
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Binuclear ruthenium complexes inhibit the fibril formation of human islet amyloid polypeptide†
Gehui Gong,
Wenji Wang and
Weihong Du *
*
Department of Chemistry, Renmin University of China, Beijing, 100872, China. E-mail: whdu@ruc.edu.cn; Fax: +86-10-62516444; Tel: +86-10-62512822
First published on 27th March 2017
Abstract
The deposition of human islet amyloid polypeptide (hIAPP) is closely correlated with type II diabetes mellitus (T2DM). hIAPP misfolding, as a significant causative factor of T2DM, can lead to the failure of islet transplant. Therefore, preventing the aggregation of hIAPP is one of the most vital factors to treat T2DM. Mononuclear Ru complexes have recently been proved to inhibit the aggregation of hIAPP. In the present work, the influences of three water-soluble binuclear Ru complexes, namely, Na2[{trans-RuCl4(DMSO)}2(μ-pyz)] (1), Na2[{trans-RuCl4(DMSO)}2(μ-pym)] (2), and Na2[{trans-RuCl4(DMSO)}2(μ-bipy)] (3), on hIAPP fibril formation with mononuclear compounds were determined and compared. Results indicated that the three binuclear Ru complexes effectively inhibited the fibrillation of hIAPP to form nanoscale particles and decreased the β-sheet component of peptides, thus reducing the cytotoxicity induced by hIAPP. Binuclear Ru complexes showed better inhibition ability than their corresponding mononuclear Ru complexes, which might be attributed to the second metal center. Our study provided new insights into the design of multinuclear Ru complexes as potential metallodrugs against T2DM.
Introduction
Abnormal folding and aggregation of proteins are linked to many protein conformational diseases, such as Alzheimer's, Parkinson's, and Huntington's diseases as well as type II diabetes mellitus (T2DM).1 T2DM is the most common form of diabetes affecting 171 million people worldwide in 2000. The number of patients with T2DM has been rapidly increasing, especially in Southeast Asia and the Western Pacific region. The morbidity and prevalence of T2DM among children have been remarkably increasing.2–4 Islet amyloid polypeptide (IAPP), also denoted as amylin, has been proved to be one of the predominant targets for the pathology of T2DM.5,6 Human islet amyloid polypeptide (hIAPP) is a 37-residue peptide hormone (KCNTATCATQR LANFLVHSSNNFGAILSSTNVGSNTY-NH2), which is secreted or synthesized by pancreatic β-cells. Normal hIAPP can help regulate glucose metabolism, suppress the release of glucagon, and control satiety.7,8 Much evidence now shows the relationship between misfolding of hIAPP and β-cell dysfunction.9 In T2DM, hIAPP undergoes misfolding from its normal α-helix structure to abnormal β-sheet, and its overexpression induces the formation of amyloid plaques in the pancreas.10.11 The early soluble oligomers produce toxicity and further accelerate apoptosis, which has been inferred to be part of the pathogenic agent related to T2DM.12,13 Therefore, a key factor in treating T2DM should be to inhibit the conformational transition of hIAPP and suppress the formation of amyloid fibrils.Numerous studies have investigated therapeutics of T2DM by preventing the aggregation of hIAPP and reducing its cytotoxicity. Many inhibitors of hIAPP aggregation include some short peptides, naturally occurring polyphenols, small organic molecules, and coordination compounds.14–17 Various metal ions are used as inhibitors of hIAPP. Copper ion has been proven to prevent the formation of β-sheet conformers, and zinc ion shows concentration-dependent promotion and inhibition of aggregation through two-site mechanism.18–20 Metal complexes, such as platinum, gold, vanadium, and ruthenium (Ru) compounds, have been applied to inhibit the aggregation of amyloid proteins.21–24 Ru complexes are potential anticancer drugs with diverse biological activities.25–29 NAMI-A, a known Ru complex [ImH][RuCl4(DMSO)(Im)], has selective antimetastatic properties and lacks adverse side effects. Thus, this compound is now being tested in the phase I clinical trials against cancer.30 In addition, several mononuclear Ru compounds, such as Ru(bipy)Cl4, and Ru(bipy)2Cl2, inhibit the aggregation and cytotoxicity of hIAPP.22 However, the application of these Ru complexes is limited by their poor water solubility. Developing effective Ru compounds, in particular, binuclear metal compounds, with better solubility is necessary. According to structural analogies, binuclear metal compounds should remain and enhance the activity of inhibiting the aggregation and toxicity of hIAPP, compared with mononuclear species. Recent studies reported that multinuclear platinum species have remarkable activity against tumor lines and different interactions with DNA, contrary to cisplatin.31–35 However, the interaction between binuclear Ru complexes and hIAPP has not yet been reported.
In the current study, a series of binuclear Ru complexes containing aromatic ligand were prepared, and their inhibitory effects on hIAPP amyloidosis were compared with the corresponding mononuclear Ru compounds (Scheme 1). These binuclear Ru complexes were Na2[{trans-RuCl4(DMSO)}2 (μ-pyz)] (1), Na2[{trans-RuCl4(DMSO)}2(μ-pym)] (2), and Na2[{trans-RuCl4(DMSO)}2 (μ-bipy)] (3). These binuclear Ru complexes behave similar to NAMI-A on the cell proliferation and cell cycle regulation in human and murine tumor cell lines,36,37 but the effects of binuclear Ru complexes on hIAPP is unclear. To compare the difference between binuclear Ru complexes and mononuclear compounds, we also tested the property of similar mononuclear Ru complexes which have the same ligands, namely, Na[{trans-RuCl4(DMSO)}(pyz)] (4), Na[{trans-RuCl4(DMSO)}(pym)] (5), and Na[{trans-RuCl4(DMSO)}(bpy)] (6). The results demonstrated that binuclear Ru complexes can effectively inhibit the aggregation of hIAPP and change the secondary structure of the peptide. By contrast, mononuclear Ru complexes were less valuable on the inhibition of hIAPP aggregation than binuclear Ru complexes. This study provides practical data on developing multinuclear Ru complexes as potential metallodrugs against T2DM and expanding their application on biomedical field.
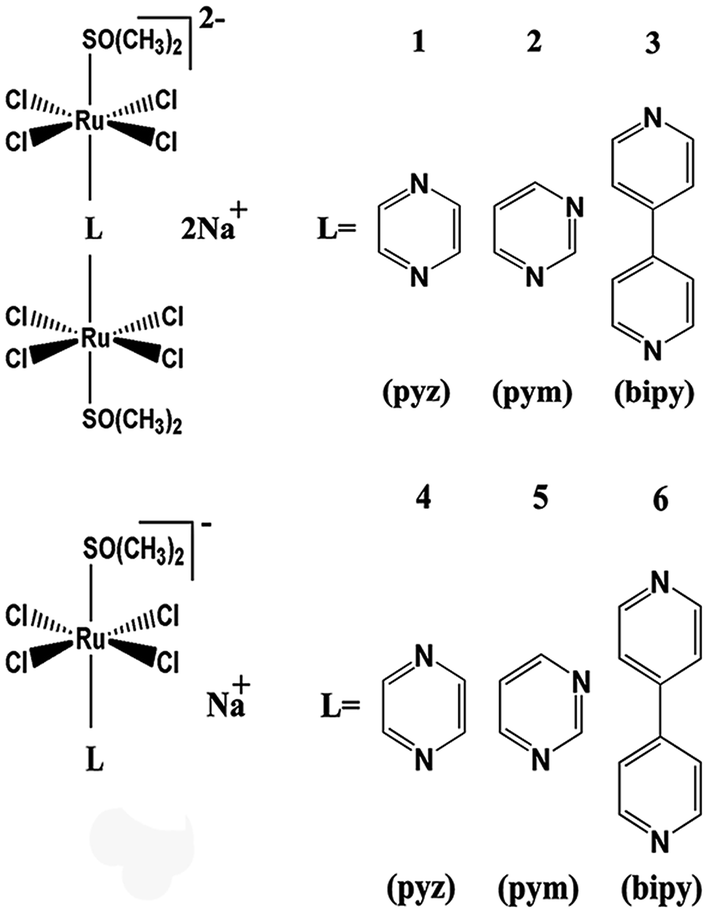 | ||
| Scheme 1 The molecular structures of Na2[{trans-RuCl4(dmso-S)}2(μ-pyz)] (1), Na2[{trans-RuCl4(dmso-S)}2(μ-pym)] (2), Na2[{trans-RuCl4(dmso-S)}2(μ-bipy)] (3), Na[{trans-RuCl4(dmso-S)}(pyz)] (4), Na[{trans-RuCl4(dmso-S)}(pym)] (5), and Na[{trans-RuCl4(dmso-S)}(bpy)] (6). | ||
Experimental methods
Materials
The full length of hIAPP (1–37) was obtained from SynPeptide Co. Ltd. (Shanghai, China). hIAPP was further identified by high-performance liquid chromatography and mass spectrometry (MS) with more than 95% purity. Lyophilized hIAPP was dissolved in hexafluoroisopropanol for 1 h to remove any preformed aggregates. Ru complexes were synthesized as previously described. 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) used to detect the toxicity of amyloid peptide was purchased from Sigma.38 All other reagents were of analytical grade.Thioflavin T (ThT) assay
The extent of hIAPP aggregation was measured by ThT assay using F-4600 spectrofluorometer (Hitachi Ltd., Japan) at room temperature. The signal of ThT fluorescence was excited at 432 nm and recorded at an emission wavelength of 485 nm for 10 s. Then, hIAPP (5 μM) was incubated with 20 μM ThT in the absence and presence of six Ru complexes in 10 mM phosphate buffer (PB) for 72 h at 310 K. In the time scale experiments of aggregation, 60 μM ThT was added into the solution of 15 μM hIAPP in 10 mM PB for 12 h. Ru complexes (5 μM) were added to 20 μM ThT in the PB to determine the effect of Ru complexes on ThT. To examine if Ru complexes could inhibit the interaction of hIAPP with ThT, 5 μM hIAPP was incubated with 20 μM ThT in 10 mM PB for 48 h at 310 K. Then, the sample was mixed with different concentration of Ru complexes, and incubated for 0 h, 12 h, 24 h, 48 h, and 72 h at 310 K. The reported data were obtained from the mean value of three repeated spectra.Atomic force microscopy (AFM)
The morphology of hIAPP was observed by acquiring the AFM images in tapping mode with a silicon tip under ambient conditions and a scanning rate of 1 Hz and a scanning line of 512 using the Veeco D3100 instrument. Samples were prepared by mixing 100 μM hIAPP with or without multiple concentrations of Ru complexes and then incubated for 72 h at 310 K. Before determination, the sample was diluted to the final concentration of 5 μM for hIAPP. For the time-dependent disaggregation experiments, 100 μM hIAPP was incubated for 48 h at 310 K alone in order to form hIAPP fibrils, then it was diluted to the concentration of 5 μM. The sample of 5 μM aged hIAPP was mixed with 25 μM Ru complexes and then incubated for 0 h, 12 h, or 24 h at 310 K. The final graphs used were from three repeated experiments.Transmission electron microscopy (TEM)
hIAPP (100 μM) was incubated with or without different concentrations of Ru complexes for 72 h at 310 K. After incubation, hIAPP was diluted to 5 μM, and the molar ratios of the Ru complex to hIAPP were 1, 5, and 10. Aliquots of each sample were spotted onto carbon-coated 600-mesh copper grids, negatively stained by 2% phosphotungstic acid. The TEM images were collected on a Hitachi-800 transmission electron microscope at 220 kV. The final graphs used were from three parallel experiments.Dynamic light scattering (DLS)
The particle size of the aggregated hIAPP was analyzed by the Zetasizer Nano instrument (Malvern Instruments, Worcestershire, UK). hIAPP (100 μM) was incubated in the absence or presence of Ru complexes for 72 h at 310 K, then diluted to 5 μM to remove oversized aggregates. The molar ratios of Ru complex to hIAPP were 5 and 10. The sample was centrifuged for 15 min at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm to avoid large precipitates. The supernatant sample was transferred to a fluorescence cuvette for further measurements.
000 rpm to avoid large precipitates. The supernatant sample was transferred to a fluorescence cuvette for further measurements.
UV-vis spectrophotometry
The absorption spectra were recorded on a Cary 50 UV-vis spectrophotometer (Agilent, USA) at room temperature. Ru complexes were dissolved in 10 mM PB, and the final concentration was 20 μM. Different concentrations of hIAPP were added to record the UV absorbance. The concentrations of hIAPP were selected at 0, 20, 40, and 80 μM respectively. The experiment was repeated thrice.Circular dichroism (CD) spectroscopy
All CD spectra were recorded from a Jasco J-810 spectropolarimeter (Japan Spectroscopy Co., Japan) at room temperature. hIAPP was dissolved in 10 mM PB and then incubated with different concentrations of Ru complexes for 72 h at 310 K. The final concentration of hIAPP was 50 μM for all CD measurements. The molar ratios of the Ru complex to hIAPP were 0.5, 1, and 3. The spectra were recorded within the range of 195 nm and 250 nm in a 1 mm quartz cell using a 2 nm bandwidth. The baseline was corrected for each spectrum referring to the same buffer. The final spectrum was displayed after subtracting the signal from the binuclear Ru complexes. The data were from three parallel experiments and processed by a smoothing algorithm.Cyclic voltammetry (CV)
Electrochemical measurements were performed on an Epsilon electrochemical workstation (USA). The glassy carbon electrode used as working electrode was polished by 0.3 mm alumina slurry, followed by ultrasonic bath in ultrapure water prior to the test. A platinum wire was used as the counter electrode. An Ag/AgCl electrode was conducted as the reference to measure the potential. After each scan, the working electrode was replaced to minimize possible electrodeposition of binuclear Ru complex. The supporting electrolyte was 10 mM PB. The final concentrations of the binuclear Ru complexes and hIAPP were 20 and 200 μM, respectively. The scan rate was set to 100 mV s−1. All data represented an average of three accumulated scans.Cell culture and MTT assay
INS-1 rat insulinoma cell line was purchased from Bogoo Biotech Co., Ltd. (Shanghai, China). Briefly, cells were cultured for 24 h and further incubated with 15 μM hIAPP for 72 h at 310 K in the absence and presence of Ru complex. The final concentrations of the Ru complexes were 1.5, 15, and 75 μM respectively. Then, 10 μL of MTT was added into the cells for detection. The absorbance of cells was measured at 570 nm by a UV spectrophotometer. Data were calculated referring to untreated control value.Results
Synthesis of Ru complexes
Three binuclear Ru complexes, namely, Na2[{trans-RuCl4(DMSO)}2(μ-pyz)] (1), Na2[{trans-RuCl4(DMSO)}2(μ-pym)] (2), and Na2[{trans-RuCl4(DMSO)}2(μ-bipy)] (3), were synthesized and identified according to previously reported methods.39 Complex 1 was synthesized as follows. [(DMSO)2H+][trans-Ru(DMSO)2Cl4]− (1.12 g) was dissolved in a mixture of 50 mL of ethanol and of 0.7 mL of water, and then added with 175 mg of NaCl. The product Na[trans-Ru(DMSO)2Cl4] was deposited immediately and collected by filtration. The filtrates were washed with cold acetone and diethyl ether and vacuum dried. Na[trans-Ru(DMSO)2Cl4] (0.2 g) was dissolved in 2 mL of DMSO. Pyrazine (0.019 g) was dissolved in 5 mL of acetone and then was added with magnetic stirring. The orange solution was filtered. After several hours at room temperature, red-orange microcrystals of the product were collected by filtration, washed with cold acetone and diethyl ether, and vacuum dried. The final yield was 45.5%. Complex 2 was synthesized by adding pyrimidine instead of pyrazine using the method similar to the preparation of complex 1. The final yield was 44.2%. Complex 3 was synthesized by adding 4,4-bipyridine instead of pyrazine with the method similar to the preparation of complex 1. The final yield was 65.0%. Fig. S1† shows the UV spectra of the three complexes which are consistent with previous reported. Fig. S2† shows the IR spectra which are also consistent with previous reports.Three mononuclear Ru compounds, namely, Na[{trans-RuCl4(DMSO)}(pyz)] (4), Na[{trans-RuCl4(DMSO)}(pym)] (5), and Na[{trans-RuCl4(DMSO)}(bpy)] (6) were synthesized and identified according to previously reported methods.39 For complex 4, the method of synthesis was similar to that described for the binuclear Ru complexes, but the ratio of pyrazine to Ru used was 5:1. The final yield was 43.5%. Complex 5 was synthesized by adding pyrimidine instead of pyrazine with method similar to that described for complex 1. The final yield was 43.1%. Complex 6 was synthesized by adding 4,4-bipyridine instead of pyrazine also using the method similar to that described for complex 1. The final yield was 46.7%. Fig. S3† shows the IR spectra which are consistent with previous reports.
hIAPP aggregation affected by Ru complexes
ThT assay was used to indicate the effects of Ru complexes on hIAPP aggregation, according to the change in ThT fluorescence intensity (Fig. 1A–C). In the absence of Ru complexes, the ThT fluorescence intensity was high, reflecting the fibrillation of hIAPP. Fluorescence intensity drastically decreased after co-incubation with binuclear Ru complexes. Moreover, the influence of Ru complexes on hIAPP aggregation was concentration-dependent (Fig. S4†). Moreover, the fibrillation of hIAPP had a lag phase of approximately 2 h during which β-sheet oligomer species formed, following a growth phase during which fibrils elongated quickly, then a steady plateau (Fig. 1D).40 The system finally reached total equilibrium after 8 h. However, addition of binuclear Ru complexes resulted in a significant delay for the lag time and evident decrease in the fluorescence intensity. These results suggested an effective inhibition of the compounds on the amyloid fibril formation of hIAPP. In addition, fluorescence experiments of binuclear Ru complexes with ThT were used to clarify whether binuclear Ru complex and ThT interact (Fig. S5†). The results showed that the intensity of ThT fluorescence was reduced in various degrees after addition of Ru complex 1, 2, or 3. This phenomenon implied that these complexes might interact with ThT, and the results of ThT assay were disturbed to a certain extent. The ability of Ru complexes to inhibit the interaction between hIAPP and ThT was examined (Fig. S6†). The results demonstrated that Ru complexes did not effectively disturb the binding of hIAPP with ThT. Therefore, morphology was used to verify the inhibition of Ru complexes on hIAPP aggregation.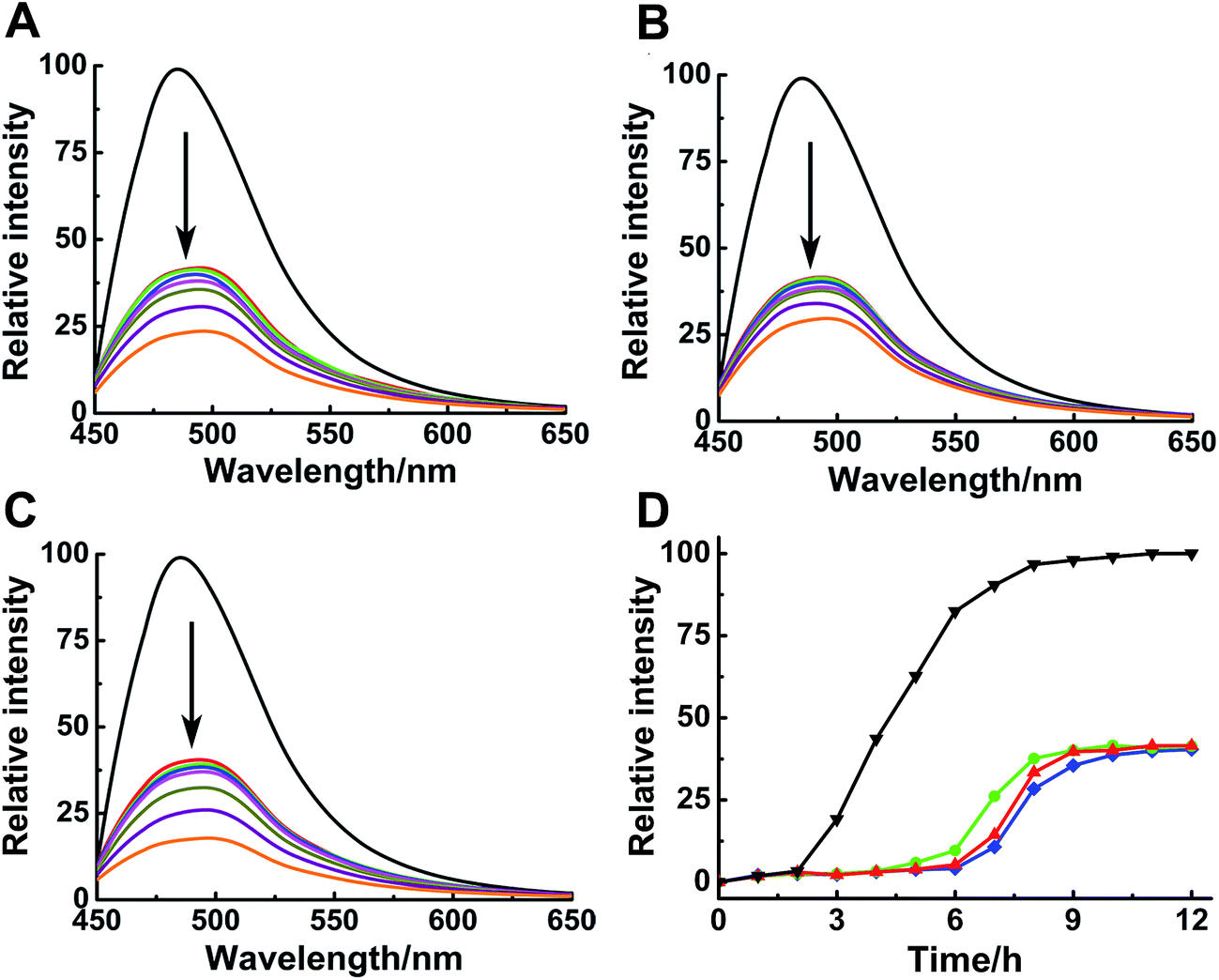 | ||
| Fig. 1 ThT fluorescence assay of 5 μM hIAPP with different concentration of Ru complexes 1 (A), 2 (B) and 3 (C). The concentration of Ru complexes was 0, 1, 2, 3, 5, 15, 30 and 50 μM (from top to bottom). The concentration of ThT was 20 μM. ThT fluorescence monitored at 484 nm during the aggregation of 15 μM hIAPP in the absence (black) and presence of 1 (red), 2 (green), and 3 (blue) (D). The molar ratio of Ru complex to hIAPP is 0.2. | ||
Morphological analysis
Although ThT assay indicates the inhibition of Ru complex on hIAPP, AFM may directly show the morphological change of amyloid peptide induced by the metal complexes. The morphology of hIAPP incubated with binuclear Ru complexes was monitored by AFM (Fig. 2A–I). When hIAPP was incubated alone for 72 h at 310 K, numerous fibrils were observed in the AFM image (Fig. S7A†). The fibrils became short and slender in the presence of equivalent amounts of binuclear Ru complex. No visible fibrils appeared, and a number of spherical particles existed when the binuclear Ru complex was increased to 5- or 10-fold. The effect of complex 3 was more obvious than those of complexes 1 and 2.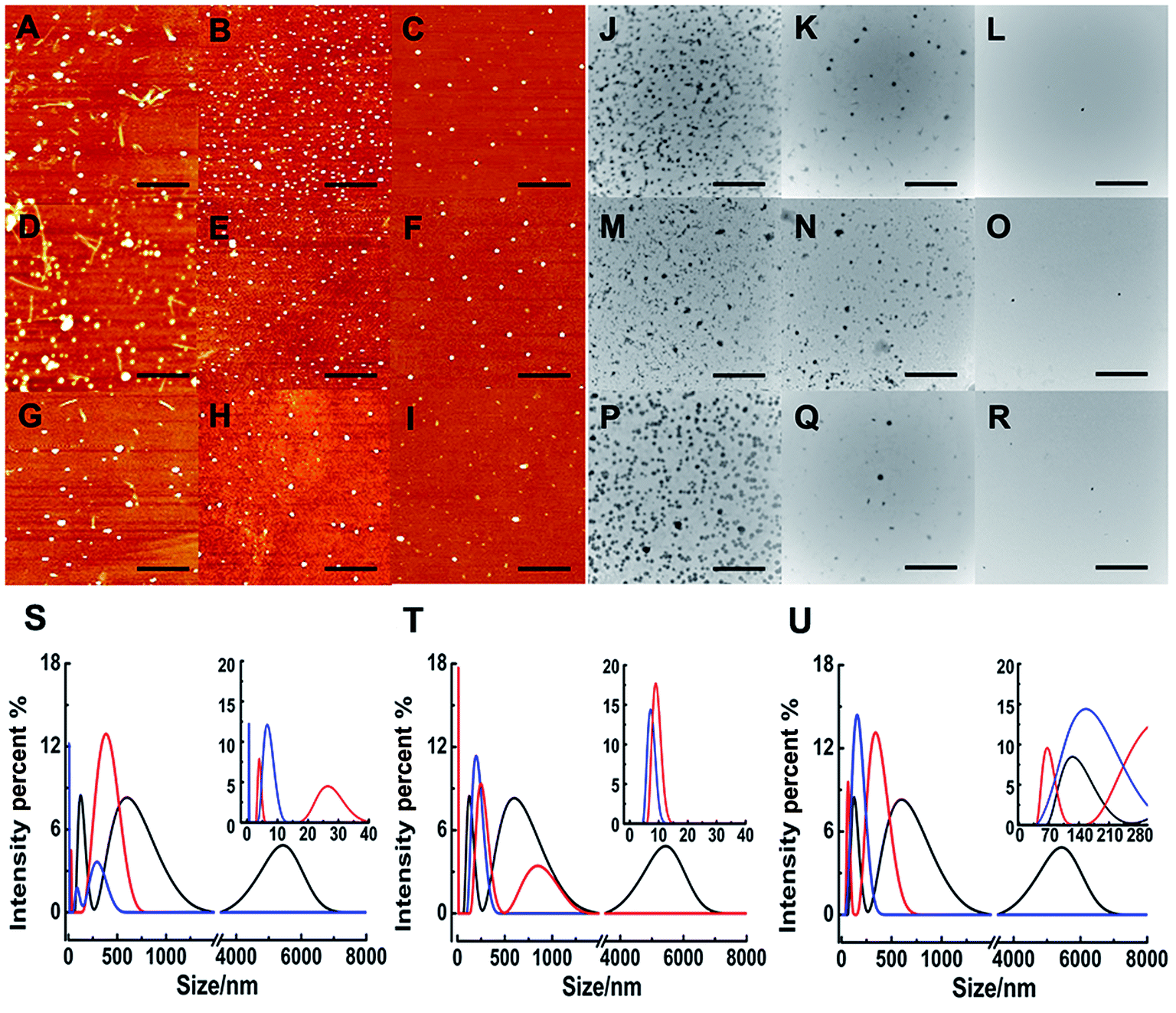 | ||
| Fig. 2 AFM images of 5 μM hIAPP in the presence of different concentration of complexes 1 (A–C), 2 (D–F), and 3 (G–I). The molar ratio of Ru complex to hIAPP is 1 (A, D, G) 5 (B, E, H), and 10 (C, F, I) respectively. The scale bar is 3 μm. TEM images of 5 μM hIAPP in the presence of complexes 1 (J–L), 2 (M–O), and 3 (P–R). The molar ratio of Ru complex to hIAPP is 1 (J, M, P), 5 (K, N, Q), and 10 (L, O, R) as well. The scale bar is 500 nm. DLS analysis of the multimodal size distribution of hIAPP (5 μM) aggregates in the absence (black) and presence of 5 (red) and 10 equivalents (blue) of 1 (S), 2 (T) and 3 (U). | ||
Fig. 3 shows the time-dependent AFM detection for the disaggregation of aged hIAPP by complexes 1–3. Disaggregation was evident after 12 h of incubation of aged hIAPP with Ru complexes. The fibrils completely disappeared, which illustrates the disaggregation ability of binuclear Ru complexes. After 24 h of incubation, the spherical particles became much smaller and sparser. The heights of the fibrils and granular aggregates along some selected lines were analyzed to clarify the results (Table 1). The lines in each graph represented a mean height. For complex 1, the height of the fibrils was 9.41 nm on line a, whereas the heights of lines b and c were 62.97 and 40.87 nm, respectively. For complex 2, the heights of lines d and e were 63.54 and 44.71 nm, respectively. The heights of lines f and g for complex 3 were 51.69 nm and 39.25 nm, respectively. These data demonstrate the difference between the smooth fibrils and granular aggregates, and the results were similar to previous study.41 More importantly, the heights of granular aggregates became lower with time, revealing disaggregation.
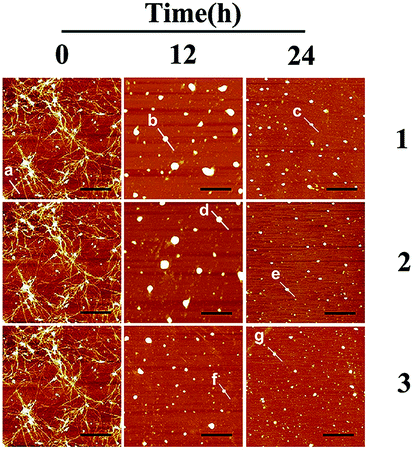 | ||
| Fig. 3 AFM images of time-dependent disaggregation of 5 μM hIAPP incubated with 25 μM Ru complex for 12 h or 24 h at 310 K. | ||
The morphology of hIAPP aggregates in the absence and presence of binuclear Ru complexes were also displayed by TEM (Fig. 2J–R). After 72 h of incubation at 310 K, the TEM image of hIAPP showed typical amyloid fibrils (Fig. S7B†). However, no fibril was found, and oligomers formed in the presence of equivalent amounts of binuclear Ru complexes. Oligomers became attenuated and even vanished with the increase in binuclear Ru complexes to 5-/10-fold. The repeated TEM images confirmed that there had almost no hIAPP aggregates under the same conditions (Fig. S8†). The results essentially agree with those of the ThT assay and AFM images, which demonstrate that binuclear Ru complexes can inhibit the formation of hIAPP fiber.
Particle size distribution
As a sensitive and powerful tool, DLS experiments were used to show the particle size distribution of amyloid peptide aggregation (Fig. 2S–U). After incubation of 5 μM hIAPP alone, the maximum particle size of hIAPP reached nearly 6000 nm. When 5-fold molar excess of complex 1 was added, the particle size decreased significantly between 5 nm and 400 nm (Fig. 2S†). The effects were more distinct within the size scope between 0 nm and 300 nm when 10-fold molar excess of Ru complexes added. Similarly, for complex 2, the size scope was 10–850 nm or 10–200 nm when 5- or 10-fold molar excess of the compound was added (Fig. 2T). For complex 3, the size scope was 65–340 nm or nearly 145 nm at different molar excesses as mentioned above (Fig. 2U). This phenomenon indicated that the size distribution scope of hIAPP decreased from micron scale to nanoscale after incubation with Ru complexes. The analysis of particle size distribution further revealed a remarkable inhibitory effect of Ru complexes on hIAPP aggregation.Interactions of Ru complexes with hIAPP
Binding properties of Ru complexes with hIAPP can be studied by UV-vis spectra,42–44 The UV spectra of Ru complexes had typical absorption bands as reported previously (Fig. 4A–C). With the addition of hIAPP, the absorbance wavelengths were slightly shifted. Meanwhile, the isoabsorptive points appeared remarkably in the spectra. The isoabsorptive points were at 310 and 392 nm for complex 1 (Fig. 4A), and they were at 330 and 430 nm for complex 2 (Fig. 4B). The points were at 290 and 333 nm in the UV absorption spectra of complex 3 (Fig. 4C). These results proved the formation of new binding complex species, suggesting the binding of Ru complexes to hIAPP.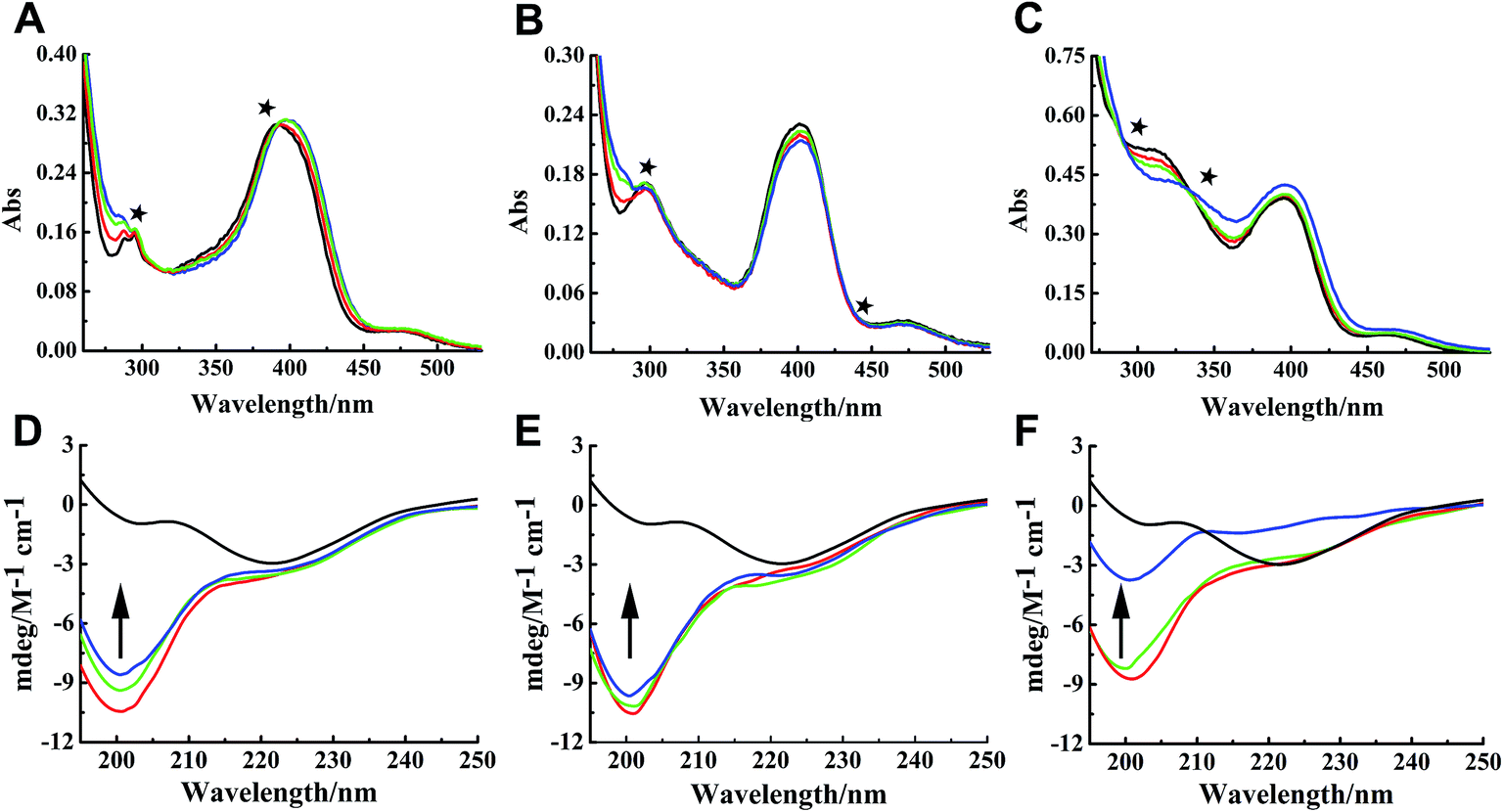 | ||
| Fig. 4 UV absorption spectra of complexes 1 (A), 2 (B), and 3 (C) (20 μM) in the absence (black) and presence of different concentration of hIAPP at 20 (red), 40 (green), and 80 μM (blue) respectively. The isoabsorptive points were marked by stars in the spectra. Circular dichroism spectra of 50 μM hIAPP solution in the absence (black) and presence of 0.5 (red), 1.0 (green), and 3.0 (blue) equivalents of Ru complexes 1 (D), 2 (E), and 3 (F). | ||
CD spectroscopy was employed to gain insight into the effects of Ru complexes on the conformation of hIAPP (Fig. 4D–F). After incubation of hIAPP, a notable negative absorption peak was observed at approximately 220 nm, which represented the β-sheet structure as a predominant component in the solution. When different concentrations of binuclear Ru complexes were added to hIAPP, the CD spectrum displayed a major negative signal at approximately 200 nm, whereas the negative peak at 220 nm was weakened. With the increase in binuclear Ru complexes, the intensity of the negative band at 220 nm gradually weakened, revealing the change of hIAPP secondary structure. In particular, the change was evident at high dose of complex 3 compared with other complexes. All these spectral changes revealed the conformational conversion of hIAPP, indicating that Ru complexes interact with hIAPP.
The interaction between a macromolecule and a small molecule can be performed by electroanalytical technique.45–49 The CV analysis contributes to the understanding of the binding mode between binuclear Ru complexes and hIAPP. Fig. 5 shows that all solutions containing binuclear Ru complexes had an apparent reductive peak. The reductive peak of complex 1 was at approximately −0.65 V. Addition of hIAPP caused no obvious change in the electric potential, but the intensity of electric current decreased notably. Similarly, the cyclic voltammogram of complex 2 showed the reductive peaks at −0.61 and −1.72 V. Addition of hIAPP diminished the reductive peak current. For complex 3, the reductive peak current at approximately −0.60 V was affected obviously. The data demonstrated the formation of a non-electroactive complex, and the decrease in peak current illustrated the hydrophobic interaction between hIAPP and binuclear Ru complexes.23,50
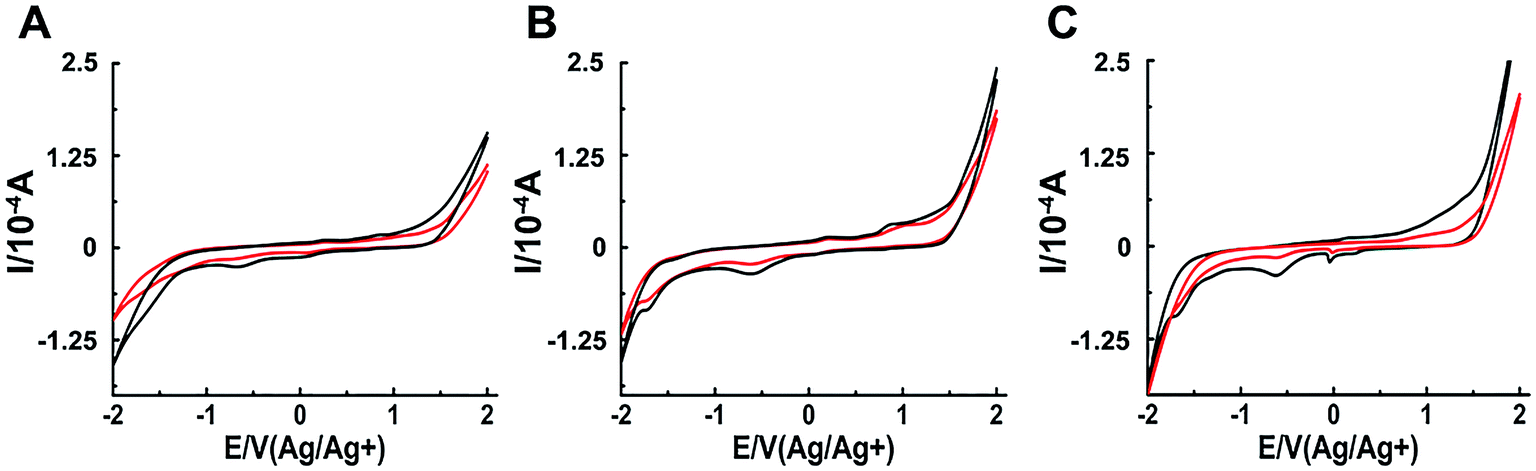 | ||
| Fig. 5 Cyclic voltammograms of 200 μM Ru complexes in the absence (black) and presence (red) of 20 μM hIAPP obtained at the glassy carbon electrode for complex 1 (A), 2 (B) and 3 (C) respectively. | ||
Comparison of binuclear and mononuclear compounds
The different inhibitions between binuclear Ru complexes and mononuclear Ru complexes were assessed by ThT assay, AFM, DLS, and CD. Fig. S9† shows the decreasing hIAPP aggregation by ThT fluorescence intensity after incubation with binuclear Ru complexes or mononuclear Ru complexes using low-dose metal compounds. AFM was also performed to study different inhibitions among the two kinds of compounds (Fig. S10†). In the presence of equivalent amounts of mononuclear Ru complexes, the short fibrils were more evident than that of binuclear Ru complexes, which implied that binuclear compounds were superior to the mononuclear compounds on the inhibition of hIAPP aggregation. The results of TEM also indicated that the inhibition of binuclear compounds were better than mononuclear compounds (Fig. S11†). Similar speculation can also be demonstrated by DLS (Fig. 6). Contrary to binuclear Ru complexes, the particle size was larger after hIAPP incubation with mononuclear Ru complexes. In CD analysis (Fig. 7), the effect of mononuclear compounds on the negative peak at 220 nm was less than that of binuclear Ru complexes.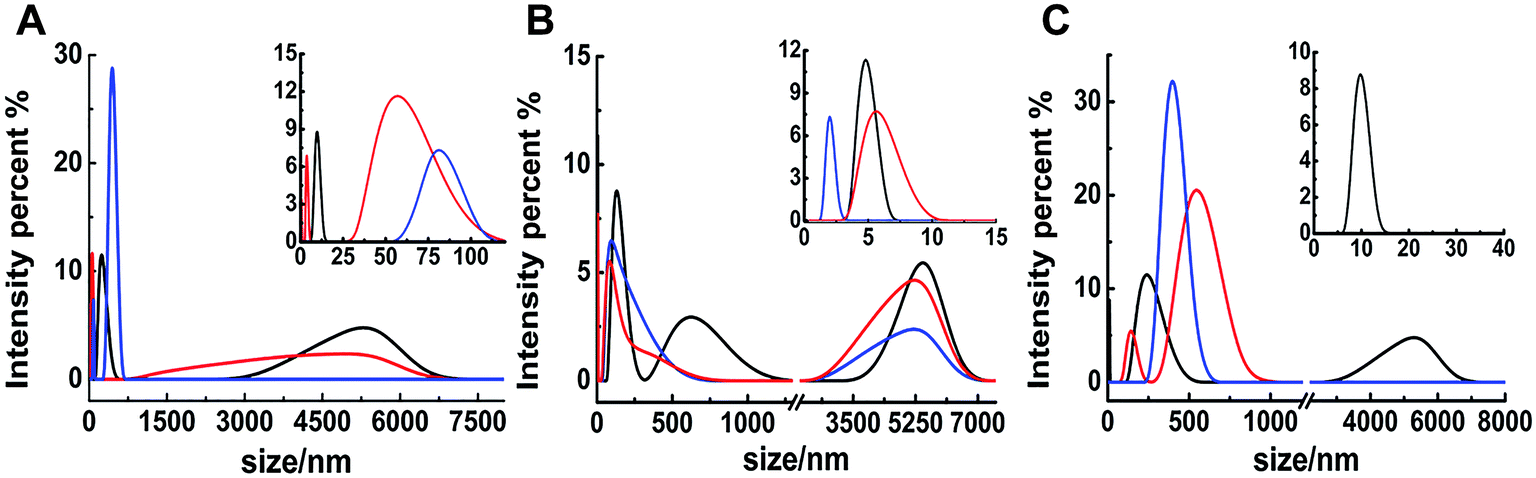 | ||
| Fig. 6 DLS analysis of the multimodal size distribution of 5 μM hIAPP aggregates in the absence (black) and presence of 5 (red) and 10 equivalents (blue) of 4 (A), 5 (B) and 6 (C). | ||
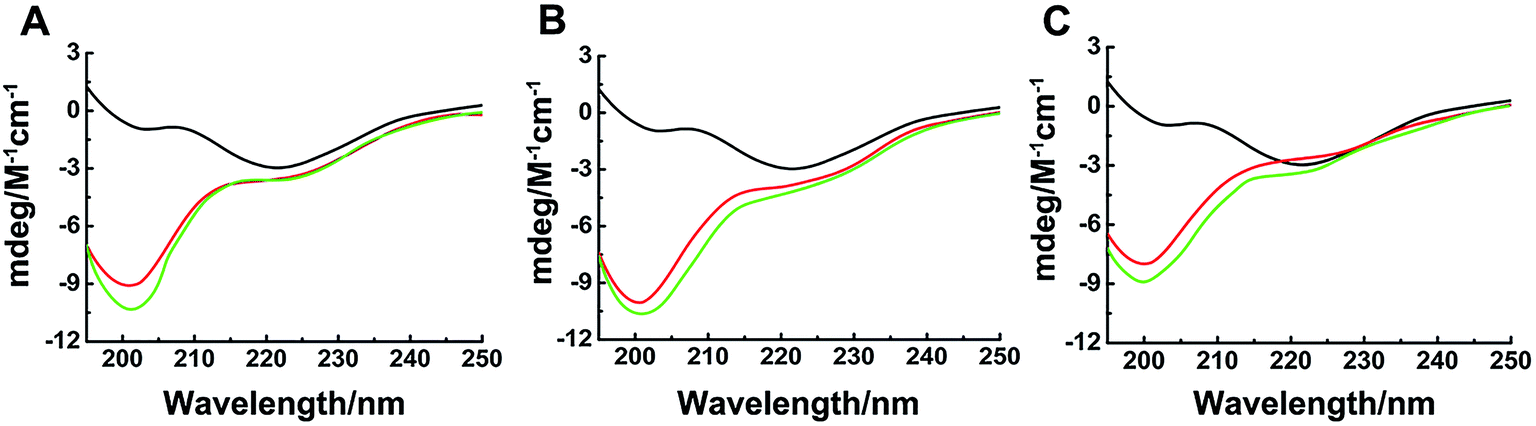 | ||
| Fig. 7 Circular dichroism spectra of hIAPP (black) in the presence of equivalent amounts of Ru complexes 1 (A), 2 (B), 3 (C) and 4 (A), 5 (B), 6 (C). The concentration of hIAPP was 50 μM. The binuclear complexes were in red, and the mononuclear complexes were in green. | ||
Comparison of single ligands and binuclear compounds
In order to verify the inhibitory effects of binuclear Ru complexes, the corresponding single ligands of these complexes were detected. The selected ligands including pyrazine, pyrimidine, and 4,4′-dipyridyl were monitored by ThT assay, DLS and AFM to act on hIAPP instead of binuclear Ru complexes. Fig. S12† shows that the fluorescence intensity was not drastically decreased after co-incubation with ligands. The morphological changes of hIAPP induced by ligands were monitored by AFM. In the presence of ligands, the fibrils evidently existed (Fig. S13†). The results of DLS was similar to that of AFM images (Fig. S14†). These results demonstrated that, compared with binuclear Ru complexes, the inhibitory effects of single ligands on hIAPP were weak.Cell viability regulated by Ru complexes
Amylin fibril formation in pancreatic islets is implicated in the pathology of T2DM with the death of pancreatic β-cells. MTT assay can be used to detect the cytotoxicity of hIAPP (Fig. 8). In the absence of Ru complexes, the incubation of hIAPP in INS-1 cells decreased cell viability to 40.3% ± 4.8%. After adding 1.5 μmol L−1 of complexes 1, 2, and 3, the cytotoxicity induced by hIAPP was significantly suppressed. Cell viability increased to 81.7% ± 6.4%, 72.1% ± 5.9%, and 89.8% ± 2.1% for complexes 1, 2, and 3, respectively. When 15 μmol L−1 of Ru complex were added, cell viability increased to 75.2% ± 2.0%, 69.8% ± 1.7%, and 81.9% ± 9.3% for 1, 2, and 3, respectively. Moreover, with the addition of 75 μmol L−1 of complexes 1, 2, and 3, cell viability increased to 72.2% ± 4.2%, 69.7% ± 1.9%, and 83.3% ± 5.7%, respectively. The data elucidated that lower amounts of Ru complexes were more effective to increase the cell viability. As the cellular system is complicated and many reactions exist, we can not clearly explain how the process was at present and further study is necessary to carry out. Meanwhile, the MTT assays showed that Ru complexes were not highly cytotoxic, especially in high dose of compounds (Fig. S15†). Furthermore, MTT assay also indicated the difference of cytotoxicity between binuclear and mononuclear compounds (Fig. S16 and 17†). In the presence of mononuclear Ru complexes, the cell viability was obviously lower than that of binuclear Ru complexes. The difference was more evident for higher amounts of Ru complexes. Therefore, these experiments can clearly show that Ru complexes indeed reduce the cytotoxicity induced by hIAPP.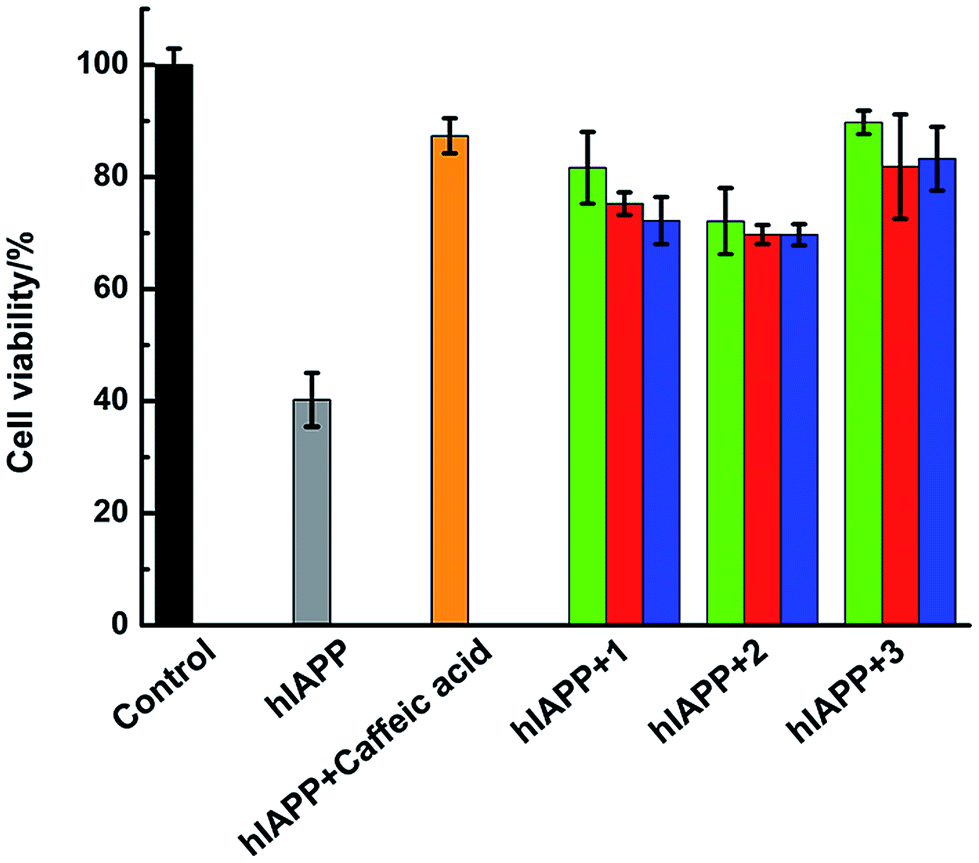 | ||
| Fig. 8 Cell viability after incubating 15 μM hIAPP without (grey) or with Ru complexes at 1.5 μM (green), 15 μM (red), and 75 μM (blue), respectively. The negative control sample of hIAPP incubated with 15 μM caffeic acid was in orange. | ||
Discussion
Effective inhibition of Ru complexes on hIAPP aggregation
Protein accumulates to form amyloid fibrils through structural transition, and this process involves β-sheet, oligomer, protofibril, and mature fibril.51,52 Hence, inhibiting the formation of amyloid fibrils is a key factor in the treatment of protein conformational diseases.23 Previous studies demonstrated that Ru complexes can effectively inhibit the aggregation of Aβ1–40, prion neuropeptide PrP106–126, and even hIAPP,23,24,41,53 indicating the advantage of Ru complexes. In the current study, we explored the interaction between binuclear Ru complexes and hIAPP and revealed their effects on hIAPP aggregation.ThT assay showed that the aggregation of hIAPP was distinctly decreased after incubation with binuclear Ru complexes. The inhibitory effect was distinct even at a low dose of the compounds compared with other reported Ru complexes inhibitors.41 Meanwhile, the aggregation kinetics also demonstrated the effects of binuclear Ru complexes on peptide fibril formation. Considering possible binding competition of ThT with the complexes, morphological analysis was used to identify the inhibition. hIAPP formed fibrils as shown in TEM and AFM images. The fibrillation was interrupted to form oligomers and then monomers with increasing binuclear Ru complexes, decreasing the size of aggregates to nanoscale as seen in DLS analysis. Although the larger species were detected in DLS experiments, the nanoscale size particles were principal and evident with the increase of the complex concentration. For the morphological data, we displayed a mean observation in different areas, which might result in the difference between various methods, while the results of ThT assay might be more macroscopic. In addition, the disaggregation measured by AFM revealed the ability of binuclear Ru complexes to scatter hIAPP fibrils. These results demonstrated that binuclear Ru complexes effectively inhibit the fibril formation of hIAPP. Furthermore, the results of AFM showed that, a number of spherical particles appeared after incubating hIAPP with complex 3, which were much less than that of complexes 1 and 2. The results of MTT assay also demonstrated that complexes 3 had a better reverse effect on peptide induced cytotoxicity. Complex 3 with a larger ligand displayed stronger inhibitory effect on hIAPP aggregation, suggesting better inhibition corresponding to the increase in the planarity of the N–N ligand.49 The aromatic ring-containing Ru complexes are indicative of better inhibition of hIAPP aggregation. For a molecule with large steric effect, some intermolecular forces such as hydrophobic interaction and van der Waals force were more plausible.22,54 The interaction of complex 3 with hIAPP might be attributed to its larger ligand steric effect.
Binding behaviors of binuclear Ru complexes with hIAPP
Ru complexes have been reported to interact with proteins, DNA, and amyloid peptides, such as Aβ1–40, PrP106–126, and hIAPP.22–24,41,55–58 The binuclear Ru complexes selected herein were used to bind DNA in anticancer study. UV absorption spectra and CD analysis were employed to identify the binding of these complexes with hIAPP. The isoabsorptive points in UV spectra might imply the formation of binding complex species between hIAPP and the compounds.42 The CD spectra can be used to detect the conformational change in the peptides.24 The β-sheet structure of hIAPP decreased at different degrees after incubation with binuclear Ru complexes, with negative signal at approximately 200 nm obviously appeared in CD spectra. The change was remarkable especially for high dose of complex 3. In addition, our CV data demonstrated that binuclear Ru complexes may interact with hIAPP through hydrophobic interaction as shown by the obvious peak current change, in accordance with previous results on Ru complexes.23,50Differences between binuclear and mononuclear compounds
Comparison of the molecular structures of selected compounds showed that each half of the binuclear Ru complexes maintains essentially the same coordination environment that characterizes NAMI-A. NAMI-A with four trans-chlorides, one S-bonded DMSO, and one heterocyclic N-ligand, had been applied to inhibit amyloid peptides.59,60 By contrast, binuclear Ru complexes are more stable in aqueous solution than general mononuclear compounds.61–63 The results based on ThT assay, AFM, DLS, and CD spectra revealed the different inhibitory abilities between binuclear and mononuclear compounds. The data elucidated that binuclear Ru complexes had better inhibitory effects than mononuclear Ru complexes on hIAPP aggregation. Complexes appear to be superior with the addition of Ru centers, even in hetero-multinuclei.64 Binuclear complexes may have more than one binding site when interacting with amyloid peptide.65 Our data were in accordance with those observed previously. Thus, the second metal center in the binuclear compounds may strengthen the role of these compounds as amyloid peptides inhibitors. Moreover, the comparison of single ligands with metal complexes on the inhibition of peptide aggregation also elucidated a crucial role of metal compounds in reversing the fibril formation of amyloid peptide.Role of Ru complexes in hIAPP-induced cytotoxicity
hIAPP was found to be a primary component of the amyloid deposits around the β-cells in patients with T2DM.66 Ru compounds have been extensively applied in biomedical field because of their low cytotoxicity and enhanced ability to pass through the blood–brain barrier.67–69 MTT assay indicated that three binuclear Ru complexes could reduce peptide-induced cytotoxicity. Among the three binuclear Ru complexes, complexes 3 showed the most remarkable effect, which might have been caused by its lower self-toxicity and appropriate molecular configuration against amyloid fibril formation. Compared with other small molecule inhibitors (such as vanadium complexes),50 the effects of binuclear Ru complexes were still distinct. The cytotoxicity are inferred to be mainly from oligomers but not fibrils.70–72 The compounds may scatter the peptide aggregates to monomer, and their ability to rescue hIAPP-induced cytotoxicity is satisfied.Conclusion
In this study, we investigated the ability of three binuclear Ru complexes for inhibiting hIAPP aggregation. The formation of hIAPP fibrils could be reversed after incubation with binuclear Ru complexes as shown by ThT assay, TEM, and AFM images. Binuclear Ru complexes effectively disaggregated the amyloid peptide fibrils into nanoscale particles. Larger ligand steric effect and enhanced flexibility of complexes were both crucial to inhibit the amyloid fibril formation. Moreover, binuclear Ru complexes could decrease the component of β-sheet and change the conformation of hIAPP by interacting with the peptide through hydrophobic interaction. MTT method proved that binuclear Ru complexes could significantly reduce the peptide-induced cytotoxicity. More importantly, binuclear compounds showed stronger inhibition on hIAPP aggregation than their corresponding mononuclear compounds. This result demonstrated the enhanced inhibition ability of Ru complexes with multiple metal centers. The study provides a strategy for designing multinuclear Ru complexes as inhibitors against amyloidosis-related diseases.Acknowledgements
We are grateful for the support of the National Natural Science Foundation of China (No. 21473251), the Fundamental Research Funds for the Central Universities and the Research Fund of Renmin University of China (RUC No. 15XNLQ04).References
- C. G. Glabe, Neurobiol. Aging, 2006, 27, 570–575 CrossRef CAS PubMed.
- P. Hossain, B. Kawar and M. E. Nahas, N. Engl. J. Med., 2007, 18, 213–215 CrossRef PubMed.
- V. N. Uversky, Cell. Mol. Life Sci., 2003, 60, 1852–1871 CrossRef CAS PubMed.
- M. Sunde and C. C. F. Blake, Q. Rev. Biophys., 1998, 31, 1–39 CrossRef CAS PubMed.
- O. Schmitz, B. Brock and J. Rungby, Diabetes, 2004, 53, 233–238 CrossRef.
- L. M. Yan, M. Tatarek-Nossol, A. Velkova, A. Kazantzis and A. Kapurniotu, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2046–2051 CrossRef CAS PubMed.
- M. Anguiano, R. J. Nowak and P. T. Lansbury, Biochemistry, 2002, 41, 11338–11343 CrossRef CAS PubMed.
- A. Mascioni, F. Porcelli, U. Ilangovan, A. Ramamoorthy and G. Veglia, Biopolymers, 2003, 69, 29–41 CrossRef CAS PubMed.
- K. Balali-Mood, R. H. Ashley, T. Hauss and J. P. Bradshaw, FEBS Lett., 2005, 579, 1143–1148 CrossRef CAS PubMed.
- P. Cao, A. Abedini and D. P. Raleigh, Curr. Opin. Struct. Biol., 2013, 23, 82–89 CrossRef CAS PubMed.
- T. A. Lutz, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 2010, 298, 1475–1484 CrossRef PubMed.
- Y. Porat, S. Kolusheva, R. Jelinek and E. Gazit, Biochemistry, 2003, 42, 10971–10977 CrossRef CAS PubMed.
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Science, 2003, 300, 486–489 CrossRef CAS PubMed.
- L. A. Scrocchi, Y. Chen, S. Waschuk, F. Wang, S. Cheung, A. A. Darabie, J. McLaurin and P. E. Fraser, J. Mol. Biol., 2002, 318, 697–706 CrossRef CAS PubMed.
- K. Jeong, W. Y. Chung, Y. S. Kye and D. Kim, Bioorg. Med. Chem., 2010, 18, 2598–2601 CrossRef CAS PubMed.
- R. Mishra, B. Bulic, D. Sellin, S. Jha, H. Waldmann and R. Winter, Angew. Chem., Int. Ed., 2008, 47, 4679–4682 CrossRef CAS PubMed.
- C. Adessi and C. Soto, Curr. Med. Chem., 2002, 9, 963–978 CrossRef CAS PubMed.
- A. K. Sharma, S. T. Pavlova, J. Kim, J. Kim and L. M. Mirica, Metallomics, 2013, 5, 1529–1536 RSC.
- S. Salamekh, J. R. Brender, S. J. Hyung, R. P. Nanga, S. Vivekanandan, B. T. Ruotolo and A. Ramamoorthy, J. Mol. Biol., 2011, 410, 294–306 CrossRef CAS PubMed.
- B. Ward, K. Walker and C. Exley, J. Inorg. Biochem., 2008, 102, 371–375 CrossRef CAS PubMed.
- H. Fukui, M. Yamamoto and T. Ando, Neuropharmacology, 1993, 32, 959–968 CrossRef CAS PubMed.
- L. He, X. S. Wang, C. Zhao, H. F. Wang and W. H. Du, Metallomics, 2013, 5, 1599–1603 RSC.
- D. S. Zhu, C. Zhao, X. S. Wang, W. J. Wang, B. H. Wang and W. H. Du, RSC Adv., 2016, 6, 16055–16065 RSC.
- X. S. Wang, B. B. Zhang, C. Zhao, Y. L. Wang, L. He, M. H. Cui, X. T. Zhu and W. H. Du, J. Inorg. Biochem., 2013, 128, 1–10 CrossRef CAS PubMed.
- C. X. Zhang and S. J. Lippard, Curr. Opin. Chem. Biol., 2003, 7, 481–489 CrossRef CAS PubMed.
- J. Reedijk, Macromol. Symp., 2008, 270, 193–201 CrossRef CAS.
- I. Kostova, Curr. Med. Chem., 2006, 13, 1085–1107 CrossRef CAS PubMed.
- G. Sava, A. Bergamo, S. Zorzet, B. Gava, C. Casarsa, M. Cocchietto, A. Furlani, V. Scarcia, B. Serli, E. Iengo, E. Alessio and G. Mestroni, Eur. J. Cancer, 2002, 38, 427–435 CrossRef CAS PubMed.
- A. Bergamo, R. Gagliardi, V. Scarcia, A. Furlani, E. Alessio, G. Mestroni and G. Sava, J. Pharmacol. Exp. Ther., 1999, 289, 559–564 CAS.
- J. M. Rademaker-Lakhai, D. van den Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717 CrossRef CAS PubMed.
- M. S. Davies, D. S. Thomas, A. Hegmans, S. J. Berners-Price and N. Farrell, Inorg. Chem., 2002, 41, 1101–1109 CrossRef CAS PubMed.
- S. Komeda, T. Moulaei, K. K. Woods, M. Chikuma, N. P. Farrell and L. D. Williams, J. Am. Chem. Soc., 2006, 128, 16092–16103 CrossRef CAS PubMed.
- A. Hegmans, S. J. Berners-Price, M. S. Davies, D. S. Thomas, A. S. Humphreys and N. Farrell, J. Am. Chem. Soc., 2004, 126, 2166–2180 CrossRef CAS PubMed.
- A. Harris, Y. Qu and N. Farrell, Inorg. Chem., 2005, 44, 1196–1198 CrossRef CAS PubMed.
- A. L. Harris, X. Yang, A. Hegmans, L. Povirk, J. J. Ryan, L. Kelland and N. P. Farrell, Inorg. Chem., 2005, 44, 9598–9600 CrossRef CAS PubMed.
- A. Bergamo, G. Stocco, B. Gava, M. Cocchietto, E. Alessio, B. Serli, E. Iengo and G. Sava, J. Pharmacol. Exp. Ther., 2003, 305, 725–732 CrossRef CAS PubMed.
- E. Alessio, E. Iengo, S. Zorzet, A. Bergamo, M. Coluccia, A. Boccarelli and G. Sava, J. Inorg. Biochem., 2000, 79, 173–177 CrossRef CAS PubMed.
- C. C. VandenAkker, M. Schleeger, A. L. Bruinen, T. Deckert-Gaudig, K. P. Velikov, R. M. A. Heeren, V. Deckert, M. Bonn and G. H. Koenderink, J. Phys. Chem. B, 2016, 120, 8809–8817 CrossRef CAS PubMed.
- E. Alessio, G. Balducci, A. Lutman, G. Mestroni, M. Calligaris and W. M. Attia, Inorg. Chim. Acta, 1993, 203, 205–217 CrossRef CAS.
- L. H. Tu and D. P. Raleigh, Biochemistry, 2013, 52, 333–342 CrossRef CAS PubMed.
- L. Ma, Y. Fu, L. Yu, X. Li, W. Zheng and T. Chen, RSC Adv., 2015, 5, 17405–17412 RSC.
- Y. Y. Wang, Y. H. Cai and C. G. Yan, Supramol. Chem., 2007, 19, 467–473 CrossRef CAS.
- Q. S. Wang, P. F. Liu, X. L. Zhou, X. L. Zhang, T. T. Fang, P. Liu, X. M. Min and X. Li, J. Photochem. Photobiol., A, 2012, 230, 23–30 CrossRef CAS.
- P. Liu, Q. S. Wang and X. Li, J. Phys. Chem. C, 2009, 113, 7670–7676 CAS.
- N. Hui, X. L. Niu, J. Y. Han, W. Sun and K. Jiao, Amino Acids, 2010, 38, 711–719 CrossRef CAS PubMed.
- M. F. Cerda, L. Luzuriaga, M. Worner and E. Mendez, Int. J. Electrochem. Sci., 2010, 5, 1618–1633 CAS.
- S. S. Kalanur, J. Seetharamappa and V. K. A. Kalalbandi, J. Pharm. Biomed. Anal., 2010, 53, 660–666 CrossRef CAS PubMed.
- M. F. Cerda, E. Mendez, G. Obal, C. Kremer, J. S. Gancheffand and A. M. C. Luna, J. Inorg. Biochem., 2004, 98, 238–244 CrossRef CAS PubMed.
- M. T. Carter, M. Rodriguez and A. J. Bard, J. Am. Chem. Soc., 1989, 111, 8901–8911 CrossRef CAS.
- L. He, X. S. Wang, C. Zhao, D. S. Zhu and W. H. Du, Metallomics, 2014, 6, 1087–1096 RSC.
- C. Zhao, X. S. Wang, L. He, D. S. Zhu, B. H. Zhang and W. H. Du, Metallomics, 2014, 6, 2117–2125 RSC.
- B. Cheng, X. Liu, H. Gong, L. Q. Huang, H. Chen, X. Zhang, C. Z. Li, M. Yang, B. J. Ma, L. H. Jiao, L. Zheng and K. Huang, J. Agric. Food Chem., 2011, 59, 13147–13155 CrossRef CAS PubMed.
- N. A. Vyas, S. S. Bhat, A. S. Kumbhar, U. B. Sonawane, V. Jani, R. R. Joshi, S. N. Ramteke, P. P. Kulkarni and B. Joshi, Eur. J. Med. Chem., 2014, 75, 375–381 CrossRef CAS PubMed.
- D. S. Zhu, G. H. Gong, W. J. Wang and W. H. Du, J. Inorg. Biochem., 2017, 170, 109–116 CrossRef CAS PubMed.
- M. I. Webb, R. A. Chard, Y. M. Al-Jobory, M. R. Jones, E. W. Y. Wong and C. J. Walsby, Inorg. Chem., 2011, 51, 954–966 CrossRef PubMed.
- J. M. Rademaker-Lakhai, D. van den Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717 CrossRef CAS PubMed.
- C. Rios-Luci, L. G. Leon, A. Mena-Cruz, E. Perez-Roth, P. Lorenzo-Luis, A. Romerosa and J. M. Padron, Bioorg. Med. Chem. Lett., 2011, 21, 4568–4571 CrossRef CAS PubMed.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS.
- A. Bergamo, G. Stocco, B. Gava, M. Cocchietto, E. Alessio, B. Serli, E. Iengo and G. Sava, J. Pharmacol. Exp. Ther., 2003, 305, 725–732 CrossRef CAS PubMed.
- A. Bergamo, B. Gava, E. Alessio, G. Mestroni, B. Serli, M. Cocchietto, S. Zorzet and G. Sava, Int. J. Oncol., 2000, 21, 1331 Search PubMed.
- A. Bergamo, G. Stocco, C. Casarsa, M. Cocchietto, E. Alessio, B. Serli, S. Zorzet and G. Sava, Int. J. Oncol., 2004, 24, 373 CAS.
- G. Ribeiro, M. Benadiba, A. Colquhoun and D. de Oliveira Silva, Polyhedron, 2008, 27, 1131–1137 CrossRef CAS.
- M. Necula, R. Kayed, S. Milton and C. G. Glabe, J. Biol. Chem., 2007, 282, 10311–10324 CrossRef CAS PubMed.
- C. M. Anderson, I. R. Taylor, M. F. Tibbetts, J. Philpott, Y. F. Hu and J. M. Tanski, Inorg. Chem., 2012, 51, 12917–12924 CrossRef CAS PubMed.
- A. Kumar, L. Moody, J. F. Olaivar, N. A. Lewis, R. L. Khade, A. A. Holder, Y. Zhang and V. Rangachari, ACS Chem. Neurosci., 2010, 1, 691–701 CrossRef CAS PubMed.
- M. J. Clarke, Coord. Chem. Rev., 2002, 232, 69–93 CrossRef CAS.
- A. Bergamo and G. Sava, Dalton Trans., 2007, 13, 1267–1272 RSC.
- G. Sava, A. Bergamo, S. Zorzet, B. Gava, C. Casarsa, M. Cocchietto, A. Furlani, V. Scarcia, B. Serli, E. Iengo, E. Alessio and G. Mestroni, Eur. J. Cancer, 2002, 38, 427–435 CrossRef CAS PubMed.
- A. Bergamo, R. Gagliardi, V. Scarcia, A. Furlani, E. Alessio, G. Mestroni and G. Sava, J. Pharmacol. Exp. Ther., 1999, 289, 559–564 CAS.
- A. Aguzzi and J. Falsig, Nat. Neurosci., 2012, 15, 936–939 CrossRef CAS PubMed.
- C. A. Ross and M. A. Poirier, Nat. Med., 2004, 10, S10–S17 CrossRef PubMed.
- L. Fioriti, N. Angeretti, L. Colombo, A. DeLuigi, A. Colombo, C. Manzoni, M. Michela, T. Fabrizio, S. Mario, C. Roberto and G. Forloni, J. Neurosci., 2007, 27, 1576–1583 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28107a |
| This journal is © The Royal Society of Chemistry 2017 |