
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Titanium tetrachloride-mediated synthesis of N-aryl-substituted azacycles from cyclic ethers†
Zunming Sun,
Shanshan Hu,
Yan Huo and
Zhihong Wang *
*
State Key Laboratory and Institute of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: zhihongwang@nankai.edu.cn
First published on 16th January 2017
Abstract
Titanium tetrachloride-mediated transformation of five- and six-membered cyclic ethers to the corresponding N-aryl-substituted azacycles is conducted in moderate to good yields under mild reaction conditions. Computational studies suggested a mechanism involving a Lewis acid-assisted ring-opening, a seven-membered metallacycle intermediate and a ring-closing process facilitated by direct participation of the metal center.
Transition metals have been making great contributions to the development of organic chemistry over the past decades. While the precious and exiguous metals such as Pd, Rh, Ir, etc. assert their superiority in high activity, earth-abundant metals have proven their value in both capability and availability. Compared with substantial studies on the chemistry of Fe, Ni, and Cu, investigations on titanium, the second most abundant transition metal in the Earth's crust, seem insufficient and “this relative lack of diversity is largely due to the need for further development rather than an inherent lack of utility”.1 Considering the existing proof of the application value of this abundant metal,2,3 the chemistry of titanium deserves more attention, both in reactivity and mechanistic studies. Seen from a mechanistic aspect, reaction pathways involving titanium-containing metallacyclic intermediates are of special interest since in those cases the metal center are usually particular, or even unsubstitutable. The formation of those intermediates concerned is in most cases from unsaturated molecules while saturated substrates are rarely used,4–8 which inspired us to explore the possibility that titanium assists the reaction of saturated molecules via the participation of metallacyclic intermediates. Herein we report the titanium-mediated transformation of five- and six-membered cyclic ethers to corresponding N-aryl-substituted azacycles.
N-substituted azacycles are important structural motifs as well as useful building blocks in organic molecules and a substantial amount of work has been done on their synthesis. Besides commonly-used methods employing dihalides or diols with primary amines,9 or using halides with N-unsubstituted azacycles,10 synthesis of N-substituted azacycles from primary amines and cyclic ethers is valuable due to the formation of water as the only waste product as well as wide availability of starting materials.11,12 The main shortcoming of this method is the requirement of drastic reaction temperature in traditional cases, while new mediators and catalysts have been exploited to facilitate the reaction, such as the introduction of dimethylaluminum-amide reagents13 and the approach via B(C6F5)3-mediated frustrated Lewis pair pathway.14
As part of our ongoing studies on titanium-mediated C–N bond formation reactions,15 we anticipated based on the existing results that the interaction between titanium reagent and primary aryl amines, when cooperated with the capability of titanium to serve as Lewis acid and promote the ring-opening of cyclic ethers,16 would be able to incorporate primary amine and cyclic ether in a metallacyclic intermediate and facilitate the C–N bond formation to provide N-substituted azacycles.
To test our anticipation, initial reaction condition screening was carried out using aniline 1a and THF 2a in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio, giving product 3a in a yield of 36% after refluxing in toluene for 24 h (Table 1, entry 1). When ten-fold excess of THF was used to make up for the loss caused by the volatility of THF, the yield was augmented to 64% (Table 1, entry 2) while a mild increase in the amount of titanium tetrachloride used also slightly improved the yield (Table 1, entry 3). However, further increase in the amount of TiCl4 or THF used did not have noticeable effect on the yield. Either pure TiCl4 liquid or its solution in dichloromethane would efficiently facilitate the reaction. The existence of titanium tetrachloride as Lewis acid turned out to be crucial while the reaction did not occur in the absence of TiCl4 (Table 1, entry 4) or when some other common Lewis acids were employed (Table 1, entry 5–10), including strongly Lewis acidic AlCl3 (Table 1, entry 6).
:2 molar ratio, giving product 3a in a yield of 36% after refluxing in toluene for 24 h (Table 1, entry 1). When ten-fold excess of THF was used to make up for the loss caused by the volatility of THF, the yield was augmented to 64% (Table 1, entry 2) while a mild increase in the amount of titanium tetrachloride used also slightly improved the yield (Table 1, entry 3). However, further increase in the amount of TiCl4 or THF used did not have noticeable effect on the yield. Either pure TiCl4 liquid or its solution in dichloromethane would efficiently facilitate the reaction. The existence of titanium tetrachloride as Lewis acid turned out to be crucial while the reaction did not occur in the absence of TiCl4 (Table 1, entry 4) or when some other common Lewis acids were employed (Table 1, entry 5–10), including strongly Lewis acidic AlCl3 (Table 1, entry 6).
|
|||
|---|---|---|---|
| Entry | 1a:2a |
Lewis acid | Isolated yieldb (%) |
| a All reactions were carried out on a 1 mmol scale.b Isolated yield based on aniline. | |||
| 1 | 1:2 |
TiCl4, 1 eq. | 36 |
| 2 | 1:10 |
TiCl4, 1 eq. | 64 |
| 3 | 1:10 |
TiCl4, 1.2 eq. | 68 |
| 4 | 1:10 |
None | NR |
| 5 | 1:10 |
Ti(OiPr)4, 1.2 eq. | NR |
| 6 | 1:10 |
AlCl3, 1.2 eq. | NR |
| 7 | 1:10 |
FeCl3, 1.2 eq. | NR |
| 8 | 1:10 |
InCl3, 1.2 eq. | NR |
| 9 | 1:10 |
ZnCl2, 1.2 eq. | NR |
| 10 | 1:10 |
SnCl4, 1.2 eq. | NR |

The scope of the reaction was then examined using the optimized reaction conditions, giving products in moderate to high yields (Scheme 1). Similar to the trend in the conversion by dimethylaluminum-amide reagents,13 electron-withdrawing halogen substituents on the aryl ring of the amines are conducive to the reaction (3b–3d), but substitution on the ortho-position is disadvantageous due to steric hindrance (3e, 3f), while electron-withdrawing substituent like nitro group (3g) and electron-donating substituent like methyl (3h) are also tolerable. Being different from the marked adverse effect of steric hindrance of 2-methyltetrahydrofuran in the B(C6F5)3-mediated synthesis,14 herein substituent on the cyclic ether did not affect the reaction substantially (3i–3k), providing N-aryl-2-methylpyrrolidines in satisfactory yields. Compared to the formation of substituted pyrrolidines, the conversion of tetrahydropyran to the corresponding 6-membered azacycles was reported to be much more challenging or even inaccessible.14 To our great delight, the titanium tetrachloride-mediated reaction herein works pretty well in this conversion when the reaction temperature was raised to the refluxing temperature of xylenes, giving N-arylpiperidines in yields up to 80% (3l–3o). Nitrogen-containing fused heterocyclic rings such as isoindolines and tetrahydroisoquinolines are also attainable by this synthetic method (3p–3r), but sometimes being accompanied by some side reactions which may be caused by the decomposition of the oxacyclic substrates under the reaction conditions.
 | ||
| Scheme 1 Scope of the reaction.a,b [a] All reactions were carried out on a 1 mmol scale at 110 °C in toluene for 5-membered cyclic ethers and at 140 °C in xylenes for 6-membered cyclic ethers. [b] Isolated yield based on amines. | ||
An intriguing fact that made us curious was the invalidity of strongly Lewis acidic AlCl3 in promoting the conversion while both Lewis acidic TiCl4 we used here and the dimethylaluminum-amide reagents generated in situ from AlMe3 could facilitate the reaction.13 To gain clues about the mechanistic details in the reaction and understand which step prevented the AlCl3-mediated conversion from happening, computational investigations were carried out while kinetic studies were done to examine the reliability of calculations.
Four possible pathways were considered in the calculational studies to gain understanding of the effect of the involvement of titanium-amide reagent (pathway A, Fig. 1), or titanium imido intermediate (pathway B), the participation of coordinated THF (pathway C), and the direct Lewis acid-promoted ring opening of oxacyclic substrate (pathway D). Comparison among the energetic requirements of the crucial transition states in each pathway suggested an energetically-favored mechanism starting with Lewis acid–base complex D. The nucleophilic attack at the α-position of the activated oxacycle and the following loss of a molecule of HCl afforded a seven-membered metallacyclic intermediate 1A. This Lewis acid-promoted ring opening of oxacyclic substrate served as the rate-determining step, with a requirement of Gibbs free energy of 26.9 kcal mol−1. The transition state for the nucleophilic attack with stereochemical inversion (TS 1D) had similar energetic requirement to that for the attack with stereochemical retention (TS 1X, see ESI†), barely suggesting stereochemical preference in this step. 1A could be in an equilibrium with an open-chain structure while 1A was slightly favored in energy by 0.8 kcal mol−1. The subsequential ring closing in the intermediate occurred via a four-membered transition state, in which the second nucleophilic attack at the α-carbon of the oxygen rendered the formation of another C–N bond. This step was assisted by the participation of titanium metal center which drew the reaction moieties closer as well as activated the oxacycle. The energy of the ring-closing transition state was about 10 kcal mol−1 lower than that of the rate-determining ring-opening step. The ring closing was followed by leaving of another molecule of HCl to give the pyrrolidine-coordinated titanium complex which would release the final product in aqueous work-up.
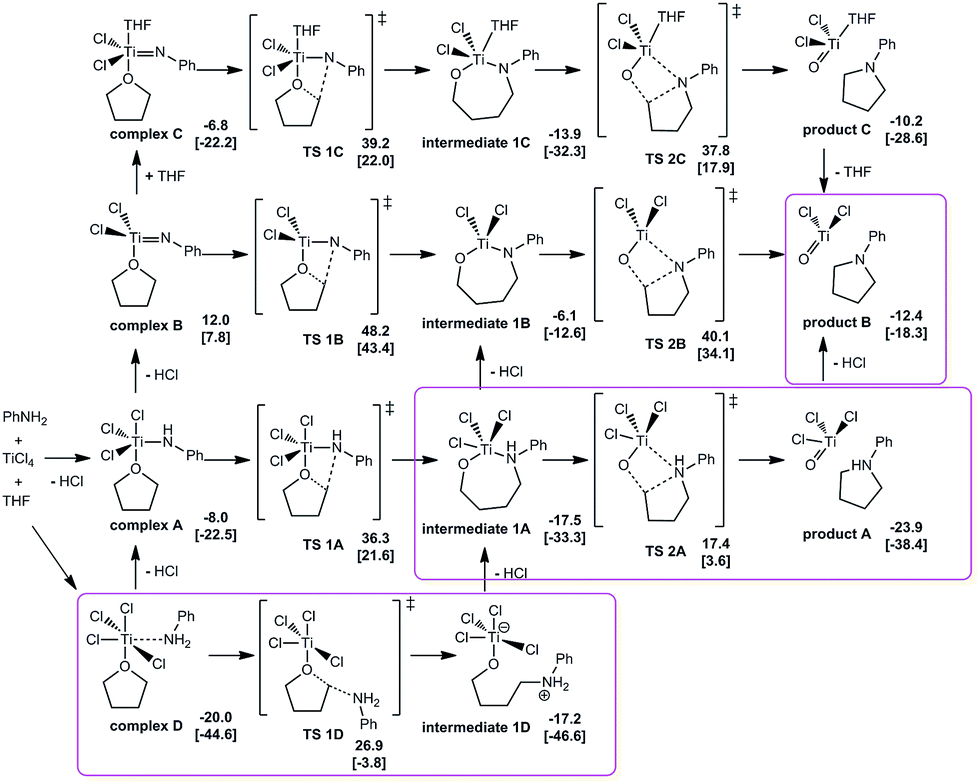 | ||
| Fig. 1 Calculations on plausible mechanism for TiCl4-mediated conversion of THF to pyrrolidine. Gibbs free energies are given in kcal mol−1 and the relative electronic energies (with ZPE) are shown in square brackets. | ||
A preliminary kinetic study was conducted for the comparison of the proposed mechanism with experimental results (Fig. 2). The reaction of 4-fluoroaniline was selected in the study owing to high yield of this substrate as well as simplification of NMR. The change in concentration exhibited pseudo-first order reaction behavior, suggesting a rate constant of 5 × 10−5 s−1 which implied an activation energy of 30 kcal mol−1 according to Eyring–Polanyi equation, which qualitatively matched with the energy requirement in the proposed mechanism.
 | ||
| Fig. 2 Concentration change of 4-fluoroaniline over time and the fitted value of pseudo-first order rate constant in the preliminary kinetic study. | ||
Based on the above calculational studies, comparisons were made among the energy profiles of some related reactions, trying to understand the mechanistic difference between the titanium-mediated and aluminium-mediated conversion of THF to pyrrolidine (Fig. 3). Reaction mediated by another commonly-used titanium Lewis acid Ti(OiPr)4 had similar energy profile to the TiCl4-mediated reaction, but with a much higher transition state energy mainly due to weaker Lewis acidity. As expected, stronger Lewis acid lowered the energy requirement for the ring-opening of oxacycle, while activation by AlCl3 made the process barrierless. But the aluminium center lacked the capability to draw the reaction moieties closer in a four-membered transition state in the ring-closing step, probably due to smaller size and lower degree of participation of d orbitals, thus the energy of the transition state was raised and rate-determining step switched to the ring-closing process. In the AlCl3-mediated case this energy requirement was as high as 42 kcal mol−1, which prevented the reaction from happening as experimentally observed.
 | ||
| Fig. 3 Energy profiles of TiCl4-mediated (black), Ti(OiPr)4-mediated (blue), AlCl3-mediated (red) and AlMe3-mediated (green) conversion of THF to pyrrolidine. Gibbs free energies are given in kcal mol−1. | ||
Conclusions
In summary, a titanium tetrachloride-mediated reaction was developed, employing non-expensive conveniently-handled titanium tetrachloride as metallic reagent, effectively converting five- and six-membered cyclic ethers to corresponding N-aryl-substituted azacycles. Calculational investigations and preliminary kinetic studies supported a mechanism consisting of a rate-determining Lewis acid-promoted ring opening of activated oxacyclic substrate, the formation of a seven-membered metallacyclic intermediate, and the ring-closing of this intermediate with the assistance of titanium metal center. The aluminium-amide-mediated reaction in literature was suggested to occur in similar processes, with ring-closing as the rate-determining step. Further investigations are ongoing in our lab to explore the addition of multiple bonds to the intermediate, which may render the ring expansion of the metallacycles.Experimental section
Synthesis of N-phenylpyrrolidine (3a)
A solution of 0.13 mL (1.2 mmol) of TiCl4 and 0.10 mL (1 mmol) of aniline in 4 mL of dry toluene was stirred at room temperature for 30 minutes. To the mixture was added 1.0 mL (10 mmol) of THF and the temperature was slowly elevated to 110 °C. After refluxing for 24 h, 10 mL of saturated NaHCO3 solution and 10 mL of dichloromethane were added and stirred for 1 h. The mixture was extracted with 3 × 10 mL of dichloromethane and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was isolated by column chromatography on silica using CH2Cl2/hexanes (1:4 v/v) as eluent to give yellow liquid (100 mg, yield 68%).
Kinetic experiments
The reaction of 4-fluoroaniline with THF was selected in the kinetic study for the simplification of NMR. Diphenylmethane was added into the reaction mixture as an internal standard for integral. To a solution of 0.13 mL (1.2 mmol) of TiCl4 in 8 mL of dry toluene was added 0.10 mL (1.0 mmol) of 4-fluoroaniline. The mixture was stirred at room temperature for 0.5 h and 2.0 mL (20 mmol) of THF was added to gain a condition of pseudo-first order reaction. The well-mixed solution was then divided into four portions for parallel experiments that were terminated after 1 h, 2 h, 3 h and 4 h respectively. After refluxing for a certain period of time, the solution was quenched with saturated NaHCO3 solution, extracted with dichloromethane and dried over Na2SO4, and the solvent was removed under reduced pressure. Proton spectra were obtained under consistent conditions and the integral of the signals at 6.95 and 6.48 ppm were adopted to measure the amount of unreacted 4-fluoroaniline, using diphenylmethane as the internal standard for integral. The concentration of 4-fluoroaniline at different reaction time were used to gain the first order rate constant.Acknowledgements
The project is funded by the Natural Science Foundation of Tianjin, China (14JCYBJC19700).Notes and references
- A. L. Odom and T. J. McDaniel, Acc. Chem. Res., 2015, 48, 2822–2833 CrossRef CAS PubMed.
- For reviews, see: (a) H. Pellissier, Adv. Synth. Catal., 2015, 357, 857–882 CrossRef CAS; (b) A. L. Odom, Dalton Trans., 2005, 225–233 RSC; (c) P. J. Walsh, Acc. Chem. Res., 2003, 36, 739–749 CrossRef CAS PubMed.
- For recent examples, see: (a) J. Streuff, M. Feurer, G. Frey, A. Steffani, S. Kacprzak, J. Weweler, L. H. Leijendekker, D. Kratzert and D. A. Plattner, J. Am. Chem. Soc., 2015, 137, 14396–14405 CrossRef CAS PubMed; (b) J. Schlüter, M. Blazejak, F. Boeck and L. Hintermann, Angew. Chem., Int. Ed., 2015, 54, 4014–4017 CrossRef PubMed; (c) R. Sun, J. Liu, S. Yang, M. Chen, N. Sun, H. Chen, X. Xie, X. You, S. Li and Y. Liu, Chem. Commun., 2015, 51, 6426–6429 RSC; (d) T. Roth, H. Wadepohl, E. Clot and L. H. Gade, Chem.–Eur. J., 2015, 21, 18730–18738 CrossRef CAS PubMed; (e) J. Joseph, P. Prethalayam, K. V. Radhakrishnan, F. Haroschik and J.-L. Vasse, Org. Lett., 2015, 17, 6202–6205 CrossRef CAS PubMed; (f) H.-T. Luu, S. Wiesler, G. Frey and J. Streuff, Org. Lett., 2015, 17, 2478–2481 CrossRef CAS PubMed; (g) M. Schrempp, S. Thiede, D. Herkommer, A. Gansäuer and D. Menche, Chem.–Eur. J., 2015, 21, 16266–16271 CrossRef CAS PubMed.
- (a) H. Mizoguchi and G. C. Micalizio, J. Am. Chem. Soc., 2015, 137, 6624–6628 CrossRef CAS PubMed; (b) N. D. Kjeldsen, E. D. Funder and K. V. Gothelf, Org. Biomol. Chem., 2014, 12, 3679–3685 RSC; (c) S. N. Greszler, H. A. Reichard and G. C. Micalizio, J. Am. Chem. Soc., 2012, 134, 2766–2774 CrossRef CAS PubMed; (d) H. A. Reichard and G. C. Micalizio, Angew. Chem., Int. Ed., 2007, 46, 1440–1443 CrossRef CAS PubMed; (e) J. Ryan and G. C. Micalizio, J. Am. Chem. Soc., 2006, 128, 2764–2765 CrossRef CAS PubMed; (f) H. Urabe, K. Mitsui, S. Ohta and F. Sato, J. Am. Chem. Soc., 2003, 125, 6074–6075 CrossRef CAS PubMed; (g) S. Okamoto, K. Subburaj and F. Sato, J. Am. Chem. Soc., 2001, 123, 4857–4858 CrossRef CAS PubMed.
- (a) Z. W. Gilbert, R. J. Hue and I. A. Tonks, Nat. Chem., 2016, 8, 63–68 CrossRef CAS PubMed; (b) C. M. Pasko, A. A. Dissanayake, B. S. Billow and A. L. Odom, Tetrahedron, 2016, 72, 1168–1175 CrossRef CAS; (c) V. Jeso, C. Aquino, X. Cheng, H. Mizogushi, M. Nakashige and G. C. Micalizio, J. Am. Chem. Soc., 2014, 136, 8209–8212 CrossRef CAS PubMed; (d) J. C.-H. Yim, J. A. Bexrud, R. O. Ayinda, D. C. Leitch and L. L. Schafer, J. Org. Chem., 2014, 79, 2015–2028 CrossRef CAS PubMed; (e) H. Fan, A. R. Fout, B. C. Beuley, M. Pink, M.-H. Baik and D. J. Mindiola, Dalton Trans., 2013, 42, 4163–4174 RSC; (f) I. A. Tonks, J. C. Meier and J. E. Bercaw, Organometallics, 2013, 32, 3451–3457 CrossRef CAS; (g) E. Barnea, S. Majumder, R. J. Staples and A. L. Odom, Organometallics, 2009, 28, 3876–3881 CrossRef CAS.
- (a) D. Yang and G. C. Micalizio, J. Am. Chem. Soc., 2012, 134, 15237–15240 CrossRef CAS PubMed; (b) M. Takahashi and G. C. Micalizio, Chem. Commun., 2010, 46, 3336–3338 RSC; (c) S. Umemura, M. McLaughlin and G. C. Micalizio, Org. Lett., 2009, 11, 5402–5405 CrossRef CAS PubMed.
- (a) F. Hermant, E. Nicolas and Y. Six, Tetrahedron, 2014, 70, 3924–3930 CrossRef CAS; (b) C. Madelaine, A. K. Buzas, J. A. Kowalska-Xis, Y. Six and B. Crousse, Tetrahedron Lett., 2009, 50, 5367–5371 CrossRef CAS; (c) J. Lee, H. Kim and J. K. Cha, J. Am. Chem. Soc., 1996, 118, 4198–4199 CrossRef CAS.
- G. Fan and Y. Liu, Tetrahedron Lett., 2012, 53, 5084–5087 CrossRef CAS.
- (a) X. Cui, X. Dai, Y. Deng and F. Shi, Chem.–Eur. J., 2013, 19, 3665–3675 CrossRef CAS PubMed; (b) G. Marzaro, A. Guiotto and A. Chilin, Green Chem., 2009, 11, 774–776 RSC; (c) C. B. Singh, V. Kavara, A. K. Samal and B. K. Patel, Eur. J. Org. Chem., 2007, 1369–1377 CrossRef CAS; (d) Y. Ju and R. S. Varma, J. Org. Chem., 2006, 71, 135–141 CrossRef CAS PubMed; (e) Y. Ju and R. S. Varma, Org. Lett., 2005, 7, 2409–2411 CrossRef CAS PubMed.
- (a) M. L. H. Mantel, A. T. Lindhardt, D. Lupp and T. Skrydstrup, Chem.–Eur. J., 2010, 16, 5437–5442 CrossRef CAS PubMed; (b) Y.-C. Teo and G.-L. Chua, Chem.–Eur. J., 2009, 15, 3072–3075 CrossRef CAS PubMed; (c) B. Sreedhar, R. Arundhathi, P. L. Reddy and M. L. Kantam, J. Org. Chem., 2009, 74, 7951–7954 CrossRef CAS PubMed; (d) K. Swapna, A. V. Kumar, V. P. Reddy and K. R. Rao, J. Org. Chem., 2009, 74, 7514–7517 CrossRef CAS PubMed; (e) C.-Y. Gao and L.-M. Yang, J. Org. Chem., 2008, 73, 1624–1627 CrossRef CAS PubMed.
- (a) D. C. Hargis and R. L. Shubkin, Tetrahedron Lett., 1990, 31, 2991–2994 CrossRef CAS; (b) R. E. Walkup and S. Searles, Tetrahedron, 1985, 41, 101–106 CrossRef CAS; (c) C. J. Olsen and A. Furst, J. Am. Chem. Soc., 1953, 75, 3026–3026 CrossRef CAS.
- Z. Amara, E. S. Streng, R. A. Skilton, J. Jin, M. W. George and M. Poliakoff, Eur. J. Org. Chem., 2015, 6141–6145 CrossRef CAS.
- B. L. Korbad and S.-H. Lee, Chem. Commun., 2014, 50, 8985–8988 RSC.
- Z. Zhang, C. Miao, C. Xia and W. Sun, Org. Lett., 2016, 18, 1522–1525 CrossRef CAS PubMed.
- (a) C. Cheng, D. Chen and Z. Wang, Chem.–Eur. J., 2013, 19, 1204–1208 CrossRef CAS PubMed; (b) Z. Sun, Q. Wang, Y. Xu and Z. Wang, RSC Adv., 2015, 5, 84284–84289 RSC.
- A. C. Gottfried, J. Wang, E. E. Wilson, L. W. Beck, M. M. B. Holl and J. W. Kampf, Inorg. Chem., 2004, 43, 7665–7670 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, full citation of Gaussian 09 and computational details. See DOI: 10.1039/c6ra27325d |
| This journal is © The Royal Society of Chemistry 2017 |