
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControllable growth of MnOx dual-nanocrystals on N-doped graphene as lithium-ion battery anode†
Zhongtao Li‡
 ,
Yuankun Wang‡,
Yan Chen and
Mingbo Wu*
,
Yuankun Wang‡,
Yan Chen and
Mingbo Wu*
State Key Laboratory of Heavy Oil Processing, School of Chemical Engineering, China University of Petroleum, Qingdao 266580, China. E-mail: wumb@upc.edu.cn; Fax: +86 532 86984615; Tel: +86 532 86984615
First published on 19th January 2017
Abstract
Nanocomposites containing Mn3O4 and MnOOH dual-nanocrystals on N-doped graphene sheets were prepared using a solvothermal method. Nanostructure and rate of dual-nanocrystals in the composites can be adjusted, having effects on their electrochemical performance. The nanohybrid NG-36 is composed of 1D MnOOH nanowires, Mn3O4 nanotetragonals and electroconductive N-doped graphene matrix. Experimental data for NG-36 reveals a high rechargeable specific capacity of 341.5 mA h g−1 at a current density 3 A g−1 after 500 cycles, resulting from synergetic effects among the components.
1. Introduction
With the increasing demand for power supplements for portable electronic devices and electric vehicles in the modern word, development of new lithium-ion battery (LIBs) materials with capacitor-like rate performance and sustainable power delivery is very desirable.1–4 On the basis of higher theoretical capacities, transition metal oxides (CoOx, FeOx, and MnOx) have received increasing attention as promising alternative anode materials compared with graphite.5–8 Among these transition metal oxides, MnOx has been intensively studied as one of the most prominent anode materials for LIBs because of its high theoretical capacity, low conversion potential, natural abundance and environmental benignity, which are desirable for practical application in LIBs.9–12Among various manganese oxides, Mn3O4 exhibits high theoretical capacity (937 mA h g−1) and electrochemical activity, which have been widely studied in electrodes for lithium batteries and/or supercapacitors.13,14 However, previous reports on Mn3O4 have suggested that this material has poor lithiation activity because of its low electrical conductivity (10−7 to 10−8 S cm−1) and poor cycle stability. Manganese oxyhydroxides, MnOOH, also have been intensively studied as potential electrocatalysts in reduction of O2 in metal–air batteries,15 and can be readily constructed as 1D nanowire by solution-based routes.16,17 However, to the authors' knowledge, reports of use of 1D MnOOH or its nanocomposites as LIB anode materials are lacking because of its low theoretical capacity.18 However, the complicated oxidation states of Mn, diversity of manganese oxides and interference from factors such as surface chemistry and morphology, mean that argument continues regarding the lithium-storage mechanisms of the manganese oxides.19
Some effective strategies have focused on resolving the intrinsic drawbacks of manganese derivation: (1) preparing nano-scaled materials with special morphologies to decrease the diffusion lengths of electrons and lithium-ions, which can improve the electroactivity and rate capability of electrode materials.20,21 (2) Constructing nanocomposites with a carbonaceous matrix to enhance electrical conductivity, as well as buffer volume expansion/contraction during the cycling process.13 Among potential carbonic supporters, N-doped graphene has been widely used in electrochemical energy storage applications.22–24 Compared with original graphene, the introduction of N atoms can effectively fix the defects of the graphene conjugated plane, thus increasing its electrical conductivity to achieve improved rate capacity.25
This study reports a facile solvothermal process for synthesizing nanohybrids with well-dispersed Mn3O4 nanoparticles and MnOOH nanowires into an N-doped graphene matrix. The content of two nanocrystals in the nanohybrid can be adjusted through modulation of reaction time and MnSO4/KMnO4 ratio during solvothermal treatment. The unique dual-crystals/N-doped graphene complex has enhanced lithium-storage capacity via the following synergistic effect: firstly, the Mn3O4 nanoparticles in the composite can effectively coordinate Li+ ions to achieve high capacity. Secondly, the 1D MnOOH nanowires can provide proper voids between Mn3O4 nanoparticles to avoid aggregation and achieve good durability, which is caused by alleviating the volume change during cycling. Thirdly, the ethanediamine N-doped graphene acts as the conductive backbone in the nanohybrids to facilitate ion and electron diffusion, resulting in progressed rate performance.
2. Experimental section
2.1. Preparation of N-doped graphene
N-Doped graphene was prepared according to previous reports.27 Briefly, 280 μL EDA was added to GO suspension (210 mL, 1 mg mL−1). Then, the mixed solution was heated at 75 °C for 6 h in a sealed glass vessel.2.2. Preparation of N-doped graphene/MnOOH/Mn3O4 hybrids
A specific amount of MnSO4 (0 mg, 36 mg for other samples) was added to the freshly prepared N-doped graphene suspension (200 mL) with vigorously stirring for 30 min, followed by slow addition of KMnO4 solution (100 mL, 0.5 g L−1) under stirring. Then, the mixed solutions were transferred to a Teflon-lined stainless steel autoclave and kept at 140 °C for 2 h. A dark precipitate was collected and washed with distilled water, and finally dried in the freeze dryer, thus, primary N-doped graphene/MnOOH/Mn3O4 hybrids were obtained. The samples with 0 mg or 36 mg MnSO4 introduced during the hydrothermal process were labeled as NG-0 or NG-36, respectively. For comparison, NG-36-12 (36 mg MnSO4 added and reacted for 12 h) was synthesized by the same method as described above.2.3. Sample characterization
The phase information of as-made samples was examined by X-ray diffraction (XRD) (X'Pert PRO MPD, Holland). The morphology and the microstructure were characterized using field emission scanning electron microscopy (SEM) (HitachiS-4800, Japan), and transmission electron microscopy (TEM) (JEM-2100UHR, Japan). The functional groups in the samples were studied by Fourier transform infrared spectrometry (FTIR) (Thermo Nicolet NEXUS 670, USA) and X-ray photoelectron spectroscopy (XPS, Thermo Scientific ESCALab250Xi). The Raman spectra were obtained from a Renishaw DXR Raman spectros system with a 532 nm laser source. The thermal properties and the contents of graphene in samples were quantitatively determined by thermogravimetric analysis (TGA, STA 409 PC Luxx, Germany).2.4. Electrochemical measurement
The working electrodes were made using 80 wt% active material, 10 wt% conductive carbon black and 10 wt% poly(vinylidene) fluoride (PVDF) binder. The mixture was dissolved in N-methyl-2-pyrrolidinone (NMP) to make black slurry. The slurry was then spread onto a copper foil current collector. After drying at 100 °C in a vacuum oven for 10 h, the collector was cut to disks typically with a diameter of 12 mm, and the average mass of the active material loading within the coin cells was around 1.5 mg for NG-0, and 1.7 mg for NG-36 and NG-36-12. Electrochemical measurements were carried out with a CR2032 coin type cell using lithium foil as the counter electrode and the reference electrode. A 1 M solution of LiPF6 dissolved in a mixture of dimethyl carbonate (DMC) and ethylene carbonate (EC) with a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was used as the electrolyte. Galvanostatic charge/discharge cycling was studied in a potential range of 0.01–3.0 V vs. Li/Li+ at room temperature using a LAND-CT2001A cycler. Cyclic voltammograms (CV) and electrochemical impedance spectra (EIS) were measured using an Ametek PARSTAT4000 electrochemistry workstation.
:1 was used as the electrolyte. Galvanostatic charge/discharge cycling was studied in a potential range of 0.01–3.0 V vs. Li/Li+ at room temperature using a LAND-CT2001A cycler. Cyclic voltammograms (CV) and electrochemical impedance spectra (EIS) were measured using an Ametek PARSTAT4000 electrochemistry workstation.
3. Results and discussion
The synthesis of N-doped graphene/MnOOH/Mn3O4 nanohybrids is illustrated in Scheme 1. In the first, ethanediamine was employed as the nitrogen source to reduce GO for preparing N-doped graphene (NG) through a solvothermal process. Then, a specific amount of KMnO4 and varying amounts of MnSO4 were introduced into vessels. After addition of the oxidant KMnO4, lots of MnOx nuclei were formed on NG sheets immediately by redox reaction between Mn2+ and MnO4−. The as-obtained mixed solution was further hydrothermally treated in an autoclave, inducing the MnOx nuclei to grow into various nanostructures and morphologies, dependent on modulation of the reaction time and MnSO4/KMnO4 ratio.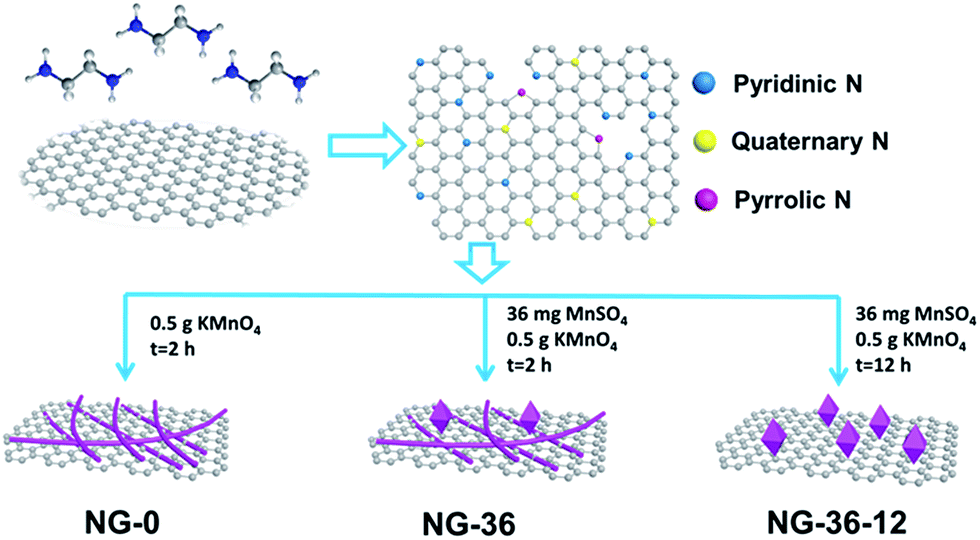 | ||
| Scheme 1 Schematic illustration of synthesized N-doped graphene/MnOOH/Mn3O4 nanohybrids. | ||
The structural characteristics of NG-x (x = 0, 36, 36-12) hybrids were determined by X-ray powder diffraction (XRD), as shown in Fig. 1. In sample NG-0 (black line), most crystals are composed of monoclinic MnOOH (manganite, P21/c(14), a = 5.300 Å, b = 5.278 Å, c = 5.307 Å, PDF# 41-1379), which adopted KMnO4 as the only Mn source. With addition of MnSO4 during synthesis of NG-36, the ratio of Mn3O4 and MnOOH increased accordingly, causing the redox reaction between MnO4− and Mn2+ to generate more Mn3O4. Both characteristic XRD peaks of tetragonal Mn3O4 (hausmannite, I41/amd, a = b = 5.7621 Å, c = 9.4696 Å, PDF# 24-0734) and monoclinic MnOOH could be identified in sample NG-36 (blue line). To further explore the growth mechanism for this synthetic route, the sample NG-36-12 was prepared by a longer hydrothermal process (after 12 h reaction), with all other conditions kept the same as in preparation of NG-36. The XRD data of NG-36-12 (red line Fig. 1) revealed that Mn3O4 and hexagonal MnCO3 (rhodochrosite, R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c(167), a = b = 4.790 Å, c = 15.694 Å, PDF# 44-1472) were indexed in the corresponding XRD. No obvious diffraction peaks of MnOOH could be observed, implying that the MnOOH phase was a metastable product and converted to other crystal after longer solvothermal treatment time. Graphene can be identified by the XRD pattern of NG (Fig. S1†) and the resultant variation indicated after the solvothermal treatment, the electrically insulating GO reduction to more conductive rGO after N-doping.26
c(167), a = b = 4.790 Å, c = 15.694 Å, PDF# 44-1472) were indexed in the corresponding XRD. No obvious diffraction peaks of MnOOH could be observed, implying that the MnOOH phase was a metastable product and converted to other crystal after longer solvothermal treatment time. Graphene can be identified by the XRD pattern of NG (Fig. S1†) and the resultant variation indicated after the solvothermal treatment, the electrically insulating GO reduction to more conductive rGO after N-doping.26
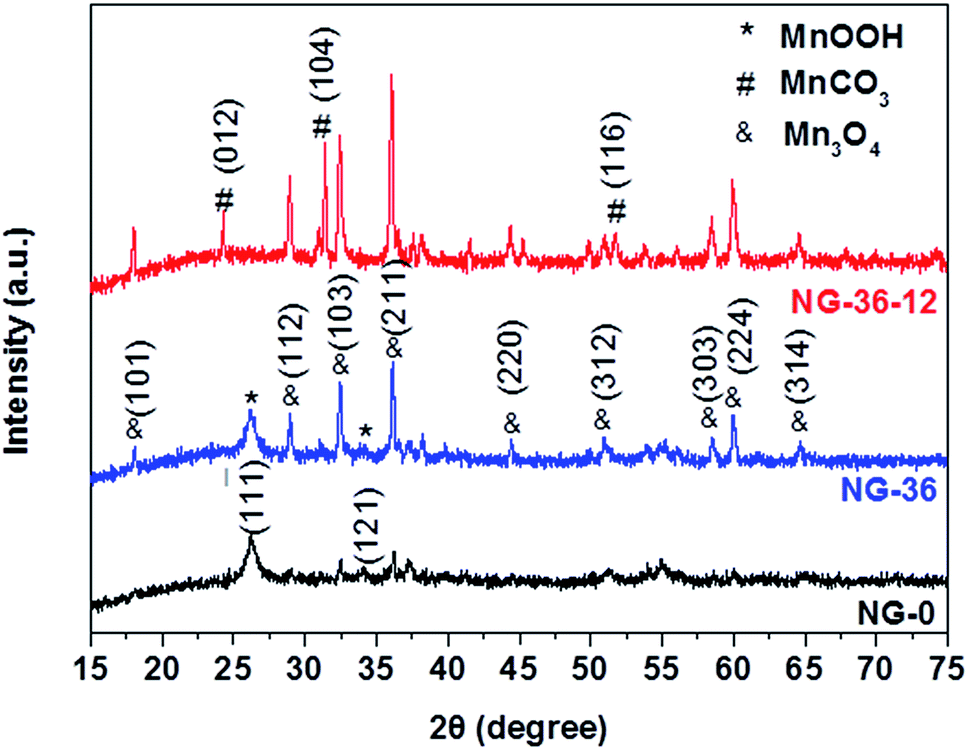 | ||
| Fig. 1 XRD patterns of NG-0, NG-36, and NG-36-12. | ||
Fourier transform-infrared spectroscopy (FTIR) was used to examine the chemical bonding in NG/MnOOH/Mn3O4 (Fig. S2†). In the spectrum of GO, four absorption peaks were observed at 1720, 1645, 1224, and 1059 cm−1, which can be ascribed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, COOH, carbonyl C–O, and epoxy C–O bonds, respectively.27 After the solvothermal treatment, most peaks of oxygen-containing groups disappeared, with some new peaks appearing at 1402, 1510, 1640, and 3430 cm−1 that can be assigned to the stretching vibrations of CN, NO, N–O, and amino groups, respectively.26,28 This represents the nucleophilic reaction between the amino groups of EDA and the oxygen-containing groups of GO during N-doping.
O, COOH, carbonyl C–O, and epoxy C–O bonds, respectively.27 After the solvothermal treatment, most peaks of oxygen-containing groups disappeared, with some new peaks appearing at 1402, 1510, 1640, and 3430 cm−1 that can be assigned to the stretching vibrations of CN, NO, N–O, and amino groups, respectively.26,28 This represents the nucleophilic reaction between the amino groups of EDA and the oxygen-containing groups of GO during N-doping.
To further understand the product properties, the Raman spectrum of NG-36 is given in Fig. 2. As shown in Fig. 2b, the two prominent peaks located at 1330 cm−1 and 1602 cm−1 can be assigned to the D band and G band of carbon, respectively.21 The intensity ratio of D band and G band (ID/IG) is measured to be 0.92 (Fig. 2b), which is greater than that of GO (0.76, Fig. 2a), indicating successful doping of nitrogen atoms in GO.26 The G band showed an obvious shift (∼12 cm−1) from 1590 (GO) to 1602 cm−1 (NG-36), which relates to the charge transfer between the carbon and N after N-doping.29,30 In addition, a minor peak centered at 645 cm−1 can be attributed to the characteristic peak of Mn3O4.23 It should be noted that the peaks arranged at 300–500 cm−1 could not be unambiguously discerned, which could be attributed to overlap of MnOOH and Mn3O4 in this area.31,32
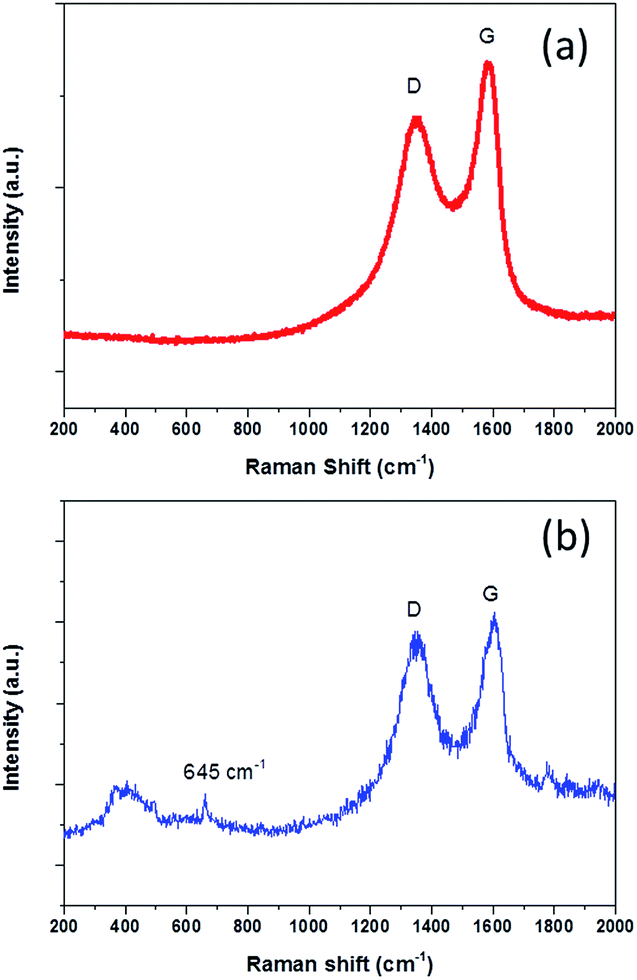 | ||
| Fig. 2 Raman spectra of (a) GO and (b) NG-36. | ||
Thermogravimetric analysis (TGA) was carried out to determine the graphene content of the NG-36 (Fig. S3†). A mass loss of about 6.8% is observed below 200 °C on the TGA curve, which may correspond to the loss of free water and physically adsorbed water, crystal water in MnOOH. The second weight loss around 41.5% after 250 °C can be attributed to combustion of graphene.
The microstructures of as-prepared samples were investigated by transmission electron microscopy (TEM, Fig. 3a–c). The panoramic views of NG-0 in Fig. 4a reveal that the nanowires are dominant over the image. The distinct lattice fringes with an interlayer spacing of 0.23 nm (inset in Fig. 3a), which repressed the (−121) plane of MnOOH, suggest that the nanowires consist of MnOOH. From Fig. 3b, two kinds of nanostructure (nanowires and nano-tetragonals) are observed in NG-36. A high-resolution TEM (HRETM) image (inset in Fig. 3b) indicates the distinct lattice fringes with 0.28 nm interlayer spacing in nanotetragonals, which can be indexed to the (211) plane of hausmannite Mn3O4. Introduction of MnSO4 clearly causes the content of tetragonals to increase. The morphologies of NG-0 and NG-36 were further verified by scanning electron microscopy (SEM). As shown in Fig. 3d and e, the wire corresponds to MnOOH with a diameter of about 30 nm and a length over 5 μm, while the average size of tetragonal Mn3O4 is 250 nm. In contrast, there are only nanotetragonal Mn3O4 were obtained in NG-36-12 (Fig. 3c and f), which is consistent with the XRD results. According to related studies, the possible chemical reaction pathways of these samples can be written as follows:
| MnO4− + 2RCOOH (GO) → MnOOH + 2RO+HCO3− + CO2 | (1) |
| 2MnOOH + Mn2+ → Mn3O4 + 2H+ | (2) |
 | ||
| Fig. 3 TEM images (a–c) and SEM images (d–f) of the NG-0 (a and d), NG-36 (b and e), and NG-36-12 (c and f). | ||
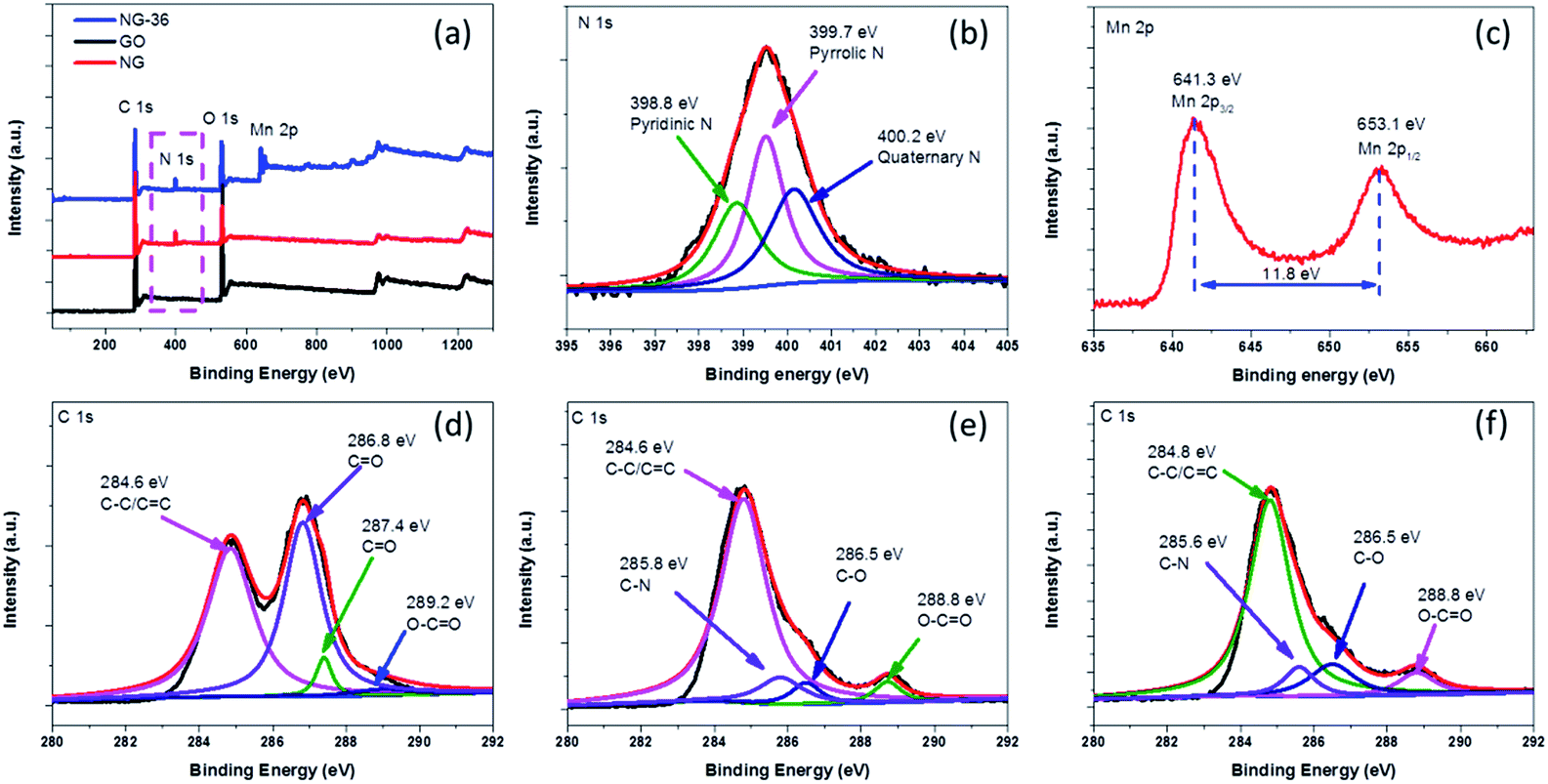 | ||
| Fig. 4 (a) XPS spectra of GO, NG, and NG-36, (b) N 1s XPS spectrum of NG-36, (c) Mn 2p XPS spectrum of NG-36, and (d–f) C 1s XPS spectra of GO, NG, and NG-36, respectively. | ||
Firstly, the carbons with some reactive groups (like carbonyl, carboxyl or hydroxyl) on GO can be readily oxidized by KMnO4 during the hydrothermal process as shown in (1). Rod-like MnOOH is formed as the product, which has been verified by XRD and electron microscopy of NG-0. When MnSO4 is involved, MnOOH can react with Mn2+ to produce tetragonal Mn3O4 as shown in (2). Reaction (2) is a thermodynamic controllable process, and long reaction time can facilitate conversion and growth from metastable MnOOH to thermodynamically stable Mn3O4. Some redox reaction between KMnO4 and graphene occurred during the long time of hydrothermal treatment. The conductive network was decomposed, resulting in deteriorated electrochemical performance of NG-36-12.
The chemical bonding states in as-prepared samples were analyzed using X-ray photoelectron spectroscopy (XPS) spectra, with the fitted data as shown in Fig. 4. As shown in Fig. 4a, for GO, only C 1s and O 1s signals were detected in the XPS spectra. After the N-doping, a new peak located at 400.0 eV was observed in both NG and NG-36. From the high resolution spectrum of N 1s (Fig. 4b), the sample mainly contains pyridinic N (N-6, 398.8 eV), pyrrolic N (N-5, 399.7 eV) and quaternary N (400.2 eV).24,26 The N content of NG and NG-36 is calculated to be 6.8 at% and 6.1 at%, respectively (Table S1†). These results further confirm that the N is incorporated into the graphene framework. These functional groups are supposed to improve the electrical conductivity and accelerate the charge transfer during cycling, as well as enhance binding between metal oxides and the carbon matrix, thus significantly enhancing the cycle stability and rate capability of LIBs. After the hydrothermal process, two new peaks appeared at ∼641.3 eV and ∼653.1 eV with a spin energy separation of ∼11.8 eV, which is in good agreement with reported data for Mn 2p1/2 and Mn 2p3/2 in MnxOy.28,33,34
As shown in Fig. S4,† GO has a C/O ratio of 2.3 and this value increases to 5.2 for NG after N-doping, implying the partial reduction of GO. The C/O ratio of NG-36 decreased to 2.9 after the hydrothermal process, which can be attributed to the result of the redox reaction among graphene, ethanediamine and KMnO4. In the high resolution C 1s spectrum of GO (Fig. 4d), four peaks are identified at 284.6 eV, 286.8 eV, 287.4 eV, and 289.2 eV, respectively, corresponding to C–C/![[C with combining macron]](https://www.rsc.org/images/entities/char_0043_0304.gif) C, C–O, O, and O–O.24,26,27 There was an obvious decrease in oxygen-containing functional groups after N doping (Fig. 4e). For NG and NG-36, new peaks appeared at ∼285.8 eV and 285.6 eV, respectively, which further confirm formation of the C–N bond during the nucleophilic reaction process.
C, C–O, O, and O–O.24,26,27 There was an obvious decrease in oxygen-containing functional groups after N doping (Fig. 4e). For NG and NG-36, new peaks appeared at ∼285.8 eV and 285.6 eV, respectively, which further confirm formation of the C–N bond during the nucleophilic reaction process.
When evaluated as an anode material in LIBs, NG-36 exhibits excellent electrochemical performance. Fig. 5a shows the cyclic voltammograms (CVs) of NG-36 for the 1st, 2nd, and 5th cycles within the potential range of 0.01–3.0 V vs. Li/Li+ at a scan rate of 0.2 mV s−1. There are four peaks for the first cathodic sweep of NG/M that could be assigned to reduction of manganite to manganous oxide (2.0 V) and reduction of manganese oxide to metallic manganese (1.15 V), formation of solid-electrolyte interphase (SEI) film (0.89 V) on the surface of electrode materials, and formation of Li2O (0.15 V).12,23,35–38 The peak located at about 1.3 V in the first anodic sweep corresponds to the oxidation of manganese.12 An additional peak at 2.1 V is observed when Li ions are discharged, which corresponds to further oxidation of MnO to a higher oxidation state with the aid of fast Li reaction kinetics and the synergistic effects of graphene and MnOx.23 Interestingly, for the next scans, the cathodic peak converts into two peaks located at 0.25 and 0.41 V, respectively. The former could be associated with reduction of Mn2+ to metallic Mn and the latter might originate from reduction of Mn2+ to metallic Mn with the assistance of LiOH, which is usually considered to be a “catalyst” in the cycling process and can increase the reducing voltage.36,37 There is no significant change in the potentials of the oxidation peaks at 2.1 V, corresponding to the lithium extraction process and further oxidation of MnO to a higher oxidation state. However, the oxidation peak at 1.3 V demonstrates a marked negative shift, probably resulting from intimate interaction between Mn-base materials and NG. Such a shift could reduce the electrode polarization and improve the charge-transfer kinetics, thus enhancing the cycling stability and rate capability of electrode materials, similar to findings observed in a previous study.12 It is worth noting that the anodic sweeps of the second and onward cycling CV curves remained almost unchanged, indicating a relatively stable discharge/charge process with good chemical reversibility. The whole reaction process can be written as:
| γ-MnOOH + Li+ + e− → MnO + LiOH | (3) |
| γ-MnOOH + 3Li+ + 3e− → Mn + Li2O + LiOH | (4) |
| MnO + 2Li+ + 2e− ↔ Mn + Li2O | (5) |
| Mn3O4 + 8Li+ + 8e− ↔ 3Mn + 4Li2O | (6) |
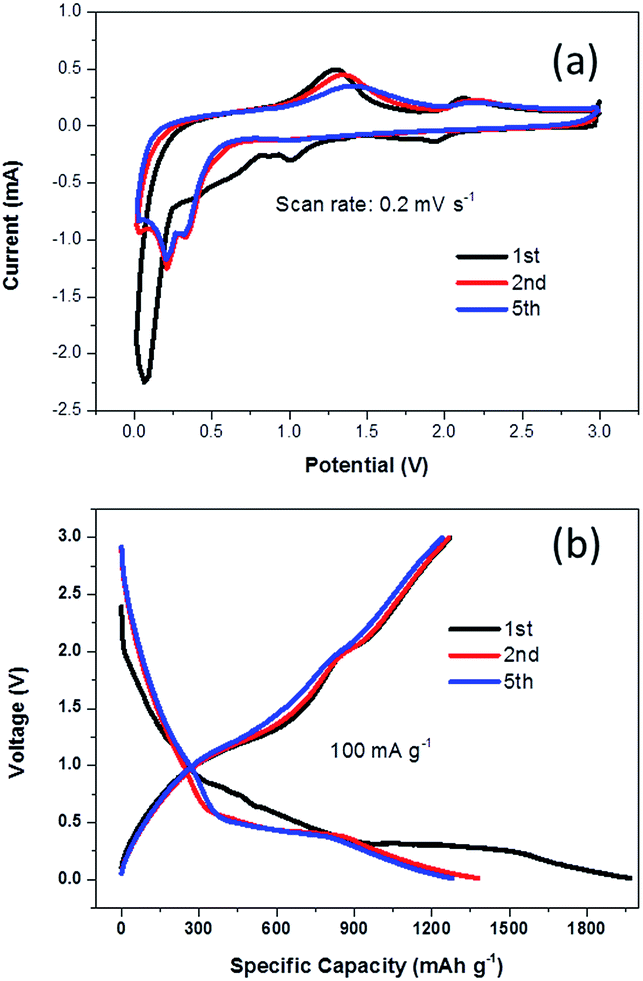 | ||
| Fig. 5 (a) Representative CV curves of the synthesized NG-36 electrode at a scan of 0.2 mV s−1 in the voltage range of 3–0.01 V vs. Li, (b) galvanostatic charge/discharge profile for the first, second, and fifth cycles of the NG/MnO2 electrode at a current density of 100 mA g−1. | ||
Fig. 5b shows the voltage versus capacity profiles of the NG-36 electrode for the 1st, 2nd, and 5th cycles at a current density of 100 mA g−1 within the same potential range. The initial discharge and charge capacities were 1967 mA h g−1 and 1270 mA h g−1, respectively, corresponding to an initial coulombic efficiency of ∼64.6%. Such capacity loss likely results from formation of a SEI layer and incomplete extraction of lithium from the active material. The discharge capacities of the 2nd and 5th cycles remain stable at 1379 mA h g−1 and 1279 mA h g−1, respectively, with the corresponding charge capacities of 1265 mA h g−1 and 1241 mA h g−1, respectively.
Fig. 6a shows cycling performance of NG-x at a current density of 1 A g−1 between 3.0 and 0.01 V. The discharge capacity of the NG-36 electrode in the second cycle is 618 mA h g−1, and the capacity is retained at 588 mA h g−1 after 140 cycles, which is much higher than that of the NG-0 and NG-36-12 anodes (277 mA h g−1 and 168 mA h g−1, respectively). Capacity retention is 95.2% for the NG-36 electrode, much better than those of NG-0 and NG-36-12 with retentions of 55.4% and 44.2%, respectively. Remarkably, even at a high current density of 3 A g−1, NG-36 still shows nearly 100% coulombic efficiency and excellent long-cyclic performance with higher capacity (342 mA h g−1) and retention (88.2%) compared with the NG-0 (98 mA h g−1, 20.9%) and NG/Mn3O4 electrodes (82 mA h g−1, 27.2%) after 500 cycles, as shown in Fig. 6b and S5.† To demonstrate the rate capabilities of these electrodes, the current density was changed from 100 to 3000 mA g−1. As shown in Fig. 6c, the NG-36 electrode shows better rate capability than do NG-0 and NG-36-12. The average discharge capacities for the NG-36 electrode are 1302, 1118, 876, 676, 543, and 395 mA h g−1, at current densities of 100, 200, 500, 1000, 2000, and 3000 mA g−1, respectively, indicating a high charge/discharge capability. Moreover, the capacity could recover close to original capacity at about 1128 mA h g−1 when the current density was reduced to 100 mA g−1, indicating the robustness structure of NG-36. The NG-0 electrode having higher cycle stability and rate performance than the NG-36-12 electrode implies that the MnOOH not only facilitates fast electron/ion transfer, but also interacts strongly with Mn3O4 and graphene to significantly enhance the rate performance and cyclic stability.
 | ||
| Fig. 6 (a) Cycling performance of the NG-0, NG-36, and NG-36-12 electrodes at a current density of 1 A g−1 for 140 cycles; (b) long-term cycling performance and coulombic efficiency of the NG-36 electrode at 3 A g−1 for 500 cycles; (c) rate performance of the NG-0, NG-36, and NG-36-12 electrodes at various current densities; (d) Nyquist plots of the NG-0, NG-36, and NG-36-12 electrodes before cycling from 100 kHz to 10 mHz. | ||
To further understand the kinetics of the electrochemical reactions for the as-prepared electrode materials, electrochemical impedance spectroscopy was carried out. As shown in Fig. 6d, the Nyquist plots of all three electrodes depict a semicircle in the high-medium frequency region corresponding to the ohmic resistance (Rs) and the charge transfer resistance (Rct) at the interface, and an inclined line located in the low frequency region representing the diffusion resistance.26 It can be seen clearly from Fig. 6d that the NG-0 electrode possesses the lowest electrical resistance compared with the NG-36 and NG-36-12 electrodes, indicating that MnOOH can facilitate fast electron/ion transfer, improving the electrochemical reaction kinetics, thus resulting in better rate capability. However, the low theoretical capacity limits its performance as anode. NG-36-12 possesses the highest electrical resistance (Fig. 6d) because of decomposition of the conductive network after long-term hydrothermal treatment with KMnO4.
4. Conclusion
In summary, N-doped graphene/MnOOH/Mn3O4 (NG-36) composites with dual-nanocrystal structure in a high electronic conductivity N-doped graphene matrix can be controllably synthesized via a facile solvothermal process. To further understand the relationship between chemical structure and electrochemical performance, various samples were prepared using different components and nanostructures. Among these samples, NG-36, which is composed of MnOOH nanowire, Mn3O4 nanotetragonals and N-GO, delivered the highest specific capacity, better cycling stability and outstanding rate performance. The experimental data indicate some synergetic effects among NG-36 components to achieve the improved performance: (1) the Mn3O4 nanoparticles in the composite could effectively coordinate Li+ ions to achieve high capacity. (2) MnOOH nanowires could provide proper voids between Mn3O4 nanoparticles to avoid aggregation, which is caused by alleviating the volume change during cycling. (3) The N-doped graphene acts as the conductive backbone to facilitate ion and electron diffusion, resulting in progressed rate performance. The material described herein has potential applications for facile preparation of an environmentally friendly, low cost and high performance anode for LIBs.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21572269, 21302224); Qingdao science and technology plan (15-9-1-108-jch) and the Fundamental Research Funds for Central Universities (15CX08005A).References
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- Z. Yang, J. Ren, Z. Zhang, X. Chen, G. Guan, L. Qiu, Y. Zhang and H. Peng, Chem. Rev., 2015, 115, 5159–5223 CrossRef CAS PubMed.
- K. Liang, T. Y. Cheang, T. Wen, X. Xie, X. Zhou, Z. W. Zhao and C. C. Shen, et al., J. Phys. Chem. C, 2016, 120, 3669–3676 CAS.
- Z. Li, Y. Wang, H. Sun, W. Wu, M. Liu, J. Zhou, G. Wu and M. Wu, J. Mater. Chem. A, 2015, 3, 16057–16063 CAS.
- M. Zhang, R. Li, X. Chang, C. Xue and X. Gou, J. Power Sources, 2015, 290, 25–34 CrossRef CAS.
- S. Qiu, G. Lu, J. Liu, H. Lyu, C. Hu, B. Li, X. Yan, J. Guo and Z. Guo, RSC Adv., 2015, 5, 87286–87294 RSC.
- Y. Fu, H. Gu, X. Yan, J. Liu, Y. Wang, J. Huang, X. Li, H. Lv, X. Wang, J. Guo, G. Lua, S. Qiu and Z. Guo, Chem. Eng. J., 2015, 227, 186–193 CrossRef.
- S. Qiu, H. Gu, G. Lu, J. Liu, X. Li, Y. Fu, X. Yan, C. Hu and Z. Guo, RSC Adv., 2015, 5, 46509–46516 RSC.
- Y. Sun, X. Hu, W. Luo, F. Xia and Y. Huang, Chem.–Eur. J., 2012, 23, 2433–2444 Search PubMed.
- Y. Wang, S. Wang, T. Zhao, Y. Chen, Z. Li, W. Wu and M. Wu, Chem. Eng. J., 2016, 306, 336–343 CrossRef CAS.
- Z. Cai, M. Yan, C. Han, L. He, K. M. Hercule, C. Niu, Z. Yuan, W. Xu, L. Qu, K. Zhao and L. Mai, Nano Lett., 2015, 15, 738–744 CrossRef CAS PubMed.
- N. Wang, J. Yue, L. Chen, Y. Qian and J. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 10348–10355 CAS.
- I. Nam, N. D. Kim, G. P. Kim, J. Park and J. Yi, J. Power Sources, 2013, 24, 56–62 CrossRef.
- C. Wang, L. Yin, D. Xiang and Y. Qi, ACS Appl. Mater. Interfaces, 2012, 4, 1636–1642 CAS.
- P. C. Li, C. C. Hu, H. Noda and H. Habazaki, J. Power Sources, 2015, 298, 102–113 CrossRef CAS.
- H. Zhong, Y. Yang, F. Ding, D. Wang, Y. Zhou and H. Zhan, Chem. Commun., 2015, 51, 164–6168 RSC.
- X. Lou, X. Wu and Y. J. Zhang, J. Alloys Compd., 2013, 550, 185–189 CrossRef CAS.
- H. Zhong, Y. Yang, F. Ding, D. Wang, Y. Zhou and H. Zhan, Chem. Commun., 2015, 51, 6164–6167 RSC.
- X. Fang, X. Lu, X. Guo, Y. Mao, Y. S. Hu, J. Wang, Z. Wang, F. Wu, H. Liu and L. Chen, Electrochem. Commun., 2010, 12, 1520–1523 CrossRef CAS.
- K. Ding, H. Gu, C. Zheng, L. Liu, L. Liu, L. Liu, X. Yan and Z. Guo, Electrochim. Acta, 2014, 146, 585–590 CrossRef CAS.
- J. Zhou, R. Xu, C. Yin, Z. Li, W. Wu and M. Wu, RSC Adv., 2016, 6, 62005–62010 RSC.
- Z. Li, G. Wu, S. Deng, S. Wang, Y. Wang, J. Zhou, S. Liu, W. Wu and M. Wu, Chem. Eng. J., 2016, 283, 1435–1442 CrossRef CAS.
- Y. Xu, X. Zhu, X. Zhou, X. Liu, Y. Liu, Z. Dai and J. Bao, J. Phys. Chem. C, 2014, 118, 28502–28508 CAS.
- F. Zheng, Y. Yang and Q. Chen, Nat. Commun., 2014, 5, 5261–5270 CrossRef CAS PubMed.
- S. Liu, Y. Dong, Z. Wang, H. Huang, Z. Zhao and J. Qiu, J. Mater. Chem. A, 2015, 3, 19659–19661 Search PubMed.
- Z. Wang, Y. Dong, H. Li, Z. Zhao, H. B. Wu, C. Hao, S. Liu, J. Qiu and X. W. Lou, Nat. Commun., 2014, 5, 5002–5009 CrossRef CAS PubMed.
- Z. Li, G. Wu, D. Liu, W. Wu, B. Jiang, J. Zheng, Y. Li, J. Li and M. Wu, J. Mater. Chem. A, 2014, 2, 7471–7477 CAS.
- L. Wang, Y. Li, Z. Han, L. Chen, B. Qian, X. Jiang, J. Pinto and G. Yang, J. Mater. Chem. A, 2013, 1, 8385 CAS.
- A. M. Rao, P. C. Eklund, S. Bandow, A. Thess and R. E. Smalley, Nature, 1997, 388, 257–259 CrossRef CAS.
- Z. Li, J. T. Zhang, Y. M. Chen, J. Li and X. W. Lou, Nat. Commun., 2015, 2, 8850–8857 CrossRef PubMed.
- X. Li, S. Xiong, J. Li, X. Liang, J. Wang, J. Bai and Y. Qian, Chem.–Eur. J., 2013, 19, 11310–11319 CrossRef CAS PubMed.
- S. Z. Huang, Y. Cai, J. Jin, J. Liu, Y. Li, Y. Yu, H. E. Wang, L. H. Chen and B. L. Su, Nano Energy, 2015, 12, 838–844 Search PubMed.
- J. Y. Liao, D. Higgins, G. lui, V. Chabot, X. Xiao and Z. Chen, Nano Lett., 2013, 13, 5467–5473 CrossRef CAS PubMed.
- L. Mai, F. Dong, X. Xu, Y. Luo, Q. An, Y. Zhao, J. Pan and J. Yang, Nano Lett., 2013, 13, 740–745 CrossRef CAS PubMed.
- G. Zhao, X. Huang, X. Wang, P. Connor, J. Li, S. Zhang and T. S. Irvine, J. Mater. Chem. A, 2015, 3, 297–303 CAS.
- S. Ni, X. Lv, T. Li, X. Yang and L. Zhang, J. Mater. Chem. A, 2013, 1, 1544–1547 CAS.
- H. Zhong, Y. Yang, F. Ding, D. Wang, Y. Zhou and H. Zhan, Chem. Commun., 2015, 51, 6164–6167 RSC.
- K. Zhang, P. Han, L. Gu, L. Zhang, Z. Liu, Q. Kong, C. Zhang, S. Dong, Z. Zhang, J. Xu, H. Yao, G. Cui and L. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 658–664 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: XRD patterns, FT-IR spectrum and the C/O ratio of GO, NG, and NG-36; the cycling performance NG-0 and NG-36 at a current density of 3 A g−1. See DOI: 10.1039/c6ra27297e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |