
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile preparation of 1,3,5,7-tetranitro-1,3,5,7-tetrazocane/glycidylazide polymer energetic nanocomposites with enhanced thermolysis activity and low impact sensitivity†
Teng Chena,
Wei Jiang *a,
Ping Du*b,
Jie Liua,
Gazi Haoa,
Han Gaoa,
Lei Xiaoa and
Xiang Kea
*a,
Ping Du*b,
Jie Liua,
Gazi Haoa,
Han Gaoa,
Lei Xiaoa and
Xiang Kea
aNational Special Superfine Powder Engineering Research Center of China, School of Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: superfine_jw@126.com; Fax: +86 25 84315042; Tel: +86 25 84315042
bSchool of Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: dp1314@163.com
First published on 17th January 2017
Abstract
1,3,5,7-tetranitro-1,3,5,7-tetrazocane/glycidylazide polymer (HMX/GAP) nanocomposites were successfully prepared via a facial sol–gel supercritical method. The as-synthesized HMX/GAP nanocomposites were characterized by X-ray diffraction (XRD), Raman, and Fourier-transform infrared spectroscopy (FT-IR), which indicated that HMX were successfully trapped in GAP gel skeleton. Scanning electron microscopy (SEM) images revealed that the GAP gel skeleton possessed nano-porous structure, which made it possible to load HMX particles. The thermal decomposition behaviours of GAP, raw HMX and HMX/GAP nanocomposites were determined by differential thermal analyser (DTA). The results indicated that GAP aerogel could promote the decomposition of HMX and enhance the interaction between GAP and HMX. The kinetic, thermodynamic and thermal stability parameters, such as activation energy (Ea), per-exponent factor (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) A), activation heat (ΔH≠), activation free energy (ΔG≠), activation entropy (ΔS≠), critical temperature of thermal explosion (Tb) and the self-accelerating decomposition temperature (TSADT) were calculated according to DTA analysis. The calculated results implied that HMX/GAP showed much lower activation energy than raw HMX. Similarly, the HMX/GAP presented a much lower Tb and TSADT than raw HMX. According to the impact sensitivity tests, the mechanical sensitivities of HMX/GAP nanocomposites were significantly lower than those of raw HMX.
A), activation heat (ΔH≠), activation free energy (ΔG≠), activation entropy (ΔS≠), critical temperature of thermal explosion (Tb) and the self-accelerating decomposition temperature (TSADT) were calculated according to DTA analysis. The calculated results implied that HMX/GAP showed much lower activation energy than raw HMX. Similarly, the HMX/GAP presented a much lower Tb and TSADT than raw HMX. According to the impact sensitivity tests, the mechanical sensitivities of HMX/GAP nanocomposites were significantly lower than those of raw HMX.
1. Introduction
With the development of modern munition systems, the demands on the properties of energetic materials become crucial. In order to satisfy these requirements, developing materials with higher energy density, higher energy release rate and lower sensitivity become critical.1 At present, fabrication of nano- or micro-scale energetic materials is the most convenient and efficient way to improve the energy release rate and combustion reaction of energetic materials.2–8 It is known that the energy release and combustion properties of energetic materials are closely related to their microstructure and dimension. Compared with micron-sized energetic materials, the nano-sized energetic materials show enhanced properties, such as higher burning rates, lower impact sensitivity and higher energy release rate.9–11 When the particle size decreases, more molecules are exposed on the surface, which makes the specific surface area and free surface energy increase. More specifically, the heat and the mass transfer length scales are drastically reduced, hence, the reactant activities of nano- or micro-scale energetic materials are significantly higher than macro-scale bulk materials.12,13Great achievements in the field of nanoscience and nanotechnology have enabled fabrication of nano-sized materials, which can be widely used in many areas for their extraordinary properties arising from their nanoscale dimension. Inspired by these achievements, intensive researches on nano-energetic materials are receiving more and more attention. Up till now, numerous studies have been carried out to prepare nanometre energetic materials, such as nano-RDX (hexahydro-1,3,5-trinitro-1,3,5-triazine),14,15 nano-HMX (1,3,5,7-tetranitro-1,3,5,7-tetrazocane),16,17 nano-CL-20 (2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane),18 nano-HNS (2,2′,4,4′,6,6′-hexanitro-stilbene),19 nano-NTO (3-nitro-1,2,4-triazol-5-one),20 and nano-TATB (1,3,5-triamino-2,4,6-trinitrobenzene).21
Over the past few years, various techniques have been developed to prepare nano-energetic materials, such as solvent/non-solvent recrystallization method,22 sol–gel method, mechanical grinding method,23 spraying evaporation method,16 supercritical fluid techniques (rapid expansion supercritical solution process or gas antisolvent process),24 and freezing evaporation method.25 Nevertheless, most of these methods have limitations. For example, by solvent/non-solvent recrystallization method, the size of energetic particles cannot be controlled precisely and the size distributions are not uniform. By comparison, sol–gel method can be regarded as a promising technique for fabricating nanoscale energetic materials, due to the convenience of ambient conditions preparation, easy control over the stoichiometry and homogeneity of composites. In a typical sol–gel process, a gel network comes into formation and the energetic particles can be restricted within the network, which can effectively prevent the agglomeration of the recrystallized particles. Different kinds of nanocomposite energetic materials have been prepared successfully by sol–gel method, including metal oxide-matrix (Fe2O3, MoO3 and CuO),26–28 inorganic non-metal oxide-matrix (SiO2)29–31 and organic RF (resorcinol-formaldehyde)-matrix32,33 nanocomposite energetic materials. It can be concluded that all of these gel matrix are non-energetic components, which can weaken the energy release of nanocomposite energetic materials. The existing of inert gel matrix will restrict the application of these materials, due to the poor energy performance of gel matrix. Hence, using an energetic matrix to substitute a non-energetic matrix can be an alternative method to resolve this problem.
Glycidylazide polymer (GAP) is a typical energetic azide polymer, which incorporates one or several –N3 groups in their monomer.34 In comparison with inert gel matrix, GAP exhibits unique properties including high nitrogen content, positive enthalpy of formation, good thermally stability and insensitivity.35,36 Thus it offers desirable properties for nanocomposite energetic materials and feasible route to manufacture novel energetic materials with higher energy release rate, higher burn rate and lower mechanical sensitivity, which can broaden the applications of nanocomposite energetic materials in modern ammunition.
In this work, HMX/GAP nanocomposites were prepared by a modified sol–gel-supercritical method. The thermal decomposition behaviours were investigated by differential thermal analysis. The kinetic, thermodynamic parameters and mechanical sensitivity were also evaluated. The HMX particles were restricted in the GAP matrix and the particle sizes were decreased to nanoscale. Moreover, the as-synthesized HMX/GAP nanocomposites showed enhanced thermolysis activity and reduced impact sensitivity.
2. Experimental
2.1 Materials
Glycidylazide polymer (GAP, Mn = 2980, hydroxyl value of 0.63 mmol g−1) was provided by the 42nd Institute of the Fourth Academy of China Aerospace Science and Technology Corporation, 1,3,5,7-tetranitro-1,3,5,7-tetrazocane (HMX) was purchased from Gansu Yinguang Chemical Co., Ltd., 1,4-butyrolactone, hexamethylene diisocyanate (HDI) and ditinbutyl dilaurate (DBTDL) were purchased from Aladdin Chemical Co., Ltd. All the materials were used as received without further purification.2.2 Preparation of HMX/GAP nanocomposites
The schematic diagram for synthesis of HMX/GAP nanocomposites is illustrated in Fig. 1. Firstly, GAP was dropped into a weighing bottle and dissolved in 10 mL 1,4-butyrolactone under vigorous magnetic stirring to form a dark yellow solution. Afterwards, stoichiometric amount of HMX, HDI and DBTDL were added into the solution with stirring until the system became homogeneous. Then, the bottle was sealed tightly and placed in water bath at 40 °C for gelation. During this period, the –OH groups in GAP were cross-linked with the isocyanate groups of HDI to generate gel matrix. The gelation time varied from 30 min to 1 d when the mass ratios of HMX in the HMX/GAP nanocomposites increased. The wet gels were aged for 4 days in order to obtain reinforced monolithic gels. Subsequently, carbon dioxide supercritical fluid (SF) extraction method was applied to dry the wet gels.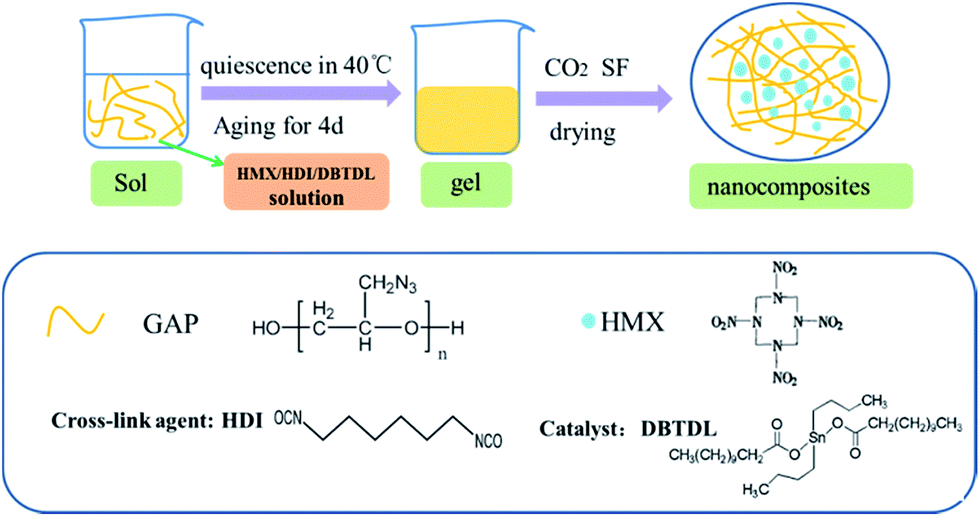 | ||
| Fig. 1 Schematic diagram of the HMX/GAP nanocomposites formation process. | ||
The synthesis procedures of the porous and strengthened gels were optimized by modifying the GAP concentration, types of crosslinking agents and solvents, catalyst dosage. A series of experiments were performed and it could be concluded that the GAP concentration and the mass ratio of GAP/HDI/DBTDL were key factors that affect the structure of the gels. All the as-synthesized gels mentioned below were prepared under the optimal conditions. The GAP concentration was no less than 8.5%, solvent was 1,4-butyrolactone, crosslinking agent was HDI, and the mass ratio of GAP/HDI/DBTDL was 9:1:0.42. For a typical formulation: 1.53 g of GAP were dissolved in 10 mL 1,4-butyrolactone, then 0.3 g of HMX, 0.17 g of HDI and 80 μL DBTDL were added into the solution. Different mass fractions of HMX in HMX/GAP nanocomposites were prepared. The mass ratios of the HMX in the HMX/GAP were 0.15, 0.30, 0.45 and 0.60, and the as-synthesized samples were denoted as HMX0.15, HMX0.30, HMX0.45, HMX0.60, respectively.
The drying method played an important role in the fabrication of energetic nanocomposites. In order to maintain the porous structure of gel skeleton and insure HMX were trapped in the matrix on the nano-scale, SF equipment was utilized to gain the complete dried HMX/GAP nanocomposite materials (the extraction temperature was 45 °C and the pressure was 120 bar) and then the nanocomposite materials were used for further characterization.
2.3 Characterization and tests
The structure of the resulting samples were detected by X-ray diffraction (XRD, Bruker Advance D8) using monochromatic Cu Kα radiation (λ = 0.154 nm) at 40 kV and 30 mA, the scanning range of 2θ was between 10° and 80°. Scanning electron microscopy (SEM, JEOL JSM-7500) was performed to characterize the morphology of the samples. Infrared spectra of the GAP gel, raw HMX and HMX/GAP nanocomposites were recorded by Bruker Vector 22 Fourier-transform infrared (FT-IR) spectrometer to detect the chemical bonds at room temperature. Raman scattering spectra were performed to characterize the difference of the resulting samples. The porous properties of the as-synthesized samples were measured by the nitrogen physical adsorption of at −196 °C. Prior to the experiment, the sample was degassed under vacuum at 90 °C for 5 h. The specific surface areas of the resultant were estimated using the Brunauer–Emmett–Teller (BET) equation. The pore size distribution was obtained from the adsorption branch according to Barrett–Joyner–Halenda (BJH) method.The thermal characteristics of the as-synthesized samples were investigated using differential thermal analyser (DTA) at heating rates of 5, 10, 15, and 20 °C min−1 under nitrogen flow (40 mL min−1). The impact sensitivity tests were conducted with a WL-1 type impact sensitivity instrument according to the Chinese GJB-772A-97 standard method 601.2. The test conditions were 5 kg of drop hammer and 30 mg of sample for each drop. The impact sensitivity was expressed by special height (H50) which represents the drop height of 50% explosion probability. The critical impact energy (I50) was calculated according to the equation of I50 = mgH50, where m represents the weight of drop hammer, g represents gravitational acceleration (g = 9.8 N kg−1).
3. Results and discussion
The morphologies of the resulting samples were measured by SEM, as depicted in Fig. 2. Fig. 2(a) shows the images of the GAP aerogel without the addition of HMX. A network structure which is cross-linked by GAP is observed. It is clear to see that plentiful porous are created among the network structure, which indicates that GAP aerogel possess a relative large surface area. Fig. 2(b) indicates that the raw HMX particles are in micro-grade. Fig. 2(c–f) reveals that the HMX particles in HMX/GAP composites are in nanoscale, ranging from approximate 100 nm to 500 nm. When HMX were added to GAP, the morphologies of HMX/GAP nanocomposites are different from GAP aerogel. The crosslinked network structures become inconspicuous and the amount of pores is much less, which are caused by the recrystallization of the HMX in GAP skeleton. With the increase of the amount of HMX, the particle sizes increase. However, compared with the raw materials of HMX, the average particle size of HMX in HMX/GAP descends, due to the restriction of GAP matrix.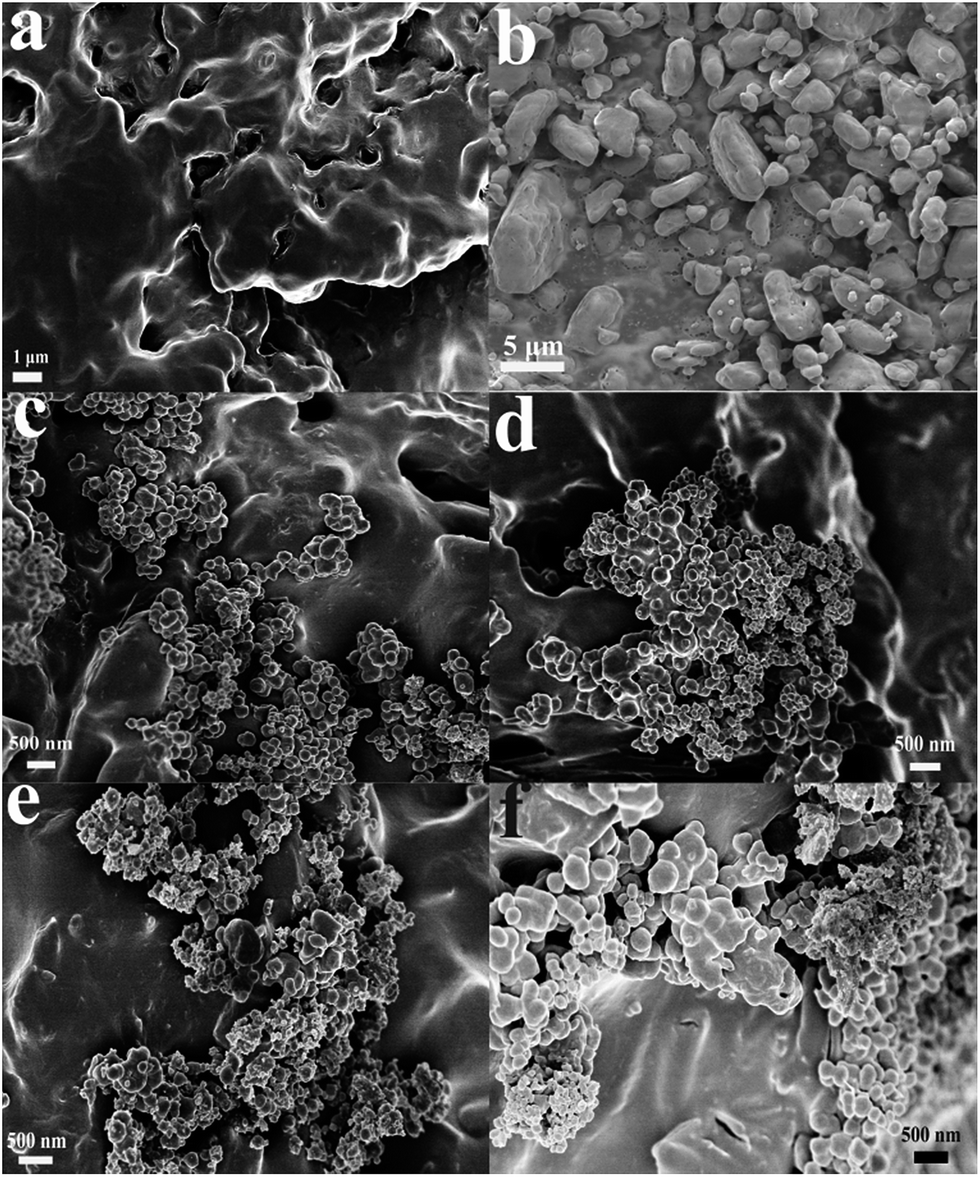 | ||
| Fig. 2 SEM images of GAP aerogel (a), raw HMX (b), HMX0.15 (c), HMX0.30 (d), HMX0.45 (e) and HMX0.60 (f). | ||
In order to identify the crystallinity of the GAP aerogel, raw HMX and HMX/GAP nanocomposites, XRD analysis was performed, and the diffraction pattern are shown in Fig. 3. It can be observed that a dispersing diffraction peak appears in GAP aerogels, which reveals that it is amorphous. Due to relatively low content of GAP gel matrix, dispersing diffraction peak for GAP aerogels disappears in the diffraction patterns of the as-synthesized HMX/GAP nanocomposites. The diffraction angles of the as-synthesized HMX/GAP nanocomposites are located in the same position compared with those of raw HMX.
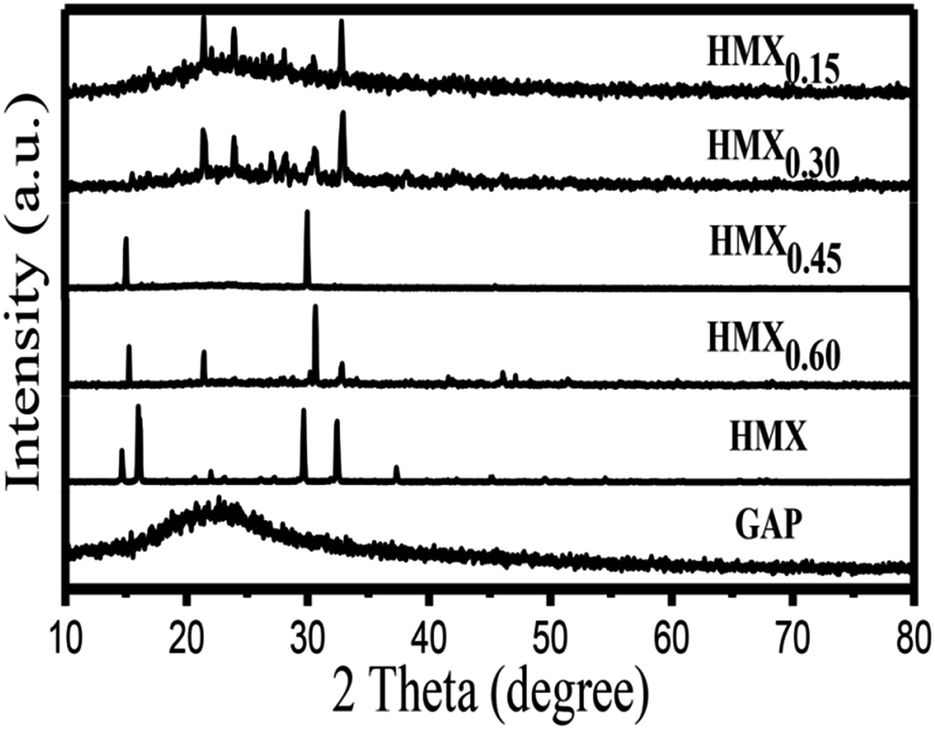 | ||
| Fig. 3 XRD patterns of GAP, HMX and HMX/GAP nanocomposites. | ||
However, the intensities of diffraction peaks increase with the increase of the content of HMX in GAP, which indicate that the crystal phases of HMX come into being during the drying process and HMX particles are embedded in GAP matrix.
The functionalized groups of HMX/GAP nanocomposites were detected by FT-IR spectrum. Fig. 4 shows the FT-IR spectra of GAP, raw HMX and HMX/GAP nanocomposites which were recorded between 4000 cm−1 and 500 cm−1. In the GAP aerogel (Fig. 4(a)), the peaks observed at 1721 cm−1, 1278 cm−1 and 1076 cm−1 are attributed to the shearing vibration of –N–H, stretching vibrations of –C–N and –C–O, which indicate that the –OH groups in GAP reacted with the –NCO groups in HDI to form homogeneous monolithic gels during the sol–gel process. The peak at 2084 cm−1 corresponds to the azide groups (–N3).37,38 Fig. 4(b) shows the FT-IR spectrum of raw HMX. It can be observed that the peak located at 3038 cm−1 is the asymmetric stretching vibration of C–H in the ring of HMX. Peaks at 1539 cm−1, 1257 cm−1, and 940 cm−1 can be assigned to the stretching vibrations of N–NO2, C–N–C and the ring vibrations of HMX, respectively. The spectra of as-prepared HMX/GAP nanocomposites are shown in Fig. 4(c). It can be observed that the characteristic peaks of GAP (–N3) and HMX (N–NO2, C–N–C and ring vibration of HMX) are appeared in the HMX/GAP nanocomposites, indicating that HMX were successfully cured in the GAP gel skeleton.
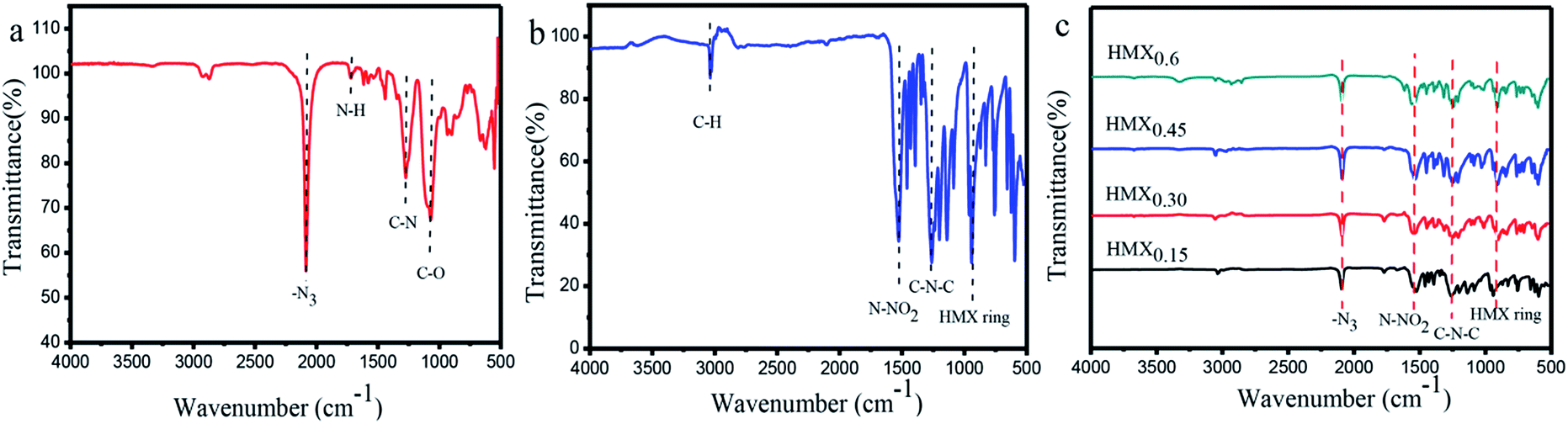 | ||
| Fig. 4 FT-IR spectra of GAP (a), HMX (b) and HMX/GAP nanocomposites (c). | ||
The Raman spectra of raw HMX and the as-prepared HMX/GAP nanocomposites are shown in Fig. 5. In view of HMX, several species of molecular vibrational mode can be observed, which are ascribed to NO2 deformation, NO2 wag, ring stretching, symmetric NO2 stretching and N–N stretching, C–H deformation, NO2 asymmetric stretching, respectively.39 Besides, it can be observed that characteristic peaks of HMX are appeared in HMX/GAP nanocomposites, indicating that HMX were embedded in the GAP gel matrix. It is to be mentioned that the bands at 1167 cm−1 and 1191 cm−1 indicate the HMX crystals are in the beta phase.40 All the Raman spectra of the of the as-prepared HMX/GAP nanocomposites present the two bands, which further confirm the crystal phase transformation of HMX did not take place in the sol–gel process.
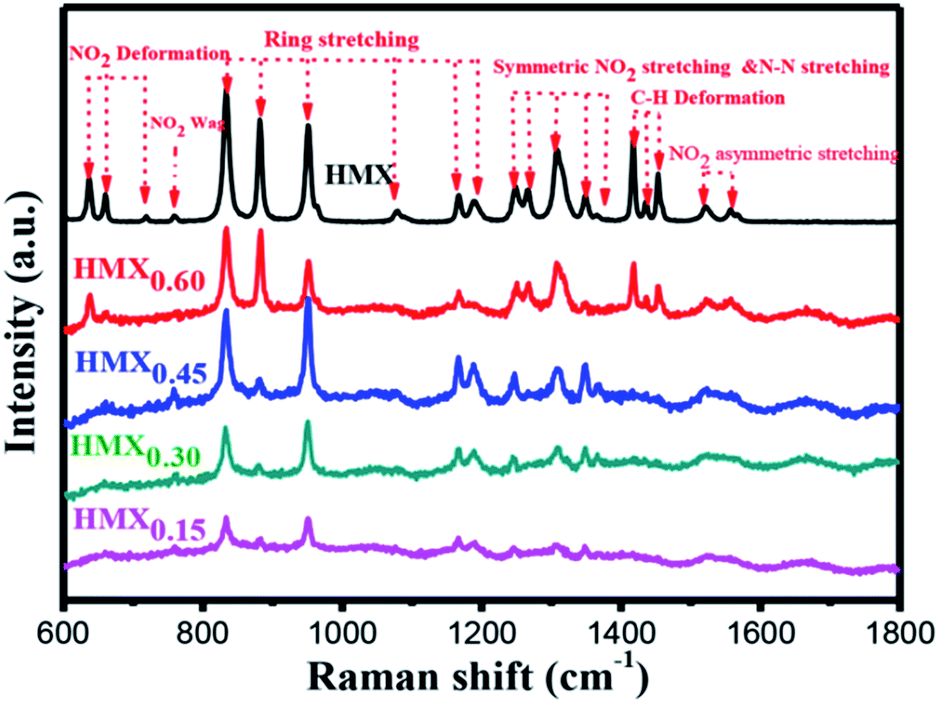 | ||
| Fig. 5 Raman spectra of GAP, HMX and HMX/GAP nanocomposites. | ||
The specific surface area and the porosity distribution characteristic of the as-synthesized HMX/GAP nanocomposites were investigated by N2 adsorption–desorption at 77 K using N2 as adsorbent. The N2 adsorption–desorption isotherms and porosity distribution curves of GAP aerogel, HMX0.15 and HMX0.45 nanocomposites are shown in Fig. S1,† respectively. The specific surface areas of GAP aerogel, HMX0.15 and HMX0.45 nanocomposites are 48.6, 18.2 and 7.9 m2 g−1, respectively, which are calculated by Brunauer–Emmett–Teller (BET) method. The specific surface area of HMX0.15 accounts for 37.4% of GAP aerogel, whereas, HMX0.45 accounts for 16.3% of GAP aerogel. It can be concluded that the specific surface areas of the nanocomposites decrease significantly with the increase of the mass ratio of HMX. The HMX particles recrystallize and occupy a certain amount of pores of in the GAP gel matrix, thus the specific surface areas decrease. The porosity distribution curves of GAP aerogel, HMX0.15 and HMX0.45 nanocomposites, obtained by Barrett–Joyner–Halenda (BJH) method, are inserted in Fig. 6. The average pore sizes of GAP aerogel, HMX0.15 and HMX0.45 nanocomposites are 10.51, 13.37 and 14.59 nm, respectively. Indicating that the distances among HMX/GAP nanocomposites are much larger than those of GAP aerogel. This phenomenon can be ascribed to the addition of HMX. When HMX particles are added, the concentration of GAP is diluted and the collision activity of the GAP colloidal particles is reduced to a certain degree. The GAP colloidal particles need to be gathered enough to form monolithic gel, thus, the pore-size distribution has a wide range and the pore-size increased when increasing the ratio of HMX.
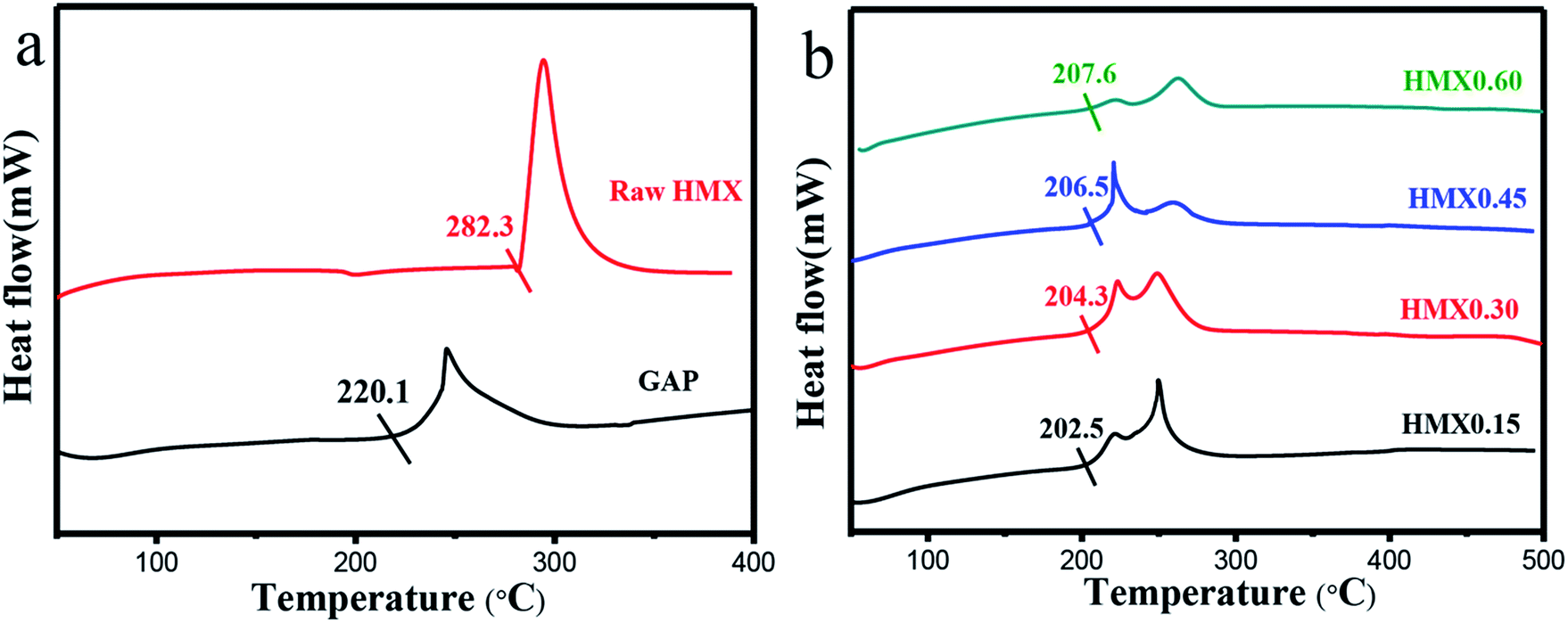 | ||
| Fig. 6 DTA curves of GAP and raw HMX (a), HMX/GAP nanocomposites (HMX0.15, HMX0.30, HMX0.45 and HMX0.60) (b). | ||
The thermal decomposition characteristics of the as-prepared HMX/GAP nanocomposites were investigated by DTA measurements at a heating rate of 20 °C min−1. Fig. 6 shows the DTA curves of raw HMX, GAP and the as-prepared HMX/GAP nanocomposites.
As shown in Fig. 6(a), it can be seen that HMX presents a strong exothermic peak, and the onset decomposition temperature (Tonset) is about 282.3 °C. Similarly, a decomposition peak can be observed for pure GAP gel, and the Tonset temperature is about 220.1 °C.
Fig. 6(b) shows the DTA curves of HMX/GAP nanocomposites. Compared with that of raw HMX, the Tonset temperatures of HMX0.15, HMX0.30, HMX0.45, and HMX0.60 are reduced by 79.8 °C, 78.0 °C, 75.8 °C, and 74.7 °C, respectively. Besides, Tonset temperatures of HMX/GAP nanocomposites are lower than that of pure GAP aerogel. It can be concluded that the onset decomposition temperature increases with increasing the mass ratio of HMX in HMX/GAP nanocomposites. As mentioned above, the HMX particles recrystallized in GAP skeleton and the mean grain size decreased to nanoscale, which is caused by the limitation of GAP matrix. It is known that the thermal decomposition characteristics of energetic materials are related on the particle size.41 Due to the size effect and surface effect of nano particles, the heat release rate for HMX/GAP is enhanced, thus, the onset decomposition temperature decreases.
To further investigate the thermal decomposition kinetic and thermodynamic of the as-prepared HMX/GAP nanocomposites, DTA tests with different heating rate were performed and the DTA curves are shown in Fig. S2.† It can be observed that all of the DTA curves for HMX/GAP nanocomposites present two peaks, which are ascribed to the low-temperature decomposition (LTD) and high-temperature decomposition (HTD), respectively, though the heating rate is varied. Each peak represents an individual decomposition reaction. In order to give a comprehensive assessment, both of the LTD and HTD kinetic parameters for HMX/GAP nanocomposites are calculated from the plot of exothermic peak temperature dependence as a function of heating rate. In order to make a distinction between the calculated parameters of LTD and HTD, they are marked as HMX-L and HMX-H.
The relationship between the decomposition temperature and heating rate can be dipicted by Kissinger correlation42 and Arrhenius equation.
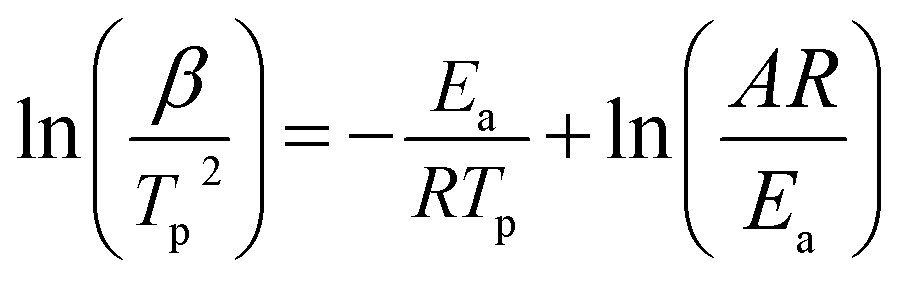 | (1) |
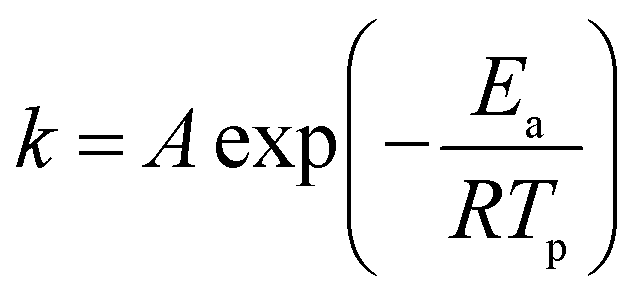 | (2) |
Where β is the heating rate (K min−1); Tp is the temperature of exothermic peak in DTA curves (K); R is the ideal gas constant (8.314 J mol−1 K−1); Ea is the activation energy (J mol−1); A is the pre-exponential factor (s−1); k is the reaction rate constant.
According to the Kissinger equation (eqn (1)), the term ln(β/Tp2) varies linearly with 1/Tp yielding the kinetic parameters of activation energy from the slope of straight line. Fig. 7 shows the experimentally measured ln(β/Tp2) versus 1/Tp of GAP, HMX and HMX/GAP nanocomposites. Based on these plots and Arrhenius equation (eqn (2)), the activation energy (Ea) and the pre-exponential factor (ln(A)) of GAP, HMX and HMX/GAP nanocomposites can be calculated, the results are summarized in Table 1. It can be inferred that the kinetic parameters for both LTD and HTD show the same tendency. The HMX0.15 shows the lowest Ea, and the values of Ea increase with the increase of HMX. For raw HMX, the activation energy of HTD is calculated to be 255.9 kJ mol−1, whereas, the HTD activation energy for HMX0.15, HMX0.30, HMX0.45, and HMX0.60 decrease to 188.1 kJ mol−1, 210.2 kJ mol−1, 225.7 kJ mol−1 and 239.7 kJ mol−1, respectively.
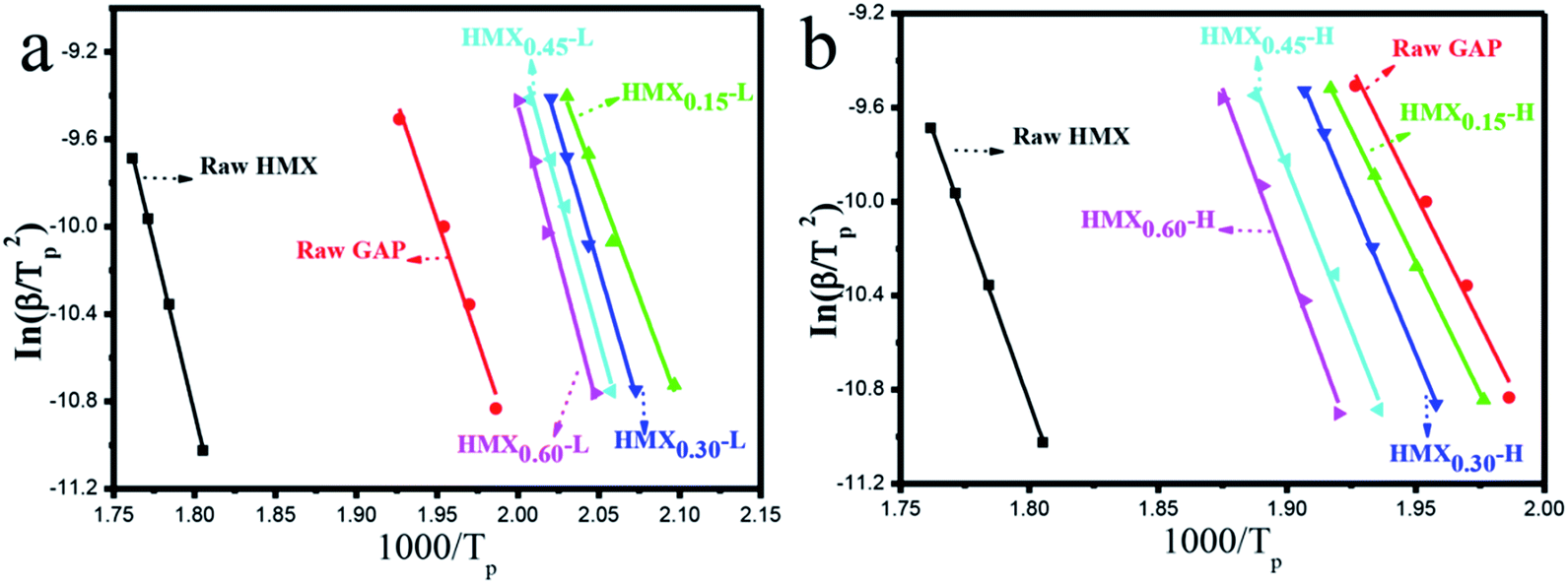 | ||
| Fig. 7 Dependence of ln(β/Tp2) on 1/Tp for raw GAP, raw HMX and HMX/GAP nanocomposites. Scatter points are experimental data and lines denotes the linear fitting results. | ||
| Sample | Ea (kJ mol−1) | ln(A) |
|---|---|---|
| HMX | 255.9 | 54.9 |
| GAP | 178.0 | 41.8 |
| HMX0.15-L | 169.4 | 41.9 |
| HMX0.30-L | 217.7 | 53.7 |
| HMX0.45-L | 223.2 | 54.9 |
| HMX0.60-L | 232.5 | 57.0 |
| HMX0.15-H | 188.1 | 43.9 |
| HMX0.30-H | 210.2 | 48.8 |
| HMX0.45-H | 225.7 | 52.0 |
| HMX0.60-H | 239.7 | 54.8 |
The thermodynamic parameters including the activation enthalpy (ΔH≠), the activation entropy (ΔS≠), and the activation Gibbs free energy of (ΔG≠) can be obtained derived from the DTA data according to eqn (3–5) and the calculated results are listed in Table 2.
| ΔH≠ = Ea − RTp | (3) |
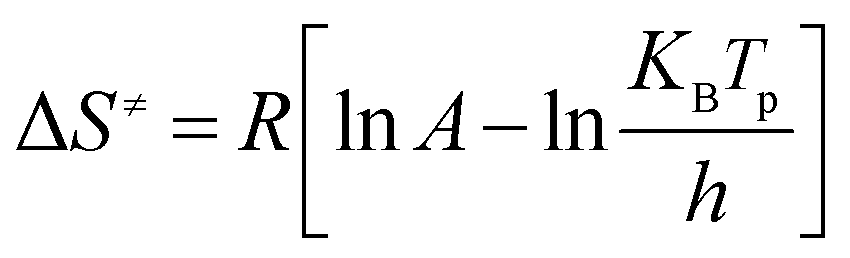 | (4) |
| ΔG≠ = ΔH≠ − TpΔS≠ | (5) |
| Sample | ΔH≠ (kJ mol−1) | ΔS≠ (J mol−1 K−1) | ΔG≠ (kJ mol−1) |
|---|---|---|---|
| HMX | 251.4 | 206.5 | 138.2 |
| GAP | 173.9 | 79.2 | 134.6 |
| HMX0.15-L | 165.7 | 99.2 | 121.3 |
| HMX0.30-L | 213.9 | 178.8 | 132.9 |
| HMX0.45-L | 219.3 | 188.6 | 131.3 |
| HMX0.60-L | 228.6 | 206.1 | 132.2 |
| HMX0.15-H | 184.0 | 135.0 | 117.3 |
| HMX0.30-H | 206.0 | 156.4 | 127.1 |
| HMX0.45-H | 221.4 | 182.9 | 127.2 |
| HMX0.60-H | 235.4 | 205.4 | 128.8 |
Where KB and h are the Boltzmann (KB = 1.381 × 10−23 J K−1) and Planck constants (h = 6.626 × 10−34 J s−1), respectively; the activation enthalpy (ΔH≠) represents the reaction heat that molecules to absorb or release in the process of chemical reaction. The calculated values of ΔH≠ for all samples are positive, indicating that molecules need to absorb energy from outside to make the reaction happen. The smaller the value of ΔH≠ is, the more easily the reaction happens. The values of ΔS≠ for all samples are also positive, indicating that the confusion degrees of the activated complexes are higher than those of the reactant molecules. The more decomposition products lead to the greater ΔS≠. Therefore, the ΔS≠ is directly proportion to the content of HMX which is the main decomposition reactant. The nonnegative values of ΔG≠ indicate that the thermal decomposition do not occur spontaneously. All of the thermodynamic parameters imply that the energetic nanocomposites cannot be activated unless their molecules absorb adequate energy, which are greater than the calculated values of ΔH≠. Hence, the energetic nanocomposites are stable and can be stored safely under normal circumstances.
The critical initiation temperature (Tb), which is defined as the lowest temperature for a specific charge that may be heated without undergoing thermal runaway, is an important parameter in view of the evaluation of thermal stability and storage safety for energetic materials. Tb can be calculated from inflammation theory and appropriate thermokinetic parameters namely the activation energy, pre-exponential factor, and heat of reaction. The critical initiation temperature (Tb) for GAP, HMX and HMX/GAP are calculated based on eqn (6) and (7).
| Tp0 = Tp − (aβ + bβ2 + cβ3) | (6) |
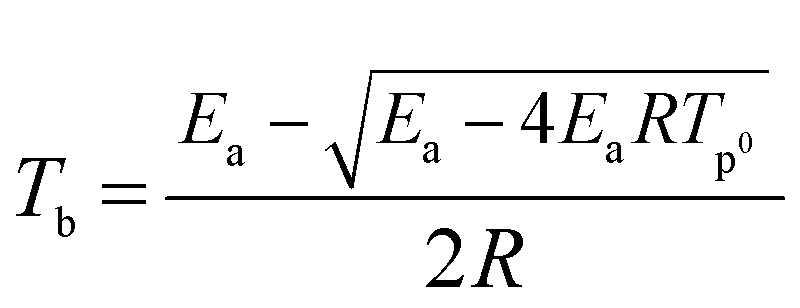 | (7) |
Where a, b and c are coefficients; β is the utilized heating rate; Tp0 is the extrapolated onset temperature when β → 0.
The self-accelerating decomposition temperature (TSADT) is also an important indicator for the thermal safety of explosives. The TSADT is defined as the minimum ambient temperature for a self-heating chemical at which the self-accelerating decomposition reaction starts in a specified commercial package during a period of just one week. TSADT is normally applied to evaluate thermal safety of hazardous materials and can provide a standard for the safe temperatures of hazardous materials during production, transportation and storage thereof. The TSADT can be predicted by some analytical apparatus including the accelerating rate calorimeter (ARC) alone or in combination with a thermal activity monitor (TAM), a heat flux calorimeter (C80D) and differential thermal analysis (DTA).43 In this study, TSADT was predicted by using DTA. The self-accelerating decomposition temperature (TSADT) for GAP, HMX and HMX/GAP are calculated according to eqn (8) and the results are also listed in Table 3.
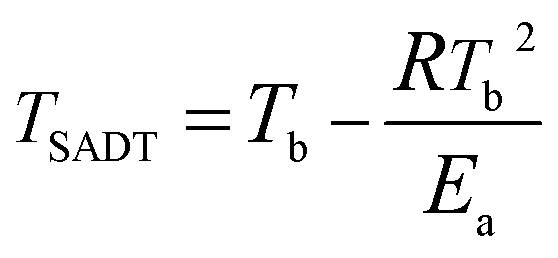 | (8) |
| Sample | Tp0 (°C) | Tb (°C) | TSADT (°C) |
|---|---|---|---|
| HMX | 270.5 | 280.5 | 262.9 |
| GAP | 222.7 | 234.7 | 222.7 |
| HMX0.15-L | 174.8 | 185.1 | 174.8 |
| HMX0.30-L | 187.0 | 195.4 | 188.3 |
| HMX0.45-L | 193.3 | 201.7 | 193.3 |
| HMX0.60-L | 194.5 | 202.6 | 194.5 |
| HMX0.15-H | 220.7 | 232.0 | 220.7 |
| HMX0.30-H | 231.3 | 241.8 | 231.3 |
| HMX0.45-H | 242.0 | 252.2 | 242.0 |
| HMX0.60-H | 246.0 | 255.7 | 245.9 |
It can be observed that the Tb and TSADT of the HMX/GAP nanocomposites are lower than that of raw HMX, implying that the thermal stability of the HMX/GAP nanocomposites are worse than that of the raw HMX.
Impact sensitivity is also a key parameter to evaluate the safety performance of energetic materials. The impact sensitivities of raw HMX and HMX/GAP nanocomposites were tested and the calculated values of H50 are listed in Table S1.† As mentioned above, the critical impact energy (I50) was calculated according to the equation of I50 = mgH50, and the results are shown in Fig. 8. It can be observed that the values of I50 increase with the decrease of the content of HMX in HMX/GAP nanocomposites. Compared with that of raw HMX (I50 = 9.60 J), the values of I50 for HMX0.15, HMX0.30, HMX0.45 and HMX0.60 are 14.75 J, 13.97 J, 12.84 J and 11.66 J, respectively, indicating the HMX/GAP nanocomposites exhibit relatively low sensitivities. The main reason for the reduced sensitivity of HMX/GAP nanocomposites is because HMX particles are capped by porous GAP matrix, which can dissipate parts of external stimuli. Thus, it was difficult to form a large hot spot. In addition, HMX trapped in GAP gel is distributed in nano-scale, which could also reduce the amount of hotspots due to the nano-effect of HMX.
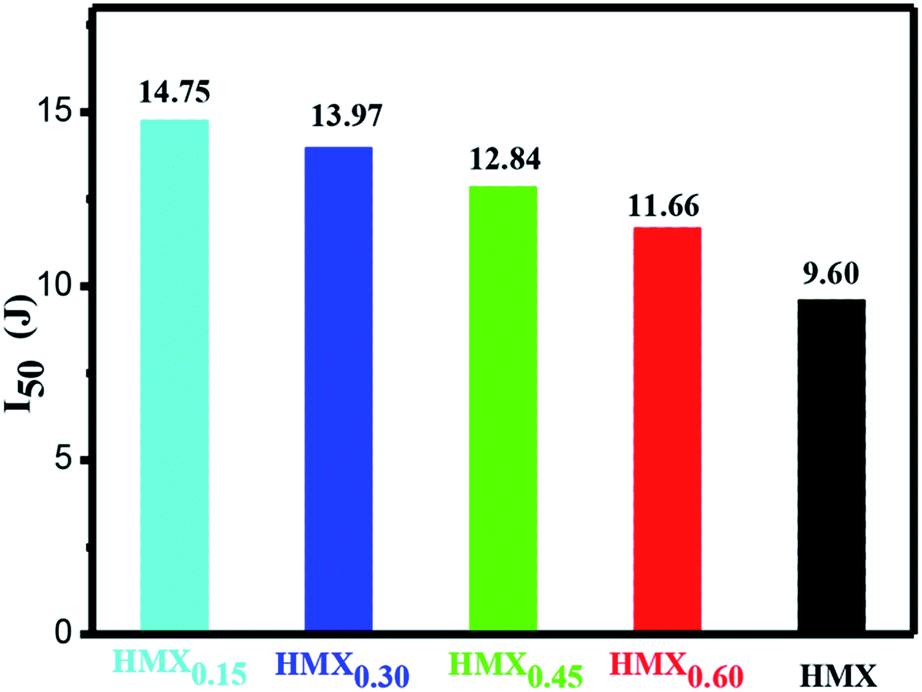 | ||
| Fig. 8 Impact sensitivity (I50) of HMX and HMX/GAP nanocomposites. | ||
It is commonly believed that detonation of explosives is firstly initiated at hot-spots,44 which are caused by defects such as voids and interfaces. When bringing external stimulation to explosives, the voids are subjected to adiabatic compression, resulting in a remarkable increase of the temperature inside the pores. When the temperature surpasses the critical temperature, hot spots create. The surrounding explosive particles are heated by the hot spots, which result in the decomposition of vicinal explosive particles. When the release heat accumulates to a certain extent, massive explosive particles are initiated, and then explosion occurs. Generally speaking, the explosion process proceeds in two crucial steps, as illustrated in Fig. 9.
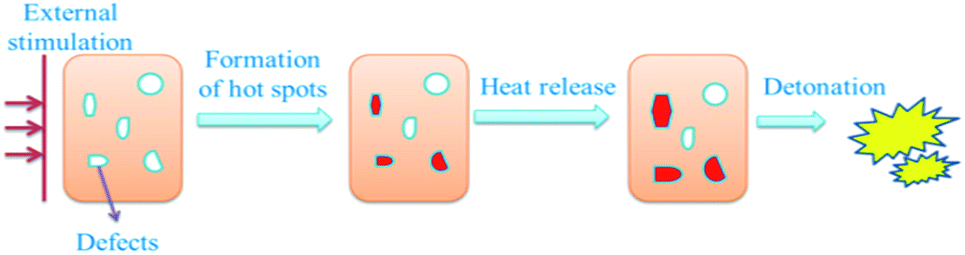 | ||
| Fig. 9 Illustration of explosion process explicated by hot-spots growth mechanism. | ||
Firstly, the hot spots are promptly formed inside the defects of the explosive; secondly, sustainable exothermic reaction within the hot spots will lead to deflagration or detonation. During this period, the size of the voids has an important influence on detonation properties. The hot spots tend to be formed more easily and the released heat is greater when the size of the voids increases.45 As the particle size of the explosive decrease to the nano-scale, the size of the voids in the nanoparticles decreases comparatively. The probability of hot spots generation is reduced and less heat will be released, the explosive nanoparticles are hard to be ignited. Consequently, the nanocomposites exhibit lower sensitivities in mechanical sensitivity tests.
4. Conclusion
In conclusion, HMX/GAP nanocomposites with high reactivity and low sensitivity have been successfully prepared by a modified sol–gel-supercritical method. The average particle sizes of HMX particles in HMX/GAP nanocomposites are estimated in nano-scale, which indicate that HMX particles are much smaller than those of raw HMX particles and GAP gel matrix could effectively prevent the HMX particles from aggregating. The as-synthesized HMX/GAP nanocomposites exhibit high thermal reactivity, and the onset decomposition temperature gradually decreases with the decrease of the content of HMX in HMX/GAP nanocomposites. Compared with that of raw HMX, the HTD temperatures of HMX0.15, HMX0.30, HMX0.45, and HMX0.60 are decreased by 79.8 °C, 78.0 °C, 75.8 °C, and 74.7 °C, respectively. The results of calculated kinetic and thermodynamic parameters indicate that the thermal reactivity of HMX/GAP nanocomposites increases as the content of HMX decrease in HMX/GAP nanocomposites. Nevertheless, the mechanical sensitivities of HMX/GAP nanocomposites are obviously lower than those of raw HMX, which shows the opposite trend in comparison with the thermal reactivity. The facile, convenient preparation method illustrated in this study can be a very prospective and scalable technology for manufacturing of other energetic nanocomposites with high reactivity and low sensitivity, which may broaden a new application in modern weapon systems.Acknowledgements
This work was financially supported by the Natural Science Foundation of China (Project No. 50972060 and No. 51606102), the Weapon Research Support Fund (62201070804), Qing Lan Project, Environmental Protection Scientific Research Project of Jiangsu Province (2016056), a Project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, the Shanghai Aerospace Science and Technology Innovation Fund (SAST2015020), Basic Product Innovation Technology Research Project of Explosives.Notes and references
- A. Freche and J. Aviles, Insensitive RDX, Insensitive Munitions and Energetic Materials Technology Symposium, San Antonio, Texas, USA, November 27-30, 2000, pp. 18–22 Search PubMed.
- G. X. Zhang, B. L. Weeks and X. Zhang, Cent. Eur. J. Eng., 2012, 2, 336–346 CAS.
- G. X. Zhang, H. Sun, J. M. Abbott and B. L. Weeks, ACS Appl. Mater. Interfaces, 2009, 1, 1086–1089 CAS.
- D. D. Dlott, Mater. Sci. Technol., 2006, 22, 463–473 CrossRef CAS.
- X. Zhang and B. L. Weeks, Thin Solid Films, 2014, 550, 135–139 CrossRef CAS.
- Y. Q. Yang, S. F. Wang, Z. Y. Sun and D. D. Dlott, J. Appl. Phys., 2004, 95, 3667–3676 CrossRef CAS.
- R. W. Armstrong, V. B. Baschung, D. W. Booth and M. Samirant, Nano Lett., 2003, 3, 253–255 CrossRef CAS.
- C. Rossi, K. Zhang, D. Estève, P. Alphonse, P. Tailhades and C. Vahlas, J. Microelectromech. Syst., 2007, 16, 919–931 CrossRef CAS.
- A. Pivkina, P. Ulyanova, Y. Frolov, S. Zavyalov and J. Schoonman, Propellants, Explos., Pyrotech., 2004, 29, 39–48 CrossRef CAS.
- B. C. Tappan and T. B. Brill, Propellants, Explos., Pyrotech., 2003, 28, 223–230 CrossRef CAS.
- X. Y. Zhang, D. B. Liu and C. X. Lv, Propellants, Explos., Pyrotech., 2005, 6, 438–441 Search PubMed.
- J. J. Granier and M. L. Pantoya, Combust. Flame, 2004, 138, 373–383 CrossRef CAS.
- J. A. Puszynski, J. Therm. Anal. Calorim., 2009, 96, 677–685 CrossRef CAS.
- R. Kumar, P. F. Siril and P. Soni, Propellants, Explos., Pyrotech., 2014, 39, 383–389 CrossRef CAS.
- B. D. He, V. Stepanov, H. W. Qiu and L. N. Krasnoperov, Propellants, Explos., Pyrotech., 2015, 40, 659–664 CrossRef CAS.
- H. W. Qiu, V. Stepanov, T. M. Chou, A. Surapaneni, A. R. D. Stasio and W. Y. Lee, Powder Technol., 2012, 226, 235–238 CrossRef CAS.
- B. Risse, F. Schnell and D. Spitzer, Propellants, Explos., Pyrotech., 2014, 39, 397–401 CrossRef CAS.
- X. D. Guo, G. O. Yang, J. Liu, Q. Li, L. X. Wang, Z. M. Gu and F. S. Li, J. Energ. Mater., 2015, 33, 24–33 CrossRef CAS.
- J. Y. Wang, H. Huang, W. Z. Xu, Y. R. Zhang, B. Lu, R. Z. Xie, P. Y. Wang and N. Yun, J. Hazard. Mater., 2009, 162, 842–847 CrossRef CAS PubMed.
- G. C. Yang, F. D. Nie, J. S. Li, Q. X. Guo and F. Q. Zhao, J. Energ. Mater., 2007, 25, 35–47 CrossRef CAS.
- G. C. Yang, F. D. Nie, H. Huang, L. Zhao and W. T. Pang, Propellants, Explos., Pyrotech., 2006, 31, 390–394 CrossRef CAS.
- X. L. Song, Y. Wang, C. W. An, X. D. Guo and F. S. Li, J. Hazard. Mater., 2008, 159, 222–229 CrossRef CAS PubMed.
- J. Liu, W. Jiang, Q. Yang, J. Song, G. Z. Hao and F. S. Li, Def. Technol., 2014, 10, 184–189 CrossRef.
- V. Stepanov, L. N. Krasnoperov, I. B. Elkina and X. Zhang, Propellants, Explos., Pyrotech., 2005, 30, 178–183 CrossRef CAS.
- B. Huang, Z. Q. Qiao, F. D. Nie, M. H. Cao, J. Su, H. Huang and C. W. Hu, J. Hazard. Mater., 2010, 184, 561–566 CrossRef CAS PubMed.
- K. Gao, G. P. Li, Y. J. Luo, L. Wang, L. H. Shen and G. Wang, J. Therm. Anal. Calorim., 2014, 118, 43–49 CrossRef CAS.
- T. M. Tillotson, A. E. Gash, R. L. Simpson, L. W. Hrubesh, J. H. Satcher Jr and J. F. Poco, J. Non-Cryst. Solids, 2001, 285, 338–345 CrossRef CAS.
- G. Davin, Cent. Eur. J. Energ. Mater., 2010, 7, 126–128 Search PubMed.
- S. V. Ingale, P. U. Sastry, A. K. Patra, R. Tewari, P. B. Wagh and S. C. Gupta, J. Sol-Gel Sci. Technol., 2010, 54, 238–242 CrossRef CAS.
- R. J. Chen, Y. J. Luo, J. Sun and G. P. Li, Propellants, Explos., Pyrotech., 2012, 37, 422–426 CrossRef CAS.
- S. V. Ingale, P. B. Wagh, R. Tewari and S. C. Gupta, J. Non-Cryst. Solids, 2010, 356, 2162–2167 CrossRef CAS.
- F. D. Nie, J. Zhang, Q. X. Guo, Z. Q. Qiang and G. Y. Zeng, J. Phys. Chem. Solids, 2010, 71, 109–113 CrossRef CAS.
- B. C. Tappan and T. B. Brill, Propellants, Explos., Pyrotech., 2003, 28, 72–76 CrossRef CAS.
- M. M. Li, F. S. Li, R. Q. Shen and X. D. Guo, J. Hazard. Mater., 2010, 186, 2031–2036 CrossRef PubMed.
- J. S. You, J. O. Kweon, S. C. Kang and S. T. Noh, Macromol. Res., 2010, 18, 1226–1232 CrossRef CAS.
- A. N. Nazare, S. N. Asthana and H. Singh, Energy Mater., 1992, 10, 43–63 CrossRef CAS.
- G. P. Li, M. H. Liu, R. Zhang, L. H. Shen, Y. Z. Liu and Y. J. Luo, Colloid Polym. Sci., 2015, 293, 2269–2279 CAS.
- J. S. Youa, S. C. Kanga, S. K. Kweonb, H. L. Kimc, Y. H. Ahnc and S. T. Noha, Thermochim. Acta, 2012, 537, 51–56 CrossRef.
- Y. Wang, W. Jiang, D. Song, J. Liu, X. D. Guo, H. Y. Liu and F. S. Li, J. Therm. Anal. Calorim., 2013, 111, 85–92 CrossRef CAS.
- D. S. Moore and K. Y. Lee, J. Raman Spectrosc., 2007, 38, 1221–1224 CrossRef CAS.
- W. P. King, S. Saxena and B. A. Nelson, Nano Lett., 2006, 6, 2145–2149 CrossRef CAS PubMed.
- H. E. Kissinger, Anal. Chem., 1957, 29, 1702–1706 CrossRef CAS.
- M. Malow and K. D. Wehrstedt, J. Hazard. Mater., 2005, 120, 21–24 CrossRef CAS PubMed.
- J. E. Field, N. K. Bourne, S. J. P. Palmer and S. M. Walley, Philos. Trans. R. Soc., A, 1992, 339, 269–283 CrossRef CAS.
- L. Borne, J. Mory and F. Schlesser, Propellants, Explos., Pyrotech., 2008, 33, 37–43 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27780b |
| This journal is © The Royal Society of Chemistry 2017 |