
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The remarkable journey of catalysts from stoichiometric to catalytic quantity for allyltrimethylsilane inspired allylation of acetals, ketals, aldehydes and ketones
Jatin J. Lade
 a,
Sachin D. Pardeshi
a,
Kamlesh S. Vadagaonkar
ab,
Kaliyappan Murugan
c and
Atul C. Chaskar
*a
a,
Sachin D. Pardeshi
a,
Kamlesh S. Vadagaonkar
ab,
Kaliyappan Murugan
c and
Atul C. Chaskar
*a
aNational Centre for Nanosciences and Nanotechnology, University of Mumbai, Mumbai 400098, India. E-mail: achaskar25@gmail.com; Tel: +91 7507375261
bDepartment of Dyestuff Technology, Institute of Chemical Technology, Matunga, Mumbai-400019, India
cNational Taiwan University, Taipei, Taiwan
First published on 23rd January 2017
Abstract
The Hosomi–Sakurai reaction has received enormous attention since its inception and will continue to do so, owing to its atom-economical carbon–carbon bond forming ability. The remarkable feature of this reaction is the use of simple achiral starting materials which leads to the product having a chiral carbon centre with two active functionalities which serve as the guiding stone towards easy synthesis of natural products/bioactive heterocyclic scaffolds/drug candidates. The journey which started with the use of stoichiometric amounts of catalyst has now reached catalytic quantity and that too with economical and ecological aspects. In this review we have emphasized on this journey with regard to Lewis acids, metal triflates, Brønsted acids, heterogeneous catalysts and easily available inexpensive catalysts such as iodine. Our objective is to brief the readers with in-depth knowledge on the development and recent trends in the use of novel catalysts so as to provoke research in this fascinating and enormously useful area which will lead to the development of environmentally benign and economically viable protocols to a greater extent.
 Jatin J. Lade | Jatin Jitendra Lade received his bachelor and Master degrees from University of Mumbai in 2013 and 2015 respectively. Subsequently, he joined for the Ph.D. under the supervision of Dr Atul Chaskar at National Centre for Nanosciences and Nanotechnology, University of Mumbai. His main research interest is the design and development of novel methodologies for the synthesis of organic materials for Organic light emitting diodes. |
 Sachin D. Pardeshi | Sachin Pardeshi received his bachelor and Master degrees from University of Mumbai in 2007 and 2009 respectively. Subsequently he joined as Lecturer at Vidya vikas college, Mumbai from 2009 to 2011. Then he moved to S.I.C.E.S. College, Mumbai as lecturer from 2013 to 2014. Following this he joined for the Ph.D. under the supervision of Dr Atul Chaskar at National Centre for Nanosciences and Nanotechnology, University of Mumbai. His main research interest is “Studies in design and synthesis of C–C and C–X bond formation via C–H activation/functionalization”. |
 Kamlesh S. Vadagaonkar | Kamlesh Vadagaonkar received his bachelor and master degrees from University of Pune in 2004 and 2006, respectively. Subsequently, he worked as a senior research chemist in TCG Lifesciences Pvt. Ltd. (formerly Chembiotek Research International Pvt. Ltd.), Pune from 2006 to 2010. Then he joined for the Ph.D. under the supervision of Prof. Prakash M. Bhate and Dr Atul C. Chaskar at Institute of Chemical Technology (formerly UDCT), Mumbai. His research focuses on the design and development of new synthetic methodologies via C–H activation/functionalization and synthesis of organic materials for optoelectronic applications. |
 Kaliyappan Murugan | Kaliyappan Murugan obtained his bachelor and master from University of Madaras and Bharathidasan University, respectively. He completed his Ph.D. in July 2011 at National Dong Hwa University, Hualien, Taiwan. Since August 2011 he has worked as a postdoctoral research Fellow at National Taiwan University, Taipei, Taiwan. His research interests include new processes related to drug development and pharmaceuticals, asymmetric synthesis and organometallics. |
 Atul C. Chaskar | Atul Chaskar received his bachelor and master degrees from University of Pune in 1995 and 1997, respectively. He earned his Ph.D. from University of Mumbai in 2006. Following this, he was awarded a postdoctoral fellowship by the Italian government in 2008 to conduct research in the laboratory of Prof. Elena Vismara at Politecnico Di Milano. Subsequently in 2010 he moved to National Taiwan University and joined the group of Prof. Ken-Tsung Wong. He joined the Institute of Chemical Technology in 2013 as a CSIR Pool Scientist. Since 2014 he is working as an Associate Professor at National Centre for Nanosciences and Nanotechnology, University of Mumbai. His research areas include the synthesis of organic materials for optoelectronic and photovoltaic applications, development of C–H functionalization/activation inspired new synthetic approaches and nanocatalysis. |
1. Introduction
Organic synthesis is one of the most important branches of chemistry which primarily involves the construction of carbon–carbon bond(s) and carbon–heteroatom bond(s) and also cleavage of these bonds. Among these bond-forming and bond–cleavage reactions, carbon–carbon bond formation is one of the most fundamental reactions in organic chemistry due to its significant role in building several classes of carbon frameworks. The invention and development of novel processes for the synthesis of complex molecules has remained an enduring challenge in organic chemistry. Thus, various useful carbon–carbon bond-forming reactions have been developed, and their applications also have been extensively studied during the last century.Present-day synthetic methods involving C–C bond formation require and even demand certain regulations and guidelines such as use of simple and readily available starting materials, operational simplicity, possibility of automation, favorable economic factors and low environmental impact. For these reasons, the creation of molecular diversity and complexity from simple and readily available substrates is one of the major current challenges of organic synthesis. Therefore, development of synthetic strategies which facilitate the construction of C–C bonds in economical and ecological manner are serving as a continuous research task for researchers in organic chemistry. The allylation of carbonyl compounds is one of the most widely explored C–C bond forming reactions.1 Among the numerous methods reported for allylation of diverse substrates,1–4 Hosomi–Sakurai reaction has received huge attention over the past decade for its ability to form carbon–carbon bond with two active functionalities and generate chiral carbon centre from easily available achiral materials under mild reaction conditions.5 Readily available, non-toxic and inexpensive allylsilanes were proved to be alternatives to allylstannanes and have been extensively explored for the synthesis of homoallylic compounds (Scheme 1).6 Beside this, allyltrimethylsilane exhibits superior reactivity and enantioselectivity than its counterparts, electron-deficient allylsilanes allyltrichlorosilane and allyltrimethoxysilane.
 | ||
| Scheme 1 | ||
The enantioselective allylations of acetals, ketals, aldehydes and ketones offer optically active homoallyl ethers/alcohols whose further synthetic elaboration result in easy access to natural products/bioactive heterocyclic scaffolds/drug candidates.
In the initial endeavours traditional Lewis acids such as titanium tetrachloride (TiCl4), boron trifluoride etherate (BF3·OEt2), tin tetrachloride (SnCl4), niobium pentachloride (NbCl5), etc. had been used in stoichiometric or near-stoichiometric amounts. Higher concentration of these acids accounted for formation of tight coordination of alcohol oxygen atom with them and poor efficiency of silicon transfer to oxygen. Then, scientists also used various rare earth metal triflates such as scandium triflate (Sc(OTf)3), lanthanum triflate (La(OTf)3), ytterbium triflate (Yb(OTf)3), zirconium triflate (Zr(OTf)4), etc. as Lewis acid catalysts but moisture sensitivity and high cost of these catalysts limited their use. In addition to these catalysts, inexpensive and water-tolerant catalysts such as iodine, ferric chloride (FeCl3), indium(III) chloride (InCl3), etc. were also used. Eventually various Brønsted acids and heterogeneous catalysts too were used by researchers to accomplish environmentally benign and economically viable synthesis of homoallyl alcohols/ethers. Although, worthful protocols have been reported, catalytic modification of the reaction has remained a never ending challenging task, especially for one-pot synthesis of active building blocks.
2. Allylation of acetals and ketals
2.1 Lewis acid catalysed allylation
In the initial attempts for the synthesis of regiospecific homoallyl ethers by using allylsilane and acetals, Hosomi et al. used titanium as a promoter.7 In the presence of stoichiometric amount of TiCl4, allylsilane reacted with numerous aliphatic, alicyclic and aromatic acetals at −78 °C and produced corresponding homoallyl ethers in good yields (Scheme 2).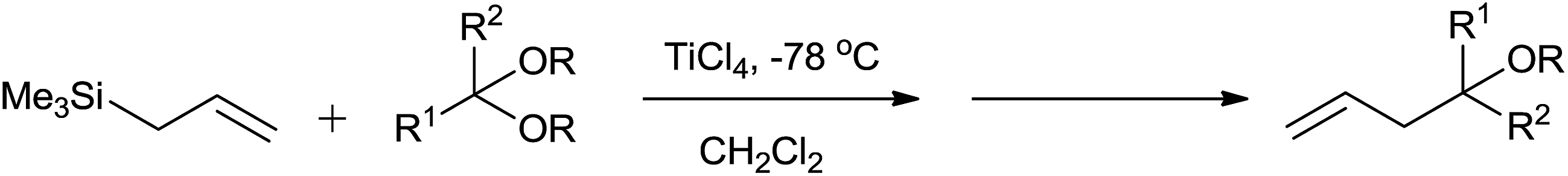 | ||
| Scheme 2 | ||
However, allyltrimethylsilane on reaction with triethyl orthoformate offered diallyl ether instead of a homoallyl acetal (Scheme 3).
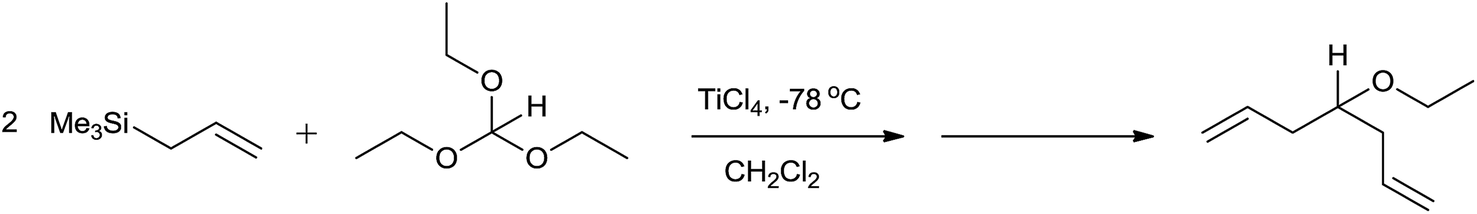 | ||
| Scheme 3 | ||
Inspired with aforementioned results of allylation, Hosomi et al. had carried out allylation of α,β-unsaturated acetals by using allylsilane in the presence of Lewis acids.8 Nature of Lewis acid, reaction temperature and structure of α,β-unsaturated acetals directed the mode of reaction. Stoichiometric use of TiCl4 promoted the reaction to yield diallylated compounds whereas aluminium trichloride (AlCl3) or BF3·OEt2 catalysed reaction gave selectively monoallylated compounds at −78 °C in dichloromethane (CH2Cl2).
In the presence of TiCl4, allylation took place at the acetal carbon (4) or β-carbon (5) or at both (4 and 5) (Scheme 4). Nature of β-substituent in acetal governed the ratio of 4 and 5 in the product. Methyl and n-propyl substituted acetals gave selectively 1,4,8-trienes (5). However, phenyl substituted acetal produced mixture of 4 and 5 at −78 °C in CH2Cl2. It is intriguing to note that the formation of monoallylated product was not observed in the presence of TiCl4 at −78 °C whereas, AlCl3 and BF3·OEt2 catalysed reaction offered monoallylated product (3) selectively under the same condition. Presumably, this could be attributed to different coordination abilities of Lewis acids. TiCl4 can coordinate to both alkoxy groups of acetals while AlCl3 and BF3·OEt2 can coordinate with only one (Fig. 1). In other words, TiCl4 activates both of the alkoxy groups whereas AlCl3 and BF3·OEt2 activate only one. Nevertheless, the second allylation took place smoothly with TiCl4.
 | ||
| Scheme 4 | ||
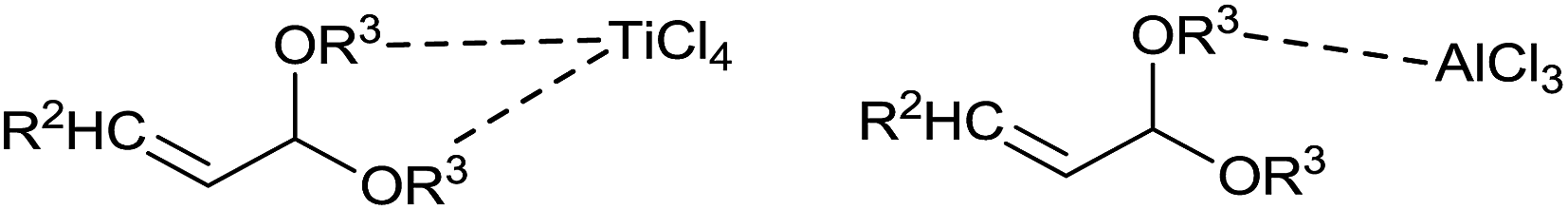 | ||
| Fig. 1 Coordinate complexes of TiCl4 and AlCl3. | ||
Ojima et al. had investigated the reaction of allylsilane with bifunctional molecules comprised of keto carbonyl and acetal functionalities.9 They had revealed that the structure of substrate and the nature of Lewis acid exerts effect on chemoselectivity of the reaction. The allylation of β-keto acetals resulted in formation of hydroxyacetal exclusively in the presence of stoichiometric amounts of AlCl3 or TiCl4 (Scheme 5). Moderate to low yields of products were obtained at −18 °C to 0 °C in dichloromethane.
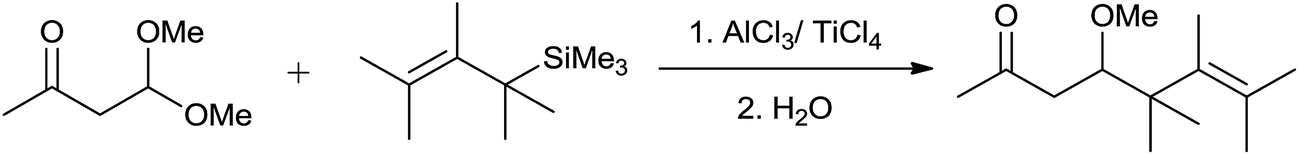 | ||
| Scheme 5 | ||
On the contrary, Lewis acid nature dependent chemoselectivity was observed in case of α-keto acetals. AlCl3 promoted allylation took place at the carbonyl group only to give homoallylalcohols exclusively whereas, TiCl4 promoted reaction led to the formation of bis homoallylicdiol monomethyl ethers as it had activated both the functional groups simultaneously (Scheme 6).
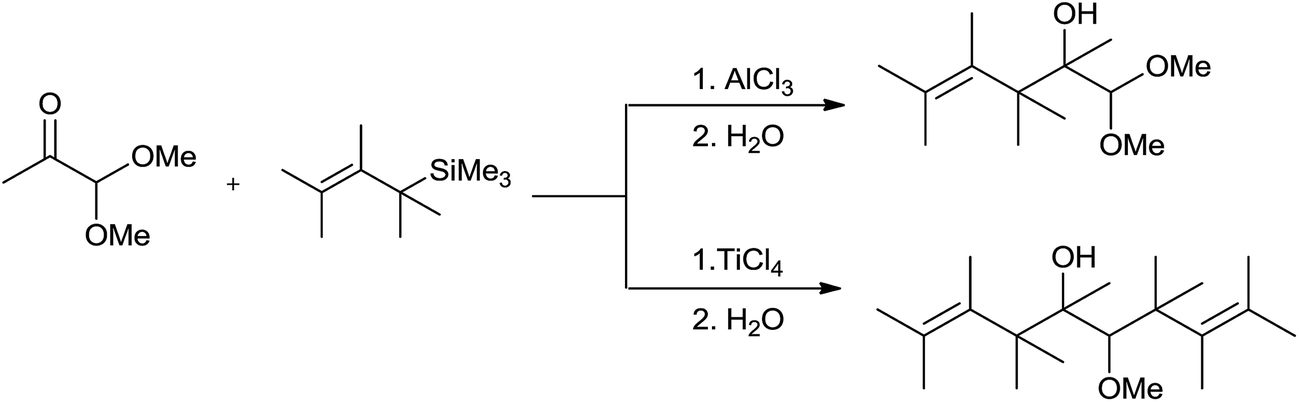 | ||
| Scheme 6 | ||
Although, when α-keto acetal was treated with two equivalents of allylsilane and one or two equivalents of AlCl3 only homoallyl alcohol was formed, whereas enhancement in the yield of diallylated product was observed in the presence of two equivalents of an allylsilane and TiCl4 at 5–20 °C.
Interestingly, use of trimethylsilyltrifluoromethanesulfonate (TMSOTf) in catalytic amount accomplished homoallylation of acetals at low temperature (−78 to −40 °C) to produce homoallyl ethers in high yields.10 The stereoselectivity of TMSOTf catalysed reaction can be understood better by using 4-tert-butylcyclohexanone acetal, which on allylation offered mixture of stereoisomers (Scheme 7). In the mixture, the ratio of equatorial to axial allyl isomer was found to be 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. However, the catalyst TMSOTf was unable to give the allylation products of carbonyl compounds viz. benzaldehyde, benzophenone, acetophenone and 4-tert-butylcyclohexanone.
:3. However, the catalyst TMSOTf was unable to give the allylation products of carbonyl compounds viz. benzaldehyde, benzophenone, acetophenone and 4-tert-butylcyclohexanone.
 | ||
| Scheme 7 | ||
Further to this the chemoselective allylation of acetals could be analysed in ionic liquids such as butylmethylimidazoliumhexafluorophosphate [bmim][PF6] and butylmethylimidazoliumtriflate [bmim][OTf] upon their reaction in the presence of 5–20 mol% of trimethylsilyltrifluoromethanesulfonate (TMSOTf).11 The homoallyl ethers were obtained in good quantities at room temperature (Scheme 8).
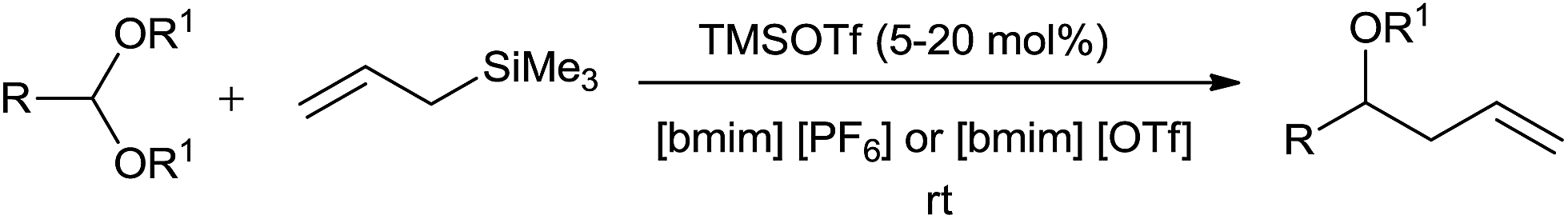 | ||
| Scheme 8 | ||
Unlike dichloromethane, a commonly used solvent for allylation, non-volatile and non-flammable ionic liquids are easy to recover and recycle. This various functional group compatible protocol offered monoallylated product of cinnamaldehyde dimethyl acetals unlike TiCl4 and aluminium tribromide (AlBr3). Along with this, the chemoselective allylation of 2,2′-diethoxyacetophenone in [bmim][OTf] produced homoallyl ether with a good yield of 78%, thereby indicating selective allylation of acetals over the carbonyl group (Scheme 9). The rate of this reaction in CH2Cl2 was found to be very slow and the product recovery was also very less (>40%). Notably, the reaction did not proceed with 5 mol% bismuth triflate (Bi(OTf)3) in CH2Cl2, thereby highlighting the influence of ionic liquid on the reaction outcome. The ionic liquid [bmim][PF6] was quantitatively recycled and successively reused upto four cycles of allylation of p-chlorobenzaldehyde dimethyl acetal.
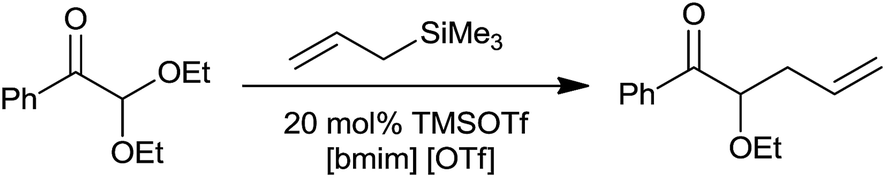 | ||
| Scheme 9 | ||
Iodotrimethylsilane (Me3SiI) plays an important role in the synthesis of various homoallylethers with regiospecific trans allyl group from acetals and allylsilanes when it is used in catalytic quantity (10 mol%) at −78 to −40 °C in dichloromethane (Scheme 10).12
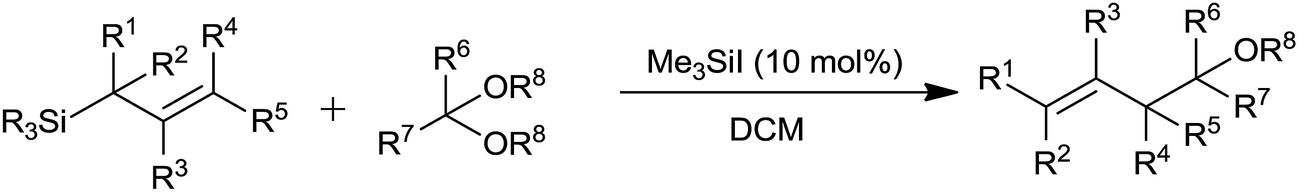 | ||
| Scheme 10 | ||
Furthermore, α,β-unsaturated acetals were formed by regioselective monoallylation at the acetal carbon while carbon–carbon double bonds remained unreacted. Though functionalized allylsilanes enhanced the yield of the product, steric hindrance around silicon and allyl groups retarded the rate of reaction. High yield of the regioselective products could be attributed to the ability of iodotrimethylsilane to form oxonium ion at low temperature when reacted with acetals. The oxonium ion further on bimolecular nucleophilic displacement with allylsilane offered homoallylether (Fig. 2).
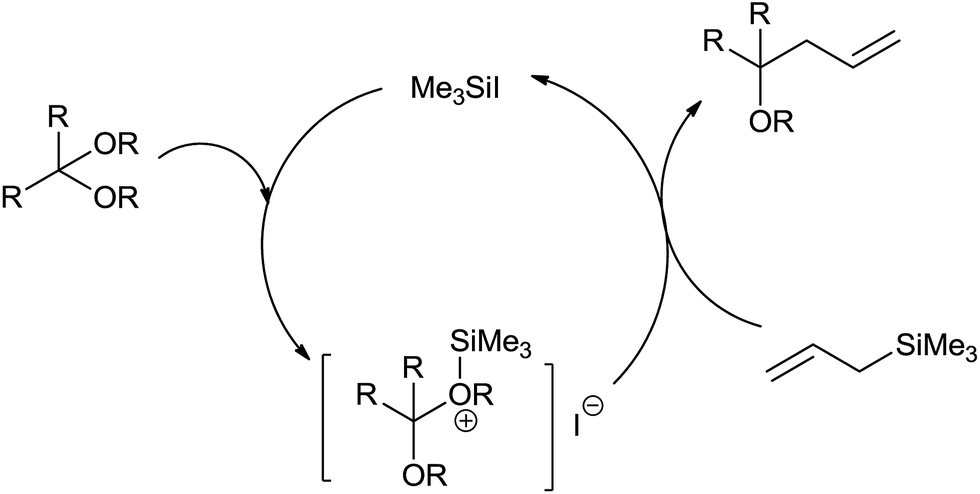 | ||
| Fig. 2 Catalytic cycle of the allylation. | ||
Mukaiyama et al. disclosed the regio and stereoselective synthesis of homoallyl ethers.13 They had used trityl perchlorate (TrClO4) and diphenylboryl triflate (Ph2BOTf) catalysts at −23 °C and −78 °C, respectively in CH2Cl2. An enhancement in diastereoselectivity was observed at lower temperature. Both the catalysts exhibited promising performance and afforded the product in good to excellent amounts. Triphenylmethyl carbenium ion of trityl perchlorate and diphenyl boron cation of diphenylboryl triflate causes activation of acetals. When allyltrimethylsilane was used, allylation of benzaldehyde dimethyl acetal occurred at γ-carbon of allylsilane while moderate diastereoselectivity was observed in the presence of crotyltrimethylsilane and cinnamyltrimethylsilane, respectively. In the presence of TrClO4 (γ-γ-dimethylallyl) trimethylsilane reacted with (3,3-dimethoxypropyl) benzene and produced homoallyl ether in 57% yield which did not happen in the presence of TMSI (Scheme 11).
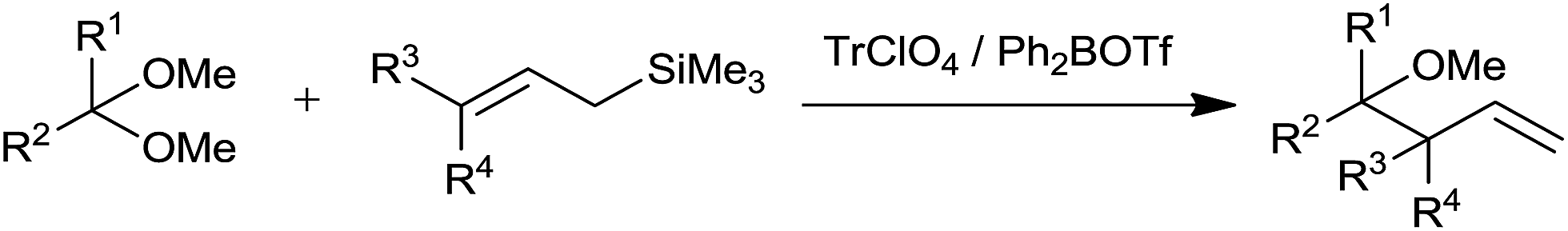 | ||
| Scheme 11 | ||
The mechanical and stereochemical aspects of Hosomi–Sakurai reaction were studied and explained by Denmark et al. Allylsilane exhibited Lewis acid dependent cyclization stereochemistry.14 TMSOTf catalysed reaction showed highly selective syn allylation while TiCl4 catalysed did not show such selectivity for allylation, thereby clearly indicating the involvement of Lewis acid in bond formation step, which is a stereochemistry determining step. Moreover, this observation is supported by the influence of SnCl4 on stoichiometry of the product (Table 1).
| Entry | Reagent | Temp (°C) | % syn | % anti | Yield, % (mass recovery) |
|---|---|---|---|---|---|
| 1 | Me3SiOTf | −70 | 96 | 4 | 100 |
| 2 | TfOH | −70 | 96 | 4 | 62 (74) |
| 3 | Ti(OiPr)2C2 | −20 | 87 | 13 | 21 (91) |
| 4 | AlCl3 | −20 | 86 | 14 | 33 (78) |
| 5 | BCl3 | −70 | 82 | 18 | 57 (73) |
| 6 | BF3·OEt2 | −20 | 77 | 23 | 95 (100) |
| 7 | TiCl4 | −90 | 47 | 53 | 55 (58) |
| 8 | SnCl4 (1.0 equiv.) | −70 | 45 | 55 | 35 (60) |
| 9 | SnCl4 (0.5 equiv.) | −60 | 71 | 29 | 81 (81) |
Interestingly, the structure of acetal and nature of Lewis acid governed the stereochemistry of homoallyl ether. The reaction proceeded to form product through either SN2 type mechanism via complexation of Lewis acid with acetal or SN1 type mechanism via formation of oxocarbenium ion. In case of methyl, ethyl and isobutyl acetals reaction followed the SN2 path resulting in syn product as major while isopropyl acetals react via SN1 path, as ready ionization was favoured owing to its inherent strain which leads to anti product as major one (Scheme 12) (Table 2).
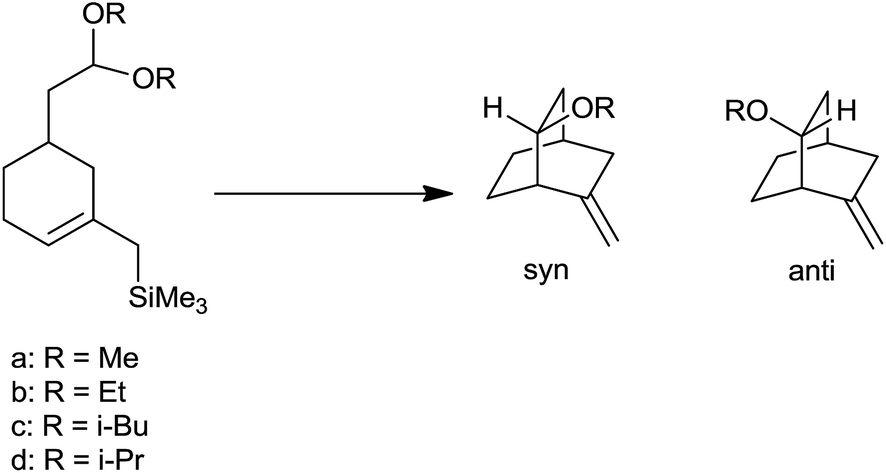 | ||
| Scheme 12 | ||
| Substrate | R | % syn | % anti |
|---|---|---|---|
| 1a | Me | 96 | 4 |
| 1b | Et | 92 | 8 |
| 1c | iBu | 90 | 10 |
| 1d | iPr | 38 | 62 |
Monoallylation of acetals and ketals was accomplished by using allyltrimethylsilane in the presence of 0.5 mol% titanium complex [TiCp2(CF3SO3)2] in nitromethane as well as dichloromethane.15 Nitromethane was found to be the solvent of choice as it accelerated the rate of the reaction by dissociating triflato ligands which easily bind to the oxygen atom of the electrophile. The lower reactivity observed for ketal as compared to acetals was accounted for steric hindrance.
With respect to enormous applications of silicon based reagents, Achla Trehan et al. successfully prepared trimethylsilyl bis(fluorosulfonyl) imide [TMSN(SO2F)2] and effectively used it for allylation of acetals.16 [TMSN(SO2F)2] exhibited superior catalytic activity than the TMSOTf, presumably due to the downfield silicon signal. Acetals of the aromatic as well as conjugated aldehydes underwent allylation in the presence of 5 mol% of [TMSN(SO2F)2] at −78 °C in dichloromethane and the corresponding homoallyl ethers were obtained in good to excellent yield within 30–50 min.
Gihani and Heaney had carried out the allylation of benzaldehydes dimethyl acetal by using trifluoromethanesulfonic (triflic) acid (TfOH).17 In the presence of bis-trimethylsilyl acetamide (BTMSA), 83% of homoallyl ether of benzaldehyde dimethyl acetal was obtained and the yield was increased upto 99% in the presence of bis-trimethylsilyl urea (BTMSU).
Admiring a high ionizing power of sulfur dioxide (SO2) owing to its electron pair acceptor ability, Mayr et al. had used liquid SO2 as Lewis acid solvent for allylation of acetals.18 Appreciable factor is recyclability of recondensed SO2 after reaction, which avoids aqueous workup. Homoallyl ethers of respective aliphatic and aromatic acetals were produced in good yields ranging from 83–98% by using 1.2 equivalent of allyltrimethylsilane in liquid SO2 at −60 °C. In addition to this, 1,1-dimethoxyheptane underwent allylation at 20 °C when a tiny crystal of I2 was added. It is worth to note that other allylsilanes also reacted with benzaldehydes dimethyl acetal in liquid SO2 at −60 °C and −20 °C, respectively.
With regard to the electron transfer ability of aminium salts, Hasegawa et al. successfully used tris(p-bromophenyl)aminiumhexachloroantimonate [(p-BrC6H4)3NSbCl6−] for the allylation of acetals with allyl trimethylsilane.19 The reaction progressed through the formation of oxonium intermediate and homoallyl ethers were obtained in good to excellent yields within 10–120 min.
Furthermore, triarylpyrylium salts catalysed photochemical as well as thermal allylation of acetals is also reported by Hasegawa and co-workers (Scheme 13).20
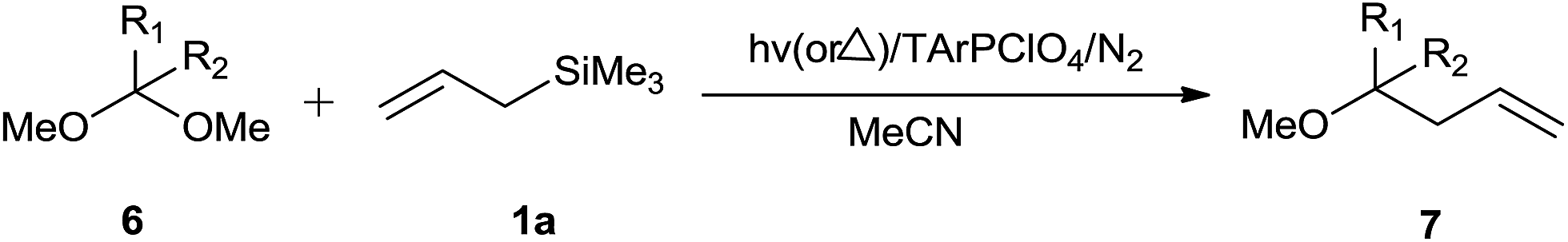 | ||
| Scheme 13 | ||
2,4,6-Triphenylpyrylium perchlorate (TPPClO4, 3 mol%) catalysed photochemical reaction worked well at λ > 360 nm, yielding product in the range of 51–99%. Thermal reaction in the presence of the same catalyst offered the product in 24–99% yield (Table 3). As compared to thermal reaction, photochemical reaction was much faster. Control experiments had revealed that photochemical reaction proceeded through photoinduced electron transfer (PET) and perchlorate anion had a pivotal role in the promotion of reaction. The initial single electron transfer (SET) took place between the singlet excited TPPClO4 and allyl silane, leading to the generation of silyl perchlorate which apparently promoted cationic reaction (Scheme 14).
| Sr. no. | Comp. | Photoreaction | Thermal reaction | ||
|---|---|---|---|---|---|
| Time (min) | Yield (%) | Time (min) | Yield (%) | ||
| 1 | 7a | 5 | 90 | 120 | 78 |
| 2 | 7b | 10 | 80 | 300 | 74 |
| 3 | 7c | 10 | 99 | 300 | 99 |
| 4 | 7d | 60 | 78 | 600 | 93 |
| 5 | 7e | 60 | 51 | 600 | 64 |
| 6 | 7f | 20 | 83 | 300 | 24 |
| 7 | 7g | 20 | 65 | 600 | 25 |
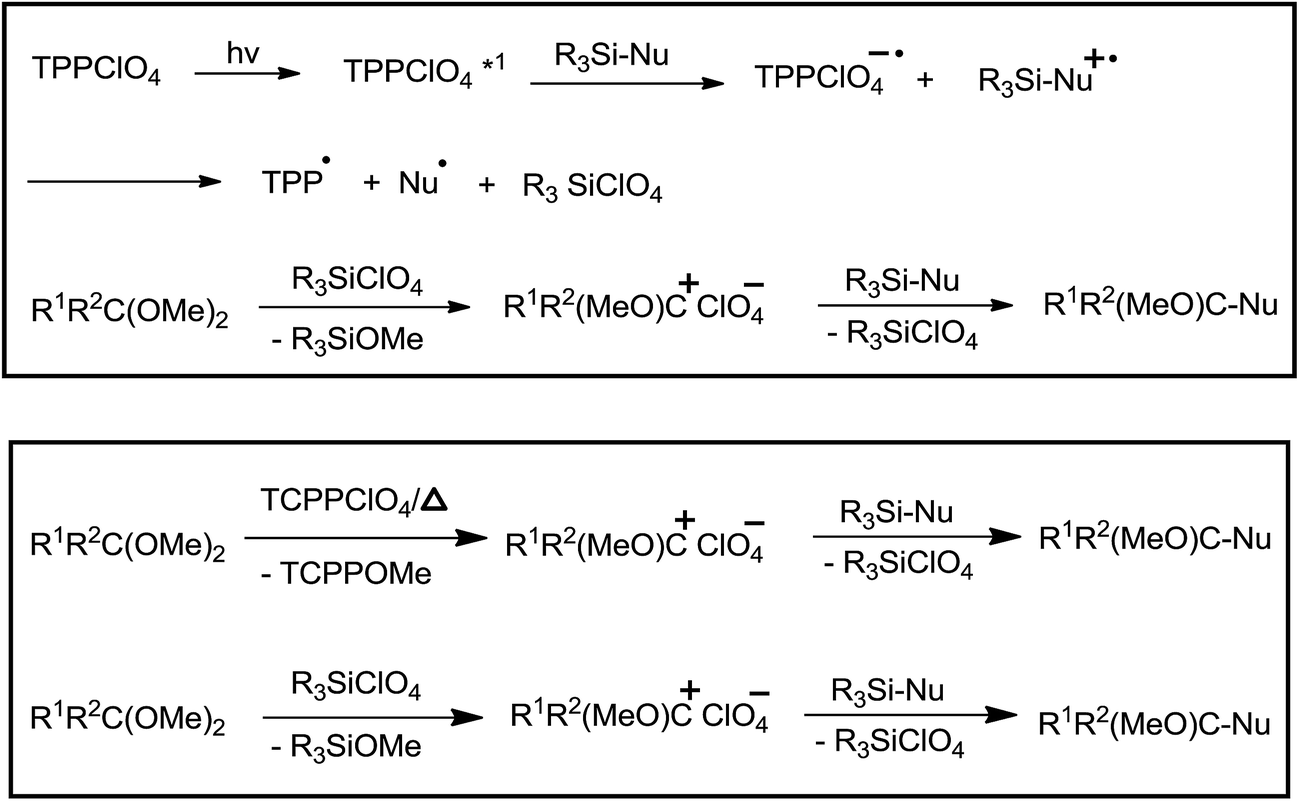 | ||
| Scheme 14 | ||
A monoallylation of cyclohexanone dimethylketal in the presence of trimethylsilyl bis(trifluoromethanesulfonyl) amide (TMSNTf2) is faster than its counterparts TMSOTf and TMSN(SO2F)2.21 This is because of the super silylating ability of TMSNTf2 which is evidenced by its downfield silicon signal than that of TMSOTf and TMSN(SO2F)2 (Table 4).
| Catalyst (eq.) | Chemical shift of Si (ppm) | Temp. (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|
| TMSOTf | 43.54 | −60 to −45 | 18 | 81 |
| TMSN(SO2F)2 | 44.92 | −78 | 0.75 | 91 |
| TMSNTf2 | 57.2 | −78 | 0.25 | 91 |
Bismuth(III) salts have emerged as promising catalysts in organic synthesis owing to their low toxicity.22 Interestingly, bismuth bromide effectively catalysed the allylation of aldehydes and acetals at room temperature with 5–20 mol% concentration. Its catalytic efficiency is better than bismuth(III) chloride (BiCl3), antimony(III) chloride (SbCl3) and comparable with TMSOTf and TMSI. Furthermore, the C-allylation of methyl 2,3,4,6-tetra-o-benzyl α-D-glycopyranoside was achieved at room temperature in CH3CN by using higher concentration (50 mol%) of bismuth(III) bromide (BiBr3) with high stereoselectivity (α/β = 8/1) (Scheme 15). Higher catalyst concentration is attributed to the partial deprotection of the benzyl groups accompanying allylation.
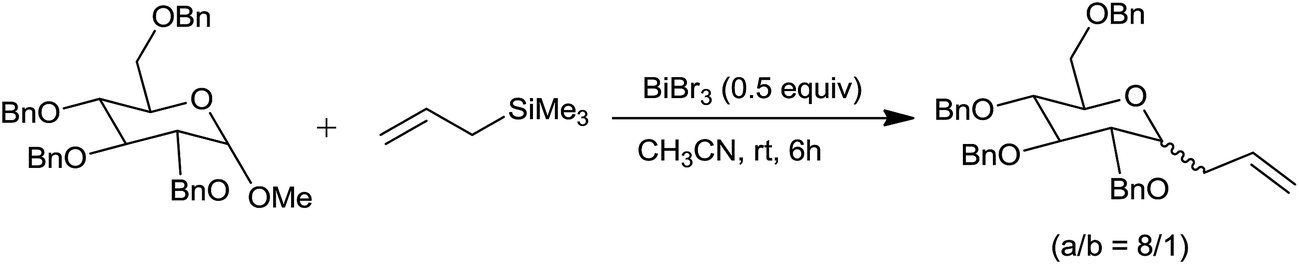 | ||
| Scheme 15 | ||
Scandium triflate (5 mol%) effectively catalysed allylation of acetals and gem-diacetates with sub-stoichiometric amount at ambient temperature in dichloromethane.23 It was found to be effective due to its high oxophilic nature and ability to form strong but labile bonds with oxygen donor ligands. Methoxy substituted aldehyde diacetates and aliphatic acetals offered low to moderate yields of the products owing to their inherent lower reactivity. The recovery of catalyst and further reuse without considerable loss of activity are the prominent features of this protocol. Allylation of ketone dimethylacetals and diacetates did not occur under present reaction conditions. Yb(OTf)3 and yttrium triflate (Y(OTf)3) were also screened for allylation but they exhibited inferior performance to that of Sc(OTf)3.
Likewise, bismuth triflate also efficiently catalysed the allylation of acetals by using allyltrimethylsilane.24 The reaction proceeded smoothly at room temperature in dichloromethane with low catalyst loading (1.0 mol%). This nontoxic catalyst worked well at little hydrous condition too. In contrast to TiCl4 initiated reaction, Bi(OTf)3 catalysed reactions offered selectively monoallylated ethers in quantitative yields. Whereas, Bi(OTf)3 was not effective for allylation of 2,2-diethoxyacetophenone and benzaldehyde.
Allylation of acetals with allyltrimethylsilane have been accomplished using 2 mol% of FeCl3 at −20 °C.25 The corresponding homoallyl methyl ethers were obtained in good to excellent yields, irrespective of the nature of substituents present on the aromatic dimethyl acetals. In addition, α,β-unsaturated dimethyl acetals and aliphatic dimethyl acetals were also subjected to allylation which on completion of the same formed products in quantitative yields at −20 °C and room temperature, respectively. Eventually homoallyl ethyl ether and homoallyl benzyl ether were also obtained in high yields from their respective acetals (Scheme 16).
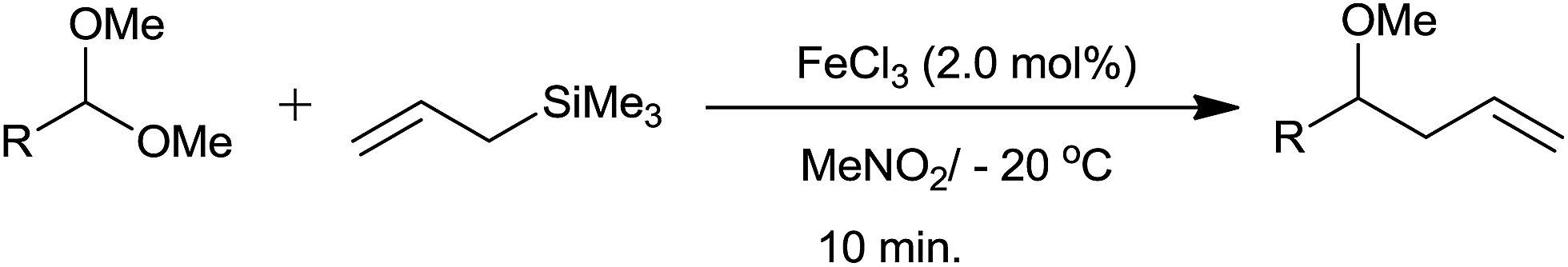 | ||
| Scheme 16 | ||
Jung and Maderna26 had explored and explained the aluminium bromide (AlBr3) catalysed allylation of acetals and cyclic ketals in the presence of trimethylaluminium (AlMe3) as a desiccant which scavenges hydrobromic acid (HBr) formed during the course of reaction. The best result was observed when 100 mol% of AlBr3/AlMe3 with the stoichiometric ratio of 6:1 has been used at −78 °C for 2 h. Yield of the product seemed to be directly proportional to the amount of catalyst i.e. product yield has decreased with the decrease in mol% of catalyst besides this product formation occurred at high temperature with a prolonged reaction time.
Indeed, substantial enhancement in the reaction rate and yield of the product were observed on addition of catalytic amount of copper(I) bromide (CuBr). The acceleration in rate of reaction could be mainly due to the formation of Al–Cu species. Allylation catalysed by CuBr/AlBr3/AlMe3 show chemoselective nature. In cyanoacetal, allylation took place selectively at acetal functionality while para-methoxy substituted acetal did not allylate as it formed para-quinonemethide cation which easily got polymerized.
Soon after to overcome these difficulties encountered with above protocols, Jung et al. developed functional group tolerant, strong Lewis acid free and microwave heating assisted method in the presence of stoichiometric CuBr.27 The reaction proceeded only in the presence of dichloromethane and 1,2-dichloroethane, mostly because of their non-binding to CuBr during the course of reaction. 1,2-Dichloroethane was the solvent of choice due to its higher boiling point. Homoallyl ethers were obtained in quantitative yields when acetals with electron donating substituents on their aromatic ring were used, while acetals with electron withdrawing substituents on their aromatic ring produced moderate yields of the products. However, aliphatic acetals and cyclic ketals afforded homoallyl ether in low yield. Unlike the routine normal reaction condition, during this procedure CuBr acts as a considerably mild but efficient Lewis acid that leads to formation of the oxocarbenium ion by using oxygen atom of the acetal.
Niobium pentachloride–silverperchlorate (NbCl5–AgClO4) catalysed allylation of acetal have been reported by Nishida et al.28 This catalyst system with the concentration of 0.5 mol% of each has worked well for aromatic as well as aliphatic acetals at 0 °C to room temperature and offered the products in good yields (84–98%). When niobium pentachloride was used as the only catalyst for allylation, it resulted in poor yield of homoallyl ether because of its strong Lewis acidic character which interrupted the establishment of catalytic cycle.
Nevertheless, a range of homoallyl ethers can be successfully synthesised by using an inexpensive and relatively non-toxic iron(III) p-toluenesulfonate Fe(OTs)3·6H2O as a catalyst.29 The synthesis could be accomplished in acetonitrile at room temperature but quantity of catalyst required for efficient synthesis varies from 2 mol% to 10 mol%.
2.2 Brønsted acid catalysed allylation
Kampen et al.30 have developed sulfonic Brønsted acid with 2 mol% catalyst loading in acetonitrile catalysed allylation of acetals by using allyltrimethylsilane. Various Brønsted acids such as diphenyl phosphate (PPA) (8), camphorsulfonic acid (CSA) (9), p-toluenesulfonic acid (p-TsOH) (10) and 2,4-dinitrobenzenesulfonic acid (DNBA) (11) were used for this reaction. Among these DNBA (Fig. 3) showed the best catalytic activity with 2 mol% catalyst loading in acetonitrile at room temperature. Acetals on reaction with Brønsted acid DNBA formed oxonium ion which on subsequent reaction with allyltrimethylsilane and methanol, respectively offered the homoallylic ether in high yield. Notably, aromatic acetals with electron-rich as well as electron-poor aryl substituents offered excellent yields while branched and unbranched aliphatic acetals formed the products in good to excellent yields. Interestingly, functional groups such as benzyl ether, alkyl bromide, nitrile and esters of aliphatic acetals were found to be tolerant to allylation in the presence of 2% DNBA. Further α,β-unsaturated acetals and ketals also offered good yields of monoallylic ethers. Neither hydrolysis nor aldolization was observed. Moderate yields of allylic ethers of few aliphatic acetals was attributed to their volatility. Unlike TiCl4 catalysed reaction, regiospecific homoallylated product formation was observed when allylation of cinnamaldehyde was carried out in the presence of DNBA. It is also found to be equally efficient for allylation of ketals and benzyl acetal.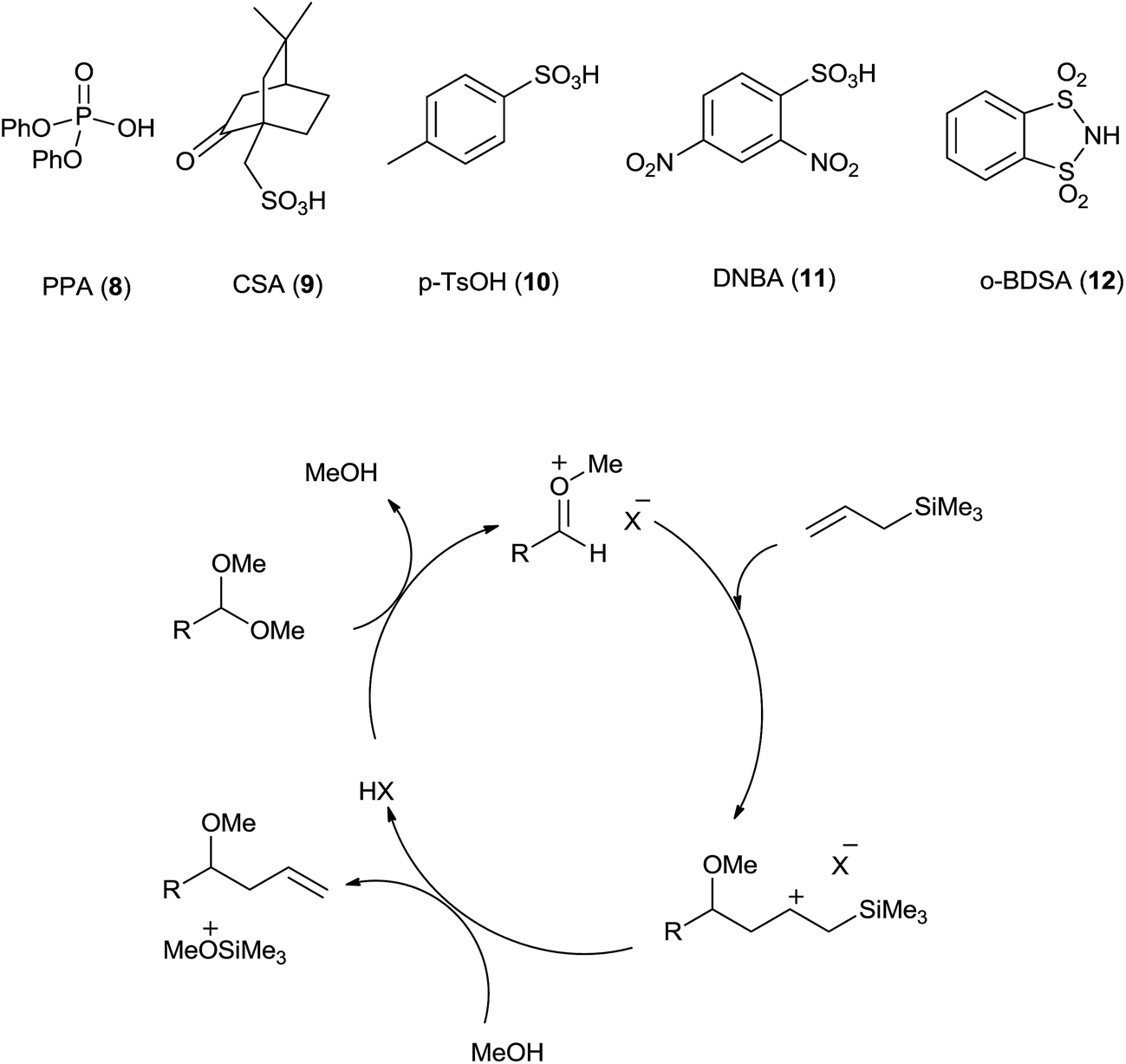 | ||
| Fig. 3 Catalytic cycle for allylation of acetal. | ||
Barbero and co-workers demonstrated allylation of acetals and ketals by using allyl trimethylsilane in the presence of 5 or 10 mol% of o-benzenedisulfonimide (BDSA) (12).31 Solvent-free condition and reusability of catalyst are some of the characteristic features of this protocol. The recovered BDSA was successfully used for three consecutive cycles without considerable loss of its catalytic activity or decrease in the yield of homoallyl ether. Moreover, the allylation of acetals could be realised by using 1-phenyl-2-(trimethylsilyl) acetylene in the presence of o-benzenedisulfonimide (20–30 mol%) in acetonitrile (Scheme 17). Lower yields of products could be accounted for the formation of less stable vinylic carbocation during the course of the reaction.
 | ||
| Scheme 17 | ||
Silicomolybdic acid supported on silica gel (10 wt%, 2.0 mol%) has proven itself to be a green catalyst for homoallylation.32 It catalysed homoallylation of acetals, ketals and acylals when 1.5 equivalents of allyl trimethylsilane was used. Anisaldehyde dimethyl acetal produced a mixture of monoallyl and diallyl products with the ratio of 74:19 when 1.5 equiv. of allyltrimethyl silane was used. However, it has then changed to 92:5 when 1.0 equiv. of allyltrimethyl silane was used. This is the first report on ring-opening allylation of cyclic acetal under catalytic and ambient conditions. It is intriguing to note that in case of allylation of acylals, less hydrolysis was observed when acetonitrile was replaced by dichloromethane as a solvent. Homoallyl esters are important with respect to their further easy manipulation.
Synthesis of silica supported triphenyl(3-sulfopropyl)phosphonium (TPSP) functionalized phosphotungstic acid (PW-Si/TPSP) with the loading of 10–30% of phosphotungstic acid on silica (w/w) was important and worth to note as they can be eventually used for selective monoallylation of acetals.33 In this combination, though self-induced phase separation of TPSP was a fascinating feature, polar solvents in the combination suppress solubility of TPSP. The Keggin structures of heteropoly anions remained unaltered while incorporation of phosphotungstic acid into the fumed silica takes place, as shown by 29Si-NMR, FT-IR and XRD. Ammonia-TPD and pyridine-FTIR were used to establish acidic strength and Brønsted acidic sites, respectively and with this the acidity order of PW > PW–TPSP > 30PW-Si/TPSP was established. For allylation of benzaldehyde dimethyl acetal PW-Si/TPSP was proved to be the best catalyst than amberlyst 15, PW, 30PW-Si, H2SO4–TPSP, PW–MIMBS, PW–TPSP etc. Highest yield of the allylated product was obtained with 30% PW loading at 0.050 g quantity of the catalyst in acetonitrile. Quantitative yield of the allylated product was observed in the presence of various substituents viz. methoxy, chloro, piperonaldehyde, 1-naphthaldehyde etc. Besides, the catalyst has remained stable for five recycling runs and offered the product with negligible loss in its yield.
2.3 Miscellaneous
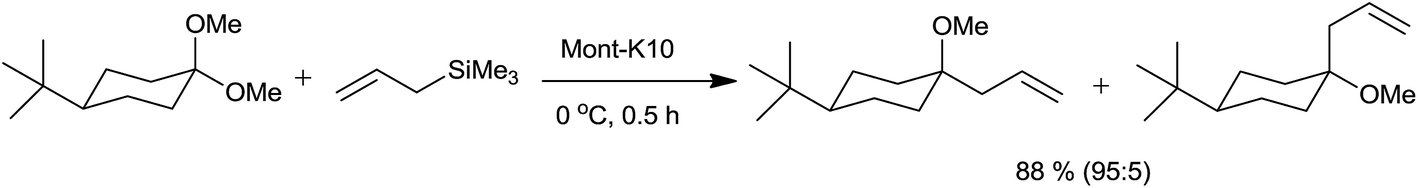
The present catalyst research has drifted towards the use of sustainable technologies such as heterogeneous catalysis. The catalysts have found and will continue to find extensive uses in a myriad of synthetic contexts as their separation and recycling is very easy.Onaka and co-workers34 have used various solid acid catalysts for allylation of acetals by using allylsilanes (Table 5). Aluminium cation exchanged montmorillonite Al-Mont and proton exchanged montmorillonite H-Mont, Mont-K10 catalysed reaction produced high yield of the product without formation of any by-product. Being heterogeneous catalysts, they were separated by just filtration after completion of reaction. Yield of the product was significantly affected during drying when montmorillonites were used as catalyst due to the less thermal stability of montmorillonites than zeolites and silica alumina (Scheme 18).
| Entry | Solid acid | Drying method of solid acid | Conditions | Mol% of homoallyl ether | Mol% of unreacted acetal | |
|---|---|---|---|---|---|---|
| Temp/°C | Time/h | |||||
| a A: pre-dried at 25 °C/0.5 Torr for 16 h and stored in a desiccator then dried at 120 °C/0.5 Torr for 3 h in a reaction vessel. B: pre-dried at 400 °C in air for 3 h and stored in a desiccator then dried at 350 °C/0.5 Torr for 10 min in a reaction vessel. C: washed with MeOH and dried at 25 °C/0.5 Torr. | ||||||
| 1 | Na-Mont | A | 0 | 0.5 | 0 | 86 |
| 2 | Al-Mont | A | 0 | 0.5 | 95 | 1 |
| 3 | H-Mont | A | 0 | 0.5 | 87 | 11 |
| 4 | Mont-K10 | A | 0 | 0.5 | 85 | 14 |
| 5 | Na-TSM | A | 0 | 0.5 | 0 | 86 |
| 6 | Al-TSM | A | 0 | 0.5 | 48 | 44 |
| 7 | HY | B | 25 | 2 | 43 | 30 |
| 8 | CaY | B | 25 | 2 | 30 | 30 |
| 9 | Silica–alumina | B | 25 | 2 | 58 | 16 |
| 10 | γ-Alumina | B | 25 | 2 | 0 | 89 |
| 11 | Amberlist-15 | C | 25 | 2 | 47 | 44 |
| 12 | Nafion-117 | — | 25 | 2 | 51 | 33 |
 | ||
| Scheme 18 | ||
Cyclohexane dimethyl acetal is exclusively allylated from the equatorial position. Similarly, the predominant equatorial allylation of 4-tert-butylcyclohexane dimethyl acetal is anticipated due to its superior stability (Scheme 19).
 | ||
| Scheme 19 | ||
In 2010, Ito and Asami et al.35 designed environmentally benign protocol of Hosomi–Sakurai homoallylation of acetals by using mesoporous aluminosilicate Al-MCM-41. As MCM-41 is having large uniform pores (2–10 nm) and a high surface area, the incorporation of metals, such as Ti, Sn and Al into it enhanced its catalytic activities. Amorphous silica–alumina (SiO2–Al2O3) and aluminium-free MCM-41 were ineffective for homoallylation of acetal. However, the optimization study revealed that the concentration of aluminum in MCM-41 plays a detrimental role. When its concentration was reduced, the product yield was alleviated from 86% to 96% (Scheme 20, Table 6).
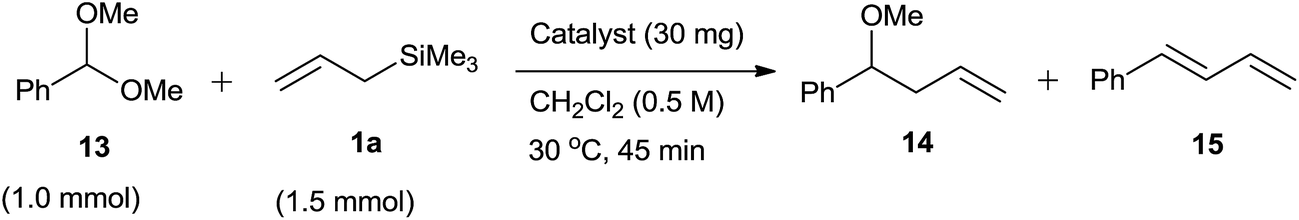 | ||
| Scheme 20 | ||
| Entry | Catalyst | Si/Al | 3a (%) | 4 (%) |
|---|---|---|---|---|
| 1 | Al-MCM-41 | 26 | 86 | 7 |
| 2 | SiO2–Al2O3 | 31 | 0 | 0 |
| 3 | MCM-41 | ∞ | 0 | 0 |
| 4 | Al-MCM-41 | 34 | 91 | 4 |
| 5 | Al-MCM-41 | 48 | 96 | Trace |
Under the optimized reaction conditions various aromatic and aliphatic acetals formed the respective homoallyl ethers in quantitative yields. Nature of the substituent on aromatic ring did not affect the rate of reaction as well as yield of the product. Naphthaldehydes and (E)-cinnamaldehyde dimethyl acetals also were allylated and the products were obtained in good yields. Interestingly, benzaldehyde diethyl acetal also gave homoallylethylether in a appreciable yield. Furthermore, allylation of cyclohexanone dimethyl ketal also proceeded well, albeit at a high concentration of allyltrimethylsilane (3 equiv.) (Scheme 21).
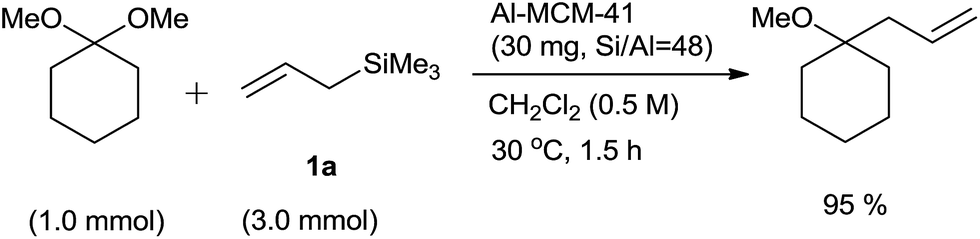 | ||
| Scheme 21 | ||
Al-MCM-41 exhibited chemoselectivity for acetal over aldehyde and consequently formation of only 4-methoxy-4-phenylbut-1-ene was observed (Scheme 22).
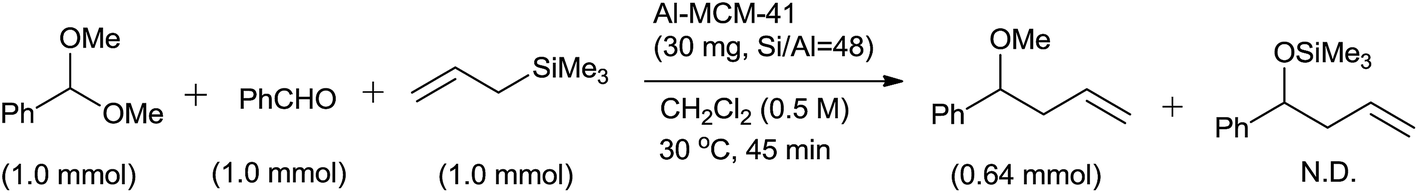 | ||
| Scheme 22 | ||
3. Allylation of aldehydes and ketones
Though the allylation of acetals and ketals has been extensively explored, several acetals and ketals are not commercially available and must have to be prepared from the corresponding aldehydes and ketones. Nevertheless, carbonyl compounds directly offer homoallyl alcohols or ethers within one step.3.1 Lewis acid catalyzed allylation
Hosomi and Sakurai36 had successfully synthesized γ,δ-unsaturated alcohols from diverse aliphatic, alicyclic and aromatic carbonyl compounds by using allyltrimethyl silane in presence of titanium tetrachloride in dichloromethane. A regiospecific transposition of allylic part took place during the course of reaction and carbon–carbon bond formation occurred exclusively at a γ-carbon of allylsilanes (Scheme 23).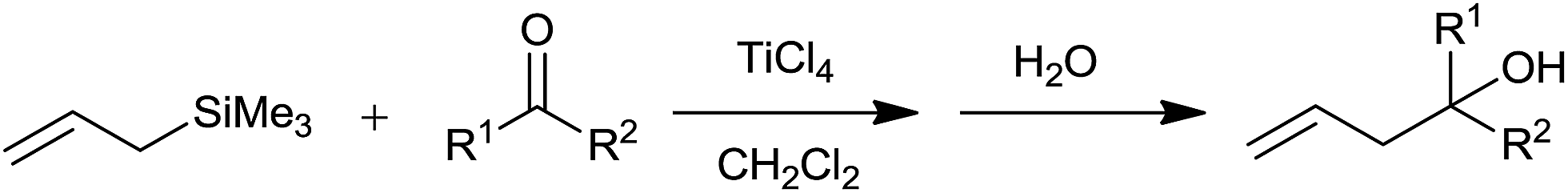 | ||
| Scheme 23 | ||
The reaction between but-3-en-2-yl trimethylsilane and equimolar carbonyl compound offered a mixture of two stereoisomeric alcohols with trans isomer as a major product while 1-phenyl allyltrimethylsilane offered exclusively trans alcohol. Nevertheless, both the stereoisomers of but-2-en-1-yl trimethylsilane and trans isomer of 3-phenylallyltrimethylsilane on reaction with aldehydes or ketones in the presence of TiCl4 produced solely trans alcohols. Looking at the yield of alcohols, TiCl4 was found to be superior than AlCl3, SnCl4 and BF3·OEt2.
Diverse aldehydes underwent allylation with allyltrimethylsilane in the presence of stoichiometric quantity of aluminium trichloride at −25 °C in dichloromethane.37 The corresponding homoallyl alcohols were obtained after the hydrolysis of silyl ether intermediate in water and methanol.
Nucleophilic affinity of fluoride ion to a silicon atom and high silicon–fluoride bond energy attracted the interest of Hosomi et al. to use tetra-n-butylammonium fluoride (TBAF)38 as a catalyst for the allylation of diverse aldehydes and ketones in THF at reflux temperature (Scheme 24). Notably, functional groups such as nitriles epoxides and esters remained unaffected.
 | ||
| Scheme 24 | ||
Fluoride ion in TBAF attacked on the silicon atom of allyltrimethylsilane and formed trimethylfluorosilane along with allylic anion species. This allylic anion species underwent chemoselective addition with carbonyl compound and formed allylated alkoxide which on reaction with the trimethylfluorosilane afforded silyl ether, which subsequently produced homoallyl alcohol upon hydrolysis (Fig. 4).
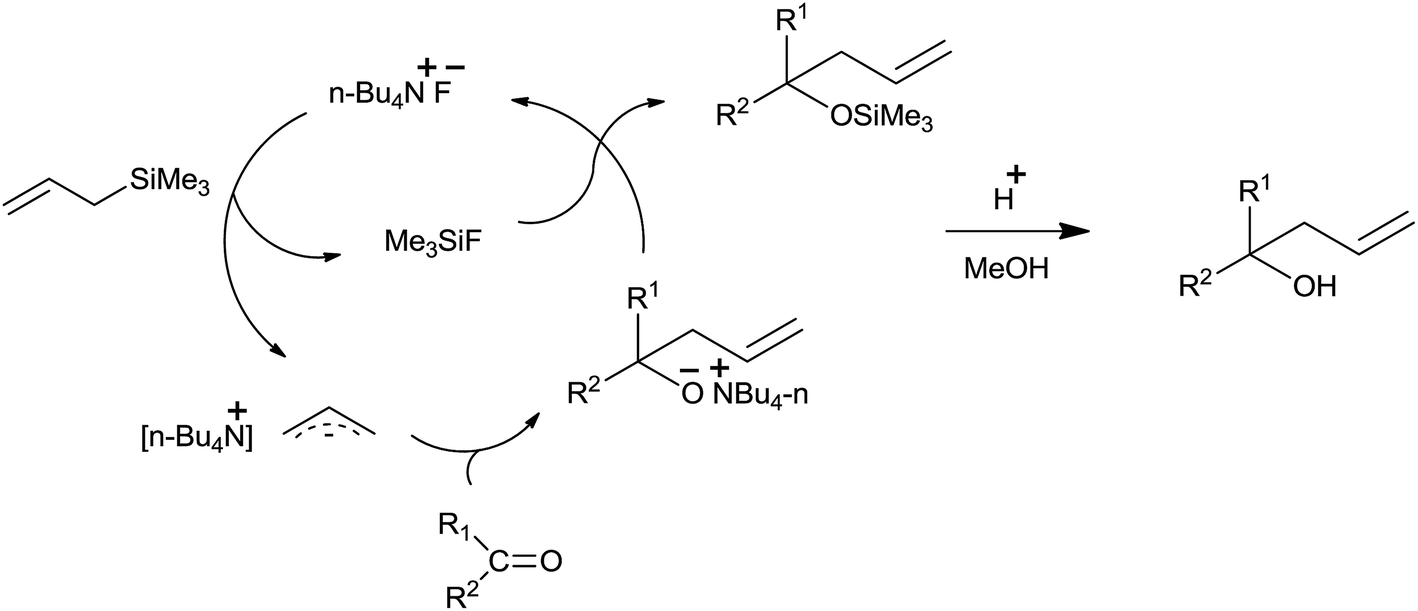 | ||
| Fig. 4 Catalytic cycle for TBAF catalyzed allylation. | ||
Various aldehydes and ketone produce corresponding alcohols in moderate to good yields. Further, the allylation in case of alkyl substituted allyltrimethylsilane occurred solely at the less substituted carbon. However, simultaneous conjugate addition as well as 1,2-addition to carbonyl group took place when α,β-enones reacted with the allyltrimethylsilane. Though catalytic TBAF substantially accelerated/stimulated the allylation reaction, potassium fluoride even in the presence of 18-crown-6 failed to do the same.
Sakurai and co-workers39 had accomplished allylation of carbonyl compounds by using catalytic quantity of trimethyl silyl iodine in the presence of alkoxysilane in dichloromethane. Interestingly, iodine catalyzed homoallylation of benzaldehydes in the presence of methanol was found to be a one-pot synthetic protocol.
Diphenylboryl triflate had effectively catalyzed allylation of benzaldehyde in dichloromethane at 0 °C and yielded the corresponding alcohol in 83% yield.13
Mukaiyama et al.40 had achieved one-pot asymmetric allylation of aldehydes by using diphenylboryl triflate (Ph2BOTf) catalyst with high stereoselectivity. Although, highest yield of the allylation product of 3-phenyl propanol by using 1-phenyl-1-trimethylsilyloxyethane as a chiral source at −23 °C in dichloromethane in the presence of catalyst TrClO4 was obtained, the method suffered from poor stereoselectivity (Scheme 25). However, good yield and best stereoselectivity (96:4) were observed when Ph2BOTf was used as a catalyst at −78 °C in toluene.
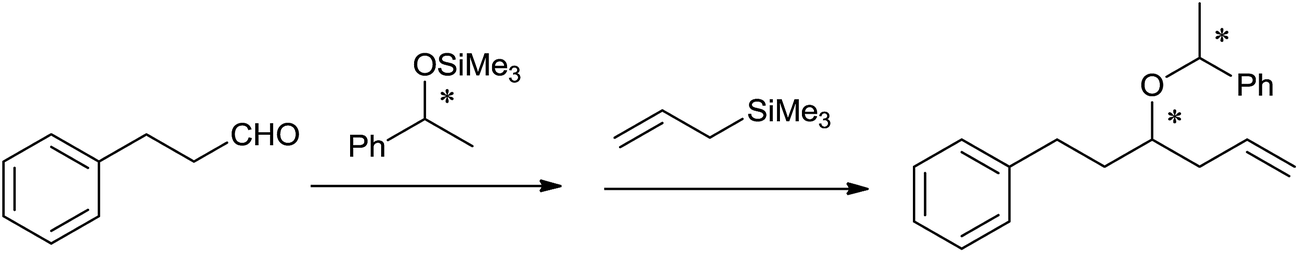 | ||
| Scheme 25 | ||
Additionally, when various aldehydes were allylated by using Ph2BOTf as a catalyst, aliphatic aldehydes produced homoallyl ethers with high stereoselectivity and the same was diminished for the benzaldehydes due to an easy formation of the benzylic oxocarbenium ion which had favored the SN1 transition state instead of SN2.
In another context, Markó et al.41 synthesized homoallylic ethers from carbonyl compounds via trimethylsilyl ethers and allyltrimethylsilane in the presence of trimethylsilyl triflate TMSOTf.
In this three components reaction, silyl ether was a co-reactant and reaction proceeded through the formation of oxonium cation 19, which eventually trapped by allyltrimethylsilane to produce diversely substituted ethers (Scheme 26). Interestingly, aliphatic as well as aromatic aldehydes reacted smoothly to afford the corresponding product with good yields. Nevertheless, ketones formed tertiary homoallylic ethers. Though the presence of silyl ether is indispensable, the reaction is independent of its nature. The reaction exhibited significant level of diastereoselectivity. Nature of the aromatic group present in silyl ether governed the ratio of diastereomers (Scheme 27).
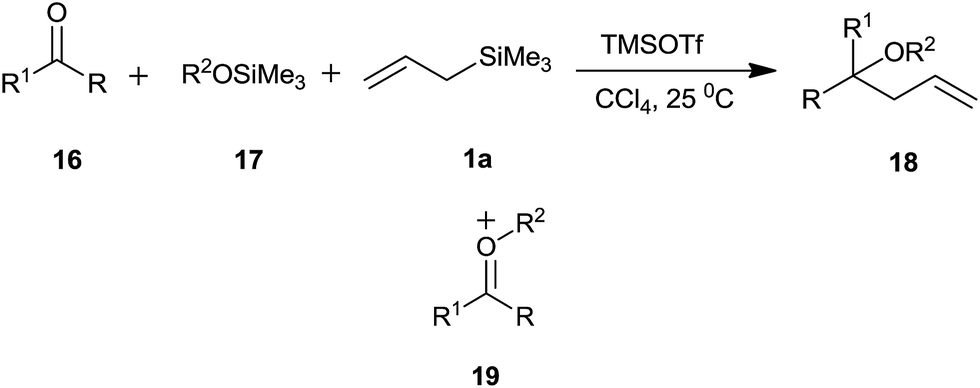 | ||
| Scheme 26 | ||
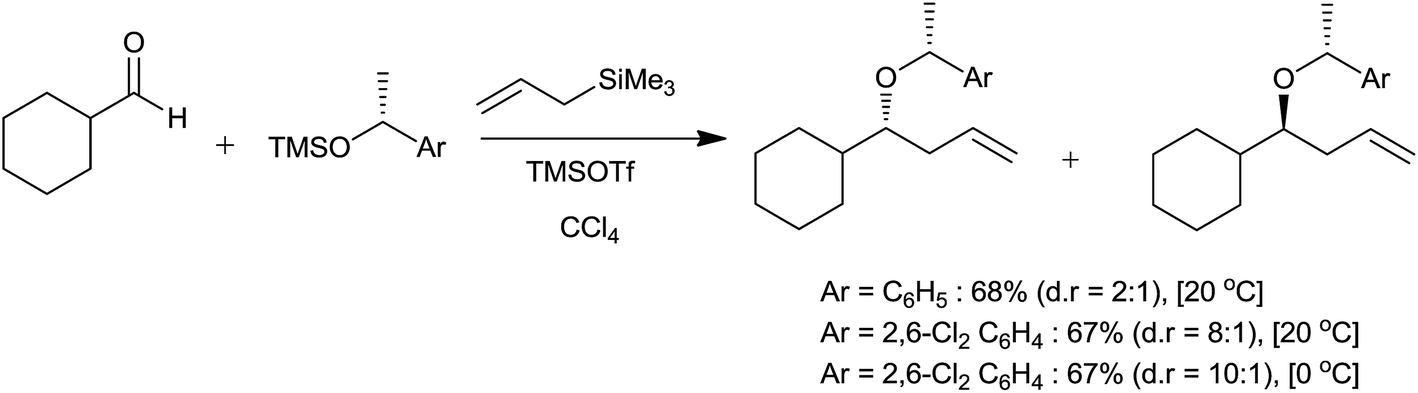 | ||
| Scheme 27 | ||
In order to support the earlier mechanism proposed for superacid catalyzed allylation, Davis and Jaspars42 prepared supersilylating reagent Me3SiB(OTf)4 and successfully employed it for allylation of aldehydes (Fig. 5). Use of 0.2–1.0 mol% Me3SiB(OTf)4 worked well in dichloromethane at room temperature and offered homoallyl alcohol with 80–93% yield within 5–25 min. On the contrary, low to moderate yield of homoallyl alcohol was obtained by using 20 mol% of Me3SiI and Me3SiOTf in 45 min and 23 h, respectively.
 | ||
| Fig. 5 Proposed reaction mechanism. | ||
Bosnich and co-workers15 had productively employed organometallic complex [TiCp2(CF3SO3)2] for allylation of aldehydes. An allyltrimethylsilane reacted very finely with benzaldehyde in the presence of 0.5 mol% [TiCp2(CF3SO3)2] to produce monoallyl ether in 90% yield within 5 minutes in nitromethane. Whereas, under the same reaction conditions it failed to react with butan-2-one and cyclopentanone in the presence of nitromethane. Nitromethane proved itself as a better solvent than dichloromethane owing to its extensive dissociation of triflato ligands of catalyst. Upon dissociation titanium atom binded to oxygen atom of the electrophile and accelerated the rate of reaction.
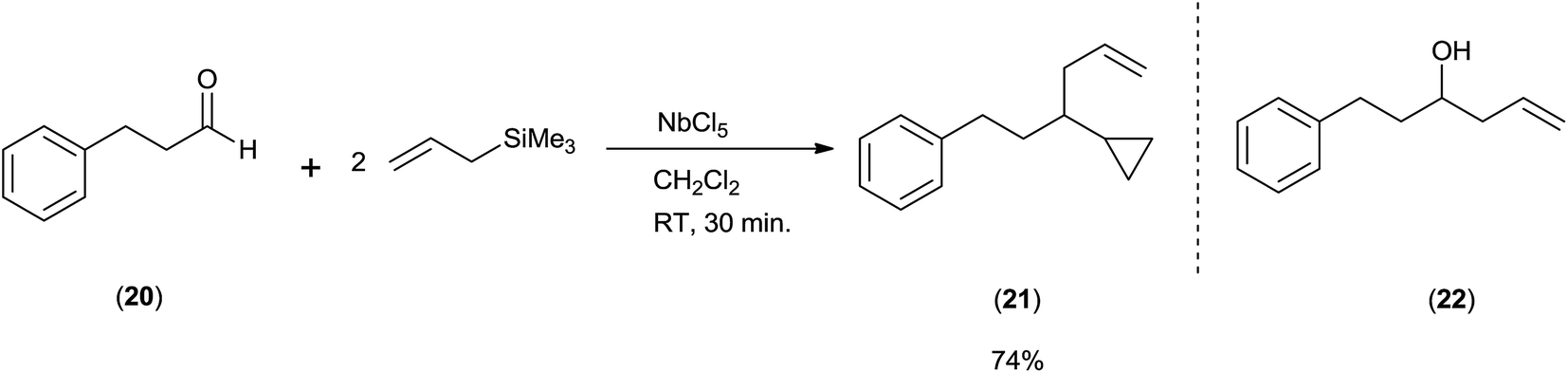
Keisuke Suzuki and co-workers43 surprisingly got cyclopropane derivative (21) instead of homoallyl alcohol (22) when they took aldehydes and allyltrimethylsilane in 1:2 ratio in the presence of NbCl5 as a catalyst at room temperature in dichloromethane (Scheme 28).
 | ||
| Scheme 28 | ||
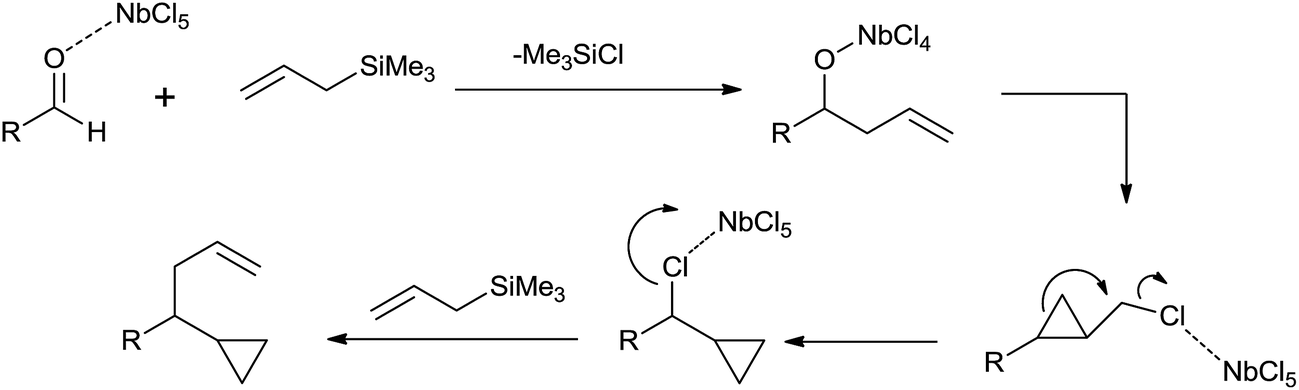
When the reaction was carried out at −20 °C for 5 min, homoallyl alcohol was obtained as the major product in 60% yield along with cyclopropane derivative (5%). However, when reaction time was increased upto 1 h, the yield of cyclopropane derivative was increased upto 69% at the cost of homoallyl alcohol (1%). Unlike TiCl4, tantalum pentachloride (TaCl5) offered cyclopropane derivative only in 48% yield. However, in the presence of ZrCl4 and hafnium tetrachloride (HfCl4), cyclopropane derivative was obtained as the major product in 47% and 48% yield while homoallyl alcohol was obtained as the minor product with 35% and 28% yield, respectively. Mechanistic investigation implies that the cyclopropyl moiety is derived from the allyl moiety which is engrafted by using allylsilanes (Scheme 29).
 | ||
| Scheme 29 | ||
The proposed mechanism is supported by ab initio molecular orbital calculations (MP216-31G*), investigated for a series of C4H7+ cationic species.
An air stable scandium triflate (Sc(OTf)3) had effectively allylated both unactivated and activated aromatic aldehydes and also aliphatic aldehydes in nitromethane.44,45 Lower catalyst loading was due to the weak binding of the homoallylic alkoxide to scandium triflate along with better efficiency of silicon transfer to oxygen. In fact only 2 mol% of the scandium triflate was required for carrying out allylation of activated and unactivated aromatic aldehydes while 10 mol% of the same was needed for the allylation of deactivated aromatic aldehydes and less reactive aliphatic aldehydes. 4-Methylbenzaldehyde after completion of allylation offered a mixture of monoallyl alcohol and diallylated product while 4-methoxybenzaldehyde offered solely the diallylated product in the presence of 10 mol% Sc(OTf)3. This was due to the electronic effect which could further be evidenced by allylation of 3-methoxybenzaldehyde in which 43% yield of monoallyl alcohol was obtained along with 12% over-addition product. Interestingly, ketones were totally unreactive with allyltrimethyl silane under these conditions and therefore chemoselective addition of allyltrimethyl silane to benzaldehyde in the presence of acetophenone was possible.
Considering nontoxic nature and commercial availability of bismuth(III) salts, Komatsu et al.22 had explored the ability of bismuth bromide (5.0 mol%) for the allylation of aldehydes in dichloromethane at room temperature. Bismuth bromide and bismuth chloride both offered good yields of allyl alcohol, while antimony(III) chloride produced only trace quantity.
Ytterbium bis(trifluoromethanesulfonyl)amide, [Yb(NTf2)3] catalyzed allylation of benzaldehydes tendered bisallylation product in 48% yield and mono-allylated trimethylsilyl ether in only trace quantity at room temperature in dichloromethane.21
Yang et al.46 had used ytterbium trifluoromethanesulfonate Yb(OTf)3 for an allylation of glyoxalate hydrates and α-keto aldehyde hydrates. Moderate to good yields of α-keto and α-ester homoallylic alcohols were observed at room temperature with catalyst loading of 5–10 mol%.
Yamamoto and co-workers47 successfully utilized a catalytic amount of co-catalyst π-allylpalladium chloride dimer 23 (5 mol%)-TBAF (0.5 equiv.) in n-hexane–THF (4:1) as a co-solvent for allylation of various aldehydes with allyltrimethylsilane (2 equiv.) at room temperature. An aromatic and alkenyl aldehydes produced corresponding homoallyl alcohols in moderate to good yields whereas poor yields were observed for aliphatic aldehydes and cyclic ketone. Interestingly, formation of bis-π-allylpalladium complex 24, took place during the course of reaction. This had occurred due to coordination of fluoride ions with allyltrimethylsilane to produce pentacoordinate allylsilicate which could then easily undergo transmetalation to Pd(II) (Fig. 6).
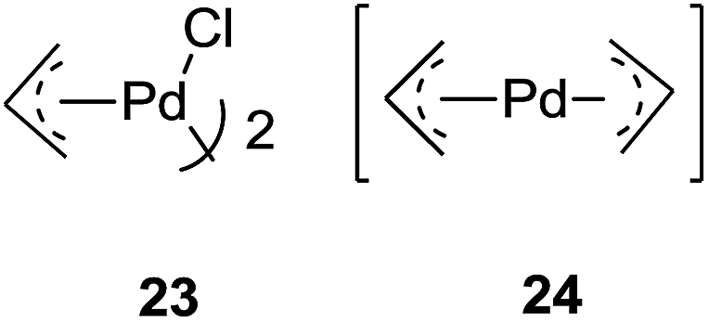 | ||
| Fig. 6 Bis-π-allylpalladium complex. | ||
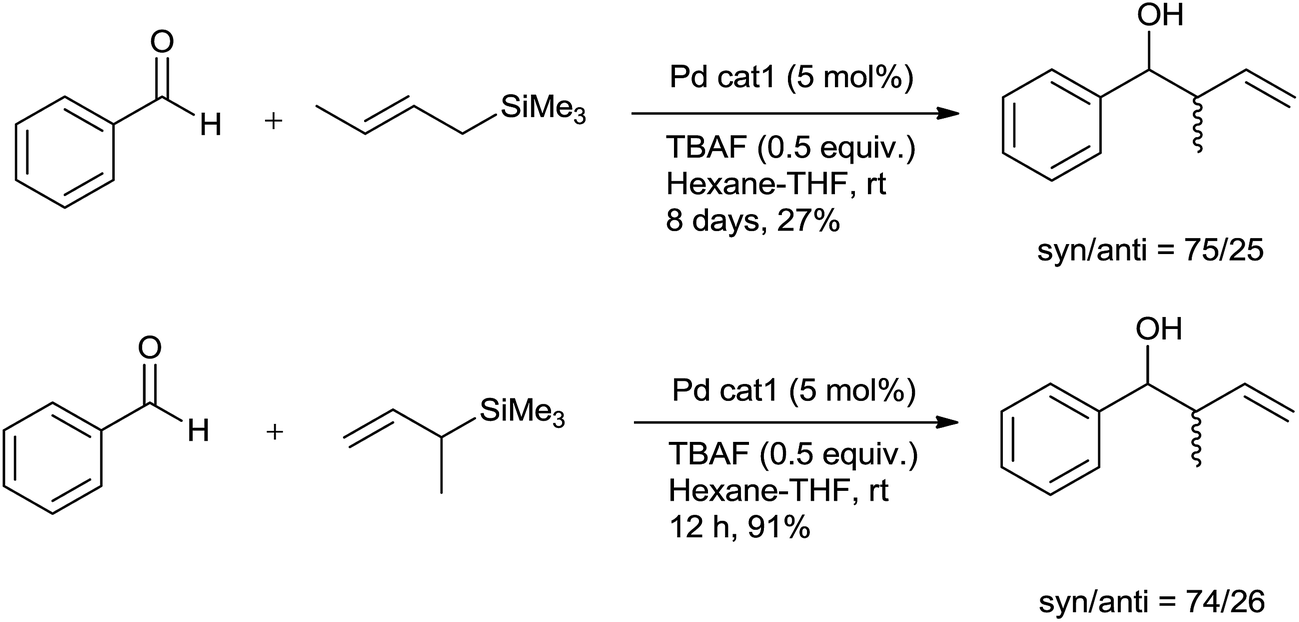
Allylation of benzaldehydes with γ-methyl allyltrimethylsilane and α-methyl allyltrimethylsilane offered γ-addition and α-addition products after 8 days and 12 h, respectively, having nearly same syn/anti diastereomer ratios (Scheme 30). This implied that transmetalation step is the rate-determining step and reaction proceeds through the formation of bis-π-allylpalladium.
 | ||
| Scheme 30 | ||
In situ generated trimethylsilanemethanesulphonate (TMSOMs) from methane sulphonic acid (MsOH) and allyltrimethylsilane had effectively catalyzed allylation of aldehydes at room temperature with only 10 mol% of catalyst loading.48 Wide range of homoallyl ethers were synthesized from aldehydes by using allyltrimethylsilane and various silyl ethers in the presence of TMSOMs.
Yadav and co-workers23 had used scandium triflate for allylation of various aromatic, aliphatic and heterocyclic aldehydes in the presence of trimethylorthoformate. The reactions occurred smoothly with 10 mol% of Sc(OTf)3 at room temperature in dichloromethane and yielded exclusively homoallyl ethers in 70–90% yield.
Considering the low Lewis acidity of the indium salts Akio Baba and co-workers49,50 used InCl3–Me3SiCl system as a strong Lewis acid catalyst for allylation of aldehydes, ketones, α-hydroxy ketones and benzoin. Both the catalysts independently failed to exhibit the catalytic activity but collectively with 5 mol% quantity they offered excellent yield of the allylation product presumably because of strong interaction between indium and silicon. This is evidenced and consequently confirmed by non-performance of AlCl3, BF3·OEt2 and TiCl4 in combination with Me3SiCl. Addition order of reactants and catalysts impressed the yield of product, as the catalyst polarised the carbonyl group as well as activities of allylsilane. Among the various chlorosilane screened, Me3SiCl was found to be the most suitable candidate for designing an efficient catalyst system along with InCl3. Mild non-protic condition was compatible for allylation of carbonyl group in the presence of nitro, ester and cyano functional groups. Nevertheless, aliphatic and sterically hindered aldehydes also offered respective homoallyl alcohols in high yields, while ketones formed homoallyl alcohols only trace amounts. It is intriguing to note that this catalyst system possessed high catalytic ability for allylation of homoallyl trimethylsilyl ethers, which could be evidenced by 25% diallylated product of p-methyl benzaldehyde and exclusively diallylated product of p-methoxy benzaldehyde. Cyclohexanone offered solely homoallyl alcohol while acetophenone tendered diallylation product preponderantly at room temperature in nitromethane. Propiophenone and p-chloroacetophenone too formed diallylated products in 49% and 55%, respectively. In the presence of this catalyst system benzoin underwent allylation at the carbonyl carbon only and afforded stereoselectively syn-diol. Notably, size of substituents on chlorosilane determined yield of the allyl alcohol and impressive selectivity with inversion of allylic unit was achieved in case of substituted allylsilanes.
Molecular iodine had effectively converted broad range of aromatic and aliphatic aldehydes into corresponding homoallyl alcohols.51 Higher yield of the product was obtained with high selectivity at 0 °C in acetonitrile within short reaction time (1 min) (Scheme 31). Iodine successfully formed the trimethyl silyl iodide on reaction with allyltrimethylsilane which as a result effectively catalyze the reaction.
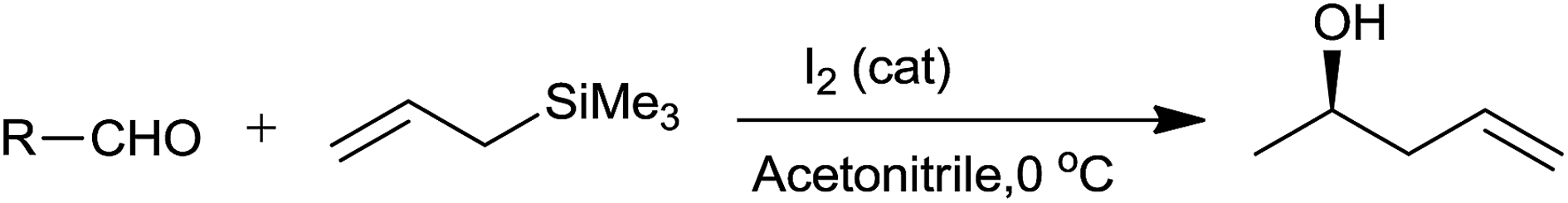 | ||
| Scheme 31 | ||
In 2002, Oriyama et al.52 had successfully used ferric chloride for allylation of various aldehydes with 1.5 equiv. allyltrimethylsilane at −20 °C in nitromethane. Benzaldehyde got over allylated (1 and 2) with two equivalent of allyltrimethylsilane in the presence of 5 mol% of anhydrous FeCl3 at room temperature (Scheme 32). While it offered 54% and 66% homoallyl alcohol with the same quantity of allyltrimethylsilane at 0 °C and −20 °C, respectively. This implied that temperature had a crucial role in the formation of product.
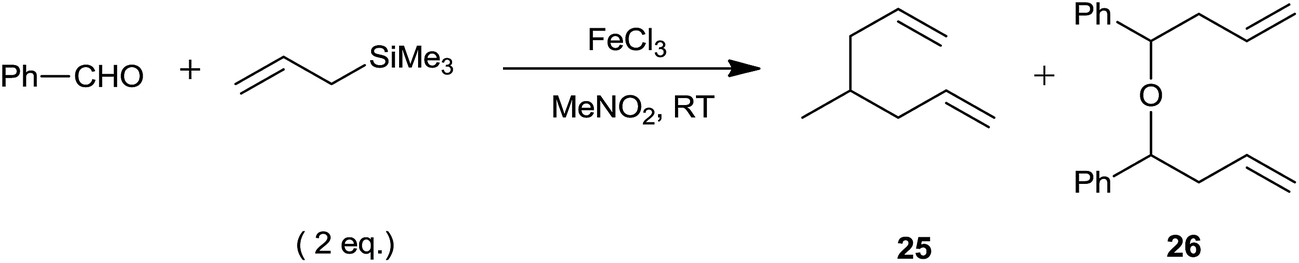 | ||
| Scheme 32 | ||
Nevertheless, the quantity (loading) of the allyltrimethylsilane and nature of solvent governed yield of the homoallyl alcohol. Thus, 92% of yield was obtained when 1.5 equiv. of allyltrimethylsilane was used in nitromethane. Interestingly, aliphatic aldehydes got allylated significantly with 5 mol% FeCl3 at room temperature even though their carbonyl groups have higher electron density than those of aromatic aldehydes. Furthermore, sterically hindered aldehydes were also allylated effectively at room temperature.
Later these authors had used the same catalyst system for one-pot synthesis of homoallyl benzyl ethers.25 In the context of two step procedure for the synthesis of homoallyl methyl ethers and their inability to undergo further synthetic manipulation owing to aliphatic ether linkage, preparation of homoallyl benzyl ethers gained more importance. A low loading of FeCl3 (2 to 5 mol%) was sufficient for the synthesis of homoallyl benzyl ethers from various aromatic as well as aliphatic aldehydes by using benzyloxytrimethylsilane (BnOTMS) at −20 °C and room temperature, respectively (Scheme 33). The reaction was chemoselective and the groups such as ketone and ester remained unaffected during the course of reaction.
 | ||
| Scheme 33 | ||
Three-component one-pot synthesis of homoallyl ethers and homoallyl acetates from aldehydes was facilitated by bismuth triflate (Scheme 34).53
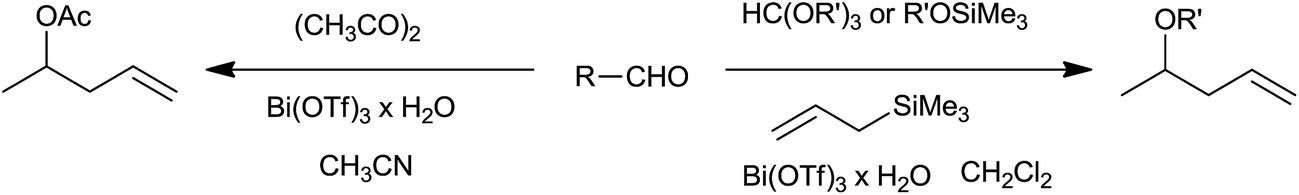 | ||
| Scheme 34 | ||
In the initial attempts trialkylorthoformate was used along with allyltrimethylsilane to obtain corresponding homoallyl ethers. Removal of alcohol formed during in situ generation of acetal by the reaction of aldehydes with trialkylorthoformate was necessary as it deactivated allyltrimethylsilane. Allyltrimethylsilane played an instrumental role as it not only facilitated allylation but also accelerated it by shifting the aldehydes–acetal equilibrium towards acetal formation, a key in situ generated intermediate. Smooth and rapid allylation took place at room temperature. This method worked well for conjugated aromatic aldehydes. However, cinnamaldehyde offered poor yield while aliphatic aldehydes underwent polymerization. The protocol was further improved and made more generalized by using alkoxytrimethylsilanes as wide range of alkoxytrimethylsilanes offered variety of homoallyl ethers viz. allyl, benzyl, ethyl and methyl. In addition, this method required very less quantity of Bi(OTf)3 (0.1–1.0 mol%) as compared to former method (2–2.5 mol%). It is worth to note that homoallyl acetates could also be synthesized in one-pot by using aldehydes, allyltrimethylsilane and acetic anhydride in the presence of Bi(OTf)3 in acetonitrile.
Looking at the promising features such as nonvolatility, nonflammability etc. of ionic liquid, Anzalone and Mohan54 had used 3–10 mol% of trimethylsilyl triflate in ionic liquid 1-butyl-3-methylimidazolium trifluoromethane sulfonate ([bmim][OTf]) for the synthesis of homoallyl ethers from aldehydes in one-pot. An easy product separation was the added advantage in this system as aldehydes were soluble in ionic liquid while the crude product of the reaction was just partially soluble. A wide range of homoallyl ethers were synthesized by using 2.0 equivalent of trialkylorthoformate and 1.2 equivalent of alkoxysilane. Nevertheless, allyl homoallyl ethers obtained by using this protocol could be used for the synthesis of cyclic enol ethers via ring-closing metathesis reactions. Aromatic aldehydes underwent allylation with 3–6 mol% of TMSOTf and offered homoallyl ethers in good yields while aliphatic alcohol afforded homoallyl ether in moderate yield with high catalyst loading (10 mol%). Furthermore, cyclohexanone too resulted in lower yield (44%) of homoallyl ether and did not undergo complete conversion even with high catalyst loading.
Commercially available inexpensive iron(III) p-toluene sulfonate Fe(OTs)3·6H2O produced homoallyl ethers of aldehydes by using alkoxysilanes in one-pot.29 Homoallyl ethers of aromatic as well as aliphatic aldehydes were formed in moderate to good yields.
Easy deprotection of benzyl group enabled further synthetic manipulation, impressed by this Phukan et al.55 had successfully synthesized homoallyl benzyl ethers from aldehydes by using benzyloxytrimethylsilane (1.2 equiv.) in the presence of iodine (10 mol%) at 0 °C in dichloromethane readily available, cheap iodine is the better candidate for being used as a catalyst when compared to other catalysts because it does not require stringent dry conditions. In case of aromatic aldehydes, substituent independent results were observed at 0 °C by using 10 mol% of iodine in dichloromethane while aliphatic aldehyde cinnamaldehyde offered the corresponding homoallyl benzyl ether in relatively low yields (Scheme 35).
 | ||
| Scheme 35 | ||
Huh et al.56 studied the stereoselectivity of nucleophilic addition of allylsilane to 3-azidoalkanals in the presence of chelating and non-chelating Lewis acid. Chelating Lewis acid TiCl4 produced 1,3-syn product while non-chelating BF3–OEt2 gave 1,3-anti product (Scheme 36).
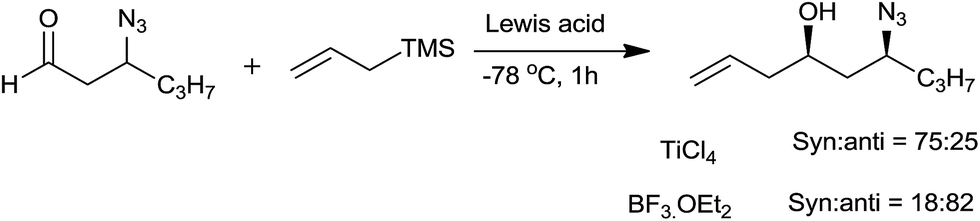 | ||
| Scheme 36 | ||
An in situ prepared complex of cerium triflate (Ce(OTf)3) (19 mol%) and chiral 2,6-bis(oxazoline-2-yl) pyridine (PyBox, 20 mol%) had been successfully used for the enantioselective allylation of aldehydes in the presence of a promoter TMSCl at room temperature.57 TMSCl promoted oxophilicity of the complex. Homoallylic alcohols were furnished in good to excellent yields with excellent enantioselectivity (upto 99%) in CH2Cl2/CHCl3 in the presence of activated molecular sieves (4 Å). Other Lewis acids such as indium(III) triflate (In(OTf)3), copper(II) triflate (Cu(OTf)2), bismuth(III) triflate Bi(OTf)3 and zinc(II) triflate (Zn(OTf)2) were also screened along with PyBox ligand for allylation of aldehydes, but they offered either low yield or lower enantioselectivity of homoallylic alcohols. Notably, the amount of PyBox ligand governed the yield as well as enantioselectivity of the product. In PyBox ligand, the substituents on the oxazoline moiety showed a crucial effect on catalytic activity. PyBox ligands bearing substituents having ability to exert larger electrostatic and steric stabilization enhanced the yield of homoallylic alcohols and their enantioselectivity. The protocol is compatible for aromatic as well as aliphatic aldehydes. Furthermore, in aromatic aldehydes nature and position of substituents influenced the yield and enantioselectivity of the products.
Zhou and co-workers58 described allylation of isatin in presence of mercury(II) perchlorate trihydrate (Hg(ClO4)2·3H2O) in dichloromethane at 25 °C. 1 mol% of Hg (ClO4)·3H2O effectively catalyzed the reaction and 98% yield of 3-allyl-3-hydroxyoxindole was obtained in 4 h. The yield was diminished to 93% and 71% when catalyst loading was reduced to 0.3 mol% and 0.1 mol%, while time was increased to 12 h and 144 h, respectively. 1.0 mol% of (S)-BINAP/Hg(ClO4)2·3H2O was employed for enantioselective allylation of isatins and excellent yield of the product was observed with 63% ee at 0 °C in THF. In addition, tandem synthesis of 3-allyl-3(thio)ether oxindoles were achieved in presence of 2 mol% of Hg(ClO4)2·3H2O in DCE at 70 °C in good yields.59 Further they had used a chiral complex of (S)-difluorophos and mercury(II) triflate (Hg(OTf)2) for allylation of isatins. 3-Allyl-3-hydroxyoxindoles were synthesized by using only 0.5–1.0 mol% of catalyst with upto 97% ee in dichloromethane at −10 °C (Scheme 37).60
 | ||
| Scheme 37 | ||
An unsubstituted isatin was allylated with 98% yield and 92% ee, while substituted isatins were allylated in the range of 75–98% yield with 78–88% ee. This implied that this protocol is substituent sensitive. Further, di(p-methoxyphenyl)phenylmethyl (DMTr) N-protected isatins were also allylated by using this catalysts system at −30 °C with good to high enantioselectivity.
Hanhan et al.61 investigated an enantioselective allylation of isatins utilizing chiral Sc(III)–Pybox catalyst along with TMSCl and sodium hexafluoroantimonate (NaSbF6) as activator and additive, respectively. The enantioselectivity was achieved upto 99% ee in presence of 0.05 mol% catalyst at room temperature and the scalability was attained at 0.1 mol% catalyst loading. The high catalyst activity is attributed to the enhanced Lewis acidity due to the interaction of TMSCl with the metal–ligand complex. Several isatins underwent allylation in yields ranging from 72 to 99% with enantioselectivity from 80 to 91% ee, though substituents on isatins affect the reaction rate. An electron-deficient allylsilanes, allyltrichlorosilanes and allyltrimethoxysilane depicted very poor/no reactivity under the similar condition. While on addition of DMF as Lewis base additive, allyltrichlorosilane offered allylation product with 74% yield but without enantioselectivity. This could be ascribed to the silicon's increased Lewis acidity and formation of the six-membered closed transition state. This catalyst system is highly effective for various other substituted allylic silanes with reference to high efficiency and selectivity. Further, crotylation of isatin was accomplished with 95% yield and 96% ee.
3.2 Brønsted acid catalyzed allylation
On the background of rapid decomposition of allylsilanes by strong protic acids, Davis and Jaspars62 had fruitfully used 0.5–3.0 mol% superacid trifluoromethanesulfoxonium tetrakis (trifluoromethanesulfonyl) boronate (TfOH2 + B(OTf)4) for allylation of aldehydes and ketones. Allyl alcohols were obtained regioselectively in high yields at room temperature in dichloromethane (Scheme 38).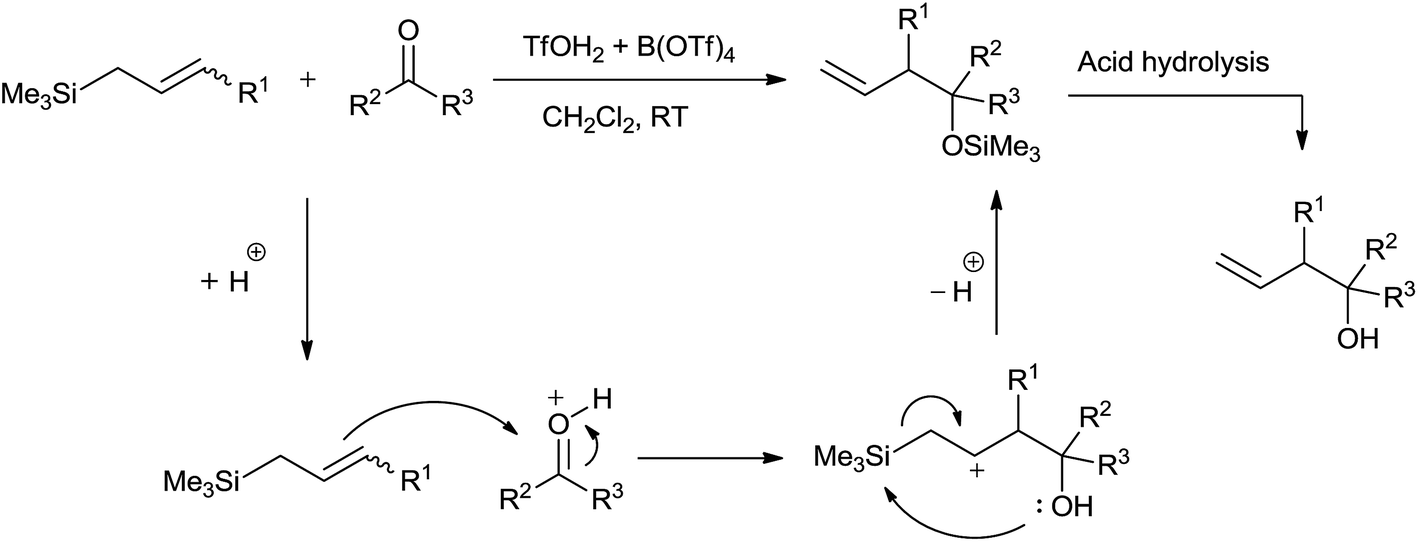 | ||
| Scheme 38 | ||
Control experiment that had been carried out shed light on mechanism of the reaction. Proton released in the reaction of superacid with allylsilane initiated the reaction. Allylsilane could not quench the proton as its counterion B(OTf)4− was highly non-nucleophilic and therefore unable to remove the Me3Si group. Homoallyl alcohols of various aldehydes were obtained in good yields. However, the same was not observed in case of crotonaldehyde and cinnamaldehyde owing to the elimination of water or trimethyl silanol (Me3SiOH), promoted by acidic condition. An enhancement in the yield of allylation of cyclohexanone was observed when temperature was changed from room temperature to −40 °C.
Gurmeet Kaur et al.63 had productively used Brønsted acid bis(fluorosulfuryl)imide [HN(SO2F)2] for allylation of aldehydes as well as ketones. Several aldehydes underwent allylation when 5 mol% HN(SO2F)2 was used at 0 °C and offered homoallyl alcohols in good to excellent yields within 5 min, except for cinnamaldehyde. While ketones were allylated at lower temperature (−40 °C), acetophenone and cyclopentanone had remained unallylated. This implied selective allylation of aldehydes in the presence of ketones. Authors have experimentally proved that HN(SO2F)2 acted only as a catalyst and neither caused protodesilylation of allylsilane nor formed TMSN(SO2F)2.
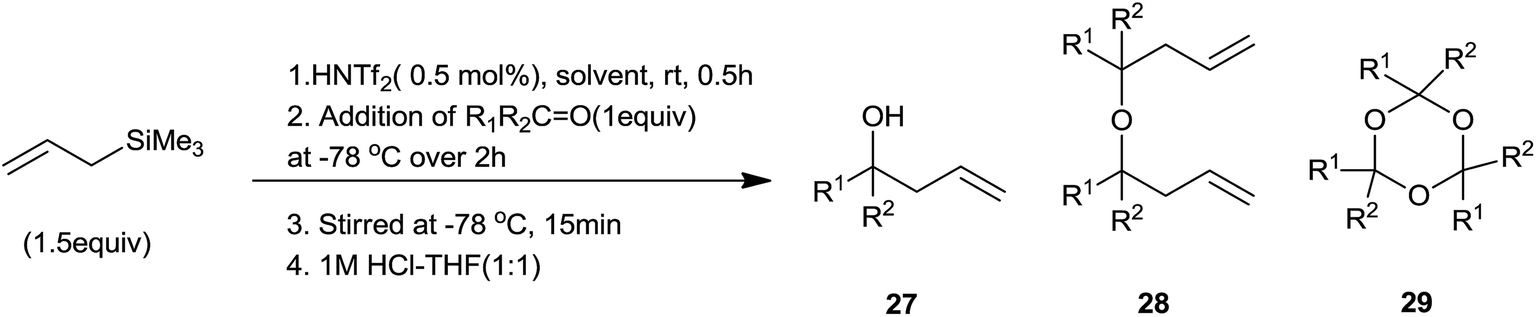
K. Ishihara et al.64 had effectively prepared Me3SiNTf2 in situ by using triflylimide (HNTf2) and eventually used it for allylation of aldehydes. A control experiment was carried out in which aldehyde was added in a drop-wise manner over a period of 2 h at −78 °C to a solution of allyltrimethylsilane and triflylimide (HNTf2) in a appropriate solvent to enhance the selectivity. Yields of allyl alcohol (27), dimeric ethers (28) and cyclic trimers of aldehyde (29) were found to be dependent on the nature of aldehyde and solvent (Scheme 39) (Table 7). Moreover, other Lewis acids such as MeAlNTf2, HN(SO2F)2, Me3SiB(OTf)4 could also be employed for this reaction but the best results were observed for Me3SiNTf2 catalyzed reaction.
 | ||
| Scheme 39 | ||
| Entry | R1R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
Solvent | Isolated yield (%) | ||
|---|---|---|---|---|---|
| 27 | 28 | 29 | |||
| a The reaction was carried out at room temperature for 15 min (step 3).b The reaction was carried out at −40 °C (step 2 and 3).c The reaction was carried out at −20 °C (step 3).d The reaction was carried out at −40 °C (step 2) and 0 °C (step 3). | |||||
| 1 | PhCHO | CH2Cl2 | 89 (27a) | 8 (28a) | 0 (29a) |
| 2 | PhCHO | Et2Oa | 98 (27a) | <2 (28a) | 0 (29a) |
| 3 | c-C6H11CHO | CH2Cl2 | 92 (27b) | 0 (28b) | <1 (29b) |
| 4 | c-C6H11CHO | PhCl | 82 (27b) | 0 (28b) | <7 (29b) |
| 5 | c-C6H11CHO | Et2O | 0 (27b) | 0 (28b) | <95 (29b) |
| 6 | PhCH2CH2CHO | CH2Cl2 | 90 (27c) | 0 (28c) | Trace (29c) |
| 7 | PhCH2CH2CHO | PhClb | 84 (27c) | 0 (28c) | Trace (29c) |
| 8 | n-C6H13COMe | CH2Cl2c | 89 (27d) | 0 (28d) | 0 (29d) |
| 9 | n-C6H13COMe | PhCld | 85 (27d) | 0 (28d) | 0 (29d) |
| 10 | c-C6H10O | CH2Cl2 | 91 (27e) | 0 (28e) | 0 (29e) |
| 11 | c-C6H10O | PhClb | 89 (27e) | 0 (28e) | 0 (29e) |
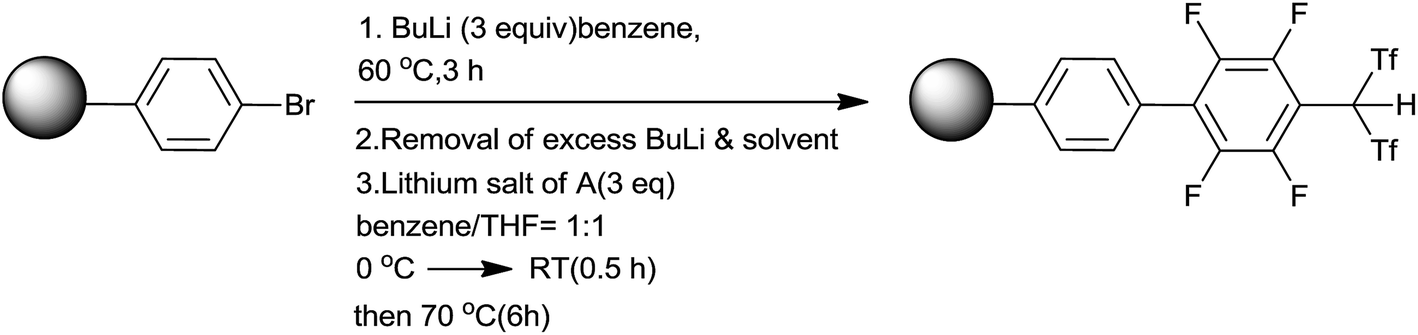
Yamamoto and co-workers65,66 had developed polystyrene bound tetrafluorophenylbis(triflyl) methane as a strong Brønsted acid catalyst and successively employed it for homoallylation of aldehydes by using allyltrimethylsilane. This catalyst possessed characteristic features like swelling in the presence of nonpolar organic solvents and superior catalytic activity than conventional resin sulfonic acids. Owing to the heterogeneous nature of the catalyst, it could be quantitatively recovered by simple filtration and reused. It was synthesized by using poly(4-bromostyrene), 2% divinylbenzene (DVB) and lithium salt of 2,3,4,5,6-pentafluorophenylbis(triflyl)methane (A) (Scheme 40).
 | ||
| Scheme 40 | ||
List and co-workers67 had reported 2,4-dinitrobenzene sulphonic acid (DNBA) catalyzed three-component (viz. aldehyde, allylsilanes and silyl ether or alcohol) Hosomi–Sakurai reaction at room temperature in acetonitrile. Versatility of the protocol could be evidenced from the fact that the reaction worked well with aromatic, polycyclic aromatic, heteroaromatic, cyclic as well as aliphatic aldehydes to offer different silyl ethers and their alcohols. The advantages of this catalyst over other catalysts include its easy availability and handling, inexpensive, nontoxic, low catalyst loading and moisture tolerant nature. Wide range of homoallyl benzyl ethers were synthesized by using benzyloxytrimethylsilane (BnOTMS) or benzyl alcohol (BnOH), as easy cleavage of benzyl group enhanced their further synthetic applications. Benzyl alcohol was less effective than benzyloxytrimethylsilane as the catalyst loading and quantity of allylsilanes required for the reaction increased from 3 mol% to 5 mol% and 2 equiv. to 3 equiv., respectively. Inspite of this, yield of the product had dropped down from 92% to 72% when benzaldehyde was used as a substrate.
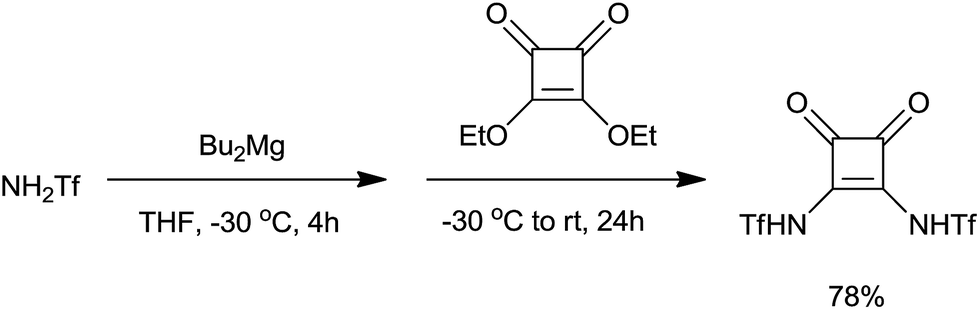
Brønsted acid N,N-bis(trifluoromethanesulfonyl)squaramide was prepared from squaric acid and was eventually used for allylation of 4-nitrobenzaldehyde (Scheme 41).68 Homoallyl alcohol was obtained in excellent yield (92%) by using 3 mol% of the catalyst at room temperature.
 | ||
| Scheme 41 | ||
Brønsted acid supported ionic liquid phase (SILP) catalyst had been expended for the allylation of aldehydes with allyltrimethylsilane which after completion of reaction led to production of homoallylic alcohols.69 The catalyst was prepared with pendant ferrocenyl group, which had been formed from 1-N-(ferrocenylmethyl) imidazole and Merrifield resin followed by anion metathesis with conc. H2SO4. Merrifield resin imparted chemical, thermal and mechanical stability to the catalyst due to its microporous structure. Ferrocene promoted unsymmetrical charge distribution on the imidazole ring which led to increase of micropolarity of Brønsted SILP catalyst. Consequently, enhancement in the rate of reaction was observed. The reaction worked well for various electron-rich and electron-deficient functional group substituted benzaldehydes and also for heteroaromatics such as furfuraldehyde and thiophene-2-aldehyde with 2 equiv. of allyltrimethylsilane. The heterogeneous catalyst was separated from reaction mixture by simple filtration and further reused for five more reaction cycles. Nearly consistent performance was experienced.
Silica supported perchloric acid had effectively promoted allylation of aldehydes.70 Optimum yields of homoallyl benzyl ethers were obtained by using acetonitrile and dichloromethane in 7:3 ratio in the presence of 2 mol% of catalyst at room temperature (Scheme 42). Nature of substituents as well as their position on aryl ring did not affect yield of the product whereas acid sensitive group like CN remained unaffected.
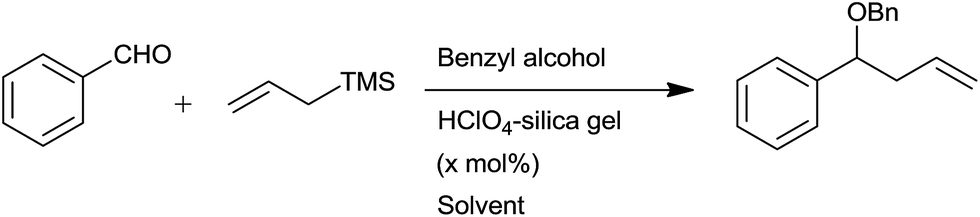 | ||
| Scheme 42 | ||
p-Anisaldehyde afforded 1:1 mixture of homoallyl ether and dialkyl derivative (Scheme 43).
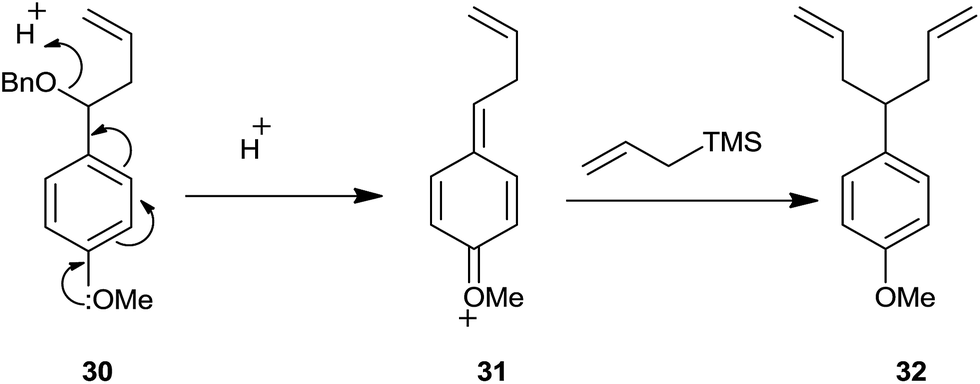 | ||
| Scheme 43 | ||
However, o-anisaldehyde exclusively offered homoallyl ether reasonably due to the intramolecular hydrogen bonding between the methoxy and benzyloxy group which may have suppressed the formation of compound 33 (Scheme 44).
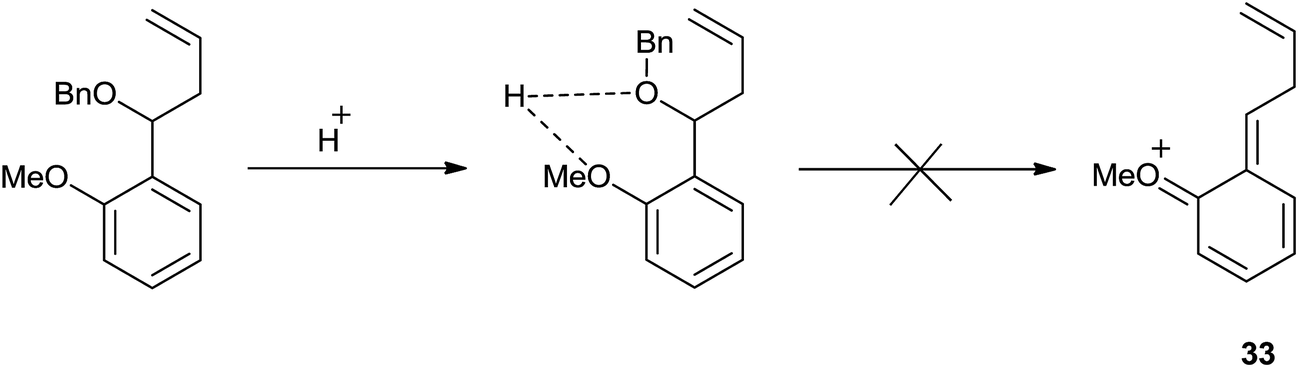 | ||
| Scheme 44 | ||
It is worth to note that polycyclic aromatic aldehydes, heteroaromatic aldehydes and α,β-unsaturated aldehydes effectively underwent allylation and offered the products in quantitative yields. However, aliphatic aldehydes could not undergo allylation presumably due to the instability of oxocarbenium ion intermediate.
Similarly, silica supported silicomolybdic acid (50 wt%) rendered homoallyl ether of various carbonyl compounds which include aromatic, polycyclic aromatic, heteroaromatic and aliphatic aldehydes as well as aliphatic and cyclic ketones.32 Low catalyst loading and its recyclability and reusability made this protocol more attractive from green chemistry perspective. The reaction worked well at room temperature as compared to high temperature, as it causes hydrolysis of acetal. Intriguingly, p-anisaldehyde offered diallyl compound exclusively, most likely because of the effective delocalization of lone pair of methoxy group. The position of carbonyl ion in heteroaromatic aldehyde governed yield of the product. While α,β-unsaturated aldehydes underwent selective allylation at 1,2-position.
Masahiro Sai and Hisashi Yamamoto71 had successfully synthesized chiral BINOL based thiophosphoramide bearing the tert-butyldiphenylsilyl (TBDPS) onto the ortho aromatic substituents and the perfluorooctyl substituent onto the sulfonyl group as a strong Brønsted acid and then used it as an effective catalyst for the asymmetric allylation of benzaldehydes. This various functional group tolerated protocol offered highly enantioselective product.
Very recently, Benjamin List and co-workers72 had wisely designed and synthesized BINOL-derived imidodiphosphorimidate (IDPi) derivatives and used for allylation of aromatic as well as aliphatic aldehydes. Notably, low catalyst loading (0.05–2.0 mol%), high yield of the homoallyl alcohols, low temperature and high enantioselectivity are the striking features of their protocol.
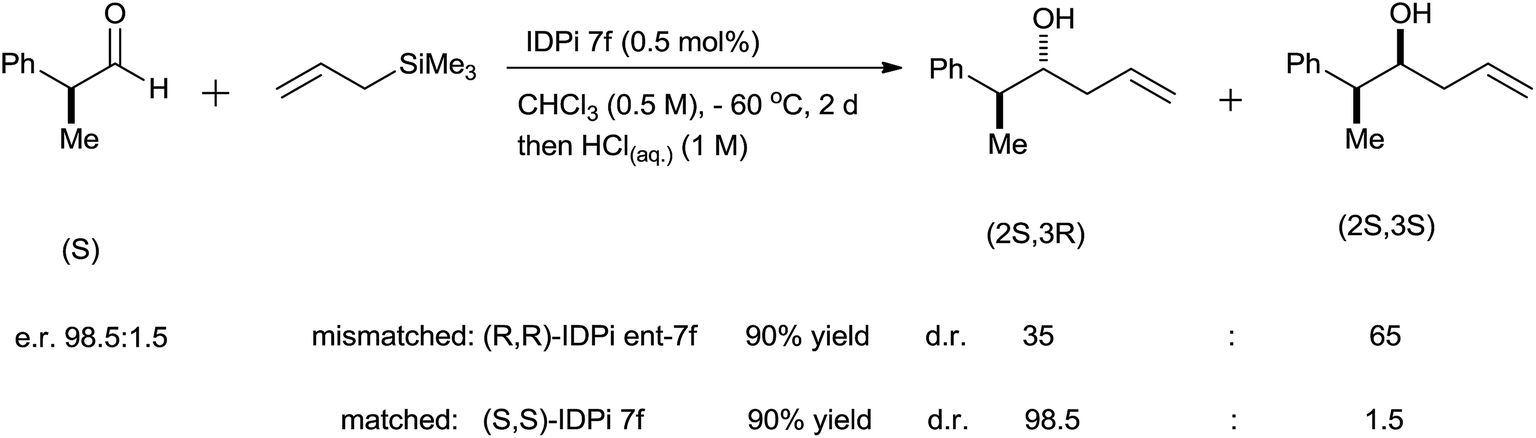
Imidodiphosphorimidate (IDPi) derivatives were simply synthesized from BINOL derivative (34), triflylphosphorimidoyl trichloride P(NTf)Cl3 and ammonia (Scheme 45). High yields of homoallyl alcohols of aromatic aldehydes with good enantioselectivity were observed at −78 °C by using 0.5 mol% of IDPi (35b) as a catalyst having 2-naphthyl substituent in the 3,3′-positions of the BINOL backbone. A lower yield was observed for ortho-substituted aromatic aldehyde in the presence of 35b whereas, enhancement in the yield was observed in the presence of 35g at elevated temperature. Besides this, higher catalyst loading (2.0 mol%) and longer reaction time were required for the allylation of α,β-unsaturated aldehyde. 6,8-Dimethylpyren-2-yl substituted IDPi (35f) catalyst offered homoallyl alcohols of aliphatic aldehydes in good to excellent yields with remarkably high enantioselectivities. 3-Phenylpropanal afforded corresponding homoallyl alcohol when 0.05 mol% of catalyst was used, whereas formation of trimethylsilyl ether was observed when catalyst loading was increased to 1 mol%. Notably, allylation of (S)-2-phenylpropanal in the presence of mismatched (R,R)-IDPi ent-35f and matched (S,S)-IDPi 35f catalysts had occurred and a diastereomeric ratio of 65:35 was observed in the presence of former catalyst while 98.5:1.5 was observed in the presence of matched (S,S)-IDPi 35f catalyst. Nevertheless, the configuration of the pre-existing stereogenic center was maintained in both cases (Scheme 46).
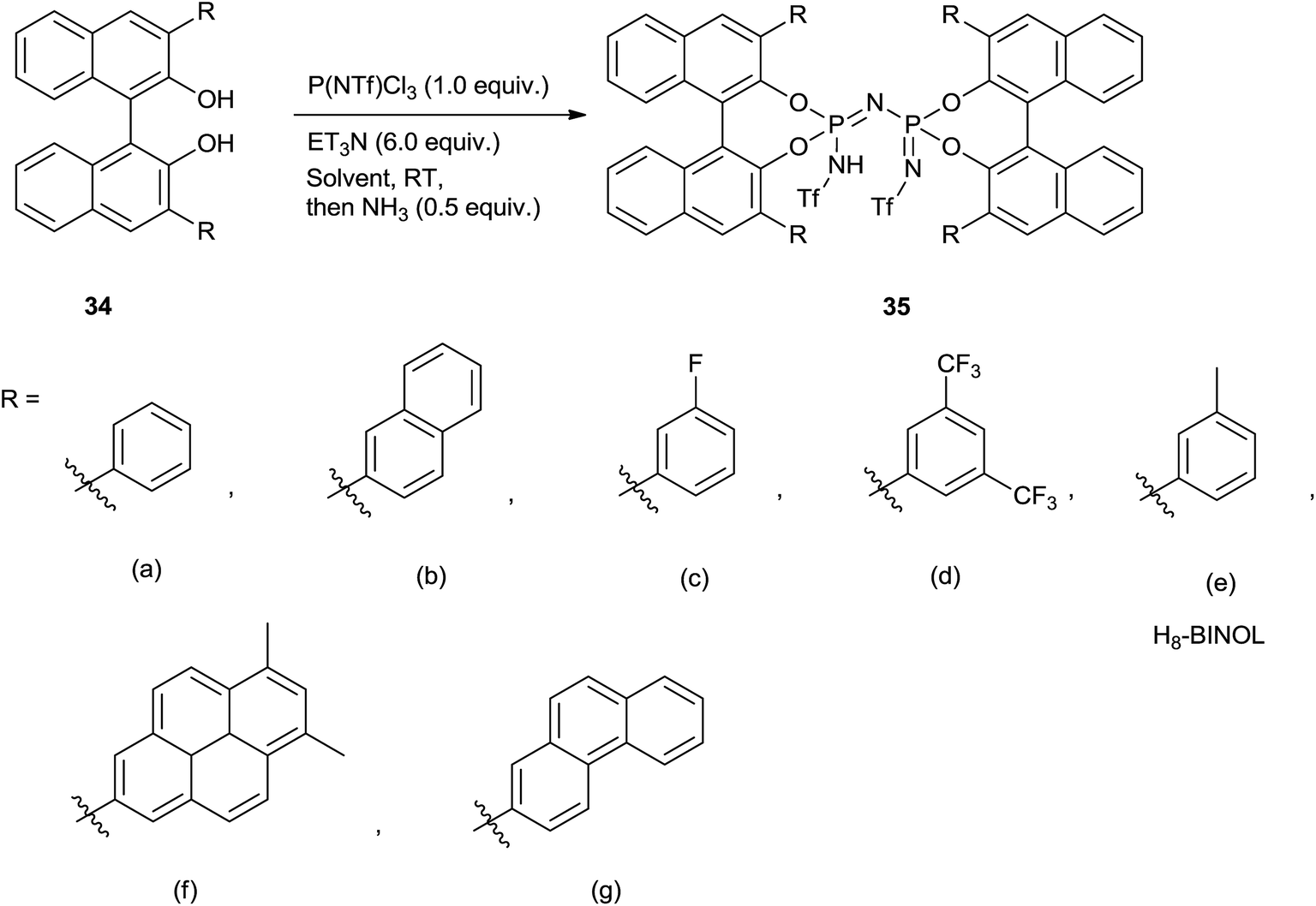 | ||
| Scheme 45 | ||
 | ||
| Scheme 46 | ||
3.3 Miscellaneous
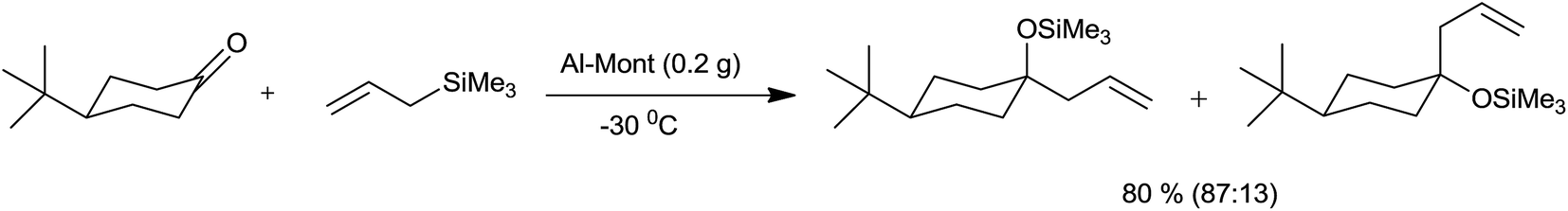
With respect to economic and environmental consideration, it is always wise to use more selective heterogeneous catalyst whose separation and recycling is very easy.Onaka et al.34 had successfully allylated aldehydes and ketones by using catalytic quantity of heterogeneous catalysts such as Al-Mont and Mont-K10 at −78 °C to 25 °C. However Me3SiOTf, Me3SiI and triphenylcarbenium perchlorate (Ph3CClO4) failed to catalyze the reaction. Notably, 4-tert-butylcyclohexanone was allylated preponderantly from equatorial direction in the presence of Al-Mont at −30 °C within 30 minutes (Scheme 47).
 | ||
| Scheme 47 | ||
Mesoporous aluminosilicate (Al-MCM-41) promoted the allylation of aromatic and aliphatic aldehydes in dichloromethane.73 It showed superior catalytic property than its counterpart silica–alumina owing to its hydrophobic mesopores having high surface area and promising interactions between organic substrate and solid surface of the catalyst. Electron-deficient aldehydes were allylated in short time with excellent yields as compared to electron-rich aldehydes. The catalyst was successfully recycled and reused upto three cycles without any considerable loss in catalytic activity.
Furthermore, Dintzner et al.74 had used activated montmorillonite K10 clay for effective addition of allyltrimethylsilane to electron-deficient aromatic aldehydes, few aliphatic aldehydes and ketones at a temperature range of 0–23 °C in dichloromethane. Homoallylic silyl ethers were obtained in good to excellent yields. Clay not only activated the carbonyl group but also protected trimethylsilyl ethers by avoiding its hydrolysis.
4. Conclusion
The mechanistically fascinating allylation of acetals, ketals, aldehydes and ketones by using allyltrimethylsilane has been extensively studied and their utility serve as an important bridge between fundamental aspects of stereochemistry and applications in target oriented synthesis. In this remarkable journey various Lewis acids, metal triflates, Brønsted acids and solid acids have been used in stoichiometric to catalytic quantity. Indeed, the ongoing quest for selective and more efficient catalytic protocol will lead to the development of processes that can operate under mild reaction conditions with greater enantioselectivity. We hope that this review will serve its intention to promote intensive research in the selective monoallylation of more complex acetals/ketals and aldehydes/ketones by using allyltrimethylsilane under mild reaction conditions without highly sensitive and toxic catalyst.Acknowledgements
Department of Science and Technology – Science and Engineering Research Board (DST-SERB), Govt. of India is gratefully acknowledged for financial support (sanction no. SB/FT/CS-147/2013). The authors thank Prof. A. K. Srivastava, Director National Centre for Nanosciences and Nanotechnology, University of Mumbai for his generous support.References
- For recent reviews, see: (a) Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 2207–2293 CrossRef CAS; (b) W. R. Roush, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, editor C. H. Heathcock, Pergamon Press, Oxford, 1991, vol. 2, pp. 1–53 Search PubMed; (c) I. Fleming, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, editor C. H. Heathcock, Pergamon Press, Oxford, 1991, vol. 2, pp. 563–593 Search PubMed; (d) C. E. Masse and J. S. Panek, Chem. Rev., 1995, 95, 1293–1316 CrossRef CAS; (e) Y. Nishigaichi, A. Takuwa, Y. Naruta and K. Maruyama, Tetrahedron, 1993, 49, 7395–7426 CrossRef CAS.
- (a) M. Yasuda, T. Fujibayashi, I. Shibata, A. Baba, H. Matsuda and N. Sonoda, Chem. Lett., 1995, 24, 167–168 CrossRef; (b) V. J. Jephcote and E. J. Thomas, Tetrahedron Lett., 1985, 26, 5327–5330 CrossRef CAS; (c) Y. Yamamoto, H. Yatagai, Y. Naruta and K. Maruyama, J. Am. Chem. Soc., 1980, 102, 7107–7109 CrossRef CAS; (d) Y. Yamamoto, H. Yatagai, Y. Ishihara, N. Maeda and K. Maruyama, Tetrahedron, 1984, 40, 2239–2246 CrossRef CAS; (e) V. J. Jephcote, A. J. Pratt and E. J. Thomas, J. Chem. Soc., Chem. Commun., 1984, 800–802 RSC; (f) A. Yanagisawa, M. Morodome, H. Nakashima and H. Yamamoto, Synlett, 1997, 1309–1311 CrossRef CAS; (g) A. Yanagisawa, H. Inoue, M. Morodome and H. Yamamoto, J. Am. Chem. Soc., 1993, 115, 10356–10357 CrossRef CAS.
- (a) C. Dong-Li and L. Chao-Jun, Tetrahedron Lett., 1996, 37, 295–298 CrossRef; (b) T.-H. Chan and C.-J. Li, J. Chem. Soc., Chem. Commun., 1992, 40, 747–748 RSC; (c) E. Kim, D. M. Gordon, W. Schmid and G. M. Whitesides, J. Org. Chem., 1993, 58, 5500–5507 CrossRef CAS; (d) C. J. Li, Chem. Rev., 1993, 93, 2023–2035 CrossRef CAS; (e) For an excellent book, see: C. J. Li and T. H. Chan, in Organic Reaction in Aqueous Media, John Wiley & Sons, Inc., 1997, p. 75 and references therein Search PubMed.
- For some leading references, see: (a) W. R. Roush, K. Ando, D. B. Powers, A. D. Palkowitz and R. L. Halterman, J. Am. Chem. Soc., 1990, 112, 6339–6348 CrossRef CAS; (b) W. R. Roush, A. D. Palkowitz and K. Ando, J. Am. Chem. Soc., 1990, 112, 6348–6359 CrossRef CAS; (c) E. J. Corey, C. M. Yu and S. S. Kim, J. Am. Chem. Soc., 1989, 111, 5495–5496 CrossRef CAS; (d) E. Corey and H.-C. Huang, Tetrahedron Lett., 1989, 30, 5235–5238 CrossRef CAS and references therein.
- (a) A. Hosomi, A. Shirahata and H. Sakurai, Tetrahedron Lett., 1978, 19, 3043–3046 CrossRef; (b) T. K. Sarkar and N. H. Andersen, Tetrahedron Lett., 1978, 19, 3513–3516 CrossRef; (c) R. Calas, J. Dunogues, G. Deleris and F. Pisciotti, J. Organomet. Chem., 1974, 69, C15–C17 CrossRef CAS; (d) G. Deleris, J. Dunogues and R. Calas, J. Organomet. Chem., 1975, 93, 43–50 CrossRef CAS; (e) E. W. Abel and R. J. Rowley, J. Organomet. Chem., 1975, 84, 199–229 CrossRef CAS; (f) A. Hosomi and H. Sakurai, Tetrahedron Lett., 1976, 17, 1295–1298 CrossRef.
- For reviews, see: (a) G. G. Furin, O. A. Vyazankina, B. A. Gostevsky and N. S. Vyazankin, Tetrahedron, 1988, 44, 2675–2749 CrossRef CAS; (b) H. Sakurai, Pure Appl. Chem., 1985, 57, 1759–1770 CrossRef CAS; (c) H. Sakurai, Pure Appl. Chem., 1982, 54, 1–22 CrossRef CAS; (d) A. Hosomi, Acc. Chem. Res., 1988, 21, 200–206 CrossRef CAS.
- A. Hosomi, M. Endo and H. Sakurai, Chem. Lett., 1976, 5, 941–942 CrossRef.
- A. Hosomi, M. Endo and H. Sakurai, Chem. Lett., 1978, 7, 499–502 CrossRef.
- I. Ojima and M. Kumagai, Chem. Lett., 1978, 7, 575–578 CrossRef.
- T. Tsunoda, M. Suzuki and R. Noyori, Tetrahedron Lett., 1980, 21, 71–74 CrossRef CAS.
- H. M. Zerth, N. M. Leonard and R. S. Mohan, Org. Lett., 2003, 5, 55–57 CrossRef CAS PubMed.
- H. Sakurai, K. Sasaki and A. Hosomi, Tetrahedron Lett., 1981, 22, 745–748 CrossRef CAS.
- T. Mukaiyama, H. Nagaoka, M. Murakami and M. Ohshima, Chem. Lett., 1985, 14, 977–980 CrossRef.
- S. E. Denmark and T. M. Willson, J. Am. Chem. Soc., 1989, 111, 3475–3476 CrossRef CAS.
- T. K. Hollis, N. P. Robinson, J. Whelan and B. Bosnich, Tetrahedron Lett., 1993, 34, 4309–4312 CrossRef CAS.
- A. Trehan, A. Vij, M. Walia, G. Kaur, R. D. Vermar and S. Trehan, Tetrahedron Lett., 1993, 34, 7335–7338 CrossRef CAS.
- M. El Gihani and H. Heaney, Synlett, 1993, 433–434 CrossRef CAS.
- H. Mayr, G. Gorath and B. Bauer, Angew. Chem., Int. Ed., 1994, 33, 788–789 CrossRef.
- M. Kamata, Y. Yokoyama, N. Karasawa, M. Kato and E. Hasegawa, Tetrahedron Lett., 1996, 37, 3483–3486 CrossRef CAS.
- M. Kamata, S. Nagai, M. Kato and E. Hasegawa, Tetrahedron Lett., 1996, 37, 7779–7782 CrossRef CAS.
- A. Ishii, O. Kotera, T. Saeki and K. Mikami, Synlett, 1997, 1145–1146 CrossRef CAS.
- N. Komatsu, M. Uda, H. Suzuki, T. Takahashi, T. Domae and M. Wada, Tetrahedron Lett., 1997, 38, 7215–7218 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy and P. Srihari, Synlett, 2001, 673–675 CAS.
- L. C. Wieland, H. M. Zerth and R. S. Mohan, Tetrahedron Lett., 2002, 43, 4597–4600 CrossRef CAS.
- T. Watahiki, Y. Akabane, A. Seiji Mori and T. Oriyama, Org. Lett., 2003, 5, 3045–3048 CrossRef CAS PubMed.
- M. E. Jung and A. Maderna, Tetrahedron Lett., 2004, 45, 5301–5304 CrossRef CAS.
- M. E. Jung and A. Maderna, J. Org. Chem., 2004, 69, 7755–7757 CrossRef CAS PubMed.
- S. Arai, Y. Sudo and A. Nishida, Tetrahedron, 2005, 61, 4639–4642 CrossRef CAS.
- M. J. Spafford, E. D. Anderson, J. R. Lacey, A. C. Palma and R. S. Mohan, Tetrahedron Lett., 2007, 48, 8665–8667 CrossRef CAS.
- D. Kampen and B. List, Synlett, 2006, 2589–2592 CAS.
- M. Barbero, S. Bazzi, S. Cadamuro, S. Dughera and C. Piccinini, Synthesis, 2010, 315–319 CAS.
- K. Murugan, S. Srimurugan and C. Chen, Tetrahedron, 2011, 67, 5621–5629 CrossRef CAS.
- S. B. Kamble, S. H. Shinde and C. V. Rode, Catal. Sci. Technol., 2015, 5, 4039–4047 CAS.
- M. Kawai, M. Onaka and Y. Izumi, Chem. Lett., 1986, 15, 381–384 CrossRef.
- S. Ito, A. Hayashi, H. Komai, Y. Kubota and M. Asami, Tetrahedron Lett., 2010, 51, 4243–4245 CrossRef CAS.
- A. Hosomi and H. Sakurai, Tetrahedron Lett., 1976, 17, 1295–1298 CrossRef.
- G. Deleris, J. Dunoguès and R. Calas, Tetrahedron Lett., 1976, 17, 2449–2450 CrossRef.
- A. Hosomi, A. Shirahata and H. Sakurai, Tetrahedron Lett., 1978, 19, 3043–3046 CrossRef.
- H. Sakurai, K. Sasaki, J. Hayashi and A. Hosomi, J. Org. Chem., 1984, 49, 2808–2809 CrossRef CAS.
- T. Mukaiyama, M. Ohshima and N. Miyoshi, Chem. Lett., 1987, 16, 1121–1124 CrossRef.
- A. Mekhalfia and I. E. Markó, Tetrahedron Lett., 1991, 32, 4779–4782 CrossRef CAS.
- A. P. Davis and M. Jaspars, Angew. Chem., Int. Ed., 1992, 31, 470–471 CrossRef.
- H. Maeta, T. Nagasawa, Y. Handa, T. Takei, Y. Osamura and K. Suzuki, Tetrahedron Lett., 1995, 36, 899–902 CrossRef.
- V. K. Aggarwal and G. P. Vennall, Tetrahedron Lett., 1996, 37, 3745–3746 CrossRef CAS.
- V. K. Aggarwal and G. P. Vennall, Synthesis, 1998, 1822–1826 CrossRef CAS.
- Y. Yang, M. Wang and D. Wang, Chem. Commun., 1997, 1651–1652 RSC.
- K. Nakamura, H. Nakamura and Y. Yamamoto, J. Org. Chem., 1999, 64, 2614–2615 CrossRef CAS PubMed.
- M. W. Wang, Y. J. Chen and D. Wang, Synlett, 2000, 385–387 CAS.
- Y. Onishi, T. Ito, M. Yasuda and A. Baba, Eur. J. Org. Chem., 2002, 2002, 1578–1581 CrossRef.
- Y. Onishi, T. Ito, M. Yasuda and A. Baba, Tetrahedron, 2002, 58, 8227–8235 CrossRef CAS.
- J. S. Yadav, P. K. Chand and S. Anjaneyulu, Tetrahedron Lett., 2002, 43, 3783–3784 CrossRef CAS.
- T. Watahiki and T. Oriyama, Tetrahedron Lett., 2002, 43, 8959–8962 CrossRef CAS.
- P. W. Anzalone, A. R. Baru, E. M. Danielson, P. D. Hayes, M. P. Nguyen, A. F. Panico, R. C. Smith and R. S. Mohan, J. Org. Chem., 2005, 70, 2091–2096 CrossRef CAS PubMed.
- P. W. Anzalone and R. S. Mohan, Synthesis, 2005, 2661–2663 CAS.
- D. Kataki and P. Phukan, Tetrahedron Lett., 2009, 50, 1958–1960 CrossRef CAS.
- C. W. Huh, C. Schroeder, G. Singh and J. Aubé, J. Org. Chem., 2011, 76, 3160–3165 CrossRef CAS PubMed.
- S. Zhao, X. Zhang, Y. Zhang, H. Yang, Y. Huang, K. Zhang and T. Du, New J. Chem., 2015, 39, 7734–7737 RSC.
- Z.-Y. Cao, Y. Zhang, C.-B. Ji and J. Zhou, Org. Lett., 2011, 13, 6398–6401 CrossRef CAS PubMed.
- F. Zhu, F. Zhou, Z.-Y. Cao, C. Wang, Y.-X. Zhang, C. H. Wang and J. Zhou, Synthesis, 2012, 44, 3129–3144 CrossRef CAS.
- Z. Y. Cao, J. S. Jiang and J. Zhou, Org. Biomol. Chem., 2016, 14, 5500–5504 CAS.
- N. V. Hanhan, Y. C. Tang, N. T. Tran and A. K. Franz, Org. Lett., 2012, 14, 2218–2221 CrossRef CAS PubMed.
- A. P. Davis and M. Jaspars, J. Chem. Soc., Chem. Commun., 1990, 54, 1176–1178 RSC.
- G. Kaur, K. Manju and S. Trehan, Chem. Commun., 1996, 581–582 RSC.
- K. Ishihara, Y. Hiraiwa and H. Yamamoto, Synlett, 2001, 1851–1854 CrossRef CAS.
- K. Ishihara, A. Hasegawa and H. Yamamoto, Angew. Chem., Int. Ed., 2001, 40, 4077–4079 CrossRef CAS PubMed.
- K. Ishihara, A. Hasegawa and H. Yamamoto, Synlett, 2002, 1299–1301 CAS.
- D. Kampen, A. Ladépêche, G. Claßen and B. List, Adv. Synth. Catal., 2008, 350, 962–966 CrossRef CAS.
- C. H. Cheon and H. Yamamoto, Tetrahedron, 2010, 66, 4257–4264 CrossRef CAS.
- G. Rashinkar, S. Kamble, A. Kumbhar and R. Salunkhe, Catal. Commun., 2011, 12, 1442–1447 CrossRef CAS.
- K. Murugan and C. Chen, Tetrahedron Lett., 2011, 52, 5827–5830 CrossRef CAS.
- M. Sai and H. Yamamoto, J. Am. Chem. Soc., 2015, 137, 7091–7094 CrossRef CAS PubMed.
- P. S. J. Kaib, L. Schreyer, S. Lee, R. Properzi and B. List, Angew. Chem., Int. Ed., 2016, 55, 13200–13203 CrossRef CAS PubMed.
- S. Ito, H. Yamaguchi, Y. Kubota and M. Asami, Tetrahedron Lett., 2009, 50, 2967–2969 CrossRef CAS.
- M. R. Dintzner, Y. A. Mondjinou and B. Unger, Tetrahedron Lett., 2009, 50, 6639–6641 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |