
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnetic Fe3O4@poly(propylene fumarate-co-ethylene glycol) core–shell biomaterials
Ana M. Díez-Pascual *a and
Angel L. Díez-Vicenteb
*a and
Angel L. Díez-Vicenteb
aAnalytical Chemistry, Physical Chemistry and Chemical Engineering Department, Faculty of Biology, Environmental Sciences and Chemistry, Alcalá University, E-28871 Alcalá de Henares, Madrid, Spain. E-mail: am.diez@uah.es; Fax: +34 91 8855088; Tel: +34 91 8856430
bAirbus Operations S. L., John Lennon s/n, 28906, Getafe, Madrid, Spain
First published on 6th February 2017
Abstract
Core–shell composites comprising a core of amino-functionalized magnetite microparticles (MagP®-NH2) covalently attached to a poly(propylene fumarate-co-ethylene glycol) P(PF-co-EG) network shell have been developed and characterized. The diameter of the core–shell structure was found to decrease while the grafting degree increased as the MagP®-NH2/(P(PF-co-EG)) ratio increased. The composites showed superparamagnetic behaviour, and those with thinner shells displayed higher magnetic susceptibility values. The microparticles significantly improved the chemical stability, biodegradability and antibacterial activity of the copolymer. Cell metabolic activity levels after exposure to the composites decreased with increasing microparticle content and incubation period. Very strong increments in stiffness and strength were attained, both under dry conditions and after soaking in a simulated body fluid. It has been found that the morphology, hence the final composite properties, can be finely tuned by varying the microparticle/copolymer weight ratio, the optimal being 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90, given that it leads to the best combination of magnetic, mechanical, rheological and antibacterial properties. These novel biocomposites also showed magnetic-field tunable mechanical properties, hence could be further used for the design of magnetic responsive scaffolds for soft-tissue replacement whose properties can be triggered by external stimuli.
:90, given that it leads to the best combination of magnetic, mechanical, rheological and antibacterial properties. These novel biocomposites also showed magnetic-field tunable mechanical properties, hence could be further used for the design of magnetic responsive scaffolds for soft-tissue replacement whose properties can be triggered by external stimuli.
Introduction
Hybrid core–shell composites have recently attracted much interest since they can be carefully designed to integrate the specific characteristics of each building block into a single multifunctional unit suitable for a number of applications ranging from catalysis, energy conversion and environmental remediation to optoelectronics and biomedicine.1–3 Among them, magnetic composites are particularly interesting, because of their magnetic responsiveness and tailorable surface. The magnetic properties of these materials allow them to be guided by non-contact forces, like an external magnetic field, and simultaneously, in situ monitoring by magnetic resonance imaging or computerized axial tomography scanning.4 Furthermore, they can be bonded to biomolecules such peptides, antibodies, drugs, etc., hence they have great potential to be used in protein purification, cell separation, medical diagnosis and targeted drug delivery.5–7Magnetite (Fe3O4) is one of the most important transition magnetic metal oxides owed to its strong magnetic properties and widespread applications in biotechnology and medicine. In particular, Fe3O4 microparticles have smooth and large-surface areas that can be used for binding proteins, enzymes, antibodies, and anticancer agents.8 They are chemically stable, nontoxic, and noncarcinogenic, and can be detected in the human body by magnetopneumographic (MPG) methods. Many techniques have been reported in the literature to synthesize Fe3O4 microparticles such as the sol–gel, microemulsion, sonochemical, ultrasonic spray pyrolysis, microwave plasma, thermal decomposition and the chemical reduction methods.9–11 A serious problem is their strong tendency to agglomerate, which is an obstacle to their potential biomedical applications. This hindrance can be overcome by subjecting the particles to chemical modification; thus, it has been demonstrated that an adequate functionalization of magnetic micro/nanoparticles enhances their colloidal stability, preventing their aggregation in solution and improving their chemical stability.12
Amongst different biodegradable synthetic polymers, poly(propylene fumarate) (PPF) has recently emerged as an important biocompatible and cross-linkable polymer for tissue engineering applications.13 This unsaturated linear polyester can be thermally or photochemically cross-linked via its fumarate double bonds, leading to a crosslinked network that satisfies a number of medical requirements such as biocompatibility, osteoconductivity, sterilizability, and handling characteristics.14–16 Further, it degradates by hydrolysis of the ester bonds, and the degradation products are non-toxic fumaric acid and propylene glycol. However, its lack of antibacterial activity and mechanical strength limit its use for certain biomedical applications. To provide antimicrobial action and improve its mechanical properties, efforts have been made in creating improved PPF-based materials by careful modulation of the polymer composition17 or addition of fillers.18,19 Polyethylene glycol (PEG), a highly hydrophilic biocompatible polyether extensively employed for the fabrication of biomaterials, has been covalently bound to PPF.20 Incorporation of PEG into PPF makes it water soluble, improves its biocompatibility and also decreases its thrombogenicity and platelet adhesion for cardiovascular applications; besides, the mechanical properties of the resulting copolymer, poly(propylene fumarate-co-ethylene glycol) (P(PF-co-EG)), can be tuned by varying the ratio between the hydrophilic PEG and hydrophobic PPF blocks.21 Nevertheless, to the best of our knowledge, P(PF-co-EG) has not been used so far for the development of core–shell composites.
In this work we describe a novel approach to synthesize core–shell structures comprising a core of amino-functionalized magnetite microparticles covalently attached to a P(PF-co-EG) network shell. The resulting core–shell biocomposites have been comprehensively analyzed to gain insight about their morphology, thermal and chemical stability, biodegradability, metabolic activity, water uptake, rheological, mechanical, magnetic and antibacterial properties. It has been found that the morphology, hence the final composite properties, can be finely tuned by varying the microparticle/copolymer weight ratio. Altogether, our results suggest that there is an optimal microparticle/copolymer weight ratio, 10:90, which leads to the best combination of properties. These novel biomaterials could be further used for the preparation of magnetic field-responsive engineered tissues whose properties can be controlled by external magnetic forces.
Experimental
Materials
MagP®-NH2, magnetite (Fe3O4) microparticles coated with a primary amine functionalized polyurethane shell, were kindly supplied by NanoMyP® (Spain). The particles have a mean diameter of 3 μm, a total amination degree of 350 μmol NH2 per g and a surface density of accessible NH2 of 10 μmol NH2 per g, and were stored at 5 °C prior to use. Diethyl fumarate (DEF) was provided by Acros Organics. Polyethylene glycol bis(carboxy methyl ether) (PEG–COOH), Mw = 600 g mol−1, was provided by Sigma-Aldrich and purified by azeotropic distillation from anhydrous benzene solution before use. N-Hydroxysuccinimide (NHS), from Aldrich, was recrystallized from an ethyl acetate/ethanol mixture and dried under vacuum prior to use. Thionyl chloride (SOCl2), N,N-dicyclohexylcarbodiimide (DCC), hydroquinone, N-vinyl-pyrrolidone (NVP), ZnCl2, benzoyl peroxide (BP), N-dimethyl-p-toluidine (DMT) and propylene glycol (PG) were also purchased from Sigma-Aldrich and used as received. All solvents used (benzene, ethyl acetate, diethyl ether, dimethylformamide (DMF)), from Aldrich, were distilled under vacuum and then dried for a few days with a Merck 4 Å molecular sieve.Synthesis of P(PF-co-EG) copolymer network
The copolymer was synthesized following a multi-stage process (Scheme 1). Firstly, a crosslinked PPF network was synthesized as reported previously18 by mixing DEF and PG in a molar ratio of 1:3 at 130 °C for 10 h, using ZnCl2 and hydroquinone as a catalyst and crosslinking inhibitor, respectively, and subsequently heated to 160 °C for 8 h. PPF was then crosslinked via free radical polymerization using NVP, BP and DMT as crosslinker, free radical initiator and accelerator, respectively.
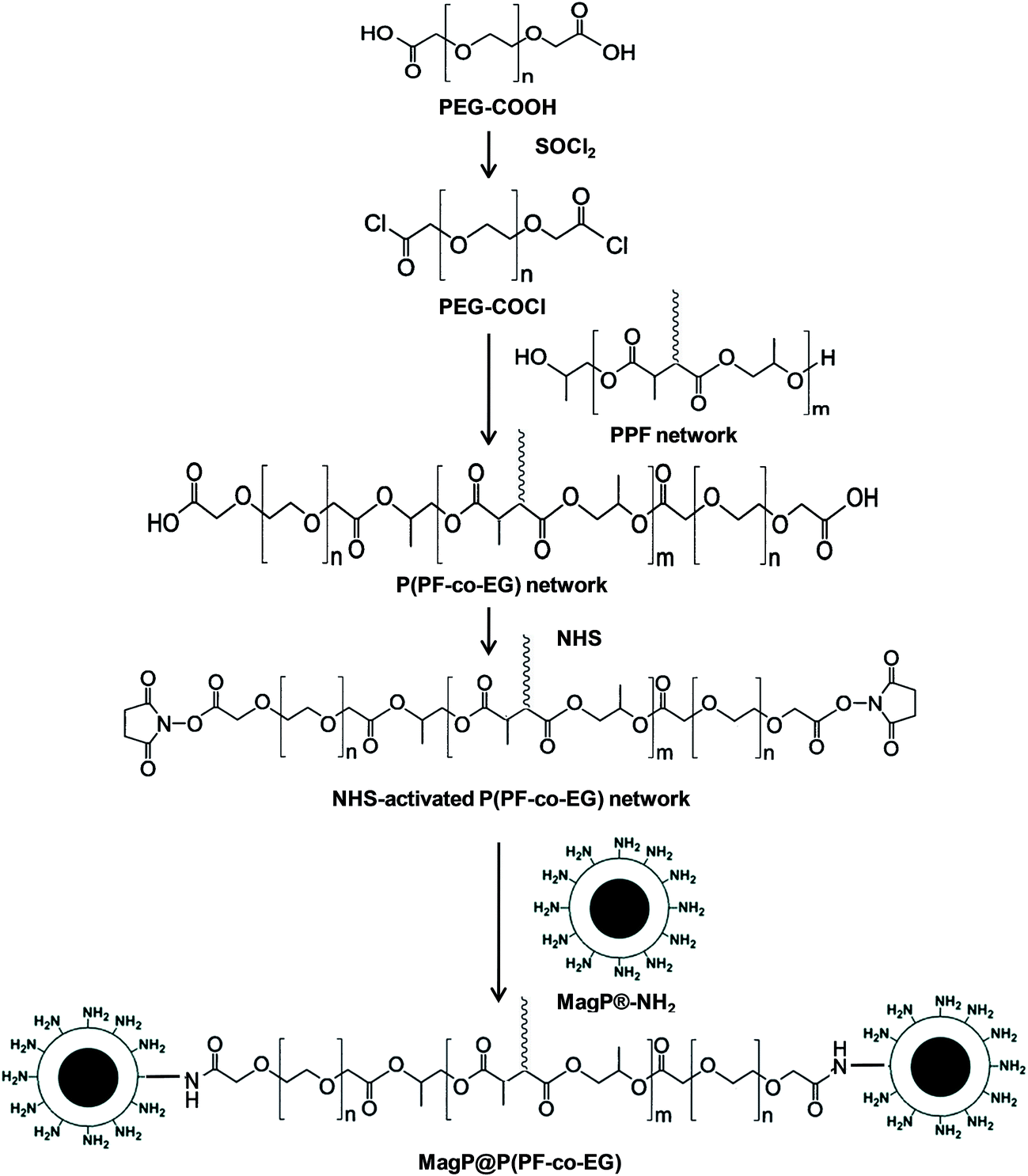 | ||
| Scheme 1 Synthesis procedure of MagP@P(PF-co-EG) core–shell composites. | ||
Separately, PEG–COOH (30 g) was dissolved in 150 mL anhydrous benzene. Subsequently, an excess of SOCl2 and three drops of DMF were added, and the reaction was carried out at 80 °C for 12 h under constant stirring. The residual SOCl2 was removed by reduced pressure distillation to yield the acyl-chloride terminated (PEG–COCl).
A solution containing the PPF network (20 g) dissolved in 100 mL anhydrous DMF was then added dropwise to a 30% (w/v) solution of PEG–COCl in anhydrous DMF placed in an ice bath. The reaction was performed at room temperature for 20 h under inert atmosphere. The product, P(PF-co-EG) network, was separated by precipitation in isopropanol, followed by filtration. Precipitation and filtration were repeated several times to remove unreacted PEG. Finally the copolymer was dried under reduced pressure. Reaction yield was 48%.
Activation of P(PF-co-EG) network with N-hydroxysuccinimide
P(PF-co-EG) network (10 g) and NHS (2 g) were dissolved in 50 mL anhydrous DMF, followed by addition of an excess DCC (4.5 g) (Scheme 1). The reaction was performed at room temperature for 24 h under inert atmosphere. Then, the precipitate was filtered and the solution containing the product (NHS-activated P(PF-co-EG) network) was purified by dissolution in ethyl acetate and precipitation with anhydrous diethyl ether. Finally, it was dried under vacuum and stored at 4 °C. Reaction yield was 60%.Grafting of NHS-activated P(PF-co-EG) network to MagP®-NH2
MagP®-NH2 were dissolved in 50 mL of phosphate buffer (pH 7.4). Separately, NHS-activated P(PF-co-EG) network was dissolved in 60 mL of anhydrous DMF and added dropwise to the MagP®-NH2 solution, in an ice bath, maintaining the temperature in the range of 5–10 °C. After 3 h of reaction, the resulting composite, MagP@P(PF-co-EG) (see Scheme 1), was isolated by filtration. Finally, the composite was rinsed several times with 1 mM HCL/phosphate buffer solution to remove the unreacted magnetic particles. Six composites were prepared with MagP®-NH2/P(PF-co-EG) initial weight ratios of 1:99; 2:98; 5:95, 8:92; 10:90 and 15:85. To simplify the nomenclature, hereafter the samples will be designated by the microparticle/copolymer weight ratio. To fabricate polymeric films, a suspension of the MagP@P(PF-co-EG) composites in chloroform (5%, w/v) was poured onto Teflon molds and dried under vacuum at 50 °C for 24 h. For the rheological characterization, composites were moulded in discs of 8 mm diameter and 1 mm thick.
Characterization techniques
The morphology of MagP®-NH2 microparticles and the core–shell composites was observed at different magnifications with a Philips Tecnai 20 FEG (LaB6 filament) transmission electron microscope (TEM) fitted with an energy dispersive X-ray analysis (EDAX) detector operating at 200 kV and with 0.3 nm point-to-point resolution. Samples were diluted in ethanol (1:10) and one drop of each suspension was cast on 200 mesh lacey grids and dried under reduced pressure. The diameter distribution of the microparticles and composites was determined based on the TEM images by measuring at least 100 particles per sample. The images were processed with Digital micrograph and ImageJ software. The chemical composition was verified by means of EDAX on the basis of the obtained high-resolution TEM (HR-TEM) images.
The extent of the grafting reaction was estimated via a spectrophotometric assay that quantifies the molar equivalents of NHS esters of methoxypolyethylene glycols derivatives.22 Briefly, the NHS ester reacts with an excess of ethanolamine in DMF at 25 °C for 15 min. Subsequently, residual ethanolamine is quantified by measuring the absorbance at 420 nm after reaction with 2,4,6-trinitrobenzenesulfonic acid (TNBS) at pH 9.2 and 55 °C for 15 min, followed by cooling to room temperature. The test was used to quantitatively assess the concentration (μmol g−1) of NHS esters in the NHS-activated P(PF-co-EG) copolymer and each MagP@P(PF-co-EG) composite, and the difference corresponds to the grafting degree (GD).
A Perkin-Elmer 1600 FT-IR spectrophotometer operating at room temperature was used to record the FTIR spectra of the samples in the wavenumber range between 4000 and 500 cm−1. The crystal structure of MagP®-NH2 and the composites was analyzed with a Bruker D8 Advance diffractometer incorporating a Cu Kα line at 1.5406 Å, in the angular range 2θ = 10–70°. A TA Instruments Thermogravimetric Analyzer (TGA Q500) was used to investigate the thermal stability of the samples. Tests were performed under a nitrogen atmosphere, in the temperature range of 0–800 °C, at a heating rate of 10 °C min−1. The magnetization of the samples was measured at room temperature using a Lake shore-7400 vibrating sample magnetometer (VSM).
The magneto-rheological properties were studied with a TA Instruments Discovery HR-3 hybrid rheometer (plate–plate system, plate diameter: 8 mm; gap: 1 mm) equipped with a magneto-rheology accessory. Oscillatory shear measurements were performed in the linear regime, with frequency sweeps from 0.01 to 10 rad s−1 at a constant strain amplitude of 1% and temperature of 25 °C. A dwell time of 10 min was allowed for all experiments to guarantee a uniform temperature before starting the measurements. All rheological measurements were carried out in triplicate and parameters (storage modulus (G′) and loss modulus (G′′)) were obtained directly from the software.
The mechanical properties of the composites were evaluated by tensile tests using dumb-bell shaped moulding specimens Type 5B (thickness = 1 mm; overall length = 35 mm), following the UNE-EN ISO 527-2 standard. An Instron 4204 tensile test machine was used operating at 23 ± 2 °C and 50 ± 5% RH, at a constant loading speed of 1 mm min−1. The following parameters were derived from the stress–strain curves according to the UNE-EN ISO 527-1 standard: the Young's modulus, calculated from the linear elastic region via Hooke's law: E = σ/ε, where σ is the applied stress and ε is the resultant strain; the tensile strength, maximum stress level reached, and the elongation at break, ratio between increased length and initial length after breakage of the tested specimen. Experiments were also performed on composites immersed in a simulated body fluid (SBF) at 37 °C for two weeks. Six to eight specimens per composite were tested for each condition. Water uptake of the composites was calculated as: [(W − W0)/W0] × 100, where W0 and W are the weight of the composite before and after immersion in SBF, respectively.
The chemical stability against acid attack was analyzed by dispersing the samples in a 0.1 M HCl solution. After 10 days their magnetic response was assessed by placing a neodymium magnet close to the vial containing the samples. The hydrolytic degradation was assessed by measuring the weight loss after immersing the samples in a SBF at 37 °C for 12 weeks. The in vitro metabolic activity was determined by the Alamar Blue assay23 using normal human dermal fibroblasts (NHDF, from Promocell). In summary, the cells were seeded in 96-well plates at a density of 5000 cells per well. After incubation for 24 h at 37 °C, the cells were exposed to 1 vol% of the composites and further incubated for periods of 12, 24, 36, 48 and 72 h; then, 10 μL of Alamar Blue were added to each well. After 2 h, the absorbance of each well was measured at 490 nm with a microplate reader. The untreated cells served as the control and their metabolic activity was set as 100%. For each sample, four measurements were performed to get an average value.
The antibacterial activity of the composites was tested against Gram-positive Staphylococcus aureus (S. aureus, ATCC 12600) and Staphylococcus epidermidis (S. epidermidis, ATCC 12228) as well as Gram-negative Escherichia coli (E. coli, ATCC 25922) and Pseudomonas aeruginosa (P. aeruginosa, ATCC 27853). Briefly, the samples were submerged in a nutrient broth of ∼2.0 × 106 colony forming units per mL (CFU mL−1). After incubation at 37 °C for 24 h, the number of viable bacteria colonies was counted manually, and the results were expressed as mean CFU per sample. The antibacterial activity was calculated as:24 log(viable cell countcontrol/viable cell countcomposite), where a beaker containing bacteria and no sample was used as control. Experiments were performed in triplicate and the average values are reported.
Results and discussion
Morphology
Fig. 1 presents typical TEM and HR-TEM images of MagP®-NH2 microparticles and the MagP@P(PF-co-EG) composite (8:92 w/w) along with their EDAX spectrum. The size distribution of the microparticles and the composites with different MagP®-NH2/P(PF-co-EG) weight ratios is also included in Fig. 1. The images of MagP®-NH2 reveal a narrow size distribution of mostly quasi-spherical particles, with diameters in the range of 1–5 μm and a mean value of 3 μm (Fig. 1k), in agreement with the supplier data. The microparticles consist in an inner core of aggregated black magnetite entities covered by a thin, uniform and bright polymeric layer (Fig. 1b and c). The HR-TEM image shown in Fig. 1d reveals that the particles are single, defect-free crystals of magnetite. The micrograph illustrates well-ordered lattice fringes with a d-spacing of 0.48 nm, which corresponds to the (111) plane of Fe3O4.25 The EDAX spectrum in Fig. 1e confirms that the microparticles are mainly composed of iron and oxygen, and reveals the presence of carbon derived from the polyurethane outermost layer.
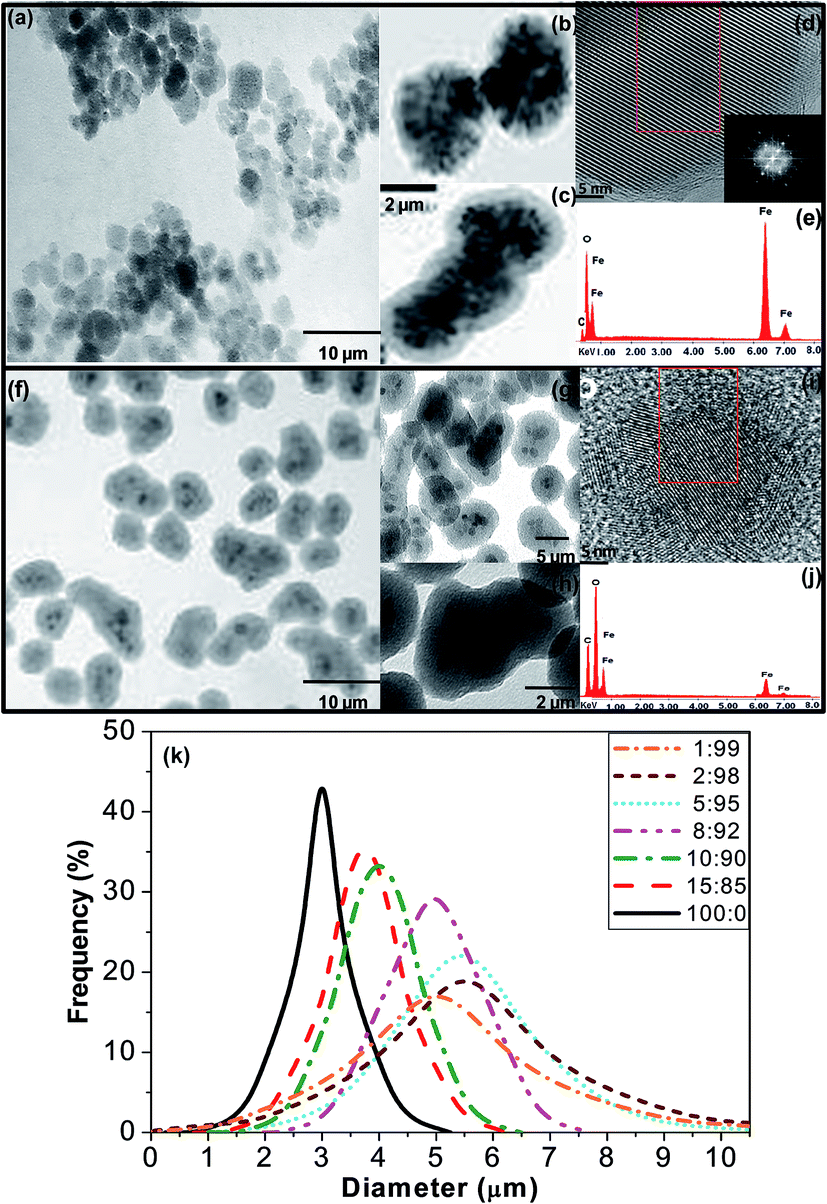 | ||
| Fig. 1 TEM images at different magnifications of MagP®-NH2 microparticles (a–c) and the MagP@P(PF-co-EG) composite (8:92 w/w) (f–h). The copolymer shell appears as a translucent thick film around the MagP®-NH2 core (g and h). HR-TEM micrograph/EDAX spectrum of MagP®-NH2 (d and e) and the indicated composite (i and j). The red squares in (d) and (i) show the area observed by EDAX. Size distribution of MagP®-NH2 and the composites with different microparticle/copolymer weight ratios (k). | ||
Upon addition of the P(PF-co-EG) network, a noticeable increase in the relative peak height of carbon is detected (Fig. 1j). Well-defined core–shell structures showing a broader size distribution and larger dimensions can be observed (i.e. between 3 and 7 μm for the 8:92 composite, Fig. 1k). The composites are less isotropic, and display a more irregular an elongated shape than the MagP®-NH2 (Fig. 1g). The copolymer shell appears as a translucent thick film around the MagP®-NH2 core (Fig. 1h), and its thickness can be estimated based on the differences in diameters of the MagP®-NH2 and the composites (i.e. typically of the order of 2–3 μm for the 8:92 composite). Interestingly, the shell diameter is larger than expected, and seems to be related to the fact that many P(PF-co-EG) shells contain several MagP®-NH2 cores (Fig. 1g and h). The size of the copolymer shell and hence, of the obtained core–shell structures varied depending on the microparticle/copolymer initial weight ratios, as shown in Fig. 1k. Higher amounts of P(PF-co-EG) yielded larger composites, thus thicker shells. For instance, changing the copolymer weight percentage from 85 to 95% caused an increase in the mean composite diameter from 3.8 to 5.5 μm. This is consistent with the results reported for magnetite–polystyrene core–shell composites, where the diameter of the ensemble decreased as the ratio of Fe3O4/styrene increased.26 Further, these observations corroborate the possibility of finely tuning the polymer shell thickness by varying its initial concentration, as shown in previous studies;27,28 controlling the thickness of the shell is of paramount importance in tailoring the final properties of core–shell biomaterials.28 However, further increase in the copolymer content did not result in a larger diameter but in a broader size distribution (Fig. 1k). On the other hand, fairly similar diameter distributions were obtained for the composites with P(PF-co-EG) weight percentages of 90 and 85%, suggesting that a saturation value may have been reached, and further decrease in the copolymer weight fraction (increase in MagP®-NH2 concentration) would not result in thinner shells. This could also be related to the fact that almost all the grafting points of the copolymer network are already anchored to microparticles, and further amount of MagP®-NH2 would not result in higher degree of grafting, as indicated by the spectrophotometric assay results (Table 1). Overall, TEM observations confirm the formation of copolymer-coated magnetite microparticles, and that the morphology, hence the final composite properties, can be fine-tuned by varying the MagP®-NH2/P(PF-co-EG) weight ratio.
| Sample codea | TGA data | Spectrophotometric assay | ||||||
|---|---|---|---|---|---|---|---|---|
| Tonset (°C) | T10 (°C) | T50 (°C) | Tmax1 (°C) | Tmax2 (°C) | Tmax3 (°C) | AGb (mg per 100 mg) | GDc (%) | |
| a MagP®-NH2/P(PF-co-EG) weight ratio.b Amount grafted expressed as mg per 100 mg of MagP®-NH2.c Grafting degree obtained from the spectrophotometric assay (see explanation in the text). Tonset: initial degradation temperature obtained at 2% weight loss; T10: temperature for 10% weight loss; T50: temperature for 50% weight loss; Tmax: temperature of maximum rate of weight loss. The subscripts 1, 2 and 3 correspond to the first, second and third degradation step, respectively. | ||||||||
| 0:100 |
276 | 304 | — | — | — | 350 | — | — |
| 1:99 |
104 | 292 | 485 | 185 | 297 | 449 | 77.5 | 8.9 |
| 2:98 |
112 | 309 | 498 | 188 | 300 | 460 | 73.8 | 16.5 |
| 5:95 |
118 | 345 | 510 | 190 | 306 | 467 | 64.1 | 37.6 |
| 8:92 |
127 | 399 | 536 | 203 | 309 | 486 | 49.9 | 56.8 |
| 10:90 |
136 | 407 | 678 | 212 | 319 | 495 | 38.2 | 78.3 |
| 15:85 |
171 | 434 | 710 | 222 | 326 | 510 | 38.4 | 76.9 |
| 100:0 |
278 | 513 | 345 | 306 | 501 | — | — | — |
FTIR study
The FTIR spectra of MagP®-NH2, PPF, P(PF-co-EG) and the composites with MagP®-NH2/P(PF-co-EG) weight ratios of 1:99, 8:92 and 15:85 are compared in Fig. 2. Qualitatively similar spectra were recorded for the rest of composites developed. The spectrum of MagP®-NH2 shows a broad peak centred at around 3370 cm−1, related to –OH and –NH stretching vibrations of surface hydroxyl and amino groups,29 and at 1625 cm−1 related to the bending vibrations of these surface moieties; further, the Fe–O stretching band characteristic of magnetite is found at 580 cm−1.30
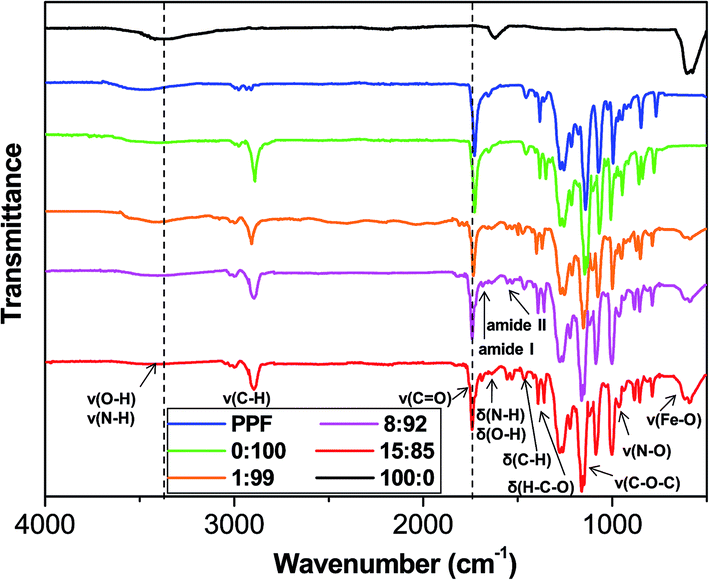 | ||
| Fig. 2 FTIR spectra of MagP®-NH2, PPF, P(PF-co-EG) and the composites with microparticle/copolymer weight ratios of 1:99, 8:92 and 15:85. The dashed lines are plotted to guide eyes. | ||
On the other hand, the IR spectrum of PPF exhibits characteristic bands at ∼3500 cm−1, ascribed to the O–H stretching, in the range of 2870–3080 cm−1, arising from C–H stretching modes, as well as at 1460 and 1350 cm−1 related to C–H bending vibrations. Besides, a very strong band appears at 1730 cm−1 due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the ester group,31 and the CC stretching appears as a very weak band at 1650 cm−1. Upon reaction with PEG–COCl, noticeable spectral changes can be observed: decrease in the intensity of the –OH stretching band, building up more C–H stretching band at 2890 cm−1, appearance of the C–O–C stretching vibration of the PEG units at 1130 cm−1 and the H–C–O in-plane bending at 1385 cm−1.18 Thus, the IR spectra corroborate that PEG–COCl has been successfully tethered to PPF, leading to the formation of the P(PF-co-EG) copolymer network.
O stretching of the ester group,31 and the CC stretching appears as a very weak band at 1650 cm−1. Upon reaction with PEG–COCl, noticeable spectral changes can be observed: decrease in the intensity of the –OH stretching band, building up more C–H stretching band at 2890 cm−1, appearance of the C–O–C stretching vibration of the PEG units at 1130 cm−1 and the H–C–O in-plane bending at 1385 cm−1.18 Thus, the IR spectra corroborate that PEG–COCl has been successfully tethered to PPF, leading to the formation of the P(PF-co-EG) copolymer network.
The spectra of the composites show the characteristic bands of both MagP®-NH2 and P(PF-co-EG). A wide band (centred at 3390 cm−1 in the 8:92 composite) related to the N–H stretching of free NH2 groups is detected, and its intensity decreases with increasing the MagP®-NH2/P(PF-co-EG) weight ratio. This band and the N–H bending (∼1640 cm−1 in the 8:92 composite) are shifted towards higher wavenumber compared to those of MagP®-NH2, which may be attributed to the grafting leading to restricted motion of N–H in stretching and bending modes.32 Further, new bands appear in the spectra that corroborate the success of the grafting reaction: the amide I band at ∼1685 cm−1, partially overlapped with the carbonyl stretching (∼1735 cm−1) and the amide II band in the range of 1540–1520 cm−1, which show increased intensity on increasing the microparticle weight fraction. The presence of non-reacted NHS esters is confirmed by two small peaks at ∼1780 and 1810 cm−1, arising from the CO stretching of the NHS moiety,33 and its N–O stretching at 960 cm−1, which intensity drops upon increasing the MagP®-NH2/P(PF-co-EG) weight ratio. Additionally, the intensity of the Fe–O stretching band rises with increasing microparticle loading. On the whole, the IR spectra support the successful coupling of the magnetite microparticles to the copolymer network, and that the extent of the grafting reaction raises upon increasing the MagP®-NH2 weight fraction.
X-Ray diffraction
The diffraction patterns of MagP®-NH2, P(PF-co-EG) and the composites with microparticle/copolymer weight ratios of 1:99, 8:92 and 15:85 are shown in Fig. 3. The diffractogram of the microparticles exhibits peaks at 2θ values of 18.6, 30.4, 35.1, 43.2, 53.7, 57.4 and 62.7°, corresponding to the (111), (220), (311), (400), (422), (511) and (440) crystal planes, respectively, of the spinel structure of Fe3O4, as identified using the ICSD database (card no. 26410). The sharpness of the peaks reveals that the microparticles are highly crystalline.
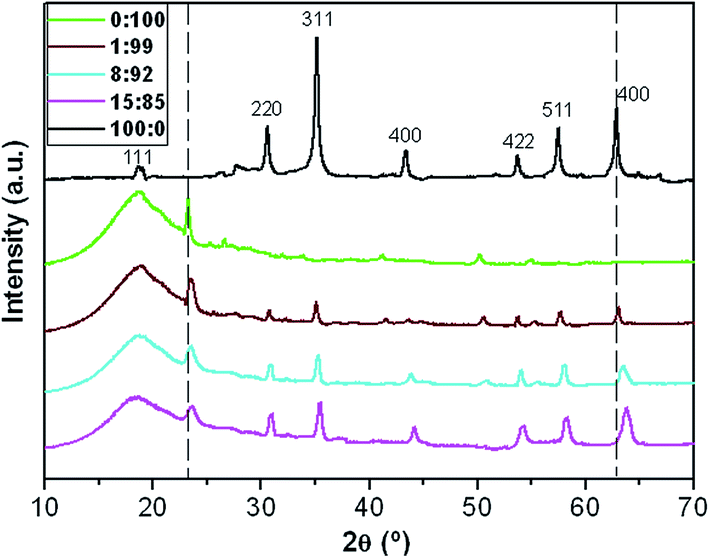 | ||
| Fig. 3 X-ray diffraction patterns of MagP®-NH2, P(PF-co-EG) and the indicated composites. The dashed lines are plotted to guide eyes. | ||
On the other hand, the diffraction pattern of P(PF-co-EG) presents a wide reflection in the range of 2θ = 10–28° arising from the PPF blocks,34 which display an amorphous character, and a diffraction peak at 2θ = 23.3° due to the reflection of the (001) plane of PEG monoclinic unit cell.18 The diffractograms of the composites show the reflections of both MagP®-NH2 and P(PF-co-EG), indicating that the crystalline structure of both components is maintained. As expected, the intensity of the peaks corresponding to the microparticles rises while that of the broad amorphous halo and the peak at 2θ ∼ 23.3° decreases with increasing the microparticle/copolymer weight ratio. However, as this ratio increases, the peaks become wider and slightly shift towards higher 2θ values. This widening of the peaks has been previously reported for other systems incorporating polymer-grafted fillers,32,35–37 hinting towards a diminution of the spatial order. Further, the broadening could also be associated to the asymmetry induced by the random attachment of the MagP®-NH2 to the polymeric chains. An analogous behaviour of small shift in the position of the peaks along with a decrease in the level of crystallinity was observed for cellulose upon polymer grafting,37 indicative that the anchoring had occurred, causing a change in the cell dimensions. The average crystallite size estimated from the (001) plane of the copolymer using the Scherrer formula38 was 36 nm, and decreased by about 28, 45 and 57% for the composites with microparticle/copolymer weight ratios of 1:99, 8:92 and 15:85, respectively. This suggests that high amounts of MagP®-NH2 reduce polymer chain mobility, hindering the growth of the copolymer crystals, therefore partially inhibiting crystallization.
Thermal stability
Fig. 4 presents the TGA curves under an inert atmosphere of the copolymer, the microparticles and the composites with different MagP®-NH2/P(PF-co-EG) weight ratios, and their characteristic degradation temperatures are summarized in Table 1. The MagP®-NH2 microparticles show a small weight loss below 300 °C related to the removal of physically and chemically adsorbed water and surface hydroxyl groups,39 and another weight loss in the range of 300–600 °C ascribed to the elimination of surface amino moieties.40 On the other hand, the P(PF-co-EG) copolymer network presents two clear degradation stages, attributed to the degradation of the PPF and PEG segments, respectively. The decomposition of the PPF blocks starts (Tonset) at about 276 °C and exhibits the maximum rate of weight loss (Tmax) at about 350 °C, while the degradation of PEG blocks takes place in the range of 380–520 °C, in agreement with former studies.18,41 At temperatures above 520 °C, the copolymer is completely decomposed.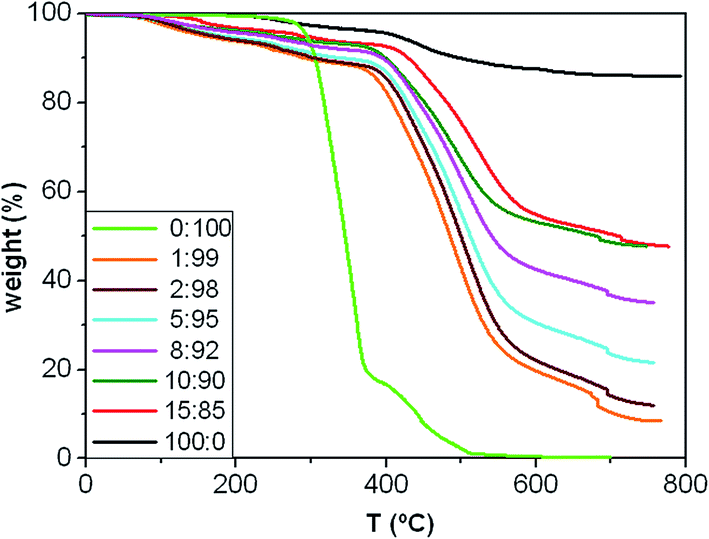 | ||
| Fig. 4 TGA thermograms under nitrogen atmosphere of MagP®-NH2, P(PF-co-EG) and the composites with different microparticle/copolymer weight ratios. | ||
On the other hand, a multi-step degradation process is observed for the composites: the mass loss in the range of 150–300 °C is attributed to the removal of remaining OH groups from the MagP®-NH2 surface together with the decomposition of NHS esters, in agreement with previous studies on β-lactam functionalized Fe3O4 nanoparticles;39 the weight loss between 300 and 400 °C corresponds to the elimination of non-reacted amino groups from the MagP®-NH2 surface, similar to that found for the microparticles, and the two last stages in the range of about 400 to about 550 °C and 550 to 700 °C are related to the decomposition of the PPF and PEG blocks, respectively. Albeit the composites start to decompose at temperatures lower than the neat copolymer, the Tmax of their polymeric network (Tmax3 in Table 1) occurs at significantly higher temperatures (i.e. an increase of about 160 °C for the 15:85 w/w composite compared to P(PF-co-EG)). Further, all the degradation temperatures raise progressively on increasing the MagP®-NH2/P(PF-co-EG) weight ratio; for instance, Tmax3 increases by ∼60 °C as the MagP®-NH2 weight fraction raises from 1.0 to 15.0 wt%, which demonstrates the thermal stabilization effect induced by the microparticles, since Fe3O4 is stable even up to 800 °C.30 Improvements in thermal stability have also been reported for polyvinylidene fluoride (PVDF)/magnetite42 and cashew gum graft poly(acrylamide)/magnetite composites,43 attributed to the inorganic nature of magnetite uniformly distributed through the matrix, which hindered the diffusion of degradation products, thus providing superior resistance to heat. Moreover, the high thermal conductivity of Fe3O4 (ref. 44) should facilitate heat dissipation within the composites, hence leading to superior thermal stability.
The TGA curves of the composites show more weight loss than MagP®-NH2, which further confirms the successful grafting of the copolymer network onto the microparticle surface; besides, the weight loss increases upon raising the P(PF-co-EG) concentration. The amount of copolymer grafted onto the microparticles can be calculated through the difference in weight of the MagP®-NH2 before and after grafting with the P(PF-co-EG) network, and the results are shown in Table 1. As expected, the amount grafted raises with increasing the copolymer weight fraction, albeit the composites with P(PF-co-EG) content of 85 and 90 wt% present similar value. Additionally, the grafting degree (GD) was estimated via a spectrophotometric assay22 that determines the concentration of NHS esters in the NHS-activated P(PF-co-EG) copolymer and each MagP@P(PF-co-EG) composite (Table 1). As can be observed, GD increases gradually upon raising the MagP®-NH2/P(PF-co-EG) weight ratio, following a quasi-linear trend, although again the 15:85 and 90:10 composites display comparable value, suggesting that GD has been leveled off. This hints that most of the NHS esters of the copolymer network are already covalently bounded to microparticles, and further amount of MagP®-NH2 would not lead to higher GD values. Similar behaviour has been reported for poly(hydroxyethyl methacrylate)-grafted magnetite nanoparticles,45 where GD saturated above a certain amount of monomer concentration.
Magnetic properties
The magnetic hysteresis loops of the microparticles and the synthesized core–shell composites at room temperature conditions are shown in Fig. 5a. The observed magnetization profiles are consistent with those of samples incorporating ferrimagnetic magnetite particles.46 Further, all the magnetization curves are reversible with almost negligible values of coercivity and remanence, which points towards a superparamagnetic behaviour that could be ascribed to the fact that the magnetite microparticles are composed of many small nanocrystals with grain sizes in the nanometer scale, which are single domain.47 The saturation magnetization (Ms) value is 73.9 emu g−1 for MagP®-NH2, and decreases gradually as the microparticle weight fraction drops, the largest drop being about 84% for the composite with 1.0 wt% MagP®-NH2 loading. Thus, the field required to saturate is considerably lower after the microparticles are anchored to the copolymer. The magnetic reduction found upon copolymer grafting can be attributed to the coating shell of P(PF-co-EG) on the microparticle surface, in agreement with the results found for other systems incorporating polymer-grafted Fe3O4 particles.12 Thus, in the composites the amount of iron is lower since magnetite is replaced by a non-magnetic polymer. Besides, the magnetic microparticles are embedded in a cross-linked network, hence cannot move easily to their axes directions, and consequently the net magnetization decreases in comparison to the bare MagP®-NH2. Further, the lower magnetization could be related to the decreased interparticle dipolar interaction, which arises from the enlarged microparticle distance upon anchoring to the polymeric chains.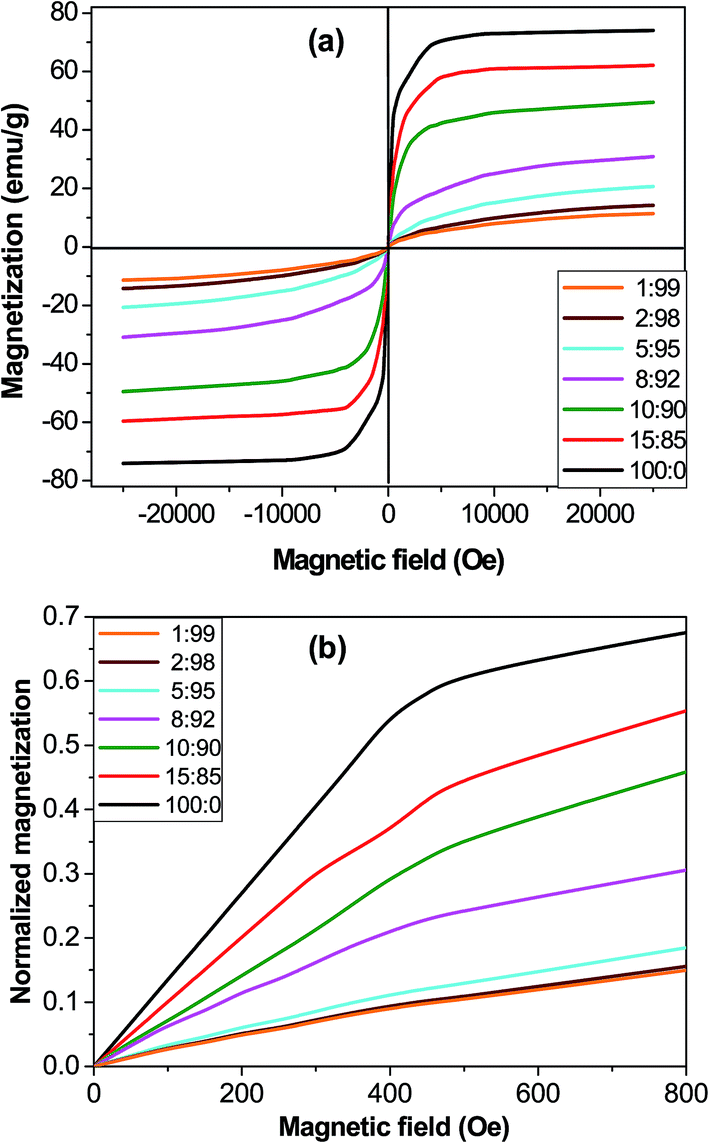 | ||
| Fig. 5 Magnetization (a) and normalized magnetization (b) versus the magnetic field for MagP®-NH2 and the composites with the indicated microparticle/copolymer weight ratios. | ||
Nonetheless, the most important advantage of using core–shell composites instead of magnetic microparticles is the opportunity of tuning the initial magnetic susceptibility (χi) by changing the microparticle/copolymer weight fraction, hence the shell thickness. In fact, the classical electromagnetism predicts higher χi for thinner shells.48 To verify this, the magnetization of the composites was first normalized by dividing by Ms and then plotted against the magnetic field (Fig. 5b). The comparison of the initial slope of the curves (χi) corroborates that composites with smaller diameters (thinner shells, Fig. 1k) display higher χi values. The observed superparamagnetic behaviour is important for potential in vivo applications such as drug delivery, since once the external magnetic field is removed, the magnetization disappears.
Magneto-rheological properties
The viscoelastic properties of the core–shell composites were investigated, and the frequency (ω) dependence of the storage modulus (G′) at a temperature of 25 °C is depicted in Fig. 6a. Qualitatively similar behaviour was found for the loss modulus, G′′ (data not shown). G′ represents the elastic, solid-like response of a material when subjected to an oscillatory mechanical strain, hence it is a measure of its mechanical strength. As can be observed, the magnitude of G′ rises with frequency and upon increasing MagP®-NH2 concentration, the increase being more pronounced in the low-frequency range. Thus, the addition of 15.0 wt% microparticle loading to the copolymer causes about 50-fold increase in modulus at 0.01 rad s−1, whilst at the frequency of 10 rad s−1 the rise is only 5-fold. Qualitatively similar phenomenon has been previously reported for other magnetite-reinforced composites, and is ascribed to a shear thinning effect.49 The increments observed herein are much higher than those predicted by the classical theory on the mechanical properties of composite materials reinforced with spherical fillers,50 and could be related to microstructural changes in the P(PF-co-EG) network due to the incorporation of the microparticles, which acted as connectors between the copolymer chains, thereby improving the adhesion between them. Further, such huge increases in G′ corroborate that the covalent grafting of MagP®-NH2 to the P(PF-co-EG) network strongly restricts the mobility of the copolymer chains; as the microparticle/copolymer weight ratio becomes higher, the grafting degree increases (Table 1), which is reflected in reduced chain motion, and consequently better mechanical properties. Similar increments upon increasing microparticle loading were obtained for G′′, related to viscous energy dissipation in the material when subjected to sinusoidal strain.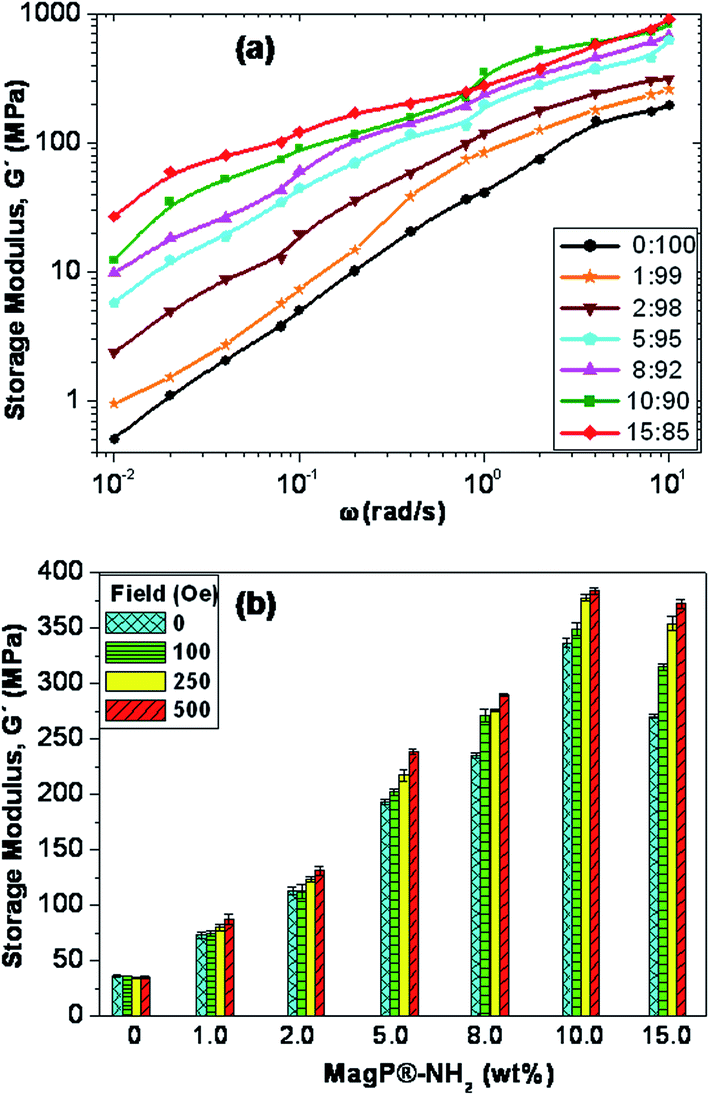 | ||
| Fig. 6 (a) Storage modulus, G′, as a function of frequency (ω) for the neat copolymer and the MagP@P(PF-co-EG) composites. (b) G′ for the indicated magnetic fields and microparticle loadings at the frequency of 1 rad s−1. | ||
More importantly, we measured the change in G′ when the intensity of the external magnetic field was increased from 0 to 500 Oe, and the results obtained for the different composites at the frequency of 1 rad s−1 are plotted in Fig. 6b. A clear rise in G′ is observed with increasing the strength of the magnetic field, the augment being microparticle concentration-dependent. Actually, the modulus of the composite with MagP®-NH2 content of 1.0 wt% only increases by about 13% within the indicated magnetic field range, whereas for that with 15.0 wt% loading the increment is around 40%. Besides, the rise in the viscoelastic modulus with the magnetic field was maintained over the whole frequency range studied. An analogous trend has been reported for elastomeric composites incorporating nano-sized iron oxide,51 where composites activated in the magnetic field reached much higher values of G′ and G′′ as a function of ω as compared to the control at zero field. Such behaviour was attributed to a specific orientation of the fillers within the polymeric matrix. In our composites, the stronger magnetic response found upon increasing MagP®-NH2/P(PF-co-EG) weight ratio could be related to the gradual enhancement in magnetic susceptibility, as observed in Fig. 5b. Thus, experimental results confirm that it is feasible to control the mechanical behaviour of these core–shell composites by the application of an external field. Stronger magnetic fields or higher MagP®-NH2 contents would result in larger modulus improvements. These field-tunable mechanical properties make these composites excellent candidates to be used in the fabrication of magnetic field-responsive tissue substitutes.
Tensile properties
With a view to use the developed composites as scaffolds for tissue-engineering, it is crucial to assess their mechanical performance. Preferably, a scaffold should have mechanical properties that match with those of the natural tissue at the anatomical site into which it is to be implanted and must have enough mechanical integrity to allow surgical handling during implantation and to function from the time of implantation to the completion of the remodeling process.52 In this regard, the static mechanical properties of the composites were investigated via tensile tests performed under both dry (23 °C and 50% RH) and physiological (37 °C in a SBF) conditions, and the results are comparatively depicted in Fig. 7. Typical stress–strain curves of P(PF-co-EG) and the composites with MagP®-NH2/P(PF-co-EG) weight ratios of 1:99, 8:92 and 15:85 are shown in the inset of Fig. 7a; clearly, the initial slope of the curve rises with increasing microparticle content, corroborating the stiffening effect induced by the MagP®-NH2. The Young's modulus (E) of the neat copolymer under dry conditions is about 52 MPa (Fig. 7a), and increases strongly with increasing MagP®-NH2 content, by a factor of about 5.5 for the composite with 10.0 wt% microparticle loading, in agreement with the results obtained from the rheological tests (Fig. 6). Qualitatively similar behaviour is found for the tensile strength (Fig. 7b), which raises by a factor of 3.8 for the abovementioned composite compared to that of P(PF-co-EG). Such strong increases are likely related to the strong covalent interactions between MagP®-NH2 and P(PF-co-EG) that restrict polymer chain movement during mechanical deformation. Due to the covalent crosslinking of the microparticles within the copolymer network, much higher load is required to deform the network compared to neat P(PF-co-EG), which is reflected in enhanced stiffness and strength. An analogous behaviour of extraordinary improvement in mechanical properties has been reported for poly(glycerol sebacate) (PGS) covalently crosslinked with carbon nanotubes (CNTs),53 where the addition of 1 wt% CNTs to PGS resulted in 5-fold increase in the tensile modulus and more than 6-fold augment in the compressive modulus, which is far superior to the mechanical properties previously reported for CNT-based nanocomposites.54,55 In our study, we attain comparable improvements at higher filler loadings (10.0 wt%), which is reasonable considering E values reported for CNTs (∼1 TPa)54 and Fe3O4 (175 GPa).56 The mechanical enhancements attained herein are considerably larger than those reported by former studies on polymer/magnetite composites (in the range of 7–200% increase with the addition of 1 to 20 wt% Fe3O4).42,43,49 Note that the tensile properties of the developed composites are in the range of those of cancellous bone (0.02–0.5 GPa), while they are significantly lower than those of cortical bone (3–30 GPa),57 which indicates their suitability for soft-tissue replacements.
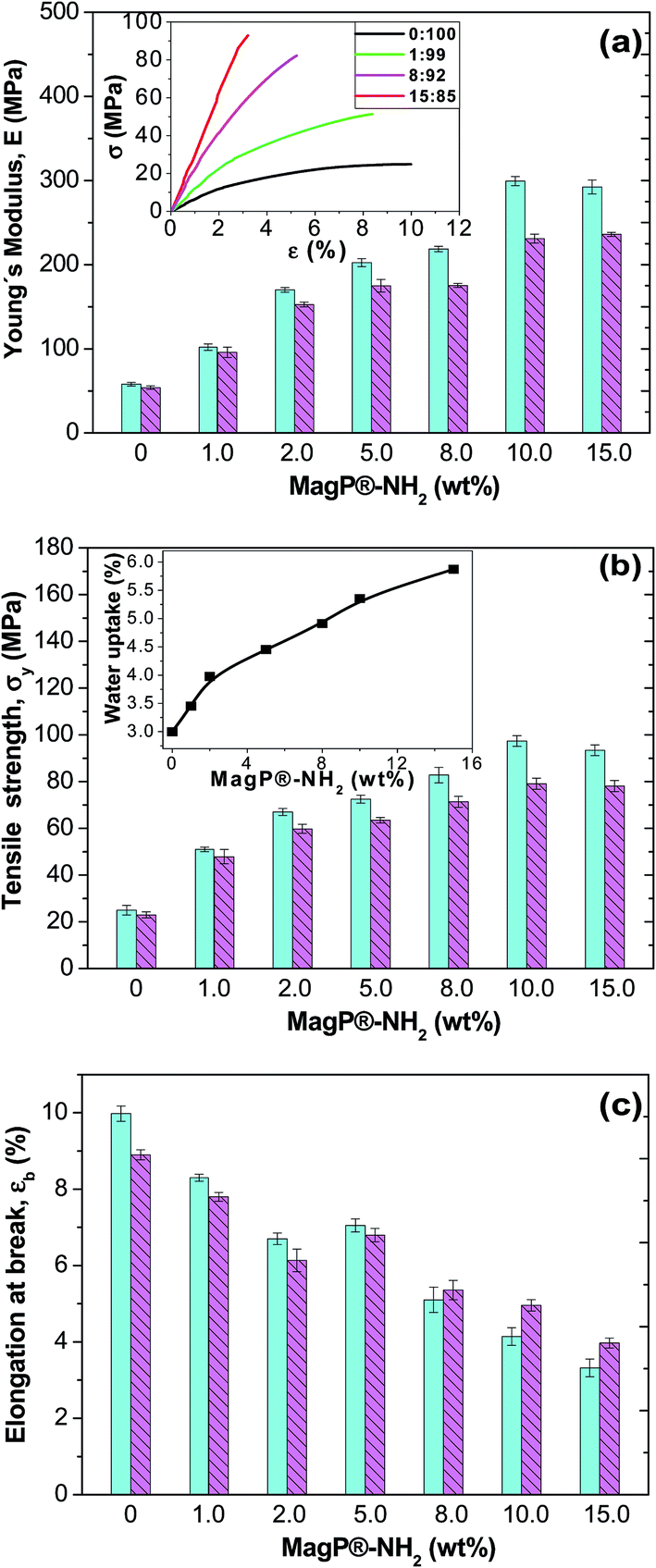 | ||
| Fig. 7 (a) Young's modulus, E, of MagP@P(PF-co-EG) composites for different microparticle concentrations. The inset shows typical stress–strain curves at 23 °C of the copolymer and the indicated composites. (b) Tensile strength of MagP@P(PF-co-EG) composites. The inset displays the water uptake as a function of microparticle loading. (c) Elongation at break of MagP@P(PF-co-EG) composites. The solid and dashed bars correspond to experiments performed at 23 and 37 °C, respectively. | ||
On the other hand, the ductility of the copolymer was reduced upon addition of the microparticles (Fig. 7c), by about 3-fold for the composite with the highest MagP®-NH2 loading. This expected behaviour should be related to the strong filler–matrix interactions that confine the ductile flow of the polymeric segments. It is worthy to note that the mechanical properties appear to level off at a microparticle content of 10.0 wt%, and further increase in MagP®-NH2 concentration does not result in additional property improvements. As discussed earlier, this may be related to the fact that nearly all the grafting points of the copolymer network are already attached to microparticles, and further amount of MagP®-NH2 does not result in higher degree of grafting (Table 1).
Focusing on the results obtained under physiological conditions, very strong increments in stiffness and strength are also observed, by up to 4.5 and 3.4-fold, respectively, for the composite with 10.0 wt% MagP®-NH2 compared to those of the neat P(PF-co-EG). Systematically, the values obtained subsequent to immersion in a SBF are lower than those achieved under dry conditions, albeit the reduction depends on the microparticle/copolymer weight ratio. For instance, the composite with 1.0 wt% MagP®-NH2 displays the smallest drops (<10%), while those with 10.0 and 15.0 wt% present the largest (in the range of 16–23%). To explain this behaviour, the water uptake of the samples was measured (inset of Fig. 7b), and it was found to rise steadily with increasing microparticle concentration, being the value of the composite with the highest loading about double that of the copolymer. This is consistent with earlier studies58 that reported that the addition of suitable amounts of magnetite can properly increase the water absorption, given that Fe3O4 is hydrophilic and can easily absorb water. Hence, the higher the water uptake, the more the absorbed water that can circulate throughout the composites, therefore reducing the degree of crosslinking of the polymeric network, and as a result, a more pronounced plasticization effect, which is reflected in lower stiffness and strength. Accordingly, the 1:99 composite, which has the lowest water uptake, should have the weakest plasticization, while the 15:85 composite would have the strongest, therefore the decrease in modulus is more pronounced. On the other hand, the elongation at break of P(PF-co-EG) and the composites with MagP®-NH2 content ≤ 5.0 wt% is lower after immersion in a SBF than under dry conditions, whereas for the rest of the composites is slightly higher. A plausible explanation for this unexpected behaviour could be that at low microparticle loadings the hydrolytic degradation that causes the shortening of the polymeric chains prevails, resulting in reduced ductility, while at high loadings the plasticizing phenomenon should overcome the degradation effect, and the consequence is an increase in ductility.
Chemical stability, biodegradation and metabolic activity
The chemical resistance of MagP®-NH2 and the composites against acid medium was assessed through evaluating their magnetic response after 10 days of immersion in a 0.1 M HCl solution. As shown in the inset of Fig. 8a, the bare microparticles lost their magnetic character, while the MagP@P(PF-co-EG) composite (8:92 w/w) retained the magnetization. Similar behaviour was found for the rest of the composites developed. The differences observed are attributed to the presence of the P(PF-co-EG) shell that protects the magnetic core from acidic attack, thus providing better chemical stability.
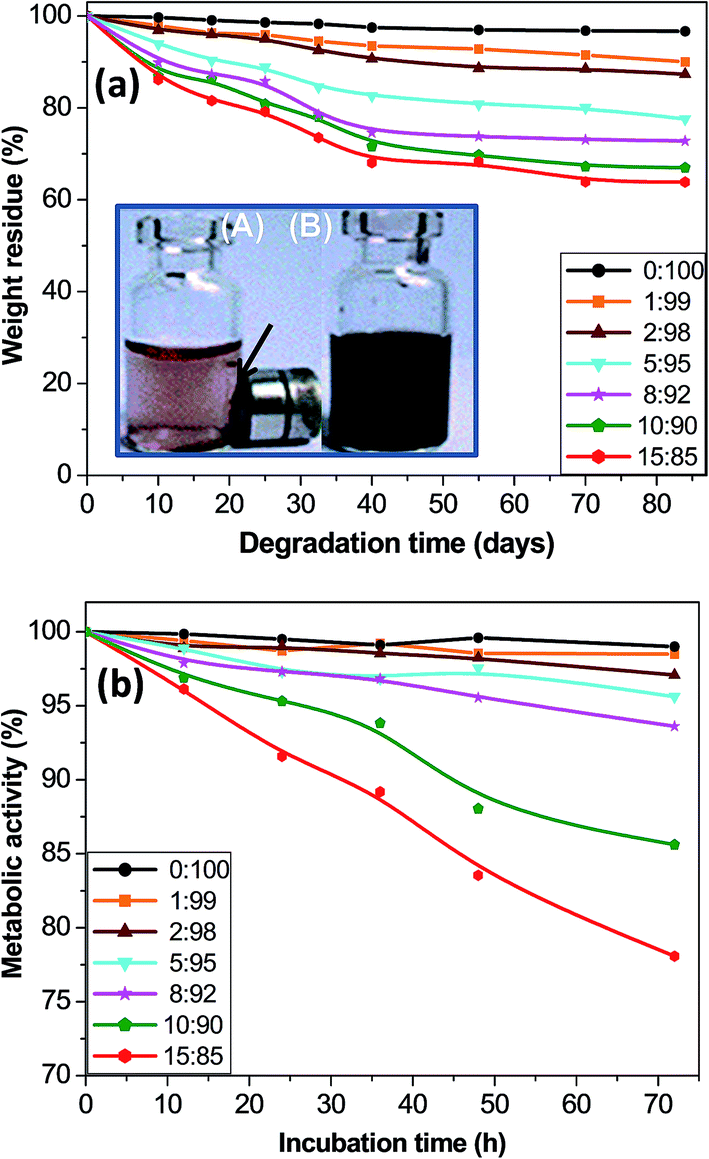 | ||
| Fig. 8 (a) Percentage of weight residue as a function of time after immersion in a SBF solution at 37 °C. The inset shows the macroscopic appearance of the MagP@P(PF-co-EG) composite (8:92 w/w) (A) and MagP®-NH2 microparticles (B) in the presence of a magnetic field after 10 days of acid treatment. (b) Metabolic activity of NHDF cells cultured in the presence of the composites versus incubation time. | ||
Biodegradability is a key parameter that has to be considered when developing materials for tissue engineering. Appropriate and controllable biodegradation rate is highly desirable to allow the material to degrade at a rate similar to tissue growth or for controlled release of biomolecules. The in vitro hydrolytic degradation of the core–shell composites was investigated in a SBF solution at 37 °C, and their percentage of weight residue as a function of time is plotted in Fig. 8a. The neat P(PF-co-EG) only shows a small weight loss of ∼3% after 12 weeks, ascribed to a random hydrolysis of the ester bonds leading to propylene glycol and fumaric acid as the two main degradation products.20 The covalent grafting onto the MagP®-NH2 surface provokes an increase in the percentage of weight loss; thus, the weight residue drops gradually with increasing the microparticle/copolymer weight ratio, and the composite with 15.0 wt% MagP®-NH2 loading only shows about 64% residue after 12 weeks. One reasonable explanation is that the polar hydroxyl and amino groups on the MagP®-NH2 surface accelerate the hydrolysis process, hence the weight loss increases as the microparticle content raises. Further, it has been demonstrated that the biodegradability greatly depends on the crystallinity and elastic modulus of the material: it increases as the crystallinity drops.59 Thus, the enhanced biodegradability upon addition of magnetite is likely related to the decrease in crystallinity, as revealed by X-ray diffraction (Fig. 3). An analogous behaviour of faster degradation rate with increasing Fe3O4 concentration has been reported for poly(3-hydroxybutyrate) (PHB)/magnetite composite fibers.60 This faster degradation is beneficial from a practical viewpoint since could enable a quicker elimination of the composites from the body.
The developed composites are promising for biomedical applications since their properties can be tuned through external magnets. In this regard, there is a plain need to identify potential cellular damages associated with these biomaterials. To get an insight about the cytotoxicity of the MagP@P(PF-co-EG) composites, the Alamar Blue assay with NHDF cells was employed, and the results for different incubation times are shown in Fig. 8b. Albeit this test measures cell metabolic activity, it has been widely used as an indicator of cell viability and toxicity in vitro.23 The highest cell metabolic activity levels (≥99% for all the incubation periods) were obtained after exposure to the neat copolymer, suggesting that this material is not toxic to human skin fibroblasts; these results are in agreement with former studies that demonstrated the good biocompatibility of PPF61 and PPF-based hydrogels21 with diverse cells. Metabolic activity values > 95% were found for NHDF cells cultured in the presence of composites with MagP®-NH2 concentration ≤ 5.0 wt%, and their cell vitality hardly changed with increasing incubation time. For cells exposed to the rest of the composites, the metabolic activity decreased as the microparticle loading and incubation time increased, which is consistent with preceding works dealing with the cytotoxicity of Fe3O4 particles, in which the toxic effect was found to be dose and time-dependent (Paracelsus).62,63 Upon exposure to composites with 8.0 and 10.0 wt% MagP®-NH2 content, cell metabolic activity data ≥ 85% were obtained for all the incubation periods studied, while for cells cultured with the MagP@P(PF-co-EG) (15:85) composite, a value < 80% was attained for an incubation time of 72 h, which is indicative of its higher cytotoxicity.
A wide number of studies64–66 have recently investigated the toxicity behaviour of Fe3O4 micro/nanoparticles, and several mechanisms have been proposed to explain their potential cytotoxicity such as membrane leakage of lactate dehydrogenase, formation of reactive oxygen species (ROS), DNA damage, impaired mitochondrial function, generation of apoptotic bodies, chromosome condensation and inflammation.65 Amongst them, the most likely by which magnetite can induce cytotoxicity is through the generation of ROS. After cell internalization, the particles are probably degraded into iron ions within the lysosomes,66 and these ions are able to cross the nuclear or mitochondrial membrane and form Fe2+ that reacts with hydrogen peroxide and oxygen, producing reactive hydroxyl radicals and Fe3+ via the Fenton reaction;66 subsequently, this radicals may damage DNA, proteins, polysaccharides and lipids. Besides, it has been found that their toxicity depends on a number of factors, namely concentration, size, synthesis process and surface modification. For instance, nanoparticles are much more cytotoxic than the micrometer sized counterparts, ascribed to their larger specific surface area that favours particle internalization in the cells and phagocytic activity.65 On the other hand, although magnetite has been shown to provoke high levels of oxidative DNA lesions,64 it seems that the toxicity can be reduced by coating the particles, leading to fewer oxidative sites, hence less DNA damage. Therefore, it seems that polymeric coatings improve the biocompatibility of magnetite particles.
Antibacterial properties
The antibacterial activity of the developed composites was investigated against endogenous bacteria like Gram-negative E. coli and P. aeruginosa or nosocomial organisms such as Gram-positive S. aureus and S. epidermidis, frequently involved in hospital-associated infections, and the results are plotted in Fig. 9. The copolymer hardly shows antimicrobial action versus any of the bacteria tested. According to the criterion on the level of antimicrobial activity mentioned in the ISO 22196:2007 standard,67 an efficient value is higher than 1.5. Thus, the composites show relatively low biocide activity (in the range of 0.12–0.74) against Gram-negative bacteria and moderate antibacterial action (in the range of 0.35–1.55) versus Gram-positive microorganisms. Only the composite with 15.0 wt% MagP®-NH2 displays efficient antibacterial action versus Gram-positive bacteria, in particular against S. epidermidis. In all cases, the biocide effect rises gradually with increasing microparticle concentration, and is systematically stronger versus Gram-positive cells. Further, the antibacterial activity of all the composites against P. aeruginosa is quite low (<0.25), while only slight differences are found between the activity towards S. aureus and S. epidermidis. These results are consistent with those reported by Behera et al.,68 who found that magnetite nanoparticles have lower activity against Gram-negative bacteria than Gram-positive ones, and that the antimicrobial action versus P. aeruginosa was almost negligible. The diverse toxicity levels observed are likely related to the different nature of the cell wall of Gram-negative and Gram-positive bacteria.69 Further, discrepancies may be ascribed to their different shape and size: S. epidermidis and S. aureus are small spherical microorganisms, whereas E. coli is a short rod-shaped and P. aeruginosa a long rod-shaped bacterium. Previous studies also suggested that Gram-negative bacteria are less sensitive towards magnetite particles than Gram-positive.70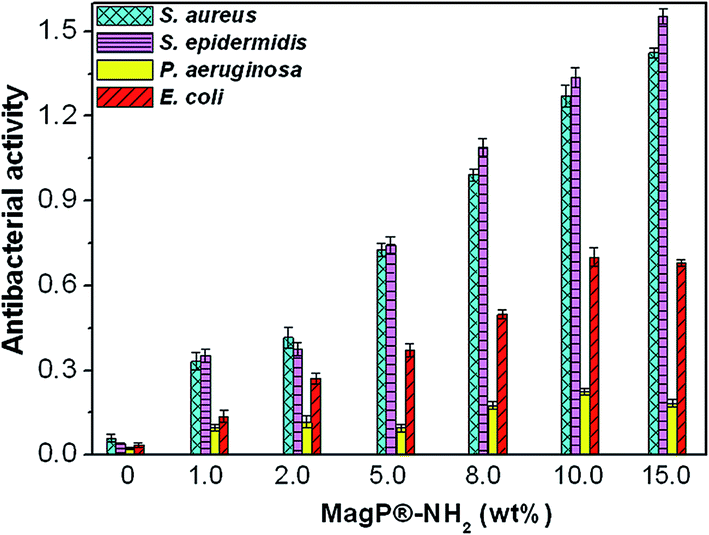 | ||
| Fig. 9 Antibacterial activity of MagP@P(PF-co-EG) composites against S. aureus, S. epidermidis, P. aeruginosa and E. coli bacteria for the indicated microparticle concentrations. | ||
Many factors have been reported to be responsible for the antibacterial activity of magnetite particles.68 The most important mechanism by which these particles show bactericidal activity seems to be via oxidative stress caused by the generation of ROS; these include radicals like O2− and –OH, hydrogen peroxide and singlet oxygen (1O2). The hydrogen peroxide produced can chemically interact with the bacteria outer bilayer and kill them. Besides, magnetite particles keep on interacting with dead bacteria once the hydrogen peroxide is generated, thus hindering further bacterial action and releasing more H2O2 to the medium. Another potential mechanism of action is the electromagnetic attraction between the metallic particles that carry positive charges and the bacteria cell wall that is negatively charged. When the attraction occurs, the microorganism becomes oxidized and dies immediately.70 Additionally, the particles release ions that can react with thiol groups of the proteins located on the bacterial cell surface, thereby leading to cell lysis.71 It has also been suggested72 that iron ions could diffuse through the cell membrane and interact with intracellular oxygen, thus generating oxidative stress and eventually causing the obstruction of the cell membrane.
In this study, the concentration of MagP®-NH2 in the composites is a key factor determining the bacterial activity inhibition. A similar concentration-dependent behaviour was previously observed by other authors70,71 who investigated the antimicrobial effects of iron oxide nanoparticles on S. aureus and E. coli. Another potential reason for the antimicrobial action observed herein is the presence of non-reacted NHS esters, since lactones are known to have a wide spectrum of antimicrobial effect.73 To verify this, the antibacterial activity of the NHS-activated P(PF-co-EG) network was also tested, and it was found to exhibit higher biocide action than the neat copolymer (i.e. in the range of 0.60–0.75 and 0.25–0.40 against Gram-negative and Gram-positive bacteria, respectively). These results hint that when the copolymer incorporating NHS esters was connected covalently to the MagP®-NH2, the antibacterial inhibition of the microparticles was improved. Therefore, there is likely an additive or synergic effect of MagP®-NH2 and the copolymer network incorporating NHS esters on bacteria killing. It is also worthy to note that the composites do not negatively influence all cells. In particular, as discussed earlier, NHDF metabolic activity decreased only slightly in the presence of composites with MagP®-NH2 content ≤ 10.0 wt%. The combination of cell metabolic activity and antibacterial tests results suggests that MagP®-NH2 microparticles could have a dual therapeutic function to maintain the human connective tissues and simultaneously inhibit bacterial infection. Further, with an appropriate external magnetic field, the developed core–shell composites might be directed to selectively kill bacteria throughout the body.
Conclusions
Magnetic core–shell composites consisting of amino-functionalized Fe3O4 microparticles (MagP®-NH2) covalently anchored to a P(PF-co-EG) network shell have been synthesized and characterized. TEM images revealed that the thickness of the copolymer shell varies depending on the microparticle/copolymer weight ratio: higher contents of P(PF-co-EG) yield thicker shells. The composites showed superparamagnetic behaviour, and the initial magnetic susceptibility increased as the shell became thinner. According to TGA and spectrophotometric analyses, the grafting degree increased upon raising the MagP®-NH2/P(PF-co-EG) weight ratio up to a microparticle concentration of 10.0 wt% and then leveled off. In addition to improved magnetic properties, the composites displayed excellent chemical and thermal stability as well as enhanced biodegradability. They also exhibited moderate inhibition against Gram-negative E. coli and P. aeruginosa as well as Gram-positive S. aureus and S. epidermidis bacteria, likely arising from a synergic effect of MagP®-NH2 and the copolymer network incorporating NHS esters. However, the metabolic activity of NHDF cells cultured in the presence of the composites diminished as the microparticle loading and incubation time increased. Unprecedented improvements in mechanical properties were also attained. In particular, the stiffness and tensile strength under dry conditions increased by factors of about 5.5 and 3.8, respectively, upon addition of 10.0 wt% MagP®-NH2 to the copolymer, and by approximately 4.5 and 3.4 in a SBF environment. Such huge increments are related to the strong covalent interactions between MagP®-NH2 and P(PF-co-EG) that restrict polymer chain movement during deformation. Further, their mechanical properties were improved when exposed to an external magnetic field. Altogether, the experimental results demonstrate that the morphology and the composite properties can be controlled by varying the microparticle/copolymer weight ratio, and that the most advantageous is 10:90, since leads to the best combination of magnetic, mechanical, rheological and antibacterial properties. These composites are excellent candidates to be used in the development of magnetic biomimetic scaffolds for soft-tissue replacement whose mechanical properties can be controlled by external stimuli. Further, they could also be suitable for other applications such as smart magnetic materials, biosensing, protein purification, cell separation, hyperthermia or targeted drug delivery.
Acknowledgements
Dr Ana Diez-Pascual wishes to acknowledge the Ministerio de Economía y Competitividad (MINECO) for a “Ramón y Cajal” Senior Research Fellowship cofinanced by the EU and for financial support (project ref. CTQ2015-66575-P).References
- K. Chatterjee, S. Sarkar, K. J. Rao and S. Paria, Adv. Colloid Interface Sci., 2014, 209, 8–39 CrossRef CAS PubMed.
- M. R. Kim, Z. Xu, G. Chen and D. Ma, Chem.–Eur. J., 2014, 20, 11256–11275 CrossRef CAS PubMed.
- P. Hu, J. V. Morabito and C.-K. Tsung, ACS Catal., 2014, 4, 4409–4419 CrossRef CAS.
- C. J. Garvey, Br. Med. J., 2002, 324, 1077–1080 CrossRef PubMed.
- W. Fang, X. Chen and N. Zheng, J. Mater. Chem., 2010, 20, 8624–8630 RSC.
- U. I. Tromsdorf, N. C. Bigall, M. G. Kaul, O. T. Bruns, M. S. Nikolic, B. Mollwitz, R. A. Sperling, R. Reimer, H. Hohenberg, W. J. Parak, S. F. Rster, U. Beisiegel, G. Adam and H. Weller, Nano Lett., 2007, 7, 2422–2427 CrossRef CAS PubMed.
- J. Park, G. von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Angew. Chem., Int. Ed., 2008, 47, 7284–7288 CrossRef CAS PubMed.
- H. Deng, X. Li, Q. Peng, X. Wang, J. Chen and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 2782–2785 CrossRef CAS PubMed.
- D. Zhang, Z. Tong, S. Li, X. Zhang and A. Ying, Mater. Lett., 2008, 62, 4053–4055 CrossRef CAS.
- Y. W. Jun, Y. M. Huh, J. S. Choi, J. H. Lee, H. T. Song, S. Kim, S. Yoon, K. S. Kim, J. S. Shin, J. S. Suh and J. Cheon, J. Am. Chem. Soc., 2005, 127, 5732–5733 CrossRef CAS PubMed.
- K. C. Huang and S. H. Ehrman, Langmuir, 2007, 23, 1419–1426 CrossRef CAS PubMed.
- W. Wu, Q. He and C. Z. Jiang, Nanoscale Res. Lett., 2008, 3, 397–415 CrossRef CAS PubMed.
- F. K. Kasper, K. Tanahashi, J. P. Fisher and A. G. Mikos, Nat. Protoc., 2009, 4, 518–525 CrossRef CAS PubMed.
- A. S. Mistry, Q. P. Pham, C. Schouten, T. Yeh, E. M. Christenson, A. G. Mikos and J. A. Jansen, J. Biomed. Mater. Res., Part A, 2010, 92, 451–462 Search PubMed.
- B. Farshid, G. Lalwani, M. S. Mohammadi, J. Simonsen and B. Sitharaman, J. Biomed. Mater. Res., Part B, 2017, 105, 406–419 CrossRef CAS PubMed.
- Z. Y. Cai, T. Zhang, L. Z. Di, D. M. Xu, D. H. Xu and D. A. Yang, Biomed. Microdevices, 2011, 13, 623–631 CrossRef CAS PubMed.
- K. Wang, L. Cai, F. Hao, X. Xu, M. Cui and S. Wang, Biomacromolecules, 2010, 11, 2748–2759 CrossRef CAS PubMed.
- A. M. Díez-Pascual and A. L. Díez-Vicente, ACS Appl. Mater. Interfaces, 2016, 8, 17902–17914 Search PubMed.
- A. M. Díez-Pascual and A. L. Díez-Vicente, RSC Adv., 2016, 6, 79507–79519 RSC.
- J. Suggs, E. J. Kao, L. L. Palombo, R. S. Krishnan, M. S. Widmer and A. G. Mikos, J. Biomater. Sci., Polym. Ed., 1998, 9, 653–666 CrossRef PubMed.
- J. Suggs, M. S. Shive, C. A. Garcia, J. M. Anderson and A. G. Mikos, J. Biomed. Mater. Res., 1999, 46, 22–32 CrossRef PubMed.
- A. Gao, X. Yang, C. Zhang, G. Long, J. Pu, Y. Yuan, H. Liu, Y. Li and F. Liao, Chem. Cent. J., 2012, 6, 142–151 CAS.
- S. N. Rampersad, Sensors, 2012, 12, 12347–12360 CrossRef CAS PubMed.
- A. M. Díez-Pascual and A. L. Diez-Vicente, J. Mater. Chem. B, 2016, 4, 600–612 RSC.
- B. Han, S. Zhang, R. Zhou, X. Wu, X. Wei, Y. Xing, S. Wang and T. Qi, RSC Adv., 2014, 4, 50752–50758 RSC.
- P. C. Hartmann and R. D. Sanderson, Macromol. Symp., 2007, 255, 24–35 CrossRef CAS.
- J. Fu, J. Wang, Q. Li, D. H. Kim and W. Knoll, Langmuir, 2010, 26, 12336–12341 CrossRef CAS PubMed.
- H. Zeng, S. Sun, J. Li, Z. L. Wang and J. P. Liu, Appl. Phys. Lett., 2004, 85, 792–794 CrossRef CAS.
- A. M. Díez-Pascual, G. Martinez and M. A. Gomez, Macromolecules, 2009, 42, 6885–6892 CrossRef.
- S. Khoee and A. A. Kavand, J. Nanostruct. Chem., 2014, 4, 111 CrossRef.
- A. M. Díez-Pascual and A. L. Diez-Vicente, RSC Adv., 2016, 6, 79507–79519 RSC.
- A. M. Díez-Pascual, G. Martinez, G. Gónzalez-Domínguez, A. Anson, M. T. Martinez and M. A. Gomez, J. Mater. Chem., 2010, 20, 8285–8296 RSC.
- A. H. Soeriyadi, B. Gupta, P. J. Reece and J. J. Gooding, Polym. Chem., 2014, 5, 2333–2341 RSC.
- K. W. Lee, S. Wang, L. Lu, E. Jabbari, B. L. Currier and M. J. Yaszemski, Tissue Eng., Part A, 2006, 12, 2801–2802 CrossRef CAS PubMed.
- A. M. Diez-Pascual and M. Naffakh, Carbon, 2012, 50, 857–868 CrossRef CAS.
- A. M. Díez-Pascual and A. L. Díez-Vicente, ACS Appl. Mater. Interfaces, 2014, 6, 3729–3741 Search PubMed.
- A. D. Tehrani and M. H. Babaabbasi, Nanocellulose Preparation, Surface Chemical Modification and its Applications, in Cellulose-Based Graft Copolymers: Structure and Chemistry, ed. V. K. Thakur, CRC Press, New York, 2015, pp. 519–552 Search PubMed.
- L. E. Alexander and R. E. Krieger, X-ray Diffraction Methods in Polymer Science, Willey, New York, 1969 Search PubMed.
- M. Esmaeilpour, A. R. Sardarian, A. Jarrahpour, E. Ebrahimi and J. Javidi, RSC Adv., 2016, 6, 43376–43387 RSC.
- A. M. Diez-Pascual and M. Naffakh, Mater. Chem. Phys., 2013, 131, 605–614 CrossRef.
- F. Bignott, M. Penco, L. Sartore, S. D'Antone, A. D'Amore and G. Spagnoli, Macromol. Symp., 2002, 180, 257–266 CrossRef.
- Z.-W. Ouyang, E.-C. Chen and T.-M. Wu, Materials, 2015, 8, 4553–4564 CrossRef.
- M. T. Ramesan and K. Surya, J. Appl. Polym. Sci., 2016, 133, 5431–5438 Search PubMed.
- J. Lis and P. O. Kellard, J. Phys. D: Appl. Phys., 1968, 1, 1117 Search PubMed.
- K. Babu and R. Dhamodharan, Nanoscale Res. Lett., 2009, 4, 1090–1102 CrossRef CAS PubMed.
- F. Márquez, G. M. Herrera, T. Campo, M. Cotto, J. Ducongé, J. M. Sanz, E. Elizalde, O. Perales and C. Morant, Nanoscale Res. Lett., 2012, 7, 210–221 CrossRef PubMed.
- S. Xuan, Y. X. J. Wang, J. C. Yu and K. C. F. Leung, Chem. Mater., 2009, 21, 5079–5087 CrossRef CAS.
- L. D. Landau and E. M. Lifshitz, Electrodynamics of Continuous Media, Pergamon Press, New York, 1984 Search PubMed.
- J. Zhu, S. Wei, J. Ryu, L. Sun, Z. Luo and Z. Guo, ACS Appl. Mater. Interfaces, 2010, 2, 2100–2107 CAS.
- S. Kasapis, Modern Biopolymer Science. Bridging the Divide between Fundamental Treatise and Industrial Application, ed. S. Kasapis, I. T. Norton and J. B. Ubbink, Elsevier, Oxford, 2009 Search PubMed.
- M. Masłowski and M. Zaborski, Elastomeric Composites with Magnetorheological and Magnetic Properties Containing Nano-sized Iron Oxide, in Nanomagnetism, ed. J. M. G. Estevez, One Central Press, Manchester, 2014, ch. 2 Search PubMed.
- F. J. O'Brien, Mater. Today, 2011, 14, 88–95 CrossRef.
- A. K. Gaharwar, A. Patel, A. Dolatshahi-Pirouz, H. Zhang, K. Rangarajan, G. Iviglia, S.-R. Shin, M. A. Hussain and A. Khademhosseini, Biomater. Sci., 2015, 1, 45–68 Search PubMed.
- A. M. Díez-Pascual, M. A. Gómez-Fatou, F. Ania and A. Flores, Prog. Mater. Sci., 2015, 67, 1–94 CrossRef.
- A. M. Díez-Pascual, M. Naffakh, C. Marco, G. Ellis and M. A. Gómez-Fatou, Prog. Mater. Sci., 2012, 57, 1106–1190 CrossRef.
- D. Chicot, J. Mendoza, A. Zaoui, G. Louis, V. Lepingle, F. Roudet and J. Lesage, Mater. Chem. Phys., 2011, 129, 862–870 CrossRef CAS.
- P. F. Costa, C. Vaquette, J. Baldwin, M. Chhaya, M. E. Gomes, R. L. Reis, C. Theodoropoulos and D. W. Hutmacher, Biofabrication, 2014, 6, 035006 CrossRef PubMed.
- M. H. Ayob, J. Efendi, L. S. Yuana, S. Chandren, H. C. Siong and H. Nura, J. Teknologi, 2014, 69, 21–23 Search PubMed.
- Y. Kumagai and Y. Doi, Polym. Degrad. Stab., 1992, 36, 241–248 CrossRef CAS.
- M.-H. Ho, S.-Y. Li, C.-Y. Ciou and T.-M. Wu, J. Appl. Polym. Sci., 2014, 131, 41070–41076 Search PubMed.
- M. O. Wang, J. M. Etheridge, J. A. Thompson, C. E. Vorwald, D. Dean and J. P. Fisher, Biomacromolecules, 2013, 14, 1321–1329 CrossRef CAS PubMed.
- A. M. Prodan, S. L. Iconaru, C. S. Ciobanu, M. C. Chifiriuc, M. Stoicea and D. Predoi, J. Nanomater., 2013, 2013, 587021 Search PubMed.
- W. J. Yang, J. H. Lee, S. C. Hong, J. Lee, J. Lee and D.-W. Han, Materials, 2013, 6, 4689–4706 CrossRef.
- N. Singh, G. J. Jenkins, R. Asadi and S. H. Doak, Nano Rev., 2010, 1, 10340 Search PubMed.
- B. Katsnelson, L. I. Privalova, T. D. Degtyareva and J. B. Beykin, NSTI Nanotechnol., 2011, 3, 537–540 CAS.
- A. S. Arbab, L. B. Wilson, P. Ashari, E. K. Jordan, B. K. Lewis and J. A. Frank, NMR Biomed., 2005, 18, 383–389 CrossRef CAS PubMed.
- P. Collins and A. Smith, Antimicrobial activity to ISO 22196. Technical report PRA ref. 09/179, March 2010 Search PubMed.
- S. S. Behera, J. K. Patra, K. Pramanik, N. Panda and H. Thatoi, World J. Nano Sci. Eng., 2012, 2, 196–200 CrossRef.
- M. T. Cabeen and C. Jacobs-Wagner, Nat. Rev. Microbiol., 2005, 3, 601–610 CrossRef CAS PubMed.
- Y. T. Prabhu, K. V. Rao, B. S. Kumari, V. S. S. Kumar and T. Pavani, Int. Nano Lett., 2015, 5, 82–92 Search PubMed.
- N. Tran, A. Mir, D. Mallik, A. Sinha, S. Nayar and T. J. Webster, Int. J. Nanomed., 2010, 5, 277–283 CAS.
- C. Lee, J. Y. Kim, W. I. Lee, K. L. Nelson, J. Yoon and D. L. Sedlak, Environ. Sci. Technol., 2008, 42, 4927–4933 CrossRef CAS PubMed.
- M. Mazur, A. Skrobiszewski, W. Gładkowski, M. Podkowik, J. Bania, J. Nawrot, T. Klejdysz and C. Wawrzeńczyk, Pest Manage. Sci., 2016, 72, 489–496 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |