
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric synthesis of C–F quaternary α-fluoro-β-amino-indolin-2-ones via Mannich addition reactions; facets of reactivity, structural generality and stereochemical outcome†
Chen Xiea,
Wanxing Shaa,
Yi Zhua,
Jianlin Han *a,
Vadim A. Soloshonokbc and
Yi Pana
*a,
Vadim A. Soloshonokbc and
Yi Pana
aSchool of Chemistry and Chemical Engineering, State Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing, 210093, China. E-mail: hanjl@nju.edu.cn
bDepartment of Organic Chemistry I, Faculty of Chemistry, University of the Basque Country UPV/EHU, Paseo Manuel Lardizabal 3, 20018 San Sebastián, Spain. E-mail: vadym.soloshonok@ehu.es
cIKERBASQUE, Basque Foundation for Science Department, Alameda Urquijo 36-5, Plaza Bizkaia, 48011 Bilbao, Spain
First published on 17th January 2017
Abstract
Reported herein is a new approach for the preparation of enantiomerically pure α-fluoro-β-amino-indolin-2-ones possessing tetrasubstituted fluorinated stereogenic centers. This method includes the detrifluoroacetylative in situ generation of tertiary enolates followed by Mannich reaction with (Ss)-sulfinylimines. The operationally convenient conditions coupled with perfect diastereoselectivity and functional substituent compatibility bode well for widespread application of this new synthetic method.
Fluoro-organic chemistry plays an essential role in furthering numerous technological advancements, most notably in the energy,1 food2 and healthcare industries.3 Consequently, the current interest in the synthesis of fluorine-containing compounds is at an all-time high, addressing the emerging scientific and practical needs.4 Thus, many challenging areas of fluorine methodology have seen important, but somewhat uneven progress.5 In particular, despite the impressive recent advances in the asymmetric synthesis of fluorinated amines/amino acids6 and derivatives containing quaternary C–F stereogenic carbon,7 and compounds possessing both of these, consecutively located, their structural features are still not well studied.8
One of the recent advances in fluoro-organic methodology is the development of detrifluoroacetylative, in situ generation of unprotected fluoro-enolates 1 (Scheme 1, eqn (1)).9 The initial study into the reactivity of enolates 1 using aldol9,10 and Mannich10,11 reactions has revealed their generally promising synthetic potential along with some problematic aspects awaiting more focused research. For example, enolates 1 easily reacts with aldehydes9,10 and highly electrophilic, CF3-containing ketones12 and imines,11 while the reactions of 1 with unactivated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN compounds still need more innovative solutions. Over the last several years, our group has contributed to this research area by the development of cyclic enolates 2 (eqn (2)) allowing preparation of structurally complex products possessing tetrasubstituted fluorinated stereogenic centers.13 The preliminary data on reactivity of tertiary enolates 2 with imines14 revealed their moderate reactivity towards highly electrophilic CF3-imines 3 affording derivatives 4 featuring the structurally challenging quaternary C–F stereogenic center next to the amino functionality. While the preparation of compounds 4 was a noticeable methodological success, one might agree that rather excessive number of fluorine atoms and relatively high configurational liability of the CH stereogenic carbon, would limit the pharmaceutical application of compounds of type 4. Therefore, to realize the synthetic potential of this methodology we have to develop the corresponding Mannich addition reactions using common, non-fluorinated imines. In this work we report the very successful implementation of this goal, disclosing the Mannich reactions between 3-fluoroindolin-2-one derived enolates 5 (eqn (3)) and non-fluorinated aldimines 6. We demonstrate that these addition reactions occur with nearly perfect (>98:2) diastereoselectivity allowing preparation of the target quaternary C–F/amino products in enantiomerically pure form.
O and CN compounds still need more innovative solutions. Over the last several years, our group has contributed to this research area by the development of cyclic enolates 2 (eqn (2)) allowing preparation of structurally complex products possessing tetrasubstituted fluorinated stereogenic centers.13 The preliminary data on reactivity of tertiary enolates 2 with imines14 revealed their moderate reactivity towards highly electrophilic CF3-imines 3 affording derivatives 4 featuring the structurally challenging quaternary C–F stereogenic center next to the amino functionality. While the preparation of compounds 4 was a noticeable methodological success, one might agree that rather excessive number of fluorine atoms and relatively high configurational liability of the CH stereogenic carbon, would limit the pharmaceutical application of compounds of type 4. Therefore, to realize the synthetic potential of this methodology we have to develop the corresponding Mannich addition reactions using common, non-fluorinated imines. In this work we report the very successful implementation of this goal, disclosing the Mannich reactions between 3-fluoroindolin-2-one derived enolates 5 (eqn (3)) and non-fluorinated aldimines 6. We demonstrate that these addition reactions occur with nearly perfect (>98:2) diastereoselectivity allowing preparation of the target quaternary C–F/amino products in enantiomerically pure form.
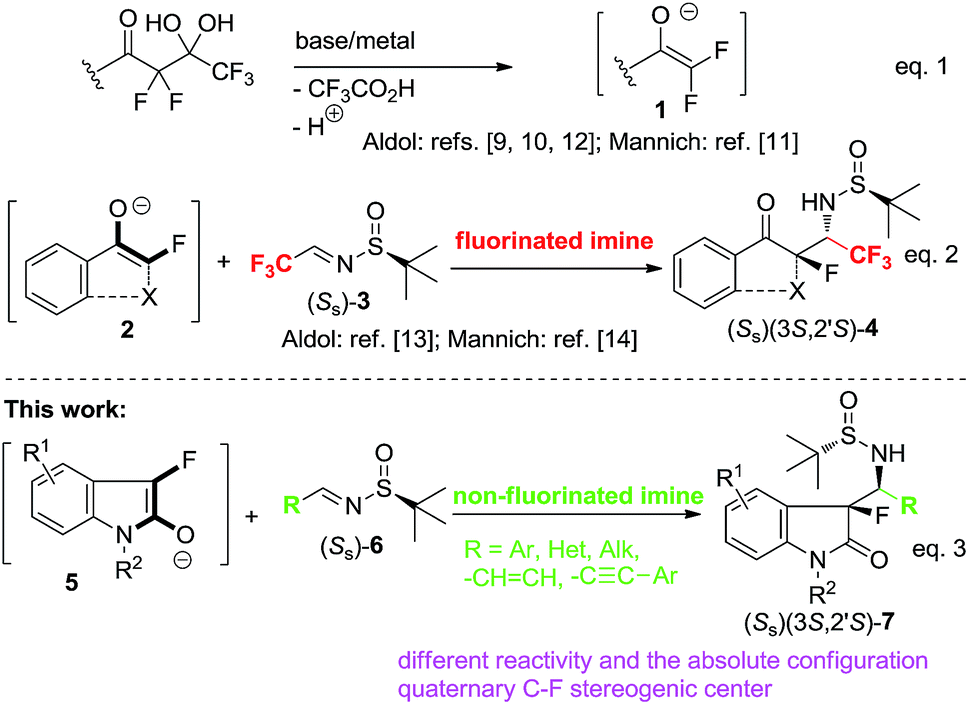 | ||
| Scheme 1 Detrifluoroacetylative reactions with CF3-imines 3 and non-fluorinated imines 6. | ||
Drawing from our experience in asymmetric Mannich additions15 and, in particular, dealing with unactivated imines,16 we selected non-enolizable and reasonably reactive imine 10a; also, bearing a trifluoromethylthio group – quite useful as the 19F-NMR reference for monitoring the reaction outcome (Table 1). As was anticipated, the reaction of imine 10a conducted under the conditions (DIPEA, 2-Me-THF, −40 °C) previously reported for the additions of CF3-sulfinylimine 3 (Scheme 1, eqn (2)), did not proceed at all. Lower reactivity of 10a was counteracted by the elevated reaction temperature. Thus, the addition was rather successful at 0 °C, affording product 11aa with moderate yield, but with quite respected diastereoselectivity (entry 1). Building on this apparent progress, we proceeded with a thorough optimization of all reaction parameters starting with the base. A selection of examples presented in entries 2–7 convincingly demonstrates that the base's basicity is not of prime importance, while its steric bulk plays rather significant role. This observation can be rationalized considering the sterically congested nature of tertiary enolate and therefore, its sensitivity towards the steric bulk of the base used. Accordingly, for the further optimization study, we selected triethylamine (entry 2) which gave the finest combination of chemical yield and diastereoselectivity.
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Yieldb (%) | Drc |
| a Reaction condition: di-ketone hydrates 1a (0.6 mmol), sulfinylimine 10a (0.5 mmol), LiBr (156.3 mg, 1.8 mmol, 3.0 equiv.), base (1.5 mmol, 2.5 equiv.) in 5 mL solvent at 0 °C for 10 min.b Isolated yields of the major diastereomers.c Determined by the 19F-NMR analysis of the crude products. | ||||
| 1 | DIPEA | THF | 54 | 97:3 |
| 2 | Et3N | THF | 88 | 97:3 |
| 3 | nPr3N | THF | <10 | n.d. |
| 4 | nBu3N | THF | <10 | n.d. |
| 5 | DABCO | THF | 92 | 96:4 |
| 6 | DBU | THF | 87 | 96:4 |
| 7 | TMEDA | THF | 76 | 96:4 |
| 8 | Et3N | 2-Me-THF | 94 | 97:3 |
| 9 | Et3N | CH3CN | 89 | >98:2 |
| 10 | Et3N | DCM | 80 | >98:2 |
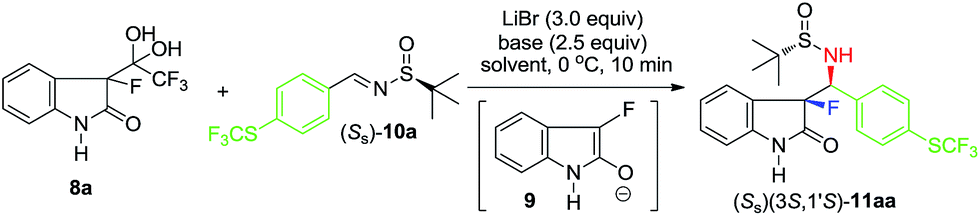
In the second round of the optimization experiments we focused on the properties of reaction solvent and its effect on the addition reaction under study; the key representative results are presented in entries 8–10. For example, the application of 2-Me-THF gave noticeably better yield (entry 8) as compared with that of the reaction conducted in THF. However, the diastereoselectivity in this reaction was still incomplete. In sharp contrast, the reactions conducted in CH3CN (entry 9) and DCM (entry 10) proceeded with nearly perfect stereochemical outcome. It should be mentioned that the used in this work notation (the diastereoselectivity of >98:2) is a very conservative estimation, indicating that the NMR signals of other diastereomeric products were not observed in the reaction mixtures. Comparing the results obtained in CH3CN (entry 9) and DCM (entry 10), one may agree, that the former, providing higher chemical yield, was selected as the optimized solvent.
Next, we proceeded to study the substrate generality of this method. First, we decided to explore the reactivity and substituent effect of sulfinylimines (Ss)-10a–r (Scheme 2) derived from aldehydes representing various structural types and classes.
 | ||
| Scheme 2 Substrate generality study of di-ketone hydrates 8 with (Ss)-10. | ||
Considering the 19 examples listed in Scheme 2, one might agree that the stereochemical outcome of the Mannich additions between di-ketone hydrates 8 and sulfinylimines (Ss)-10 is rather exceptional. In fact we failed to find an imine substrate reacting with less than >98:2 diastereoselectivity. In terms of the chemical yields, the lowest value of 64% was obtained for product 11ab and, most likely, can be attributed to a slow reaction rate due to the steric shielding of the CN group in 10b by the ortho-trifluoromethyl group. The highest, 97% yield, was registered for product 11aj, possessing para-nitro group. In general, for the compounds bearing electron-withdrawing substituents, the chemical yields were normally above 85%, while unsubstituted 11ai and para-methyl products 11ae gave a bit lower (83% and 79%) yields. The observed tendency is apparently a function of the electrophilicity/reactivity of the CN bond in sulfinylimines (Ss)-10. Generality of this reaction was further exemplified by the preparation of products 11am and 11an, bearing naphthyl and hetero-aromatic rings. Of particular interest were derivatives 11ao and 11ap featuring unsaturated moieties, which can be used for synthetic elaborations. Quite remarkably, this method was found to be readily applicable for enolizable alkyl-aldehyde derived sulfinylimines (Ss)-10q,r. Thus, products 11q,r, bearing methyl and n-butyl groups, were isolated with the same excellent (>98:2) diastereoselectivity and preparatively attractive chemical yields. Finally, N-Me β-keto-amide-hydrate 8b was reacted with benzaldehyde derived imine 10i. The addition proceeded without any observable problems giving rise to diastereomerically pure product 11bi with isolated 82% yield.
N-Methyl derivative 11bi was a logical connection to the subsequent second series of substrate generality experiments where we explored the effects of the substituents positioned on the starting β-keto-amide-hydrates 12 (Scheme 3). Imine (Ss)-10a, derived from 4-(trifluoromethylthio)benzaldehyde, was selected as standard electrophile and all reactions were conducted under the same conditions. The twelve examples shown in Scheme 3 were selected to represent the two-pronged substituent positioning on the starting di-ketone hydrates 12. The major conclusion that one can draw from these data, is the synthetically attractive generality of this method, as the nearly perfect diastereoselectivity (>98:2) and respected chemical yields of products 13a–l were maintained in all cases studied. Preparation of compounds bearing hydrolytically liable groups (13c), di-substitution (13e) and electronically conjugated N-allyl (13k), N-phenyl (13l) moieties, deserve some particular mentioning.
 | ||
| Scheme 3 Substrate generality study of di-ketone hydrates 12 with (Ss)-10a. | ||
As the last synthetic goal of this study we decided to demonstrate the deprotection procedure affording the target compounds with free amino functionality. We selected derivative 11aa, possessing unsubstituted NH group on the indolin-2-one moiety (Scheme 4). The deprotection reaction was conducted under typical mild acidic conditions17 giving α-fluoro-β-amino product (3S,1′S)-14, which was isolated with 89% yield. Furthermore, product 14 was found to be of >99% ee, strongly suggesting that compounds 11 and 13, listed in Schemes 2 and 3, are virtually diastereomerically pure.
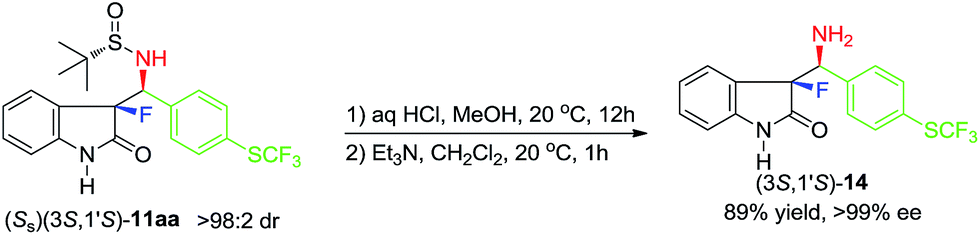 | ||
| Scheme 4 Deprotection of product (Ss)(3S,1′S)-11aa. | ||
The final objective of this research was the assignment of the absolute configuration of products 11 and 13. To this end we performed a crystallographic analysis of N-Me derivative 11bi (Scheme 2), which gave rather unexpected result. Thus, according to the X-ray data (see ESI†), the configuration of 11bi was found to be (Ss)(3S,2′S), that is in sharp contrast to the (Ss)(3S,2′S)18,19 configuration reported for the reactions of CF3-sufinylimine (Ss)-3 (Scheme 1).10,14
To rationalize this unexpected stereochemical outcome, we can propose the transition state (TS-A) presented in Fig. 1. First of all, based on the literature data,17 the observed (1′S) absolute configuration in products (Ss)(3S,2′S)-11/13, strongly suggest that sulfinylimines (Ss)-10 reacts in the corresponding s-trans geometric configuration. Furthermore, the (3S) stereochemistry of the quaternary carbon indicates that sulfinylimines (Ss)-10 approach the re-face of the corresponding tertiary enolates. Taking these experimental facts together, and complying with the principle of minimum charge separation,20 we constructed plausible TS-A leading to the products of the observed (Ss) (3S,2′S) absolute configuration. Noteworthy, the close proximity of enolate O–Li moiety and the imine nitrogen in TS-A, allows not only for the concerted oxygen–nitrogen charge transfer, but also for the additional stabilization via Li-coordination to the S–O oxygen. This feature is never observed in the case of CF3-sulfenylimines 3 (ref. 5a and 10) and may be the underlying reason of the different stereochemical outcome observed in the present study. Moreover, in TS-A the substituent R on the imine, the aromatic ring of the tertiary enolate and N-R group are perfectly located to avoid unfavorable overlaps or any other steric interactions. Accordingly, one may agree that TS-A can be quite credibly used to elucidate the remarkable substrate generality observed in these Mannich addition reactions.
 | ||
| Fig. 1 Plausible transition state TS-A accounting for the observed stereochemical outcome and s-trans geometry of sulfinylimines (Ss)-10. | ||
Conclusions
In summary, we have developed highly stereoselective detrifluoroacetylative Mannich reactions between the in situ generated tertiary enolates and sulfinyl-imines. The reactions feature nearly complete stereochemical outcome and remarkable generality accommodating several type of functional substitution on both enolates and imines. These reaction characteristics constitute a method of high synthetic value for preparation of enantiomerically pure α-fluoro-β-amino-indolin-2-ones with tetrasubstituted fluorinated stereogenic center.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21472082).Notes and references
- (a) Y. Liang, D. Feng, Y. Wu, S.-T. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792 CrossRef CAS PubMed; (b) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496 RSC.
- (a) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed; (b) T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16 CrossRef CAS.
- (a) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed; (b) W. Zhu, J. Wang, S. Wang, Z. Gu, J. L. Aceña, K. Izawa, H. Liu and V. A. Soloshonok, J. Fluorine Chem., 2014, 167, 37 CrossRef CAS; (c) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (d) K. Izawa, J. L. Aceña, J. Wang, V. A. Soloshonok and H. Liu, Eur. J. Org. Chem., 2016, 8–16 CrossRef CAS; (e) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed.
- (a) J. Han, A. E. Sorochinsky, T. Ono and V. A. Soloshonok, Curr. Org. Synth., 2011, 8, 281 CrossRef CAS; (b) K. Mikami, S. Fustero, M. Sánchez-Roselló, J. L. Aceña, V. A. Soloshonok and A. E. Sorochinsky, Synthesis, 2011, 3045 CAS; (c) N. Shibata, T. Ishimaru, S. Nakamura and T. Toru, J. Fluorine Chem., 2007, 128, 469 CrossRef CAS; (d) N. Shibata, S. Mizuta and H. Kawai, Tetrahedron: Asymmetry, 2008, 19, 2633 CrossRef CAS; (e) X. H. Xu and F. L. Qiang, Curr. Org. Chem., 2015, 19, 1566 CrossRef CAS.
- (a) H. B. Mei, C. Xie, J. L. Han and V. A. Soloshonok, Eur. J. Org. Chem., 2016, 5917 CrossRef CAS; (b) X. H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731 CrossRef CAS PubMed; (c) S. Lectard, Y. Hamashima and M. Sodeoka, Adv. Synth. Catal., 2010, 352, 2708 CrossRef CAS; (d) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS PubMed; (e) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826–870 CrossRef CAS PubMed; (f) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed; (g) S. Zhang, L. Li, Y. Hu, Y. Li, Y. Yang, Z. Zha and Z. Wang, Org. Lett., 2015, 17, 5036 CrossRef CAS PubMed.
- (a) J. L. Aceña, A. E. Sorochinsky and V. A. Soloshonok, Synthesis, 2012, 1591 CrossRef; (b) K. V. Turcheniuk, V. P. Kukhar, G.-V. Roeschenthaler, J. Luis Acena, V. A. Soloshonok and A. E. Sorochinsky, RSC Adv., 2013, 3, 6693 RSC; (c) X.-L. Qiu and F.-L. Qing, Eur. J. Org. Chem., 2011, 3261 CrossRef CAS.
- (a) X. Yang, R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5225 CrossRef CAS PubMed; (b) N. Mankad and F. D. Toste, Chem. Sci., 2012, 3, 72 RSC; (c) R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 1268 CrossRef CAS PubMed; (d) N. Shibata, J. Kohno, K. Takai, T. Ishimaru, S. Nakamura, T. Toru and S. Kanemasa, Angew. Chem., Int. Ed., 2005, 44, 4204 CrossRef CAS PubMed; (e) I. Saidalimu, S. Suzuki, E. Tokunaga, E. Tokunaga and N. Shibata, Chem. Sci., 2016, 7, 2106 RSC; (f) W. Yu, X. H. Xu and F. L. Qing, Adv. Synth. Catal., 2015, 357, 2039 CrossRef CAS; (g) Y. Hamashima, K. Yagi, H. Takano, L. Tamas and M. Sodeoka, J. Am. Chem. Soc., 2002, 124, 14530 CrossRef CAS PubMed; (h) L. Hintermann and A. Togni, Angew. Chem., Int. Ed., 2000, 39, 4359 CrossRef CAS; (i) H. Ibrahim and A. Togni, Chem. Commun., 2004, 1147 RSC.
- (a) K. Hiramatsu, T. Honjo, V. Rauniyar and F. D. Toste, ACS Catal., 2016, 6, 151 CrossRef CAS PubMed; (b) R. J. Phipps, K. Hiramatsu and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 8376 CrossRef CAS PubMed; (c) H. P. Shunatona, N. Fruh, Y. M. Wang, V. Rauniyar and F. D. Toste, Angew. Chem., Int. Ed., 2013, 52, 7724 CrossRef CAS PubMed; (d) Y. Hamashima, T. Suzukia, H. Takanoa, Y. Shimura and M. Sodeoka, J. Am. Chem. Soc., 2005, 127, 10164 CrossRef CAS PubMed; (e) B. M. Trost, T. Saget, A. Lerchen and C. I. Hung, Angew. Chem., Int. Ed., 2016, 55, 781 CrossRef CAS PubMed.
- (a) C. Han, E. H. Kim and D. A. Colby, J. Am. Chem. Soc., 2011, 133, 5802 CrossRef CAS PubMed; (b) P. Zhang and C. Wolf, Angew. Chem., Int. Ed., 2013, 52, 7869 CrossRef CAS PubMed; (c) I. Saidalimu, X. Fang, X.-P. He, J. Liang, X. Yang and F. Wu, Angew. Chem., Int. Ed., 2013, 52, 5566 CrossRef CAS PubMed.
- H. Mei, C. Xie, J. L Aceña, V. A. Soloshonok, G.-V. Röschenthaler and J. Han, Eur. J. Org. Chem., 2015, 6401 CrossRef CAS.
- (a) C. Xie, L. Wu, H. Mei, V. A. Soloshonok, J. Han and Y. Pan, Org. Biomol. Chem., 2014, 12, 7836 RSC; (b) C. Xie, L. Wu, H. Mei, V. A. Soloshonok, J. Han and Y. Pan, Tetrahedron Lett., 2014, 55, 5908 CrossRef; (c) C. Xie, L. Wu, J. Zhou, H. Mei, V. A. Soloshonok, J. Han and Y. Pan, J. Fluorine Chem., 2015, 172, 13 CrossRef CAS.
- P. Zhang and C. Wolf, J. Org. Chem., 2012, 77, 8840 CrossRef CAS PubMed.
- (a) C. Xie, L. Wu, J. Han, V. A. Soloshonok and Y. Pan, Angew. Chem., Int. Ed., 2015, 54, 6019 CrossRef CAS PubMed; (b) L. Zhang, C. Xie, Y. Dai, H. Mei, V. A. Soloshonok, Y. Pan and J. Han, J. Fluorine Chem., 2016, 184, 28 CrossRef CAS; (c) W. Sha, L. Zhang, W. Zhang, H. Mei, V. A. Soloshonok, J. Han and Y. Pan, Org. Biomol. Chem., 2016, 14, 7295 RSC.
- (a) C. Xie, Y. Dai, H. Mei, J. Han, V. A. Soloshonok and Y. Pan, Chem. Commun., 2015, 51, 9149 RSC; (b) C. Xie, L. Zhang, W. Sha, V. A. Soloshonok, J. Han and Y. Pan, Org. Lett., 2016, 18, 3270 CrossRef CAS PubMed.
- (a) H. Mei, C. Xie, L. Wu, V. A. Soloshonok, J. Han and Y. Pan, Org. Biomol. Chem., 2013, 11, 8018 RSC; (b) C. Xie, H. Mei, L. Wu, V. A. Soloshonok, J. Han and Y. Pan, RSC Adv., 2014, 4, 4763 RSC; (c) H. Mei, Y. Xiong, C. Xie, V. A. Soloshonok, J. Han and Y. Pan, Org. Biomol. Chem., 2014, 12, 2108 RSC.
- (a) L. M. Wu, C. Xie, H. B. Mei, V. A. Soloshonok, J. L. Han and Y. Pan, Org. Biomol. Chem., 2014, 12, 4620 RSC; (b) Y. L. Dai, C. Xie, L. M. Wu, H. B. Mei, V. A. Soloshonok, J. L. Han and Y. Pan, RSC Adv., 2015, 5, 3491 RSC.
- (a) C. Xie, L. Zhang, H. Mei, R. Pajkert, M. Ponomarenko, Y. Pan, G.-V. Röschenthaler, V. A. Soloshonok and J. Han, Chem.–Eur. J., 2016, 22, 7036 CrossRef CAS PubMed; (b) M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600 CrossRef CAS PubMed.
- To avoid a confusion, it should be noted that while the descriptors of the absolute configuration for products 11, 13 and those derived from CF3-imine 3 (Scheme 1) are the same, these compounds are stereochemically different due to the Cahn–Ingold–Prelog priority rules ref. 19.
- R. S. Cahn, C. K. Ingold and V. Prelog, Angew. Chem., Int. Ed., 1966, 5, 385 CrossRef CAS.
- (a) V. A. Soloshonok, C. Cai, V. J. Hruby, L. V. Meervelt and T. Yamazaki, J. Org. Chem., 2000, 65, 6688 CrossRef CAS PubMed; (b) V. A. Soloshonok, H. Ohkura, A. Sorochinsky, N. Voloshin, A. Markovsky, M. Belik and T. Yamazaki, Tetrahedron Lett., 2002, 43, 5445 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedure and full characterization data, NMR spectra of product 11, 13, 14 and X-ray analysis data for 11bi. CCDC 1457060. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra27710a |
| This journal is © The Royal Society of Chemistry 2017 |