
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coupling of anhydro-aldose tosylhydrazones with hydroxy compounds and carboxylic acids: a new route for the synthesis of C-β-D-glycopyranosylmethyl ethers and esters†
Tímea Kaszás,
Marietta Tóth,
Sándor Kun and
László Somsák *
*
Department of Organic Chemistry, University of Debrecen, PO Box 400, H-4002 Debrecen, Hungary. E-mail: somsak.laszlo@science.unideb.hu; Fax: +36 52512744; Tel: +36 52512900 ext. 22348
First published on 7th February 2017
Abstract
Cross couplings of O-peracylated 2,6-anhydro-aldose tosylhydrazones (C-(β-D-glycopyranosyl)formaldehyde tosylhydrazones) with alcohols, phenols, and carboxylic acids were studied under thermic or photolytic conditions in the presence of K3PO4 or LiOtBu. The reactions failed with EtOH, BnOH, or tBuOH, however, (CF3)2CHOH, electron poor phenols and carboxylic acids gave the corresponding C-β-D-glycopyranosylmethyl ethers and esters, respectively, representing a new access to these glycomimetic compounds.
Introduction
Metal-catalysed and metal-free cross coupling reactions have profoundly changed the way how complex organic molecules are assembled nowadays.1 Metal-free coupling reactions can be a good choice to avoid the use of expensive and toxic metals and ligands. In the last decade tosylhydrazones emerged as reactants in both metal-catalysed and uncatalysed coupling reactions2–4 for example with alcohols and phenols,5,6 carboxylic acids,7,8 amines,9–11 thiols,12–14 arylboronic acids,15 aryl triflates,16 aryl halides,17 and benzyl halides.18Despite the use of a large variety of aliphatic and aromatic tosylhydrazones in cross couplings, analogous reactions with anhydro-aldose tosylhydrazones have not yet been investigated. While tosylhydrazones can easily be obtained from aldehydes or ketones, anhydro-aldose tosylhydrazones are not readily available, and their preparation needs special methods. Thus, the reduction of glycosyl cyanides by RANEY®-nickel in the presence of NaH2PO2 with in situ trapping of the intermediate imine with tosylhydrazine yields anhydro-aldose tosylhydrazones.19–21 Synthetic utility of these compounds as carbene precursors was also examined to result in exo-glycals in aprotic Bamford–Stevens-reactions.20,22,23
Insertion of carbenes into O–H bonds is a long known transformation.24 Carbenes generated from tosylhydrazones were inserted into alcohols and phenols5,6,25–30 as well as into carboxylic acids7,8 to give the corresponding ethers and esters, respectively.
Only a few methods can be found in the literature for the synthesis of C-glycopyranosylmethyl ether and ester derivatives G (Scheme 1). Such compounds are most frequently prepared by etherification/esterification of C-glycopyranosyl methanols F obtained by ozonolysis–reduction reaction sequences (routes a and b) from C-α-D-glycopyranosyl allenes B,31,32 C-glycopyranosyl ethenes C of both α-D33,34 and β-D35 configurations, reduction of methyl (C-β-D-glycopyranosyl)formate D (route c),36 or ring opening of glycal epoxides E by the Grignard-reagent (iPrO)Me2SiCH2MgCl followed by Tamao–Kumada oxidation (route d) to give β-D-configured C-glycopyranosyl methanol derivatives G.37 By using this methodology, ether-linked glycoside mimics were synthesized from bioactive compounds such as ezetimibe38 and 4′-demethylepipodophyllotoxin39 derivatives. C-β-D-Glycopyranosyl siloxymethanes H were obtained from variously protected 1-O-acetates of mono and disaccharides in Co2(CO)8 catalyzed reactions with hydrosilane in the presence of carbon monoxide (route e).40–44 Replacement of the siloxy moiety by an acetoxy group furnished C-β-D-glycopyranosylmethyl acetates40,43 G and such compounds were also prepared by nucleophilic substitution of epimeric mixtures of C-D-glycopyranosylmethyl iodides I by nBu4NOAc (route f).45 Scheme 1 allows one to estimate the number of synthetic steps necessary to get the target compounds G from a common precursor, a suitably protected 1-O-acetyl glycose derivative A.
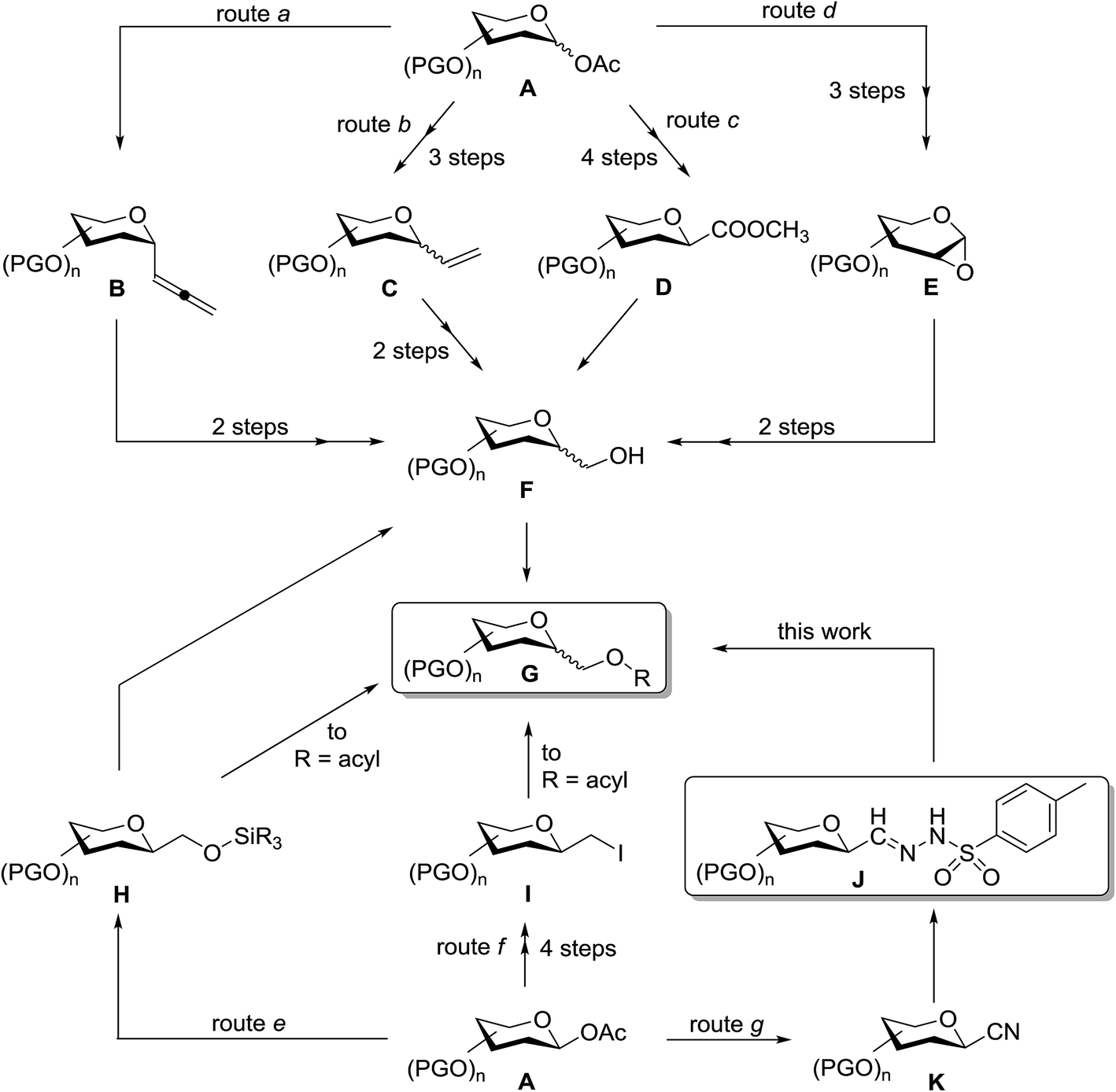 | ||
| Scheme 1 Synthetic routes toward C-glycosylmethyl ethers and esters. | ||
Given the above interest in C-glycopyranosylmethyl ethers and esters G we envisaged that cross coupling reactions of anhydro-aldose tosylhydrazones J (easily obtained from glycosyl cyanides K on route g) with alcohols, phenols or carboxylic acids may directly lead to these types of glycomimetics. Herein we disclose our trials in this field which can provide new, alternative, and shorter reaction pathways to the above compounds, and also represent the first cross couplings with anhydro-aldose tosylhydrazones.
Results and discussion
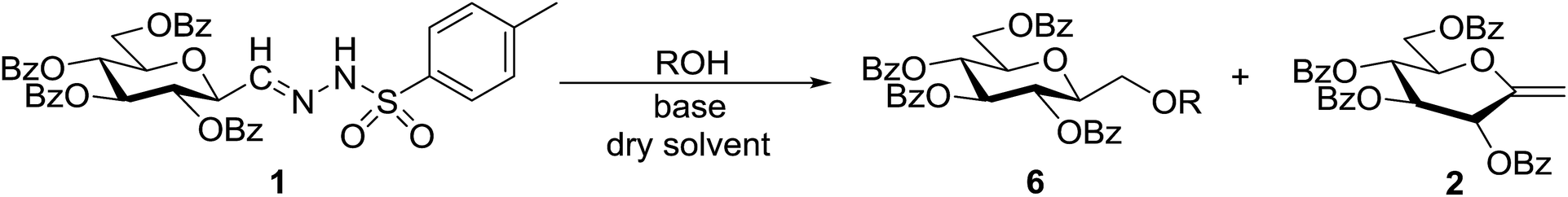
In our previous studies,20,21 carbene generation from anhydro-aldose tosylhydrazones was effected by using NaH (Table 1, entry 1). To find more easily operable bases several salts were screened with O-perbenzoylated 2,6-anhydro-D-glycero-D-gulo-heptose tosylhydrazone‡ (C-(β-D-glucopyranosyl)formaldehyde tosylhydrazone)20,21 (1) in the absence of any trapping agent to give the corresponding exo-glucal 2. The bases K2CO3, LiOtBu, and Bu4NF were not efficient enough for the reaction (Table 1, entries 2–7) since the yields of 2 were low and/or 2 was accompanied by side-products such as 3 and 4. The formation of 3 can be explained by hydrolysis of the tosylhydrazone moiety due to traces of water in the reaction mixtures followed by elimination of benzoic acid from the 1–2 positions. The liberated benzoic acid may be a partner in an insertion reaction of the carbene35 derived from 1 to give benzoate ester 4. On the other hand, the use of K3PO4 resulted in 2 as the only product in acceptable yield (entry 8), and its application in a 5-fold excess (entry 9) proved equipotent with the use of NaH (entry 1). In coupling reactions of tosylhydrazones with OH-compounds fluorobenzene was reported to be an efficient solvent,6 however, in the above reactions it did not perform better but even worse than 1,4-dioxane (entries 10 and 11). Therefore, in the further transformations mainly K3PO4 and in some cases LiOtBu in 1,4-dioxane were employed as the base.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base (equiv.) | Yielda (%) | ||
| 2 | 3 | 4 | |||
| a Isolated yields from a complex mixture which do not reflect the actual product ratios.b Literature experiment.20,21c Performed in a sealed tube, reaction temp. 110 °C.d Performed in a sealed tube, reaction temp. 100 °C. | |||||
| 1 | 1,4-Dioxane | NaH (10) | 72b | − | − |
| 2 | 1,4-Dioxane | K2CO3 (1.5) | 21 | 5 | 16 |
| 3 | 1,4-Dioxane | K2CO3 (5) | 26 | 6 | 9 |
| 4 | 1,4-Dioxane | K2CO3 (10) | 25 | 9 | 5 |
| 5 | 1,4-Dioxane | LiOtBu (5) | 24 | − | − |
| 6 | 1,4-Dioxane | LiOtBu (5) | 50c | − | − |
| 7 | 1,4-Dioxane | Bu4NF (5) | 44c | + | 14 |
| 8 | 1,4-Dioxane | K3PO4 (3) | 46 | − | − |
| 9 | 1,4-Dioxane | K3PO4 (5) | 70 | − | − |
| 10 | PhF | K3PO4 (5) | 10 | − | − |
| 11 | PhF | K3PO4 (5) | 29d | − | − |
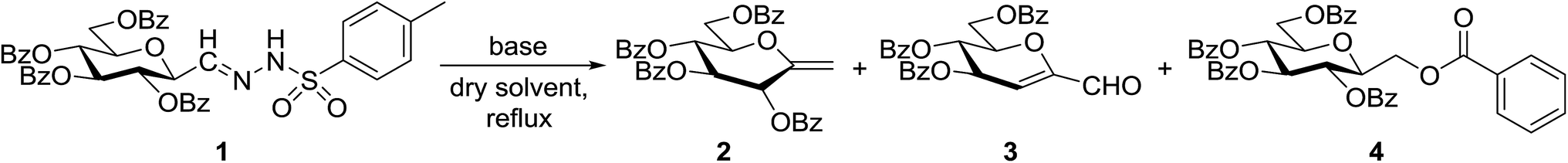
Tosylhydrazone 1, when reacted with EtOH as the solvent at reflux temperature in the presence of K3PO4 (5 equiv.), led only to decomposition whereupon no discrete product could be isolated from the reaction mixture (Table 2, entry 1). Similar experiments with tBuOH (either 20 equiv. in 1,4-dioxane shown in entry 2 or as the solvent, 10 equiv. of K3PO4) allowed exo-glucal 2 or ester 4 to be isolated in less than 30% yields, respectively. In order to avoid the possibility of failure or incompleteness of the deprotonation of 1, its Li-salt 5 was prepared (Scheme 2), and subjected to carbene generation in the presence of both EtOH or tBuOH (neat or 100–160 equiv. in 1,4-dioxane under irradiation by a 250 W mercury-vapour lamp at λmax = 365 nm at rt or under thermic conditions at reflux temperature), however, only decomposition or traces of 2 or 4 could be detected in these reaction mixtures. To check the effect of PhF,6 the reactions of 1 with tBuOH or BnOH (both 20 equiv., entries 3 and 4, respectively) in the presence of LiOtBu (1.2 equiv.) were carried out in this solvent under MW heating, however, only the formation of 2 could be observed.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R | ROH equiv. | Solvent | Base (equiv.) | Temperature (°C) | Time (h) | Yielda (%) | ||
| 6 | 2 | ||||||||
| a Isolated yields from a complex mixture which do not reflect the actual product ratios.b MW (150 W at 100 °C, 200 W at 155 °C).c Performed in a sealed tube.d With irradiation by a mercury-vapour lamp (250 W, λmax = 365 nm).e Could not be separated by column chromatography. Yields were calculated on the basis of the proton NMR spectra. | |||||||||
| 1 | CH3CH2– | Solvent | K3PO4 (5) | 78 | 3 | Decomposition | |||
| 2 | (CH3)3C– | 20 | 1,4-Dioxane | K3PO4 (10) | 80 | 3 | − | 28 | |
| 3 | 20 | PhF | LiOtBu (1.2) | 100b | 0.25 | − | 42 | ||
| 4 |  |
20 | PhF | LiOtBu (1.2) | 100b | 0.25 | − | + | |
| 5 | a | (CF3)2CH– | 20 | 1,4-Dioxane | LiOtBu (1.2) | 110c | 0.5 | 35 | 28 |
| 6 | 20 | PhF | LiOtBu (1.2) | 100b | 0.25 | 25 | 5 | ||
| 7 | b |  |
35 | 1,4-Dioxane | K3PO4 (10) | 101 | 1 | − | − |
| 8 | 33 | 1,4-Dioxane | LiOtBu (1.5) | 110c | 1 | 25 | 45 | ||
| 9 | 20 | 1,4-Dioxane | LiOtBu (1.5) | rtd | 1.5 | 8 | 33 | ||
| 10 | c | 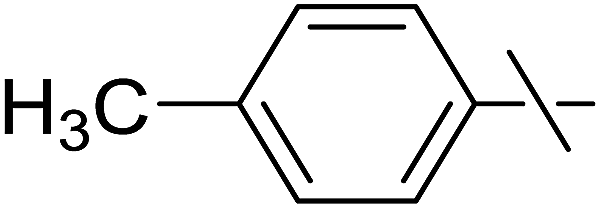 |
5 | 1,4-Dioxane | K3PO4 (2) | 110c | 0.5 | + | 42 |
| 11 | 20 | 1,4-Dioxane | LiOtBu (1.2) | 110c | 0.5 | + | 55 | ||
| 12 | d | 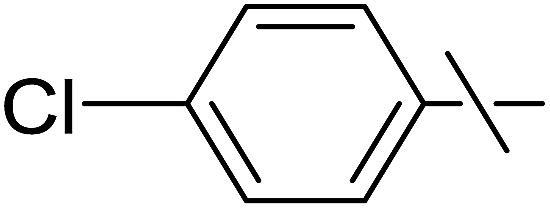 |
20 | 1,4-Dioxane | K3PO4 (5) | 110c | 1 | 20e | − |
| 13 | 5 | 1,4-Dioxane | K3PO4 (2) | 101 | 0.5 | 18e | 57e | ||
| 14 | 20 | 1,4-Dioxane | LiOtBu (1.2) | 101 | 0.5 | 30 | 13e | ||
| 15 | 20 | 1,4-Dioxane | LiOtBu (1.2) | 100b | 0.25 | 30 | − | ||
| 16 | 20 | PhF | LiOtBu (1.2) | 100c | 17.5 | 39 | − | ||
| 17 | 20 | PhF | LiOtBu (1.2) | 155b | 0.3 | 11 | − | ||
| 18 | 2 | PhF | K2CO3 (3.5) | 155b | 0.3 | 17 | + | ||
| 19 | 20 | PhF | LiOtBu (1.2) | 100b | 0.25 | 30 | + | ||
| 20 | e | 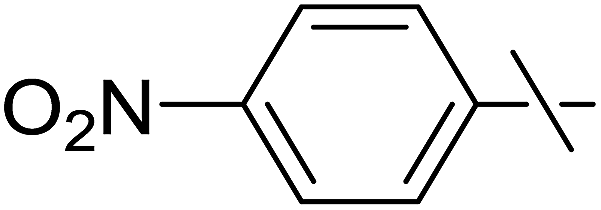 |
20 | 1,4-Dioxane | K3PO4 (10) | 110c | 0.5 | 28 | − |
| 21 | 20 | 1,4-Dioxane | LiOtBu (1.2) | 110c | 0.5 | 34 | + | ||
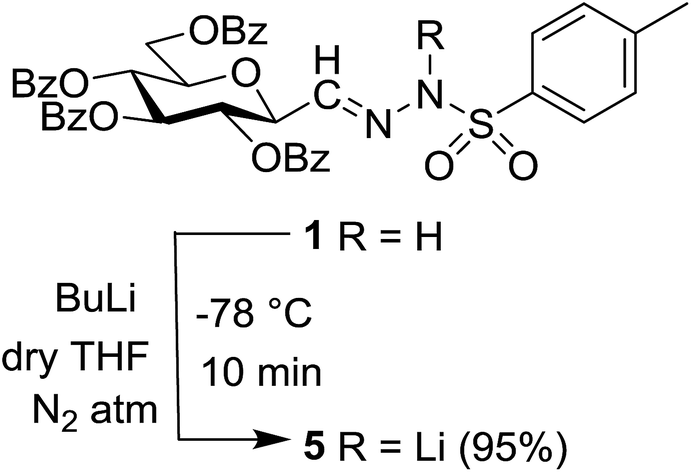 | ||
| Scheme 2 Formation of Li-salt 5 from anhydro-aldose tosylhydrazone 1. | ||
From the reaction of 1 with (CF3)2CHOH in the presence of LiOtBu the coupled product 6a could be isolated beside some exo-glucal 2 (Table 2, entries 5 and 6). The use of PhF as the solvent (entry 6) was inferior to 1,4-dioxane (entry 5) in these reactions, as well.
Next, we turned to analogous transformations with phenols (Table 2). Reaction of 1 with phenol gave a complex mixture in the presence of K3PO4 (Table 2, entry 7), but resulted in ether 6b in moderate and low yields with LiOtBu under thermic or photolytic conditions, respectively (entries 8 and 9). From the reaction of p-cresol (entries 10 and 11) exo-glucal 2 was isolated as the main product regardless of base. However, transformations with p-chloro- (entries 12–15) and p-nitro-phenol (entries 20 and 21) provided the desired ethers 6d and 6e, respectively, in moderate yields both with K3PO4 and LiOtBu. In the case of p-chloro-phenol PhF was again tried as the solvent (entries 16–19) with both bases and under conventional or MW heating, however, only a slight increase of the yield was observed with oil bath heating in a sealed tube (entry 15).
Coupling reactions of anhydro-aldose tosylhydrazones with carboxylic acids in the presence of K3PO4 were also examined (Table 3). Reactions with aliphatic carboxylic acids resulted in the desired esters 7a–e as the sole products with moderate and good yields (Table 3, entries 1–6). Coupling reactions with benzoic, 2-naphtoic, and substituted benzoic acids gave compounds 7f–l, respectively, in moderate yields (entries 7–15). Application of higher excess of carboxylic acids and the base generally increased the yields (compare entries 3–4, 9–10). Adapting the applied reaction conditions to sugar derived carboxylic acids (O-peracetylated D-galactonic acid,46 O-perbenzoylated C-(β-D-glucopyranosyl)formic acid,47 O-peracetylated C-(β-D-galactopyranosyl)formic acid,48 1,2-O-isopropylidene-3,5-O-benzylidene-D-glucofuranuronic acid49) the expected 7m–p, respectively, were isolated in good yields (entries 16–19).
|
|||||
|---|---|---|---|---|---|
| Entry | R | RCOOH equiv. | K3PO4 equiv. | Yield (%) | |
| 7 | |||||
| a 7f = 4 in Table 1. | |||||
| 1 | a | CH3– | 20 | 10 | 31 |
| 2 | b | CH3CH2– | 20 | 10 | 49 |
| 3 | c | 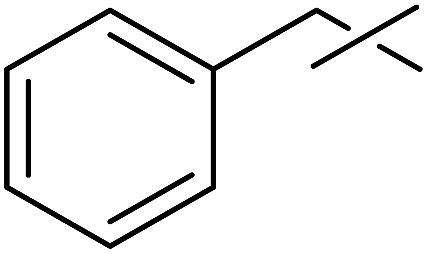 |
2 | 2 | 39 |
| 4 | 20 | 10 | 58 | ||
| 5 | d | 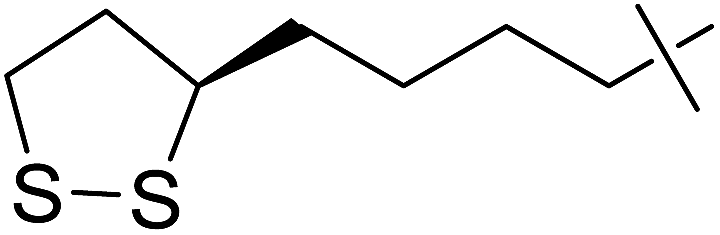 |
5 | 5 | 39 |
| 6 | e | 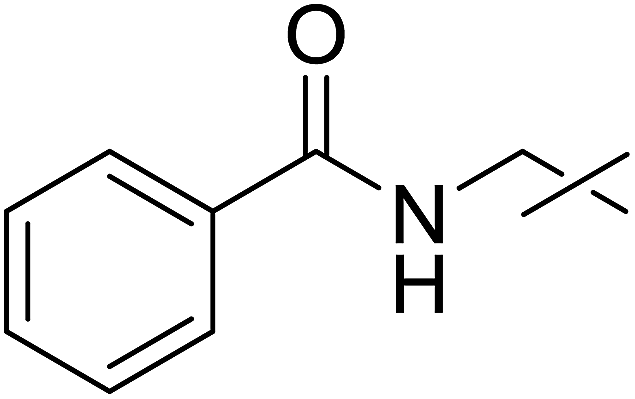 |
5 | 5 | 28 |
| 7 | fa | 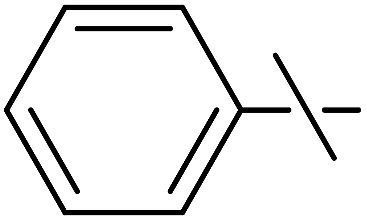 |
40 | 20 | 22 |
| 8 | g | 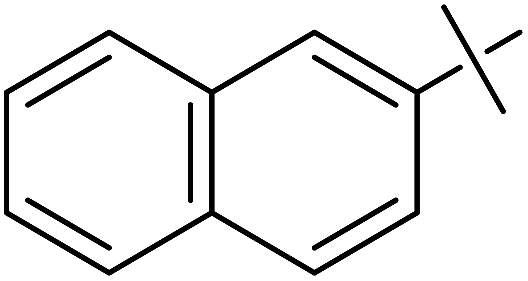 |
20 | 10 | 37 |
| 9 | h | 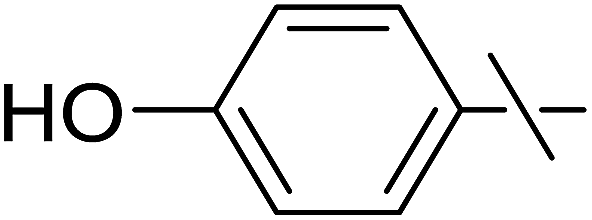 |
5 | 7 | 23 |
| 10 | 20 | 20 | 43 | ||
| 11 | i | 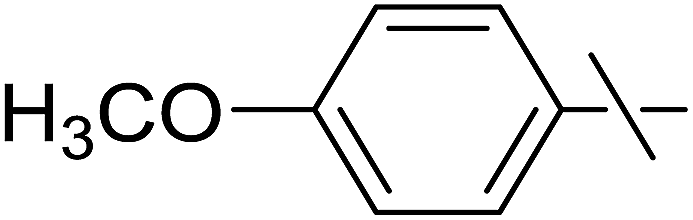 |
20 | 25 | 29 |
| 12 | j | 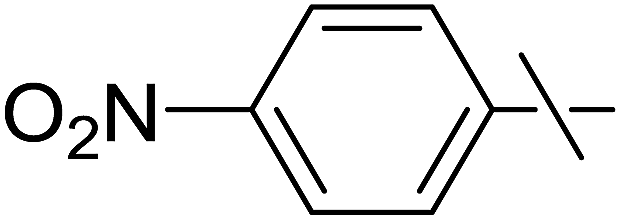 |
5 | 9 | 33 |
| 13 | 20 | 25 | 51 | ||
| 14 | k |  |
3 | 8 | 36 |
| 15 | l | 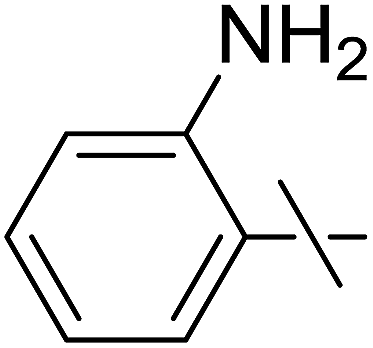 |
20 | 15 | 51 |
| 16 | m | 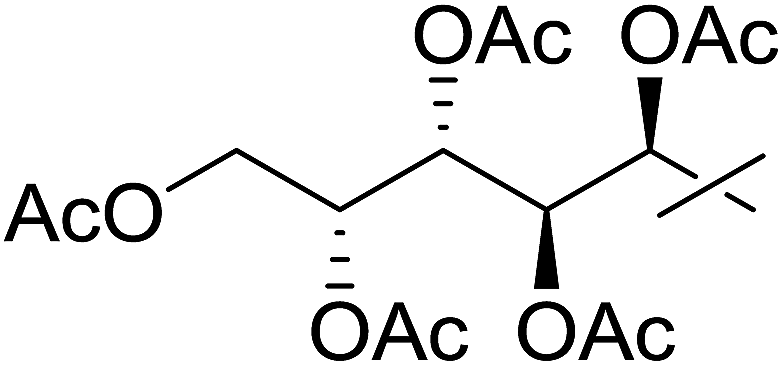 |
5 | 5 | 48 |
| 17 | n | 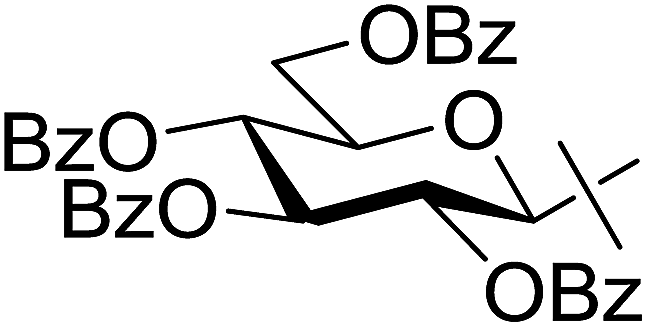 |
5 | 4 | 60 |
| 18 | o | 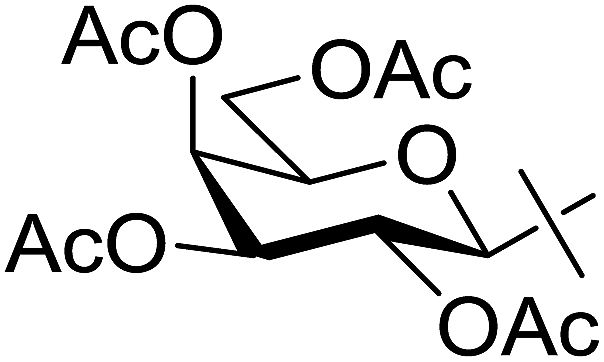 |
5 | 3 | 58 |
| 19 | p | 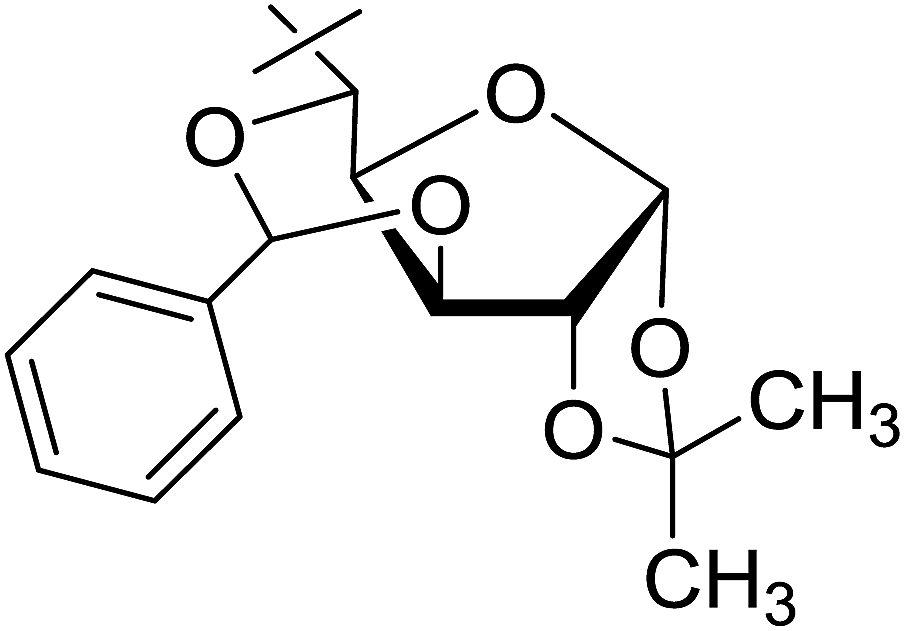 |
5 | 5 | 66 |

The examinations were extended to the D-galacto configured tosylhydrazone 8 (Table 4). The corresponding esters 9a–c derived from aliphatic carboxylic acids were isolated in moderate yields (entries 1–3), while 9d was obtained from O-perbenzoylated C-(β-D-glucopyranosyl)formic acid in good yield (entry 4).
|
|||||
|---|---|---|---|---|---|
| Entry | R | RCOOH equiv. | K3PO4 equiv. | Yield (%) | |
| 9 | |||||
| 1 | a | CH3– | 20 | 10 | 51 |
| 2 | b | CH3CH2– | 5 | 4 | 30 |
| 3 | c | 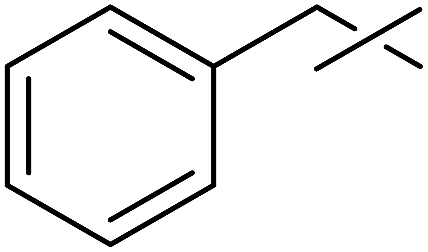 |
2 | 2 | 25 |
| 4 | d | 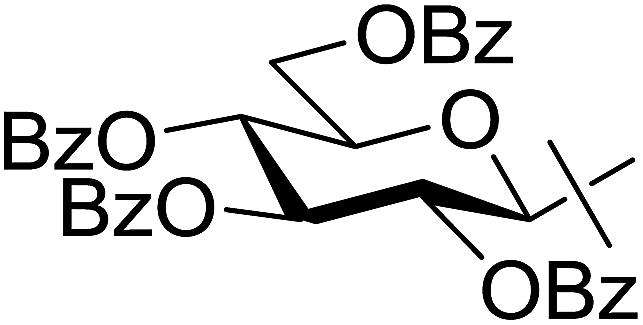 |
5 | 3 | 75 |
A comparison of the investigated reactions allows one to conclude that the acidity of the OH-bond of the coupling partners seems to be essential in terms of the yields (Table 5). While alcohols (entries 1–3), and the electron rich (and thereby less acidic) p-cresol (entry 4) did not give the expected ethers, phenol, p-Cl- and p-NO2-phenols of higher acidity (entries 5, 6, and 8) as well as carboxylic acids (entries 9–24) gave the expected coupling products. This assumption is supported by the reaction of 1 with hexafluoro-isopropanol (entry 7) which also gave the expected coupled product. It is noteworthy that 4-hydroxybenzoic acid (entry 12) reacted only at the COOH group, a finding also corroborating the role of acidity of the coupling partner. Interestingly, sugar derived carboxylic acids (entries 21–24) gave the highest yield of the products. Based on these experiences, it can be assumed that from the possible mechanistic pathways25 (Scheme 3) protonation of either the intermediate diazo compound (path a) or the carbene (path b) is more probable than the direct insertion of the carbene in the OH bond (path c).
| Entry | Reagent | Reagent equiv. | Yield of the coupled product | pKa | Ref. |
|---|---|---|---|---|---|
| a Taken from SciFinder (https://scifinder.cas.org/scifinder/view/scifinder/scifinderExplore.jsf) predicted properties calculated using Advanced Chemistry Development (ACD/Labs) Software V11.02 (© 1994–2017 ACD/Labs).b The predicted data were in the given range. | |||||
| 1 | (CH3)3COH | 20 | None | 17.0 | 51 |
| 2 | CH3CH2OH | 20 | None | 15.5 | 50 |
| 3 | 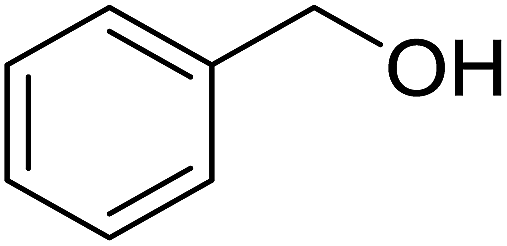 |
20 | None | 14.4a | |
| 4 | 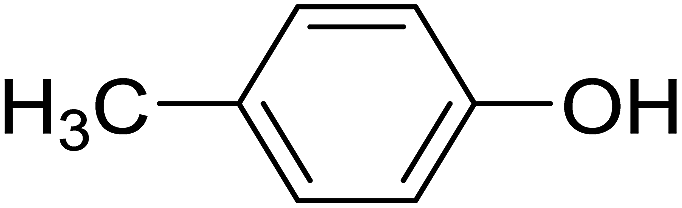 |
20 | Trace | 10.3 | 50 |
| 5 | 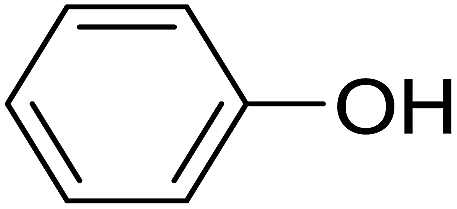 |
20 | 25 (6b) | 9.9 | 50 |
| 6 | 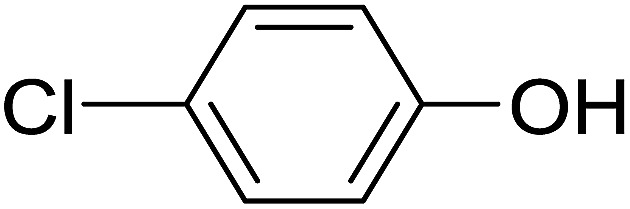 |
20 | 39 (6d) | 9.4 | 50 |
| 7 | (CF3)2CHOH | 20 | 35 (6a) | 9.3 | 51 |
| 8 | 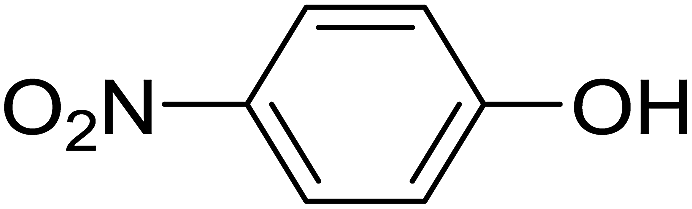 |
20 | 34 (6e) | 7.2 | 50 |
| 9 | CH3CH2COOH | 20 (with 1) | 49 (7b) | 4.9 | 50 |
| 5 (with 8) | 30 (9b) | ||||
| 10 | CH3COOH | 20 (with 1) | 31 (7a) | 4.8 | 50 |
| 20 (with 8) | 51 (9a) | ||||
| 11 | 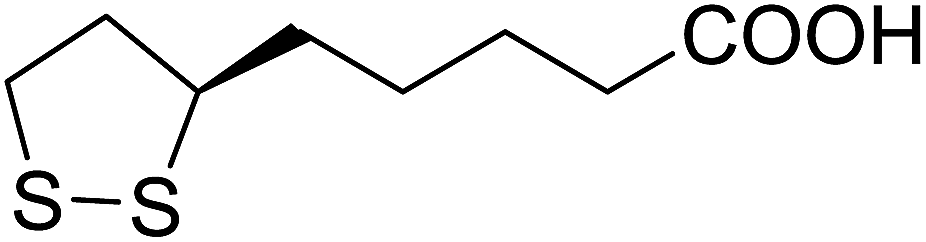 |
5 | 39 (7d) | 4.8a | |
| 12 | 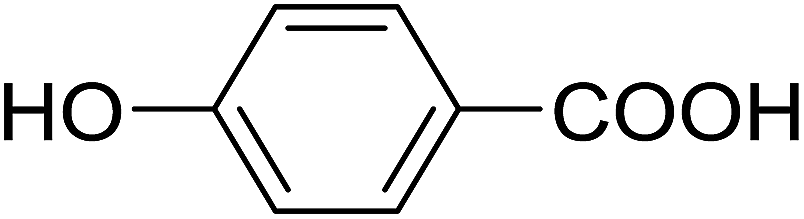 |
20 | 43 (7h) | 4.6 | 50 |
| 13 | 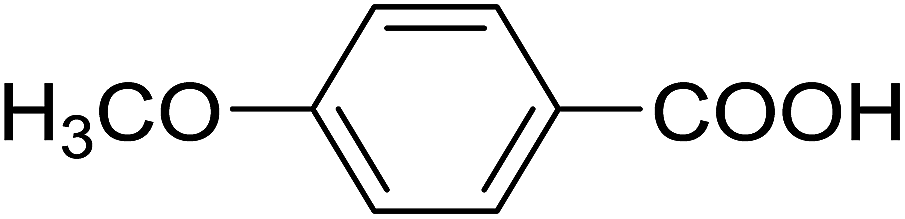 |
20 | 29 (7i) | 4.5 | 50 |
| 14 | 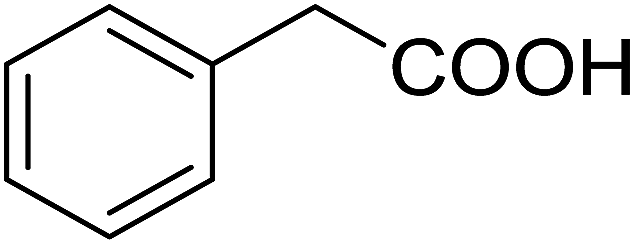 |
20 (with 1) | 58 (7c) | 4.3 | 50 |
| 2 (with 8) | 25 (9c) | ||||
| 15 | 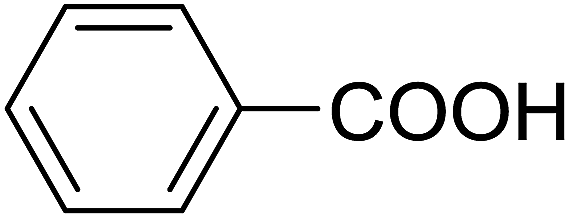 |
20 | 22 (7f) | 4.2 | 50 |
| 16 | 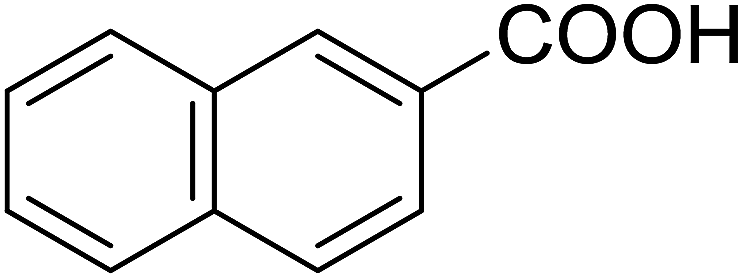 |
20 | 37 (7g) | 4.2 | 50 |
| 17 | 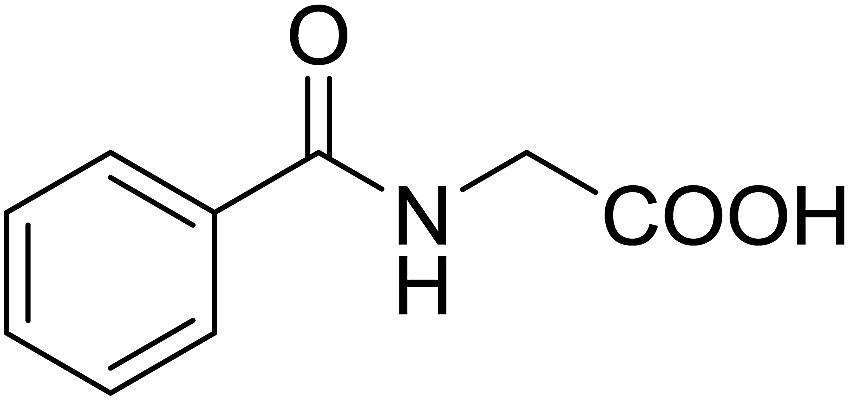 |
5 | 28 (7e) | 3.6 | 50 |
| 18 |  |
20 | 51 (7j) | 3.4 | 50 |
| 19 |  |
3 | 36 (7k) | 2.5 | 50 |
| 20 | 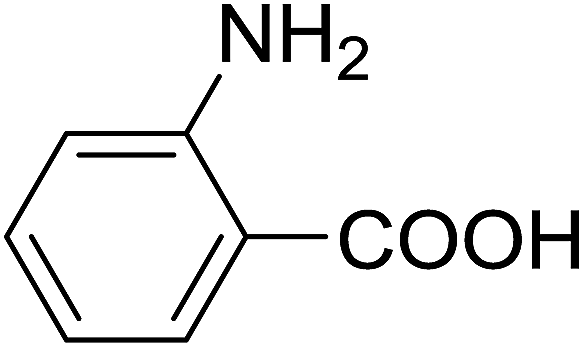 |
20 | 51 (7l) | 2.2 | 50 |
| 21 | 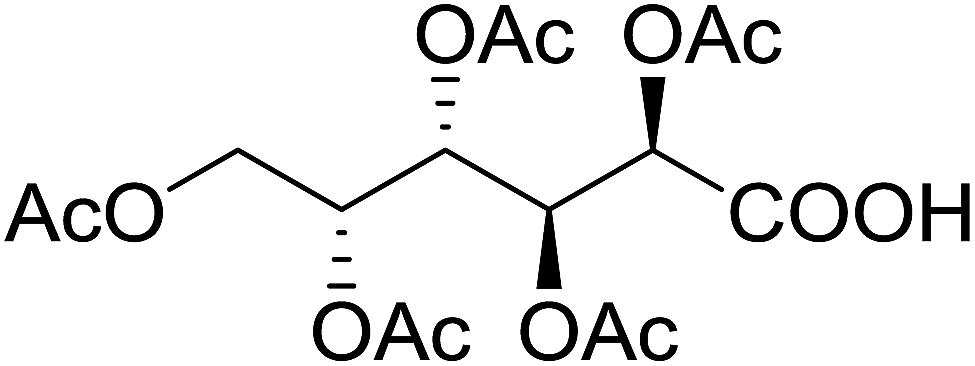 |
5 | 48 (7m) | 2.3–2.6b | |
| 22 | 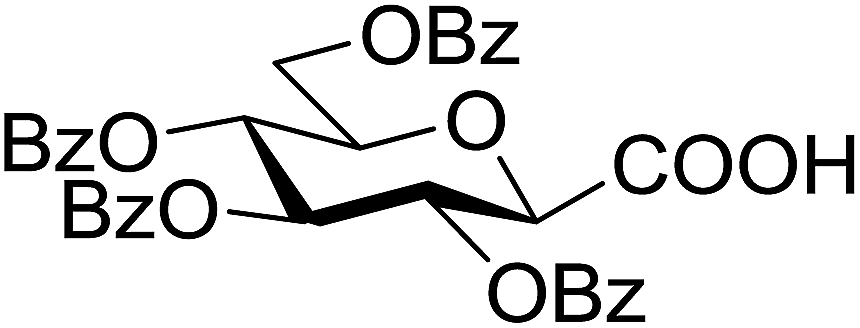 |
5 (with 1) | 60 (7n) | ||
| 5 (with 8) | 75 (9d) | ||||
| 23 | 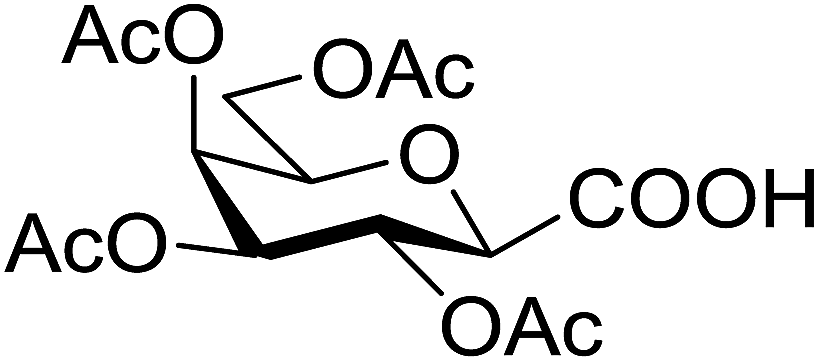 |
5 | 58 (7o) | ||
| 24 | 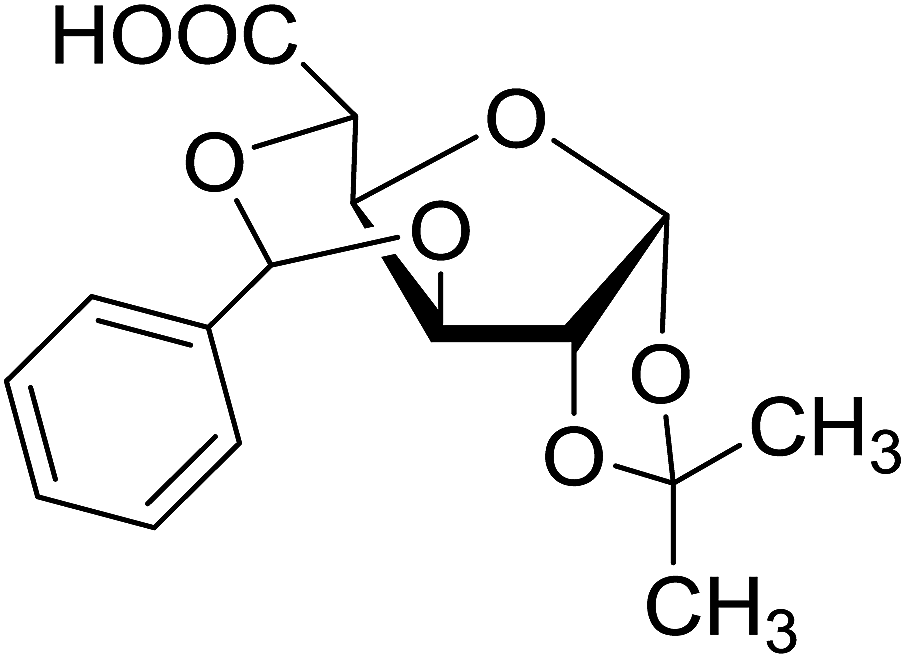 |
5 | 66 (7p) | ||
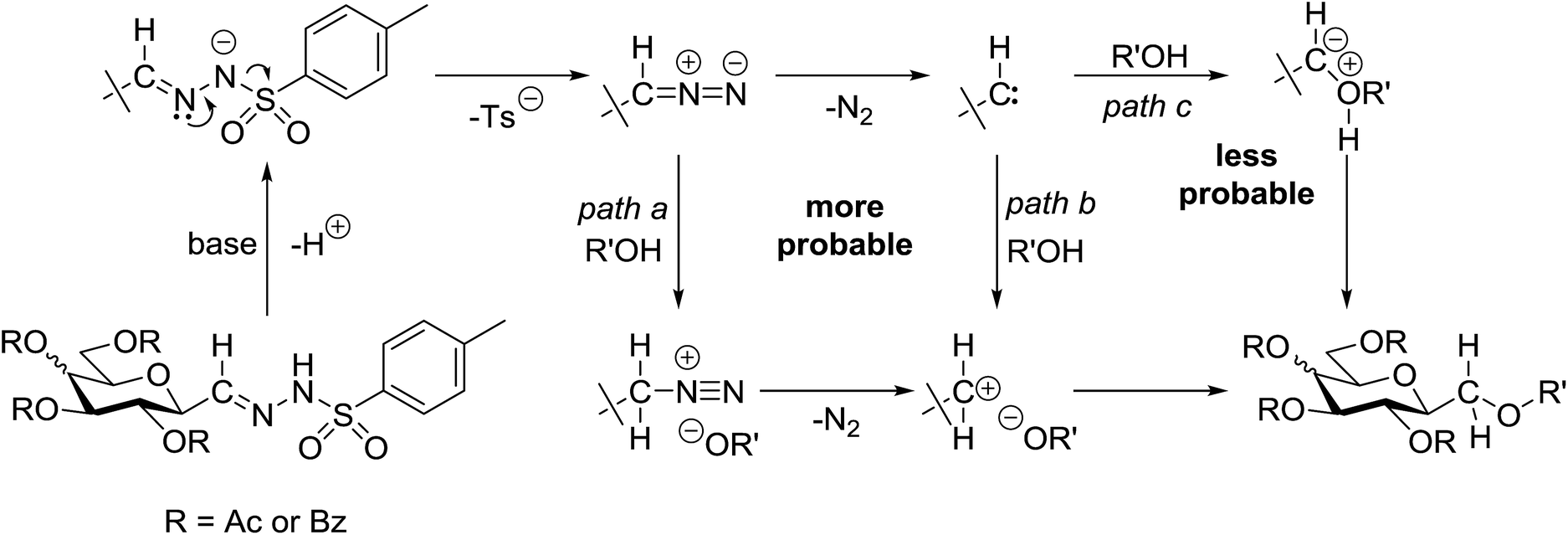 | ||
| Scheme 3 Mechanistic possibilities of the transformations. | ||
Conclusion
This study on the coupling reactions of C-(β-D-glycopyranosyl)formaldehyde (2,6-anhydro-aldose) tosylhydrazones with OH-compounds revealed that perfluoroalkanols, electron poor phenols and carboxylic acids gave moderate to good yields of the expected glycopyranosylmethyl ethers and esters, respectively, while normal alcohols and electron rich phenols furnished no coupled products. The method seems especially suitable to form glycopyranosylmethyl esters of sugar derived carboxylic acids, thereby opening a new possibility to get such kinds of disaccharide mimetics. In addition, the scope of tolerable functionalities in tosylhydrazone couplings was also extended to amino, carboxamide, and disulfide groups.Acknowledgements
This work was supported by the Hungarian Scientific Research Fund (OTKA 109450), by the European Union and the State of Hungary, co-financed by the European Social Fund in the framework of TÁMOP-4.2.4.A/2-11/1-2012-0001 ‘National Excellence Program’ (to MT) and by the University of Debrecen (5N5XBTDDTOMA320).References
- Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, 2004 Search PubMed.
- J. Barluenga and C. Valdés, Angew. Chem., Int. Ed., 2011, 50, 7486–7500 CrossRef CAS PubMed.
- Z. Shao and H. Zhang, Chem. Soc. Rev., 2012, 41, 560–572 RSC.
- Q. Xiao, Y. Zhang and J. B. Wang, Acc. Chem. Res., 2013, 46, 236–247 CrossRef CAS PubMed.
- S. Chandrasekhar, G. Rajaiah, L. Chandraiah and D. N. Swamy, Synlett, 2001, 2001, 1779–1780 CrossRef.
- J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Angew. Chem., Int. Ed., 2010, 49, 4993–4996 CrossRef CAS PubMed.
- A.-H. García-Muñoz, M. Tomás-Gamasa, M. C. Pérez-Aguilar, E. Cuevas-Yañez and C. Valdés, Eur. J. Org. Chem., 2012, 3925–3928 CrossRef.
- A. Zhou, L. Wu, D. Li, Q. Chen, X. Zhang and W. Xia, Chin. J. Chem., 2012, 30, 1862–1866 CrossRef CAS.
- A. Hamze, B. Tréguier, J.-D. Brion and M. Alami, Org. Biomol. Chem., 2011, 9, 6200–6204 CAS.
- A. Khanna, C. Maung, K. R. Johnson, T. T. Luong and D. L. Van Vranken, Org. Lett., 2012, 14, 3233–3235 CrossRef CAS PubMed.
- J. Aziz, J.-D. Brion, A. Hamze and M. Alami, Adv. Synth. Catal., 2013, 355, 2417–2429 CrossRef CAS.
- Q. Ding, B. Cao, J. Yuan, X. Liu and Y. Peng, Org. Biomol. Chem., 2011, 9, 748–751 CAS.
- Q. Ding, X. Liu, J. Yu, Q. Zhang, D. Wang, B. Cao and Y. Peng, Tetrahedron, 2012, 68, 3937–3941 CrossRef CAS.
- Y. Lin, P. Luo, Q. Zheng, Y. Liu, X. Sang and Q. Ding, RSC Adv., 2014, 4, 16855–16863 RSC.
- X. Zhao, J. Jing, K. Lu, Y. Zhang and J. B. Wang, Chem. Commun., 2010, 1724–1726 RSC.
- B. Tréguier, A. Hamze, O. Provot, J.-D. Brion and M. Alami, Tetrahedron Lett., 2009, 50, 6549–6552 CrossRef.
- J. Barluenga, P. Moriel, C. Valdés and F. Aznar, Angew. Chem., Int. Ed., 2007, 46, 5587–5590 CrossRef CAS PubMed.
- Q. Xiao, J. Ma, Y. Yang, Y. Zhang and J. B. Wang, Org. Lett., 2009, 11, 4732–4735 CrossRef CAS PubMed.
- M. Tóth and L. Somsák, Tetrahedron Lett., 2001, 42, 2723–2725 CrossRef.
- M. Tóth, K. E. Kövér, A. Bényei and L. Somsák, Org. Biomol. Chem., 2003, 1, 4039–4046 Search PubMed.
- M. Tóth, L. Somsák and D. Goyard, Preparation of 2,6-Anhydro-Aldose Tosylhydrazones in Carbohydrate Chemistry: Proven Synthetic Methods, ed. P. Kováč, CRC Press, Boca Raton, 2012, vol. 1, pp. 355–365 Search PubMed.
- M. Tóth and L. Somsák, J. Chem. Soc., Perkin Trans. 1, 2001, 942–943 RSC.
- M. Tóth, S. Kun, L. Somsák and D. Goyard, Preparation of Exo-Glycals from (C-Glycopyranosyl)formaldehyde Tosylhydrazones in Carbohydrate Chemistry: Proven Synthetic Methods, ed. P. Kováč, CRC Press, Boca Raton, 2012, vol. 1, pp. 367–375 Search PubMed.
- M. Regitz, Methoden der organischen Chemie (Houben-Weyl), Thieme, Stuttgart, 1989. vol. E19b Search PubMed.
- R. Grandi, A. Marchesini, U. M. Pagnoni and R. Trave, J. Org. Chem., 1976, 41, 1755–1758 CrossRef CAS.
- W. Kirmse, K. Kund, E. Ritzer, A. E. Dorigo and K. N. Houk, J. Am. Chem. Soc., 1986, 108, 6045–6046 CrossRef CAS PubMed.
- X. Creary, M. E. Mehrsheikh-Mohammadi and S. McDonald, J. Org. Chem., 1987, 52, 3254–3263 CrossRef CAS.
- G. Hoemberger, A. E. Dorigo, W. Kirmse and K. N. Houk, J. Am. Chem. Soc., 1989, 111, 475–477 CrossRef CAS.
- W. Kirmse and K. Kund, J. Am. Chem. Soc., 1989, 111, 1465–1473 CrossRef CAS.
- W. Kirmse and K. Kund, J. Org. Chem., 1990, 55, 2325–2332 CrossRef CAS.
- L. Kröger, D. Henkensmeier, A. Schäfer and J. Thiem, Bioorg. Med. Chem. Lett., 2004, 14, 73–75 CrossRef.
- M. I. Torres-Sanchez, C. Zaccaria, B. Buzzi, G. Miglio, G. Lombardi, L. Polito, G. Russo and L. Lay, Chem.–Eur. J., 2007, 13, 6623–6635 CrossRef CAS PubMed.
- D. Zhai, W. Zhai and R. M. Williams, J. Am. Chem. Soc., 1988, 110, 2501–2505 CrossRef CAS.
- R. Chang, T.-T. Vo and N. S. Finney, Carbohydr. Res., 2006, 341, 1998–2004 CrossRef CAS PubMed.
- C. Mayato, R. L. Dorta and J. T. Vázquez, Tetrahedron: Asymmetry, 2007, 18, 931–948 CrossRef CAS.
- K. C. Nicolaou, H. Flörke, M. G. Egan, T. Barth and V. A. Estevez, Tetrahedron Lett., 1995, 36, 1775–1778 CrossRef CAS.
- F. P. Boulineau and A. Wei, J. Org. Chem., 2004, 69, 3391–3399 CrossRef CAS PubMed.
- M. Werder, H. Hauser and E. M. Carreira, J. Med. Chem., 2005, 48, 6035–6053 CrossRef PubMed.
- P. Allevi, M. Anastasia, P. Ciuffreda and A. Scala, J. Carbohydr. Chem., 1993, 12, 209–222 CrossRef CAS.
- N. Chatani, T. Ikeda, T. Sano, N. Sonoda, H. Kurosawa, Y. Kawasaki and S. Murai, J. Org. Chem., 1988, 53, 3387–3389 CrossRef CAS.
- S. J. Spak and O. R. Martin, Tetrahedron, 2000, 56, 217–224 CrossRef CAS.
- D. C. Croll and L. R. Schroeder, J. Wood Chem. Technol., 2005, 24, 27–38 CrossRef.
- Y. Li, Z. Yin, B. Wang, X.-B. Meng and Z.-J. Li, Tetrahedron, 2012, 68, 6981–6989 CrossRef CAS.
- W. Yao, M.-J. Xia, X.-B. Meng, Q. Li and Z.-J. Li, Org. Biomol. Chem., 2014, 12, 8180–8195 CAS.
- S. Toumieux, P. Compain and O. R. Martin, Tetrahedron Lett., 2005, 46, 4731–4735 CrossRef CAS.
- C. D. Hurd and J. C. Sowden, J. Am. Chem. Soc., 1938, 60, 235–237 CrossRef CAS.
- K. Czifrák, P. Szilágyi and L. Somsák, Tetrahedron: Asymmetry, 2005, 16, 127–141 CrossRef.
- R. W. Myers and Y. C. Lee, Carbohydr. Res., 1986, 152, 143–158 CrossRef CAS PubMed.
- L. Zervas and P. Sessler, Ber. Dtsch. Chem. Ges., 1933, 66, 1326–1329 CrossRef.
- Dissociation Constants Of Organic Acids And Bases, http://sites.chem.colostate.edu/diverdi/all_courses/CRC%20reference%20data/dissociation%20constants%20of%20organic%20acids%20and%20bases.pdf, accessed January 1 2017.
- D. H. Ripin and D. A. Evans, pKa's of Inorganic and Oxo-Acids, http://evans.rc.fas.harvard.edu/pdf/evans_pKa_table.pdf, accessed January 1 2017.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27282g |
| ‡ This is the systematic name according to IUPAC carbohydrate nomenclature, however, the one in parenthesis reflects the parent sugar configuration in a more easily followable way, therefore, both names will be applied throughout this text. |
| This journal is © The Royal Society of Chemistry 2017 |