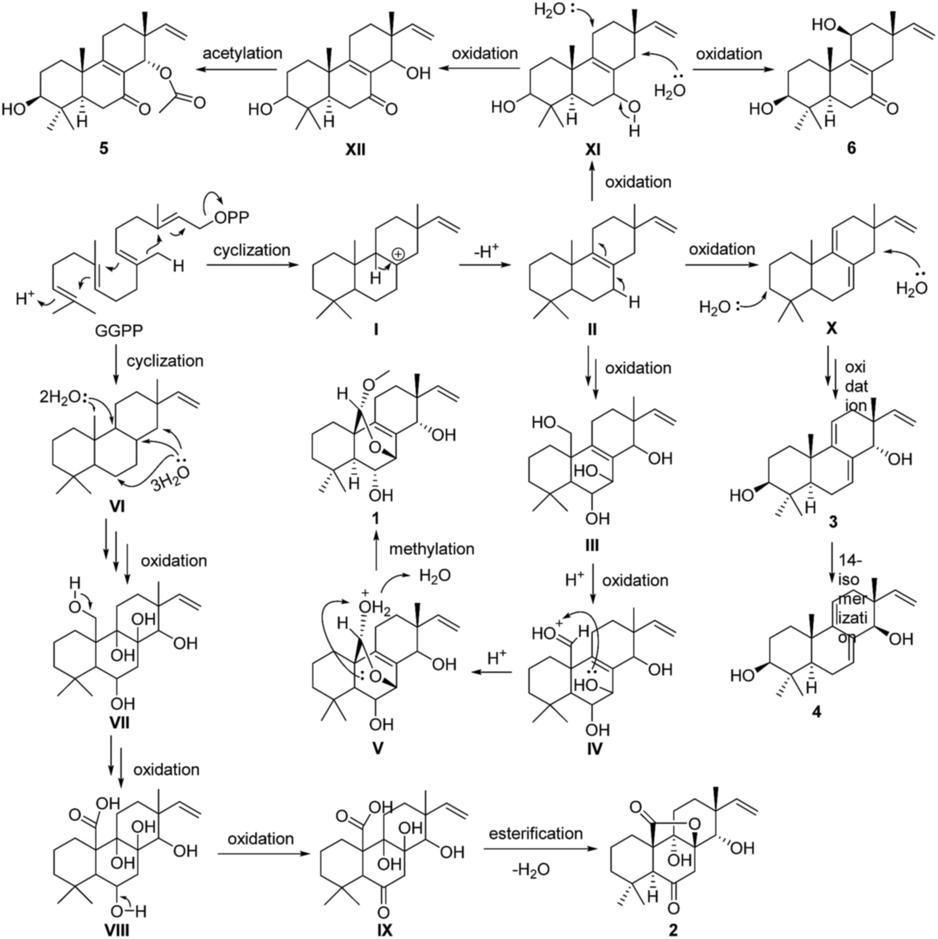 | ||
| Scheme 1 Proposed biosynthetic pathway for compounds 1–6. | ||
The isolated compounds were tested against 11 human-, and aqua-pathogenic bacteria and seven plant-pathogenic fungi. Compound 1 exhibited selective activities against Phytophthora parasitica, Fusarium. oxysporum f. sp. lycopersici, Fusarium graminearum, and Botryosphaeria dothidea with MIC values of 8.0, 4.0, 1.0, and 4.0 μg mL−1, respectively, which were comparable to that of the positive control, amphotericin B (MIC = 2.0, 1.0, 1.0, and 2.0 μg mL−1, respectively). However, other compounds didn‘t displayed potent inhibitory activity.
Conclusions
In summary, we isolated and characterized six new compounds wentinoids A–F (1–6), which are new members of highly oxygenated isopimarane-type diterpenoids. Among them, wentinoid A (1) contains unusual 20-acetal moiety and 7,20-oxa-bridged functionality, whereas wentinoid B (2) possesses a novel 8,20-lactone-bridged scaffold. A plausible biosynthesis mechanism for compounds 1–6 was proposed. The discovery of these compounds might provide further insight into the biosynthesis of isopimarane family and also provide new targets for synthetic or biosynthetic studies. Compound 1, which may prove useful as antifungal agent, exhibited potent antimicrobial activities against some plant pathogenic fungi.Experimental section
General experimental procedures
Melting points were determined with an SGW X-4 micromelting-point apparatus. Optical rotations were measured on an Optical Activity AA-55 polarimeter. UV spectra were measured on a PuXi TU-1810 UV-visible spectrophotometer. CD spectra were acquired on a JASCO J-715 spectropolarimeter. 1D and 2D NMR spectra were recorded at 500 and 125 MHz for 1H and 13C, respectively, on a Bruker Avance 500 MHz spectrometer with TMS as internal standard. Mass spectra were obtained on a VG Autospec 3000 or an API QSTAR Pulsar 1 mass spectrometer. Analytical and semi-preparative HPLC were performed using a Dionex HPLC system equipped with a P680 pump, an ASI-100 automated sample injector, and a UVD340U multiple wavelength detector controlled by Chromeleon software (version 6.80). Commercially available Si gel (200–300 mesh, Qingdao Haiyang Chemical Co.), Lobar LiChroprep RP-18 (40–63 μm, Merck), and Sephadex LH-20 (Pharmacia) were used for open column chromatography. All solvents were distilled prior to use.Fungal material
The isolation and identification of the fungal material were identical to those described in our previous report.15 The strain is preserved at the Key Laboratory of Experimental Marine Biology, Institute of Oceanology of the Chinese Academy of Sciences (IOCAS), with accession number SD-310.Cultivation
For chemical investigations, the fungal strain was dynamic fermented at a 500 L fermentator preloaded with 300 L of sterilized liquid medium containing 50% (v/v) sea water (20% potato, 2% glucose, 0.5% peptone, and 0.3% yeast extract, pH 6.0) for 7 days at room temperature.Extraction and isolation
The whole fermented cultures were filtered to separate the broth from the mycelia. The former was extracted three times with EtOAc, while the latter was extracted three times with a mixture of acetone and water (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20, v/v). The acetone solution was evaporated under reduced pressure to afford an aqueous solution, which was then extracted three times with EtOAc. Since the TLC and HPLC profiles of the two EtOAc solutions from the broth and mycelia were almost identical, they were combined and concentrated under reduced pressure to give an extract (34.7 g) for further separation.
:20, v/v). The acetone solution was evaporated under reduced pressure to afford an aqueous solution, which was then extracted three times with EtOAc. Since the TLC and HPLC profiles of the two EtOAc solutions from the broth and mycelia were almost identical, they were combined and concentrated under reduced pressure to give an extract (34.7 g) for further separation.
The combined extract was fractionated by silica gel vacuum liquid chromatography (VLC) using a stepwise gradient of a mixture of petroleum ether (PE)-ethyl acetate (EtOAc) (1:0, 50:1, 20:1, 5:1, 2:1 and 1:1) and CH2Cl2–MeOH (20:1, 10:1, 5:1 and 0:1) to yield 10 major primary fractions (Fr.1–Fr.10). Fr.4 (7.1 g) was separated by CC on Lobar LiChroprep C18 eluting with MeOH–H2O gradient to give nine subfractions (Frs. 4.1–4.9). Further purification of Fr. 4.4 by CC on silica gel with a CH2Cl2–MeOH gradient (from 50:1 to 5:1) yielded Frs. 4.4.1–4.4.9, and then Fr. 4.4.3 was purified by CC on silica gel eluted with PE-EtOAc 5:1 to afford compounds 3 (10.9 mg) and 4 (8.3 mg). Fr. 4.5 was also resolved (Frs. 4.5.1–4.5.9) by CC on silica gel eluting with a CH2Cl2–MeOH gradient (from 50:1 to 5:1). Fr. 4.5.5 was further subjected to CC on silica gel eluting with a PE–EtOAc gradient (from 5:1 to 2:1) to yield compounds 1 (14.4 mg) and 2 (8.7 mg). Fr. 4.6 was chromatographed over silica gel (CH2Cl2–MeOH 50:1–15:1) to get Fr. 4.6.8, which was then purified by preparative TLC (PE–EtOAc 5:1) to give compound 5 (Rf = 0.4, 5.8 mg) and 6 (Rf = 0.6, 3.5 mg). Fr. 4.2 was subfractioned by CC on silica gel eluting with PE–EtOAc 15:1, then further purified by Sephadex LH-20 (MeOH) to yield 7 (6.6 mg).
ε): 204 (2.07), 229 (1.63) nm. ECD (MeCN, λ [nm] (Δε), c = 1.1 × 10−3 M): 207 (−16.0). 1H and 13C-NMR: see Tables 1 and 2 ESIMS (positive): m/z 371 [M + Na]+. HRESIMS (positive): m/z 371.2203 ([M + Na]+, calcd for C21H32O4Na 371.2193).ε): 209 (2.99), 270 (2.30) nm. ECD (MeCN, λ [nm] (Δε), c = 1.1 × 10−3 M): 228 (+2.9), 301 (−0.6), 348 (+0.2) nm. 1H and 13C-NMR: see Tables 1 and 2 ESIMS (positive): m/z 371 [M + Na]+. HRESIMS (positive): m/z 371.1832 ([M + Na]+, calcd for C20H28O5Na 371.1829).ε): 238 (3.20) nm. ECD (MeCN, λ [nm] (Δε), c = 1.2 × 10−3 M): 234 (−24.6). 1H and 13C-NMR: see Tables 1 and 2 EIMS: m/z 302 [M]+, 287 [M − CH3]+, 284 [M − H2O]+, 270 [M − CH2O]+, 266 [M − 2H2O]+. HREIMS: m/z 302.2248 ([M]+, calcd for C20H30O2 302.2246).ε): 240 (3.16) nm. ECD (MeCN, λ [nm] (Δε), c = 1.2 × 10−3 M): 238 (−24.6). 1H and 13C-NMR: see Tables 1 and 2 EIMS (negative): m/z 302 [M]+, 287 [M − CH3]+, 284 [M − H2O]+, 270 [M − CH2O]+, 266 [M − 2H2O]+. HREIMS (positive): m/z 302.2242 ([M]+, calcd for C20H30O2 302.2246).ε): 242 (3.31) nm. ECD (MeCN, λ [nm] (Δε), c = 1.0 × 10−3 M): 212 (−23.2), 242 (−57.2), 332 (+6.0) nm. 1H and 13C-NMR: see Tables 1 and 2 ESIMS (positive): m/z 361 [M + H]+. HRESIMS (positive): m/z 361.2367 ([M + H]+, calcd. for C22H33O4 361.2373).ε): 250 (2.05) nm. ECD (MeCN, λ [nm] (Δε), c = 1.2 × 10−3 M): 219 (+1.9), 261 (−1.6), 340 (+0.7) nm. 1H and 13C-NMR: see Tables 1 and 2 ESIMS (positive): m/z 341 [M + Na]+. HRESIMS (positive): m/z 341.2082 ([M + Na]+, calcd. for C20H30O3Na 341.2087).Computational section
Conformational searches were performed via the molecular mechanics using MM + method in HyperChem 8.0 software, and the geometries were further optimized at B3LYP/6-31G(d) level in vacuo via Gaussian 09 software to give the energy-minimized conformers. Then, the optimized conformers were subjected to the calculations of ECD spectra using TD-DFT at B3LYP/6-31G(d) level; solvent effects of the MeCN solution were evaluated at the same DFT level using the SCRF/PCM method.19X-ray crystallographic analysis of compounds 1, 3 and 5
All crystallographic data were collected on a Bruker Smart-1000 CCD diffractometer using graphite-monochromated Cu-Kα radiation (λ = 1.54178 Å) at 293(2) K. The data were corrected for absorption using the program SADABS.20 The structures were solved by direct methods using the SHELXTL software package.21 All non-hydrogen atoms were refined anisotropically. The H atoms were located by geometrical calculations, and their positions and thermal parameters were fixed during the structure refinement. The structures were refined using full-matrix least-squares techniques.22Antimicrobial assay
Antimicrobial assay against eleven human-, and aqua-pathogenic bacteria P. aeruginosa, E. tarda, A. hydrophilia, M. luteus, E. tarda, E. coli, V. parahaemolyticus, S. aureus, V. alginolyticus, V. harveyi, and E. tarda as well as seven plant pathogenic fungi C. albicans, P. parasitica, C. nicotianae, A. alternate, F. oxysporum f. sp. lycopersici, F. Graminearum, and B. dothidea was carried out using the well diffusion method.23 Chloramphenicol and Amphotericin B were used as a positive control for the bacteria and fungi, respectively.Acknowledgements
Financial support from the Ministry of Science and Technology of China (2012AA092104) and from the Scientific and Technological Innovation Project of Qingdao National Laboratory for Marine Science and Technology (No. 2015ASKJ02) is gratefully acknowledged. B.-G. Wang acknowledges the Taishan Scholar Project from Shandong Province and X.-D. Li thanks the China Postdoctoral Science Foundation Grant (2016LH00033) for the support to the work.Notes and references
- N. N. Win, T. Ito, S. Aimaiti, H. Imagawa, H. Ngwe, I. Abe and H. Morita, J. Nat. Prod., 2015, 78, 1113 CrossRef CAS PubMed.
- K. Jiang, L. L. Chen, S. F. Wang, Y. Wang, Y. Li and K. Gao, J. Nat. Prod., 2015, 78, 1037 CrossRef CAS PubMed.
- N. N. Win, T. Ito, T. Matsui, S. Aimaiti, T. Kodama, H. Ngwe, Y. Okamoto, M. Tanaka, Y. Asakawa, I. Abe and H. Morita, Bioorg. Med. Chem. Lett., 2016, 26, 1789 CrossRef CAS PubMed.
- X. K. Xia, J. Qi, Y. Y. Liu, A. R. Jia, Y. G. Zhang, C. H. Liu, C. L. Gao and Z. G. She, Mar. Drugs, 2015, 13, 1124 CrossRef CAS PubMed.
- N. Liu, R. J. Li, X. N. Wang, R. X. Zhu, L. Wang, Z. M. Lin, Y. Zhao and H. X. Lou, J. Nat. Prod., 2013, 76, 1647 CrossRef CAS PubMed.
- R. Wang, W. H. Chen and Y. P. Shi, J. Nat. Prod., 2010, 73, 17 CrossRef CAS PubMed.
- X. N. Wang, B. P. Bashyal, E. M. K. Wijeratne, J. M. U'Ren, M. X. Liu, M. K. Gunatilaka, A. E. Arnold and A. A. L. Gunatilaka, J. Nat. Prod., 2011, 74, 2052 CrossRef CAS PubMed.
- S. Dettrakul, P. Kittakoop, M. Isaka, S. Nopichai, C. Suyarnsestakorn, M. Tanticharoen and Y. Thebtaranonth, Bioorg. Med. Chem. Lett., 2003, 13, 1253 CrossRef CAS PubMed.
- D. Pattamadilok and R. Suttisri, J. Nat. Prod., 2008, 71, 292 CrossRef CAS PubMed.
- S. Thongnest, C. Mahidol, S. Sutthivaiyakit and S. Ruchirawat, J. Nat. Prod., 2005, 68, 1632 CrossRef CAS PubMed.
- Z. Y. Wu, Y. B. Zhang, K. K. Zhu, C. Luo, J. X. Zhang, C. R. Cheng, R. H. Feng, W. Z. Yang, F. Zeng, Y. Wang, P. P. Xu, J. L. Guo, X. Liu, S. H. Guan and D. A. Guo, J. Nat. Prod., 2014, 77, 2342 CrossRef CAS PubMed.
- G. J. Zhang, Y. H. Li, J. D. Jiang, S. S. Yu, X. J. Wang, P. Y. Zhuang, Y. Zhang, J. Qu, S. G. Ma, Y. Li, Y. B. Liu and D. Q. Yu, Tetrahedron, 2014, 70, 4494 CrossRef CAS.
- A. Yılmaza, P. Çağlarc, T. Dirmencid, N. Görenc and G. Topçu, Nat. Prod. Commun., 2012, 7, 693 Search PubMed.
- P. Zhang, L. H. Meng, A. Mándi, T. Kurtán, X. M. Li, Y. Liu, X. Li, C. S. Li and B. G. Wang, Eur. J. Org. Chem., 2014, 4029 CrossRef CAS.
- X. D. Li, X. M. Li, X. Li, G. M. Xu, Y. Liu and B. G. Wang, J. Nat. Prod., 2016, 79, 1347 CrossRef CAS PubMed.
- X. Li, X. M. Li, X. D. Li, G. M. Xu, Y. Liu and B. G. Wang, RSC Adv., 2016, 6, 75981 RSC.
- R. W. Denton, W. W. Harding, C. I. Anderson, H. Jacobs, S. McLean and W. F. Reynolds, J. Nat. Prod., 2001, 64, 829 CrossRef CAS.
- A. C. Pinto and C. Borges, Phytochemistry, 1983, 22, 2011 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- G. M. Sheldrick, SADABS, Software for Empirical Absorption Correction, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXTL, Structure Determination Software Programs, Bruker Analytical X-ray System Inc., Madison, WI, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97 and SHELXS-97, Program for X-ray Crystal Structure Solution and Refinement, University of Göttingen, Germany, 1997 Search PubMed.
- S. K. S. Al-Burtamani, M. O. Fatope, R. G. Marwah, A. K. Onifade and S. H. Al-Saidi, J. Ethnopharmacol., 2005, 96, 107 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR and HRESIMS spectra of compounds 1–6. CCDC 1462858, 1462859 and 1462860 for compounds 1, 3, and 5 respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra27209f |
| ‡ X. L. and X. D. L. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |